
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA self-assembling peptide hydrogel for ultrarapid 3D bioassays†
Paola
Gagni‡
 a,
Alessandro
Romanato‡
a,
Greta
Bergamaschi‡
a,
Paolo
Bettotti
b,
Renzo
Vanna
c,
Chiara
Piotto
b,
Carlo F.
Morasso
c,
Marcella
Chiari
a,
Marina
Cretich
*a and
Alessandro
Gori
*a
a,
Alessandro
Romanato‡
a,
Greta
Bergamaschi‡
a,
Paolo
Bettotti
b,
Renzo
Vanna
c,
Chiara
Piotto
b,
Carlo F.
Morasso
c,
Marcella
Chiari
a,
Marina
Cretich
*a and
Alessandro
Gori
*a
aNational Research Council of Italy, Istituto di Chimica del Riconoscimento Molecolare (ICRM), Via Mario Bianco, 9, 20131-Milano, Italy. E-mail: alessandro.gori@icrm.cnr.it; marina.cretich@icrm.cnr.it
bNanoscience Laboratory, Department of Physics, University of Trento, Via Sommarive 14, 38123 Povo, Italy
cIstituti Clinici Scientifici Maugeri IRCCS, Via Maugeri 4, 27100, Pavia, Italy
First published on 22nd October 2018
Abstract
Biosensing analytical platforms rely on the intimate structure–function relationship of immobilized probes. In this context, hydrogels are appealing semi-wet systems to locally confine biomolecules while preserving their structural integrity and function. Yet, limitations imposed by biomolecule diffusion rates or fabrication difficulties still hamper their broad application. Here, using a self-assembling peptide, a printable and self-adhesive hydrogel was obtained and applied to fabricate arrays of localized bio-functional 3D microenvironments on analytical interfaces. This soft matrix represents a robust and versatile material, allowing fast and selective tuning of analyte diffusion, which is exploited here to run in-gel immunoassays under solution-like conditions in an unprecedented (<10 min) time frame. The developed material overcomes major limitations associated with hydrogels for bioassays, widening the prospects for easy fabrication of multifunctional bio-interfaces for high-throughput, molecular recognition assays.
1. Introduction
The array technology advent has set a new paradigm in the development of multiplexed assays for biomedical applications, with immune-sensing being a major area of interest.1–3 The success of such platforms is linked to maintaining the biomolecule 3D structure and function upon surface immobilization. In this sense, many efforts have been oriented towards developing innovative surface chemistries that balance stable binding with retained functionality of immobilized bioprobes. Such studies mostly exploit the use of polymeric coatings to engineer microarray surfaces and complementary chemoselective conjugation techniques to circumvent traditional drawbacks associated with nonselective strategies.4–8 In this scenario, hydrogels represent ideal systems bridging dry and wet conditions, as they enable biomolecules to be locally entrapped on analytical surfaces in their active form under solution mimetic conditions within the aqueous gel matrix.9,10 Landmark examples of semi-wet microarrays (both for proteins and antibodies) have been reported, highlighting additional advantages of 3D platforms, such as increased loading capacity, smaller nonspecific binding and enhanced signal-to-noise ratio. Hydrogels for (micro)arrays are commonly based on polyacrylamide, alginate, agarose and polyethylene glycol matrices. Also, low-molecular weight supramolecular hydrogelators have been exploited to build semi-wet 3D arrays11–17 and hydrogel sensors.18–20 However, some hydrogel characteristics still prevent their broad application for bioassay development. Particularly, one major issue is the ability to match target diffusion rates through the gel matrix with stable probe entrapment. Mass transport can indeed be severely limited in dense cross-linked matrices and, accordingly, extensive incubation times are required to allow large molecules to diffuse through the gel aqueous cavities.21,22 Consequently, most current applications focus on arrayed enzymes or antibodies probed with small substrates/analytes, while only few reported systems allow permeation of large biomolecules such as antibodies. In this sense, hydrogel droplet microarrays provide a more accessible format in contrast to hydrogel layered surfaces, but they can require laborious strategies for chip fabrication, especially if permeation to large molecules is desired.23 Gel viscosity is indeed a severe obstacle to direct microarray printing; therefore, in situ hydrogel formation, after spotting of constituent units, is commonly used to circumvent this issue.13,16,24–27 Often, this is accomplished by UV-triggered cross-linking, a process that can however be harmful to the functionality for sensitive biomolecules. Thus, it is highly desirable to develop robust strategies enabling user-friendly and automated array fabrication that entails few, simple and benign manipulation steps compatible with established production workflows.In recent years, self-assembling peptides have experienced widespread and different applications in nanoscience.28–32 The spontaneous bottom-up assembly of monomer peptide motifs can drive the formation of ordered supramolecular architectures in a tuneable and controlled manner, that can easily generate new functional soft materials, including peptide hydrogels.33–36 These materials are usually formed by entangled self-assembled nanofibers that bundle to form a stable network that entraps water within it. Ordered noncovalent interactions are the key driving force for the assembly process, and they can confer stunning robustness to these fibrillar nanostructures.37
In the present work, using micromolar concentrations of a self-assembling peptide we obtained a soft hydrogel matrix with controlled permeability properties for the straightforward confinement of biomolecular probes in arrayed 3D microenvironments. Using this strategy, we obtained a stable and yet highly permeable nano-scaffold that enabled us to run fluorescence immunoassays under solution-like conditions within an ultrashort timeframe (<10 min) and a pM detection limit. The supramolecular hydrogel forms rapidly and spontaneously, it is printable using a piezoelectric microarray spotter and it self-adheres onto poly(methyl methacrylate) (PMMA) slides, so that neither post-spotting cross-linking nor polymerization is required for gel formation and surface attachment, greatly simplifying the fabrication process. Overall, our platform is user-friendly, robust and cost-effective and possesses a combination of favorable features that are unique in the area of 3D multiplexed bioassays in hydrogels.
2. Results and discussion
2.1. Hydrogel design and characterization
Self-assembly of proteins and peptides is a frequently found phenomenon in biological systems.38–40 Assembly can be highly specific, originating from precisely defined and hierarchical supramolecular nanostructures. In particular, peptides are uniquely attractive self-assembling monomers with a number of favourable and distinctive features. Ease of synthesis and chemical manipulation, and virtually unlimited design possibilities to control monomer assembling properties, have indeed made peptides key components for the development of new (bio)nanomaterials.41–43 In this light, the self-assembling Q11 peptide (QQKFQFQFEQQ) is a fibril-forming amino acid stretch previously used as a versatile nanostructured scaffold for several applications.44–48 This peptide and related analogues were reported to self-organize and to form stable hydrogels within a wide millimolar concentration range (1–100 mM). While for many applications the formation of stiff hydrogels is sought for, in our design we rationalized that working at low peptide concentrations (micromolar range) was a necessary condition to obtain a low-viscosity hydrogel that could be handled for straightforward (micro)array production. Moreover, the possibility of obtaining a mildly dense yet resistant matrix was considered crucial to allow the fast diffusion of larger molecules (proteins and antibodies). We also speculated that hydrogel permeability to differently sized biomolecules could be tuned by varying the hydrogel microstructure, i.e. the concentration of the gel-forming peptide hydrogelator. Overall, our envisaged platform would entail a spontaneously forming, printable and tunable hydrogel that could be directly applied to array fabrication. Fig. 1 summarizes the designed strategy. | ||
| Fig. 1 Overview of the hydrogel bioassay strategy. (a) YF-Q11 is incubated at 40 °C to rapidly self-organize into a soft hydrogel. Protein and antibody bioprobes are then pipetted into the gel matrix and entrapped within the fibrillary network due to their size whereas (b) peptides can be immobilized by means of a co-assembly strategy with YF-Q11 that entails covalent conjugation of peptide probes to the YF-Q11 monomer via click-chemistry and addition to the gel forming matrix. (c) Hydrogels are then deposited onto analytical slides by a non-contact spotting technology to obtain 3D arrays. | ||
We tested our hypotheses in the fabrication of hydrogel microarrays using PMMA slides, a hydrophobic material that is widely used in Biological Micro-Electro-Mechanical Systems (BioMEMS).49 Upon preliminary screening of a set of Q11 analogues, we used a slightly modified Q11 sequence (Ac-YFQQKFQFQFEQQ-conh2, YF-Q11) that formed PMMA-adhesive soft hydrogels over a large micromolar concentration range (25–500 μM). Structural characterization of the new peptide by circular dichroism (CD) and ATR-FTIR analysis revealed that YF-Q11 preferentially shows a beta-sheet character (Fig. 2a–c and S2†), in accordance with the previously reported behaviour of the Q11 parent molecule.44 Spectrofluorimetric studies (Fig. 2b and S3†) using the hydrophobic fluorescent molecular probe 8-anilino-1-naphthalenesulfonic acid (ANS), widely used to sense micropolarity variation following the formation of supramolecular nanostructures, demonstrated that upon incubation at 40 °C the spontaneous assembly proceeds rapidly and reaches a stable level within a convenient operative time frame (1–4 hours).50,51 This behaviour was also observed using thioflavin (Fig. S4†). Atomic force microscopy (AFM) experiments were performed to confirm the presence of entangled fibrillar nanostructures responsible for hydrogel formation, which could be clearly detectable even at 25 μM concentration (Fig. 2d). Peptide chains hierarchically aggregate in fibrillary structures that, in turn, self-organize into larger micrometer sized bundle of fibers. The smallest detectable fibers were down to 1 nm in height (Fig. S1†), while the limited thickness of even the largest fibrils (about 10–30 nm) and their high aspect ratio are compatible with the typically observed structures assumed by the Q11 self-assembling peptide.45,52
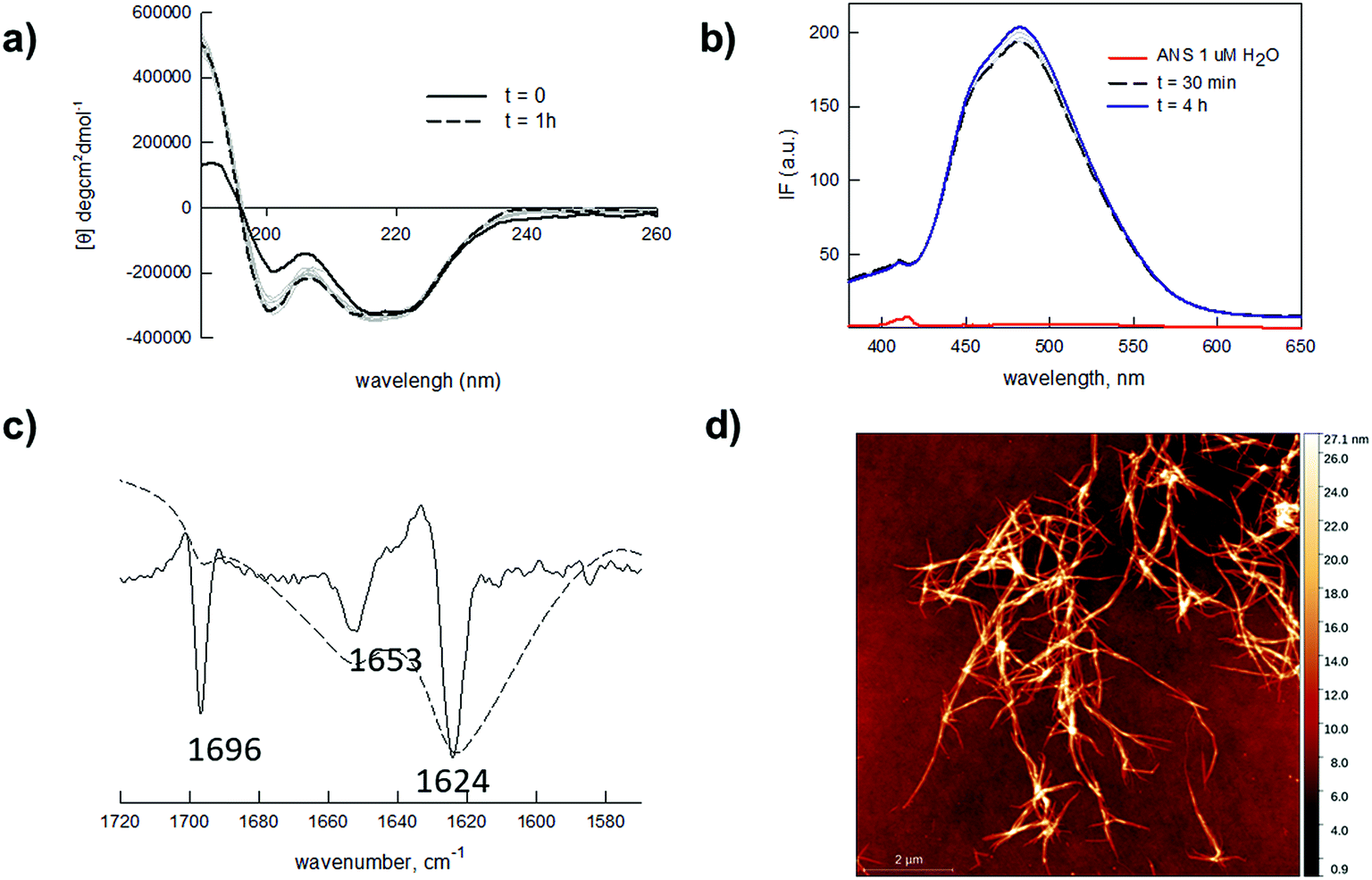 | ||
| Fig. 2 (a) CD spectra of 25 μM YF-Q11 in water upon incubation at 40 °C. Increasing traits of beta-sheet character are detectable over time. CD spectra at 50, 100 and 250 μM are reported in Fig. S2.† (b) Fluorescence spectra of ANS (1 μM) in water (red line) and in the presence of 250 μM YF-Q11 (blue and black lines) at different times (λex = 360 nm). The fluorescent signal rapidly appears and remains stable over time. (c) ATR-FTIR spectrum (dotted) and second derivative (solid). Major peaks at 1624 cm−1 and 1696 cm−1 indicate a predominant YF-Q11 beta-sheet character. A helical structure (1653 cm−1) is also present to a lower extent.53–55 (d) AFM representative image of 25 μM YF-Q11 showing the formation of entangled nanofibers. | ||
We then performed rheology characterization of the YF-Q11 hydrogel, showing that it behaves as a typical viscoelastic fluid under increasing strain excitation (Fig. 3a). The storage modulus (G′) remains constant up to approximately 0.4% strain when an oscillation at a constant frequency of 0.2 Hz is applied, after which it starts to decrease. Within such a range (LVER), changes to the gel microstructure are reversibly (instantaneously) recovered. The difference between the elastic and the viscous (G′′) modulus indicates that the peptide fibers are loosely connected. At larger shear strain, G′′ approaches G′ up to a point (at about 7%) at which the material behaves essentially like a liquid. These reduced interactions are confirmed by the limited viscosity of the material (Fig. 3b), which also displays shear-thinning properties, that make it compatible with robot-assisted spotting technology.
 | ||
| Fig. 3 (a) Measurement of the LVER (Linear Visco Elastic Region) for 250 μM YF-Q11. Oscillation amplitude table: frequency 0.2000 Hz, 1.000 × 10−4 Pa to 0.1000 Pa, 5 samples per decade. (b) Yield stress curve showing a decrease in viscosity starting at very low stress (around 0.02 Pa). This is indicative of the limited cohesive energy endowed by the material. Viscometry shear stress ramp: 0.000 Pa to 1.000 Pa, 500 samples, linear. (c) Drop shape analysis performed by contact angle measurements. YF-Q11 (25 μM) droplet volume observation by video capturing over three repeated drying/rehydration steps. (d) Sequential Raman images of two hydrogel drops (8 nL). Empty spot (left) and malachite-green labelled antibody containing spot (right) were acquired in parallel by a raster-scanning acquisition of the area of interest and by applying a 3 μm pitch in the Z direction. The false-color maps and intensities correspond to the presence of the typical Raman spectra of malachite-green used as a Raman label and automatically identified by direct classical least squares (DCLS) component analysis (see the ESI†). | ||
Interestingly, by visual inspection, spots produced by the arrayer rapidly dried out, but instantaneously rehydrated upon immersion in water regaining their 3D shape (Fig. S5†). To gain insight into this process, we used a drop shape analyser that allowed us to monitor microdroplet volume and to verify the full reversibility of this process (Fig. 3c). We confirmed that, upon an initial loss of hydrogel outer shell occurring within the first minute of incubation, microdroplets regain their initial volume over repeated cycles of drying and re-swelling. In this sense, the fibrillar matrix behaves like a supramolecular sponge.
2.2. Hydrogel allows controllable permeation to biomolecules
To assess the feasibility of the YF-Q11 hydrogel for bioassay applications, we designed an experiment providing combined information on spotted gel permeation to biomolecules while monitoring its stability under the repeated washing and drying steps typical of a bioassay protocol. To this aim we exploited dual fluorescence detection (Cy3 and Cy5) on two independent laser channels of a microarray scanner. To assess stability, a Cy3-labelled YF-Q11 peptide (0.1 μM) was co-assembled into different YF-Q11 solutions (25–500 μM range) to monitor the variation of fluorescence intensity of gel spots upon incubation of the slide in washing buffer at different times. Simultaneously, YF-Q11 spots containing Cy5-labelled biomolecules were scanned for Cy5 residual fluorescence to monitor diffusion through the gel matrix. Labelled probes were added to the hydrogels immediately before spotting to minimize nonspecific biomolecule entrapment due to the possible occurrence of hydrophobic and non-covalent interactions. To screen representative biomolecules differing in their hydrodynamic radius (Rh), we selected anti-α-lactalbumin IgG (150 kDa; Rh = 5–6.5 nm),56–58 streptavidin (53 kDa; Rh = 2.5–4 nm)59 and V5 peptide (2 kDa; estimated Rh = 1–2 nm).The whole range of YF-Q11 hydrogel concentrations tested (25–500 μM) showed good stability over time. Indeed, after 10 min incubation in a typical wash buffer (0.05 M Tris/HCl pH 9, 0.25 M NaCl, 0.05% v/v Tween 20), spot fluorescence was basically unaltered (Fig. S6†). Biomolecule-containing spots behaved similarly to empty controls, suggesting that the YF-Q11 fibrillar network integrity is not affected by the nature of entrapped biomolecules, and that stability is preserved. After 60 minutes of washing, around 15% decay in the Cy3 signal was observed (Fig. S6†). However, this is largely outbalanced by the fact that complete biomolecule diffusion occurred in approximately 2 minutes (Fig. 4a). Notably, by using different YF-Q11 concentrations (i.e. varying the hydrogel microstructure), gel permeability can be selectively tuned to control differential analyte permeation and, under selected conditions, free and fast diffusion is allowed (Fig. 4). The V5 peptide was fully permeable at all tested concentrations, while streptavidin was entrapped in the 250–500 μM range but freely diffused below 100 μM. The same concentration allowed only for partial α-lactalbumin antibody diffusion, which was instead almost quantitative at 25 μM. These tuneable diffusion properties are likely related to the combined contribution of the hydrodynamic radii of the bioprobes and the occurrence of noncovalent interactions with the hydrogel matrix. When large molecules such as IgGs are confined within a relatively dense hydrogel microstructure, as seen at higher (>100 μM) YF-Q11 concentrations, probes are mostly retained. In contrast, the highly porous hydrogel microstructure at 25–50 μM YF-Q11 is ideal to avoid bioprobe entrapment due to nonspecific interactions, and to enable unimpaired diffusion. The simultaneous monitoring on the Cy3 channel allowed us to exclude a major contribution of hydrogel matrix disruption to the observed Cy5 fluorescence decrease.
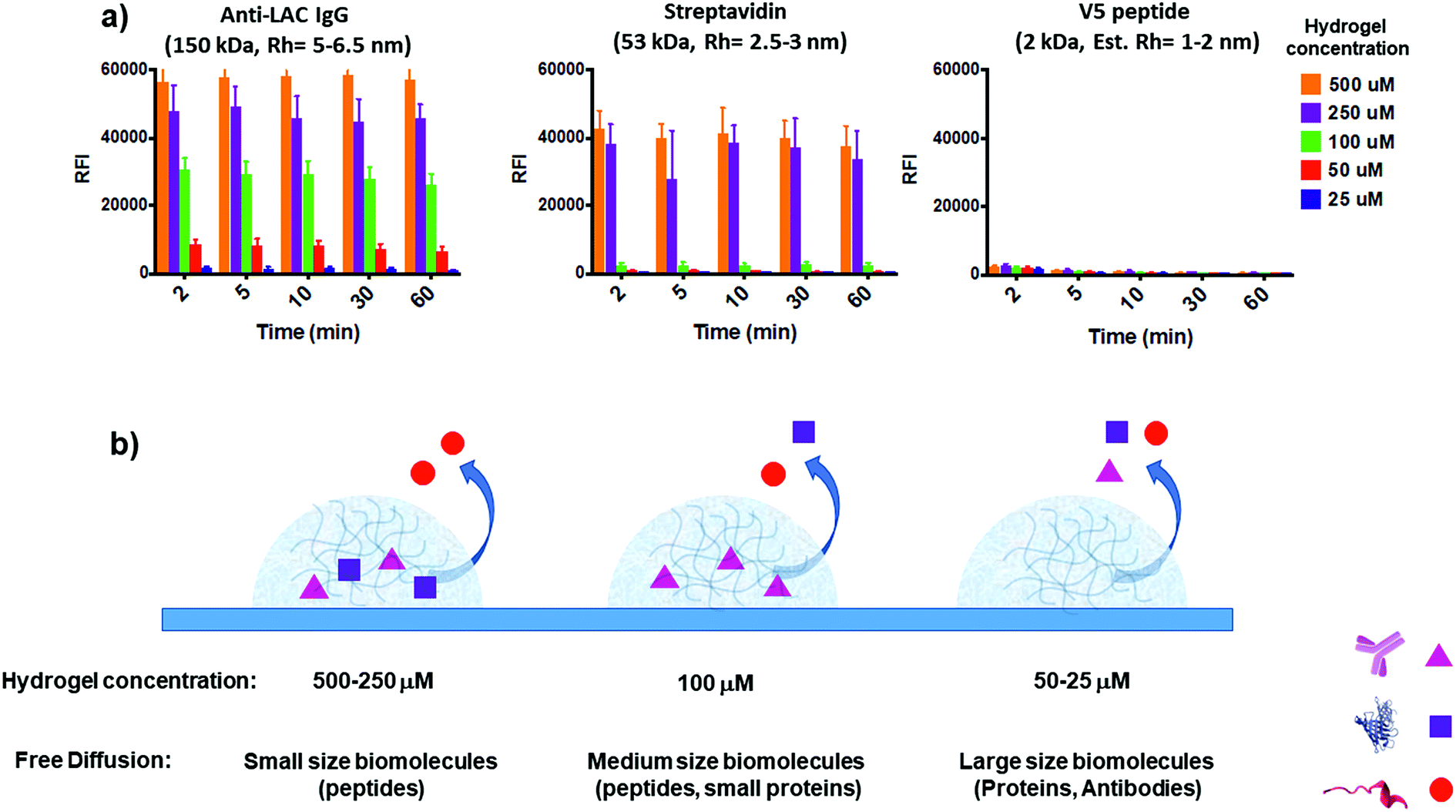 | ||
| Fig. 4 (a) Residual Cy5 fluorescence of hydrogel spotted labelled bioprobes upon increasing incubation time in 0.05 M Tris/HCl pH 9, 0.25 M NaCl, and 0.05% v/v Tween 20 buffer. Anti-LAC IgG (150 kDa; Rh = 5–6.5 nm, left) is mostly retained at >100 μM YF-Q11 hydrogel concentration, while almost complete signal loss indicates that it freely diffuses in the 25–50 μM range within 2 minutes. The hydrogel matrix is fully permeable to V5 peptide (2 kDa, right) over the entire range tested. Streptavidin (53 kDa, Rh = 2.5–3 nm, centre) shows intermediate properties, being residual fluorescence stable at >250 μM YF-Q11 hydrogel concentration. The Cy3 fluorescence channel was used to simultaneously monitor gel stability and exclude a major contribution of gel disruption to the observed Cy5 fluorescence decrease (Fig. S6†). (b) Graphical representation summary of hydrogel permeation to biomolecules. | ||
Overall, these experiments highlighted promising hydrogel features and, in view of real bioassays using complex samples, we verified that these properties were unaltered upon immersion of the slides into powdered skimmed milk prior to spotting, a common protocol used to block non-specific interactions of biomolecules that potentially occur on untreated PMMA surfaces.
To verify that the biomolecules are distributed in a 3D shell, confocal Raman imaging experiments were performed on an antibody-containing hydrogel. A malachite-labelled antibody was entrapped into a 250 μm YF-Q11 matrix and multiple images were directly captured in water using a 3 μm pitch in the Z direction (Fig. 3d and S7†). The results indicated that the labeled antibody was stably embedded in the hydrogel spots and uniformly distributed throughout the gel matrix, while the long acquisition time required for the experiment further supports hydrogel stability. As previously suggested by contact-angle experiments, the rendered images confirmed that spotted microdroplets assume a disc-like shape with a height of approximately 18 μm (8 nL spot, Fig. 3d). This is due to the low-viscosity properties of the hydrogel favoring spreading of the gel on the surface.
2.3. Hydrogel matrix allows for ultrafast immunoassays
We then verified whether these premises could translate into specific molecular recognition assays inside the hydrogel matrix. Accordingly, we designed symmetrical antibody–peptide recognition tests. In the first experimental setting, V5 and LAC peptide epitopes were immobilized within the antibody-permeable matrix (25 μM YF-Q11) by means of a co-assembly strategy (Fig. 1b). Hydrogels were then probed with Cy3-anti-V5 and Cy5-anti-LAC labelled-antibodies (Fig. 5a). In a specular assay, a 250 μM YF-Q11 concentration was used to entrap the anti-V5 and anti-LAC IgGs, which were probed by Cy3-V5 and Cy5 LAC labelled-peptides (Fig. 5b). Both experiments showed that probe-specific recognition occurred only between respective antibody–peptide pairs. Negligible fluorescence signals were detected in spots containing non-related antibodies/peptides or in empty spot controls.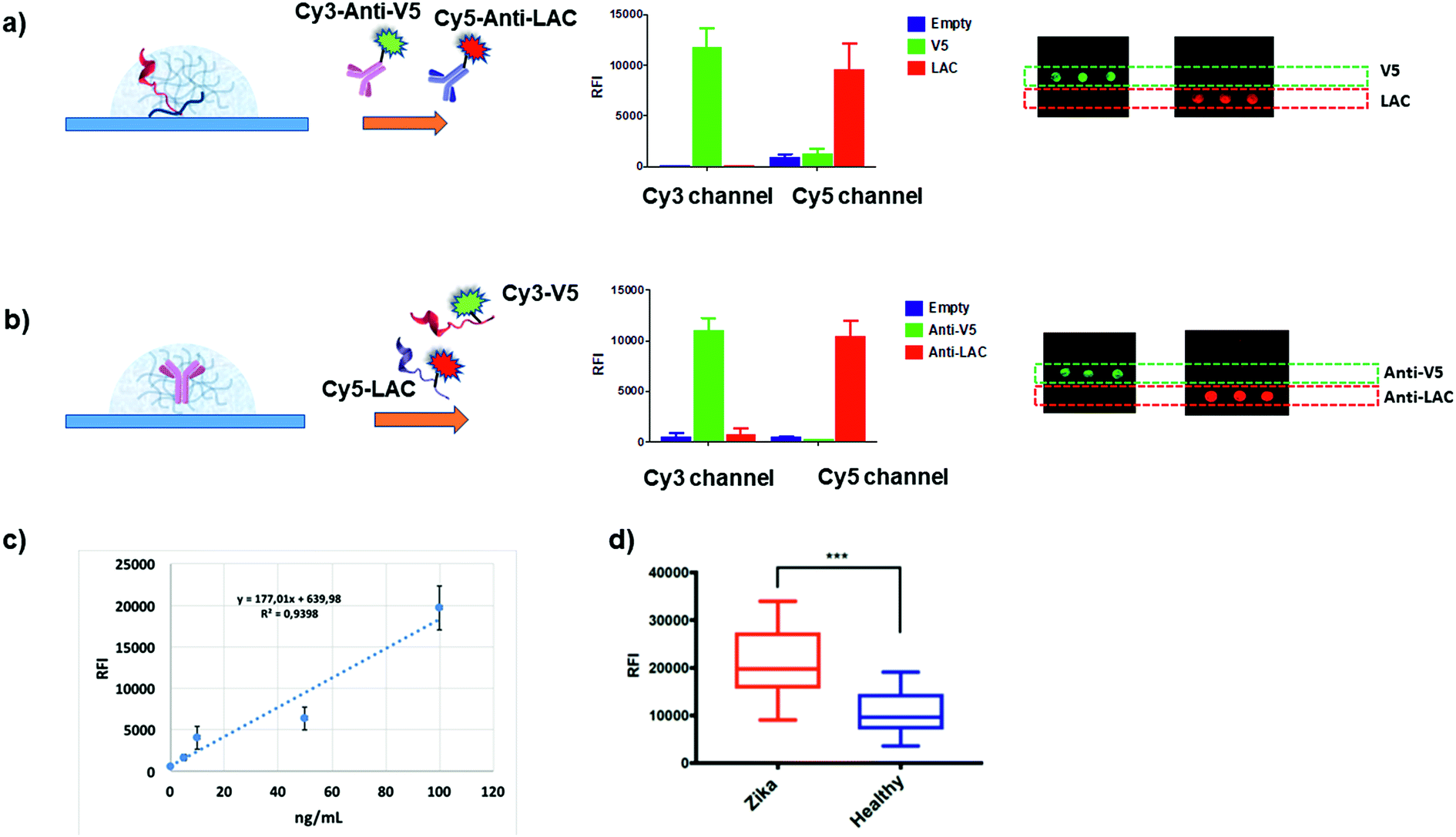 | ||
| Fig. 5 (a and b) Histograms showing the fluorescence intensities detected in the Cy3 and Cy5 channels after 5 minutes incubation of hydrogel spots with the mixture of (a) Cy3-anti-V5 and Cy5-anti-LAC IgGs within the antibody-permeable matrix (25 μM YF-Q11) and (b) Cy3-V5 and Cy5-LAC peptides in the antibody-trapping 250 μM YF-Q11. Correctly, fluorescence is detected in the appropriate channel for the specific probe–antibody pair interaction; microarray scan images are reported on the right. Inappreciable signals detected in both experiments on the empty spots reveal the absence of fluorescence noise due to fluorophore interaction with the hydrogel matrix. (c) Dose–response curve to calculate the limit of detection using 20% Q11-V5 epitope. (d) Fluorescence signal and unpaired t test (p < 0.001) for two panels of 12 ZIKV+ individuals and 12 healthy controls screened for immunoreactivity on 20% ZEp1 co-assembled with YF-Q11. Experimental details are reported in the ESI.† | ||
The ultra-short antibody incubation time (5 minutes) used in the assay was optimized by following the kinetic curves for the saturation of the fluorescence signal of the V5-antibody pair. Comparison of the saturation kinetics profiles observed for the in-gel assay and on a flat surface confirms probe immobilization capacity within the 3D scaffold, as demonstrated by the higher fluorescence response (Fig. S8†). Antibody diffusion was not hindered by the soft peptide matrix, which conversely behaves as a solution-like environment. In addition, it allowed short incubation times, disfavoured nonspecific interactions and molecular adsorption on the surface, thus reducing the fluorescence background noise. Subsequently, in order to estimate the ideal probe content of hydrogels, we tested increasing amounts (from 0% to 50%) of co-assembled V5 epitope with 25 μM YF-Q11 and we calculated the LODs (limits of detection) for different concentrations (0 to 100 ng mL−1) of anti-V5 IgG spiked into human serum (Fig. S9†). The 20% V5 peptide content provided the lowest calculated LOD of 2 ng mL−1. The R2 (0.94) of the dose–response curve demonstrated the linearity of the assay outcome in a clinically relevant antibody concentration range (Fig. 5c).
As the final benchmark for our platform, we performed a real immunodiagnostic assay to detect Zika Virus (ZIKV) infection in human serum samples, using a previously described immunoreactive epitope (ZEp1) of ZIKV NS1 protein.60 Two panels of 12 ZIKV positive individuals and 12 healthy controls were screened for immunoreactivity against ZEp1 epitope and results were evaluated by an unpaired t test (Fig. 5d). Our test effectively enabled discrimination between the two populations at a statistically significant level (p < 0.001), confirming the feasibility of our system in the context of a real serological assay for infectious diseases. It is remarkable that the incubation times for ZEp1 recognition took less than 5 minutes, while the incubation time for the secondary antibody was 2 minutes. Overall, the total time required for the assay was <10 minutes.
3. Conclusion
Herein we exploited a self-assembling peptide (YF-Q11) to obtain a soft hydrogel matrix for local confinement of biomolecules on microarray slides. The developed hydrogel showed combined favourable properties that overcome many limitations typically associated with hydrogels for bioassays. Firstly, low viscosity hydrogels obtained using micromolar YF-Q11 concentrations can be directly used for array fabrication, while being stable enough to locally confine biomolecular probes. Notably, the diffusion properties of different macromolecules (peptides, proteins, and antibodies) through the hydrogel matrix can be favourably tuned depending on YF-Q11 self-assembling monomer concentration.Under selected conditions, biomolecule diffusion properties through the hydrogel matrix closely mimic solution conditions, so that ultrafast (<10 min) immunoassays, arguably one of the most challenging bioassay settings, can be run using this platform. This was demonstrated by running a real case diagnostic immunoassay for the detection of ZIKV infection. Importantly, the hydrogel matrix did not interfere with the molecular recognition properties of the entrapped probes, nor did it show nonspecific interactions in the presence of complex biological samples such as sera. Based on these results and given the unlimited opportunities for flexible molecular design of peptides, we envisage that analogous versatile peptide hydrogel platforms may find extensive applications as modular 3D microenvironments with solution-like properties in a wide range of automated and multiplex bio-assays, including lab-on-chip devices and cell-based biosensors.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was partially funded by Regione Lombardia, project READY (Regional Network for developing diagnostic methods in rapid response to emerging epidemics and bio-emergencies) ID 229472. The CARIPLO foundation is also gratefully acknowledged for partial financial support to A. G (grant no. 2016-0472). P. G., A. R., and G. B. contributed equally to this work.References
- Y. Hu, M. Uttamchandani and S. Q. Yao, Comb. Chem. High Throughput Screening, 2006, 9, 203–212 CrossRef CAS PubMed.
- H. Andresen and C. Grotzinger, Curr. Proteomics, 2009, 6, 1–12 CrossRef CAS.
- H. Sun, G. Y. J. Chen and S. Q. Yao, Chem. Biol., 2013, 20, 685–699 CrossRef CAS PubMed.
- N. Gupta, B. F. Lin, L. M. Campos, M. D. Dimitriou, S. T. Hikita, N. D. Treat, M. V. Tirrell, D. O. Clegg, E. J. Kramer and C. J. Hawker, Nat. Chem., 2010, 2, 138–145 CrossRef CAS PubMed.
- S. B. Nimse, K. Song, M. D. Sonawane, D. R. Sayyed and T. Kim, Sensors, 2014, 14, 22208–22229 CrossRef PubMed.
- V. Romanov, S. N. Davidoff, A. R. Miles, D. W. Grainger, B. K. Gale and B. D. Brooks, Analyst, 2014, 139, 1303–1326 RSC.
- A. Gori, L. Sola, P. Gagni, G. Bruni, M. Liprino, C. Peri, G. Colombo, M. Cretich and M. Chiari, Bioconjugate Chem., 2016, 27, 2669–2677 CrossRef CAS PubMed.
- A. Gori, M. Cretich, R. Vanna, L. Sola, P. Gagni, G. Bruni, M. Liprino, F. Gramatica, S. Burastero and M. Chiari, Anal. Chim. Acta, 2017, 983, 189–197 CrossRef CAS PubMed.
- M. Ikeda, R. Ochi and I. Hamachi, Lab Chip, 2010, 10, 3325 RSC.
- C. P. Tanase, R. Albulescu and M. Neagu, Expert Rev. Mol. Diagn., 2011, 11, 461–462 CrossRef PubMed.
- S. Kiyonaka, K. Sada, I. Yoshimura, S. Shinkai, N. Kato and I. Hamachi, Nat. Mater., 2004, 3, 58–64 CrossRef CAS PubMed.
- K. M. Mabry, S. Z. Payne and K. S. Anseth, Biomaterials, 2016, 74, 31–41 CrossRef CAS PubMed.
- R. Mateen, M. M. Ali and T. Hoare, Nat. Commun., 2018, 9, 602 CrossRef PubMed.
- X. H. Dong, A. C. Obermeyer and B. D. Olsen, Angew. Chem., Int. Ed., 2017, 56, 1273–1277 CrossRef CAS PubMed.
- S. Sharma, M. Floren, Y. Ding, K. R. Stenmark, W. Tan and S. J. Bryant, Biomaterials, 2017, 143, 17–28 CrossRef CAS PubMed.
- M. Moschallski, A. Evers, T. Brandstetter and J. Rühe, Anal. Chim. Acta, 2013, 781, 72–79 CrossRef CAS PubMed.
- C. Vigier-Carrière, F. Boulmedais, P. Schaaf and L. Jierry, Angew. Chem., Int. Ed., 2018, 57, 1448–1456 CrossRef PubMed.
- D. Men, H. Zhang, L. Hang, D. Liu, X. Li, W. Cai, Q. Xiong and Y. Li, J. Mater. Chem. C, 2015, 3, 3659–3665 RSC.
- D. Men, F. Zhou, L. Hang, X. Li, G. Duan, W. Cai and Y. Li, J. Mater. Chem. C, 2016, 4, 2117–2122 RSC.
- C. Fenzl, T. Hirsch and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2014, 53, 3318–3335 CrossRef CAS PubMed.
- D. A. Zubtsov, E. N. Savvateeva, A. Y. Rubina, S. V. Pan'kov, E. V. Konovalova, O. V. Moiseeva, V. R. Chechetkin and A. S. Zasedatelev, Anal. Biochem., 2007, 368, 205–213 CrossRef CAS PubMed.
- P. T. Charles, E. R. Goldman, J. G. Rangasammy, C. L. Schauer, M. S. Chen and C. R. Taitt, Biosens. Bioelectron., 2004, 20, 753–764 CrossRef CAS PubMed.
- H. Li, R. F. Leulmi and D. Juncker, Lab Chip, 2011, 11, 528–534 RSC.
- R. Zhang, A. Liberski, F. Khan, J. J. Diaz-Mochon and M. Bradley, Chem. Commun., 2008, 1317 RSC.
- D. H. Kang, S. M. Kim, B. Lee, H. Yoon and K.-Y. Suh, Analyst, 2013, 138, 6230 RSC.
- S. Sugaya, S. Kakegawa, S. Fukushima, M. Yamada and M. Seki, Langmuir, 2012, 28, 14073–14080 CrossRef CAS PubMed.
- M. Pla-Roca, R. F. Leulmi, S. Tourekhanova, S. Bergeron, V. Laforte, E. Moreau, S. J. C. Gosline, N. Bertos, M. Hallett, M. Park and D. Juncker, Mol. Cell. Proteomics, 2012, 11, M111.011460 CrossRef CAS PubMed.
- J. Zhou, J. Li, X. Du and B. Xu, Biomaterials, 2017, 129, 1–27 CrossRef CAS PubMed.
- M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2017, 50, 2440–2448 CrossRef CAS PubMed.
- A. Altunbas, S. J. Lee, S. A. Rajasekaran, J. P. Schneider and D. J. Pochan, Biomaterials, 2011, 32, 5906–5914 CrossRef CAS PubMed.
- V. Nguyen, R. Zhu, K. Jenkins and R. Yang, Nat. Commun., 2016, 7, 13566 CrossRef CAS PubMed.
- Y. Nagai, H. Yokoi, K. Kaihara and K. Naruse, Biomaterials, 2012, 33, 1044–1051 CrossRef CAS PubMed.
- A. Méndez-Ardoy, J. R. Granja and J. Montenegro, Nanoscale Horiz., 2018, 3, 391–396 RSC.
- A. Pizzi, C. Pigliacelli, A. Gori, N. Nonappa, O. Ikkala, N. Demitri, G. Terraneo, V. Castelletto, I. W. Hamley, F. Baldelli Bombelli and P. Metrangolo, Nanoscale, 2017, 9, 9805–9810 RSC.
- D. M. Raymond and B. L. Nilsson, Chem. Soc. Rev., 2018, 47, 3659–3720 RSC.
- T.-W. Wang, K.-C. Chang, L.-H. Chen, S.-Y. Liao, C.-W. Yeh and Y.-J. Chuang, Nanoscale, 2017, 9, 16281–16292 RSC.
- S. Ling, D. L. Kaplan and M. J. Buehler, Nat. Publ. Gr., 2018, 3, 1–15 Search PubMed.
- L. Yang, A. Liu, S. Cao, R. M. Putri, P. Jonkheijm and J. J. L. M. Cornelissen, Chem.–Eur. J., 2016, 22, 15570–15582 CrossRef CAS PubMed.
- C. K. McLaughlin, G. D. Hamblin and H. F. Sleiman, Chem. Soc. Rev., 2011, 40, 5647 RSC.
- S. Mann, Angew. Chem., Int. Ed., 2008, 47, 5306–5320 CrossRef CAS PubMed.
- N. Singh, M. Kumar, J. F. Miravet, R. V. Ulijn and B. Escuder, Chem.–Eur. J., 2017, 23, 981–993 CrossRef CAS PubMed.
- E. C. Wu, S. Zhang and C. A. E. Hauser, Adv. Funct. Mater., 2012, 22, 456–468 CrossRef CAS.
- M. Rad-Malekshahi, L. Lempsink, M. Amidi, W. E. Hennink and E. Mastrobattista, Bioconjugate Chem., 2016, 27, 3–18 CrossRef CAS PubMed.
- J. H. Collier and P. B. Messersmith, Bioconjugate Chem., 2003, 14, 748–755 CrossRef CAS PubMed.
- J. S. Rudra, Y. F. Tian, J. P. Jung and J. H. Collier, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 622–627 CrossRef CAS PubMed.
- J. P. Jung, A. K. Nagaraj, E. K. Fox, J. S. Rudra, J. M. Devgun and J. H. Collier, Biomaterials, 2009, 30, 2400–2410 CrossRef CAS PubMed.
- G. A. Hudalla, T. Sun, J. Z. Gasiorowski, H. Han, Y. F. Tian, A. S. Chong and J. H. Collier, Nat. Mater., 2014, 13, 829–836 CrossRef CAS PubMed.
- C. Zhang, X. Xue, Q. Luo, Y. Li, K. Yang, X. Zhuang, Y. Jiang, J. Zhang, J. Liu, G. Zou and X. J. Liang, ACS Nano, 2014, 8, 11715–11723 CrossRef CAS PubMed.
- S. Hosseini, F. Ibrahim, I. Djordjevic and L. H. Koole, Analyst, 2014, 139, 2933 RSC.
- J. Raeburn, L. Chen, S. Awhida, R. C. Deller, M. Vatish, M. I. Gibson and D. J. Adams, Soft Matter, 2015, 11, 3706–3713 RSC.
- J. Swain, M. Kamalraj, H. S. Prakash Rao and A. K. Mishra, RSC Adv., 2014, 4, 55377–55382 RSC.
- J. H. Collier and P. B. Messersmith, Adv. Mater., 2004, 16, 907–910 CrossRef CAS.
- P. I. Haris and D. Chapman, Biopolymers, 1995, 37, 251–263 CrossRef CAS PubMed.
- N. A. Nevskaya and Y. N. Chirgadze, Biopolymers, 1976, 15, 637–648 CrossRef CAS PubMed.
- A. Nagai, D. Sato, J. Ishikawa, B. Ochiai, H. Kudo and T. Endo, Macromolecules, 2004, 37, 2332–2334 CrossRef CAS.
- K. Felgenhauer and E. Renner, Ann. Clin. Biochem., 1977, 14, 100–104 CrossRef CAS PubMed.
- J. K. Armstrong, R. B. Wenby, H. J. Meiselman and T. C. Fisher, Biophys. J., 2004, 87, 4259–4270 CrossRef CAS PubMed.
- D. Karlsson, G. Zacchi and A. Axelsson, Biotechnol. Prog., 2002, 18, 1423–1430 CrossRef CAS PubMed.
- J. L. Swift and D. T. Cramb, Biophys. J., 2008, 95, 865–876 CrossRef CAS PubMed.
- M. C. L. C. Freire, L. Pol-Fachin, D. F. Coêlho, I. F. T. Viana, T. Magalhães, M. T. Cordeiro, N. Fischer, F. F. Loeffler, T. Jaenisch, R. F. Franca, E. T. A. Marques and R. D. Lins, ACS Omega, 2017, 2, 3913–3920 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed procedures for the material synthesis, stability and biomolecule diffusion tests, and bioassays. See DOI: 10.1039/c8na00158h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |