
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Kinetic analysis of the accumulation of a half-sandwich organo-osmium pro-drug in cancer cells†
Annabelle
Ballesta
 *ab,
Frédérique
Billy
a,
James P. C.
Coverdale
c,
Ji-Inn
Song
c,
Carlos
Sanchez-Cano
c,
Isolda
Romero-Canelón
*cd and
Peter J.
Sadler
*c
*ab,
Frédérique
Billy
a,
James P. C.
Coverdale
c,
Ji-Inn
Song
c,
Carlos
Sanchez-Cano
c,
Isolda
Romero-Canelón
*cd and
Peter J.
Sadler
*c
aINSERM & Paris Sud University, UMRS 935, ATIP-Avenir Team, Campus CNRS, Villejuif, F-94807, France. E-mail: a.c.ballesta@warwick.ac.uk
bDivision of Biomedical Sciences, Warwick Medical School, University of Warwick, Coventry CV4 7AL, UK
cDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: i.romerocanelon@bham.ac.uk; p.j.sadler@warwick.ac.uk
dSchool of Pharmacy, Institute of Clinical Sciences, University of Birmingham, Birmingham B15 2TT, UK
First published on 17th September 2019
Abstract
The organo-osmium half-sandwich complex [(η6-p-cymene)Os(Ph-azopyridine-NMe2)I]+ (FY26) exhibits potent antiproliferative activity towards cancer cells and is active in vivo. The complex is relatively inert, but rapidly activated in cells by displacement of coordinated iodide. Here, we study time-dependent accumulation of FY26 in A2780 human ovarian cancer cells at various temperatures in comparison with the chlorido metabolite [(η6-p-cymene)Os(Ph-azopyridine-NMe2)Cl]+ (FY25). Mathematical models described the time evolution of FY26 and FY25 intracellular and extracellular concentrations taking into account both cellular transport (influx and efflux) and the intracellular conversion of FY26 to FY25. Uptake of iodide complex FY26 at 37 °C was 17× faster than that of chloride complex FY25, and efflux 1.4× faster. Osmium accumulation decreased markedly after 24 h of exposure. Modelling revealed that this phenomenon could be explained by complex-induced reduction of osmium uptake, rather than by a model involving enhanced osmium efflux. The intracellular osmium concentration threshold above which reduction in drug uptake was triggered was estimated as 20.8 μM (95% confidence interval [16.5, 30]). These studies provide important new insight into the dynamics of transport of this organometallic anticancer drug candidate.
Significance to metallomicsResistance and side-effects of platinum compounds are current major clinical problems and so the development of alternative metal compounds is needed. Here we studied the organo-osmium complex FY26, a potent prodrug which is active in vitro and in vivo and is not cross-resistant with platinum drugs. The cellular transport and metabolism of FY26 were investigated in human ovarian cancer cells through combined mathematical and experimental tools. Such data are likely to be important for furthering the clinical development of FY26. The presented interdisciplinary methodology guides the design of experiments, thus decreasing the costs associated to drug development. |
Introduction
Around half of all cancer chemotherapy treatments currently use platinum compounds, including cisplatin, carboplatin and oxaliplatin, introduced over 30 years ago. Despite the success of such therapies, resistance to platinum is now a clinical problem, together with drug-induced side-effects.1,2 Complexes of other precious metals may provide anticancer drugs with new mechanisms of action to overcome such resistance, and fewer side-effects. Organo–metallic complexes, and osmium(II) arene complexes in particular, have recently shown promising results.3–7In general, metallodrugs are pro-drugs which undergo ligand exchange or redox reactions before they reach the target site. For example, cisplatin contains square-planar 5d8 Pt(II) and is activated in cells by hydrolysis (aquation), in which chlorido ligands are substituted by water. The aqua adducts, and resulting positively-charged complexes, are much more reactive towards target DNA. Octahedral platinum(IV) anticancer complexes are even more unreactive, as is common with low-spin 5d8 complexes, and are activated in vivo by reduction to Pt(II). Such inertness allows some low-spin d6 complexes to reach their target sites intact and bind by outer sphere interactions, e.g. between the ligands and amino acid side-chains in an enzyme binding site, for example potent kinase inhibitors of staurosporine conjugated to Ru(II) and Ir(III).8 However, the inertness of such metal ions is dependent not only on the electronic configuration of the metal, but also on the nature of the ligands to which it is bound.
Here we study the inert iodido complex [(η6-p-cymene)OsII(N,N-dimethyl-phenylazopyridine)I]+ (FY26) which is ca. 49× more active than cisplatin, in a panel of over 800 cancer cell lines screened by the Sanger Institute, and also active in vivo.9,10FY26 is not cross-resistant with cisplatin, nor the second generation Pt(II) drug oxaliplatin, retaining potency in resistant/non-sensitive cell lines, which suggests a different mechanism of action.10–12 The complex appears to act as a redox modulator, disrupting mitochondria and inducing generation of reactive oxygen species (ROS), especially superoxide, which has been observed in both cancer cells and in vivo.10–12 Such a mechanism exploits an inherent weakness in cancer cells, as their malfunctioning mitochondria cannot cope with overproduction of ROS. This likely contributes to the increased selectivity of FY26 towards cancer cells versus normal cells compared to cisplatin.13
Surprisingly, FY26 does not readily undergo hydrolysis in the extracellular medium. However, we discovered by 131I radiolabelling that FY26 is rapidly activated inside cancer cells by displacement of its iodide ligand.14 Intriguingly, this activation appears to be mediated by attack of the intracellular tripeptide glutathione (GSH) on the azo bond of FY26 in a catalytic mechanism which weakens the Os–I bond.15 Under intracellular cytoplasmic conditions where the chloride concentration is ca. 25 mM, HPLC studies show that this reaction can lead to the generation of the chlorido complex FY25.11 We have detected the chlorido complex FY25 as a metabolite in liver microsomes after reactions with FY26 (unpublished data).
The importance of pharmacodynamic (PD) and pharmacokinetic (PK) studies in preclinical drug development based on mathematical modelling has recently been emphasised.16 As a key part of such studies, we have investigated the kinetics of the influx and efflux of both the iodido organo-osmium prodrug FY26 and its chlorido metabolite FY25 in human ovarian cancer cells. We have constructed a mathematical model which describes the time evolution of each chemical species, taking into account both cellular transport, and intracellular conversion of FY26 into FY25 (Fig. 1).
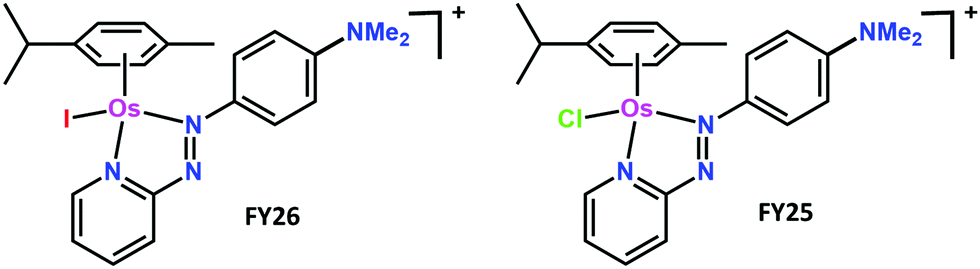 | ||
| Fig. 1 Structures of the two complexes FY26 and FY25. | ||
Experimental section
Ovarian cell line
A2780 human ovarian carcinoma cells were obtained from the European Collection of Cell Cultures (ECACC) and grown in Roswell Park Memorial Institute medium (RPMI-1640) supplemented with 10% of foetal calf serum, 1% of 2 mM glutamine and 1% penicillin/streptomycin using a 5% CO2 humidified atmosphere and passaged at approximately 70–80% confluence.Drugs and reagents
FY25 and FY26 were synthesized and characterized with purities >95% according to reported methods.11 Osmium stock solution standardization: stock solutions of FY25 and FY26 were freshly prepared in RPMI-1640 cell culture medium using 5% DMSO to aid solubilisation. An aliquot was taken and diluted using 3.6% nitric acid (containing thiourea (10 mM) and ascorbic acid (100 mg L−1) to stabilize Os in nitric acid solution).17 The resulting solution was analysed using a PerkinElmer Optima 5300DV ICP-OES. Calibration standards for Os (50–700 ppb) were freshly prepared in 3.6% nitric acid (containing 10 mM thiourea and 100 mg L−1 ascorbic acid).17 Data were acquired and processed using WinLab32 for Windows.Cellular osmium accumulation (without recovery time)
Briefly, 4 × 106 A2780 ovarian cancer cells were seeded on a Petri dish. After 24 h of pre-incubation time in drug-free medium at either 4 °C, 23 °C or 37 °C, the osmium complexes were added to give final concentrations equal to their IC50 (1.8 ± 0.1 μM and 0.15 ± 0.01 μM for FY25 and FY26, respectively). Cells were exposed to the Os(II) complexes with variable exposure time, but without recovery time in drug-free medium. Cells were then washed, treated with trypsin/EDTA and counted. Cell pellets were collected and washed again with PBS. Each pellet was digested overnight in 200 μL freshly-distilled concentrated nitric acid (72%) at 353 K. Experiments were carried out in triplicate and the standard deviations were calculated. Statistical significances were determined using Welch's unpaired t-test.Cellular osmium accumulation (with recovery time)
These experiments were carried out as above, using fixed 24 h drug exposure time and equipotent IC50 concentrations. After drug-exposure, supernatants were removed, cells were washed with PBS and fresh medium was added to each plate. Multiple recovery times in drug free medium were allowed before collecting cell pellets that were again processed as for the previous experiment.Osmium cell pellet quantification
The resulting solutions after cell digestion were diluted using doubly-distilled (MilliQ) water containing thiourea (10 mM) and ascorbic acid (100 mg L−1)17 to achieve a final working acid concentration of 3.6% v/v HNO3. Osmium (189Os) was quantified using an Agilent 7500 series ICP-MS in no-gas mode with an internal standard of 166Er (50 ppb). Calibration standards (0.1–1000 ppb) were freshly prepared in 3.6% nitric acid supplemented with thiourea (10 mM) and ascorbic acid (100 mg L−1) to stabilize osmium in nitric acid solution.7,17 Osmium concentrations were first expressed in ng per million cells using cell counts from biological experiments. Experiments were carried out as triplicates and standard deviations were calculated. Data were acquired and processed using ICP-MS-TOP and Offline Data Analysis (ChemStation version B.03.05, Agilent Technologies, Inc.). The osmium content of cells was converted to intracellular molar concentration assuming a single cell volume of 1 pL.18,19 See ESI,† for full numerical data.Mathematical modeling
Mathematical models are based on ordinary differential equations computing the time evolution of intracellular and extracellular FY25 and FY26 concentrations (see ESI,† for details). All models include two physiological compartments corresponding to the extracellular medium and the intracellular medium of one million cells. The volume of the culture medium was set to 6 mL and that of the intracellular compartment was estimated as 1 μL assuming a single cell volume of 1 pL.18,19 Parameter estimation consisted in a weighted least-square approach using the CMAES algorithm for the minimization task. All individual data points were fitted by iteratively running the estimation procedure and updating initial conditions with the current best-fit parameters until reaching convergence. Model identifiability was performed using the profile likelihood method20 (see ESI†). Parameter 95% confidence intervals were derived from the likelihood profiles. All computations were carried out in Matlab (Mathworks, USA).Results
FY25 and FY26 cellular transport
First, A2780 human ovarian cancer cells were treated with chlorido complex FY25 or iodido complex FY26 for 72 h at physiological temperature (37 °C) using equipotent IC50 concentrations in this cell line (1.5 μM and 0.15 μM respectively). The intracellular concentration of Os in treated cells was subsequently determined using ICP-MS after acidic digestion of cell pellets. During exposure to FY25, Os accumulated in A2780 cells in the course of the first 24 h. After that time, the intracellular Os concentration decreased gradually for the next 48 h (Fig. 2A). Similarly, treatment of ovarian cancer cells with FY26 resulted in an intracellular accumulation of Os over the first 12 h and then an overall decrease in Os concentration over the following 48 h of exposure. Intracellular Os concentration versus time profiles are very similar for exposure to either FY25 or FY26 (Fig. 2A).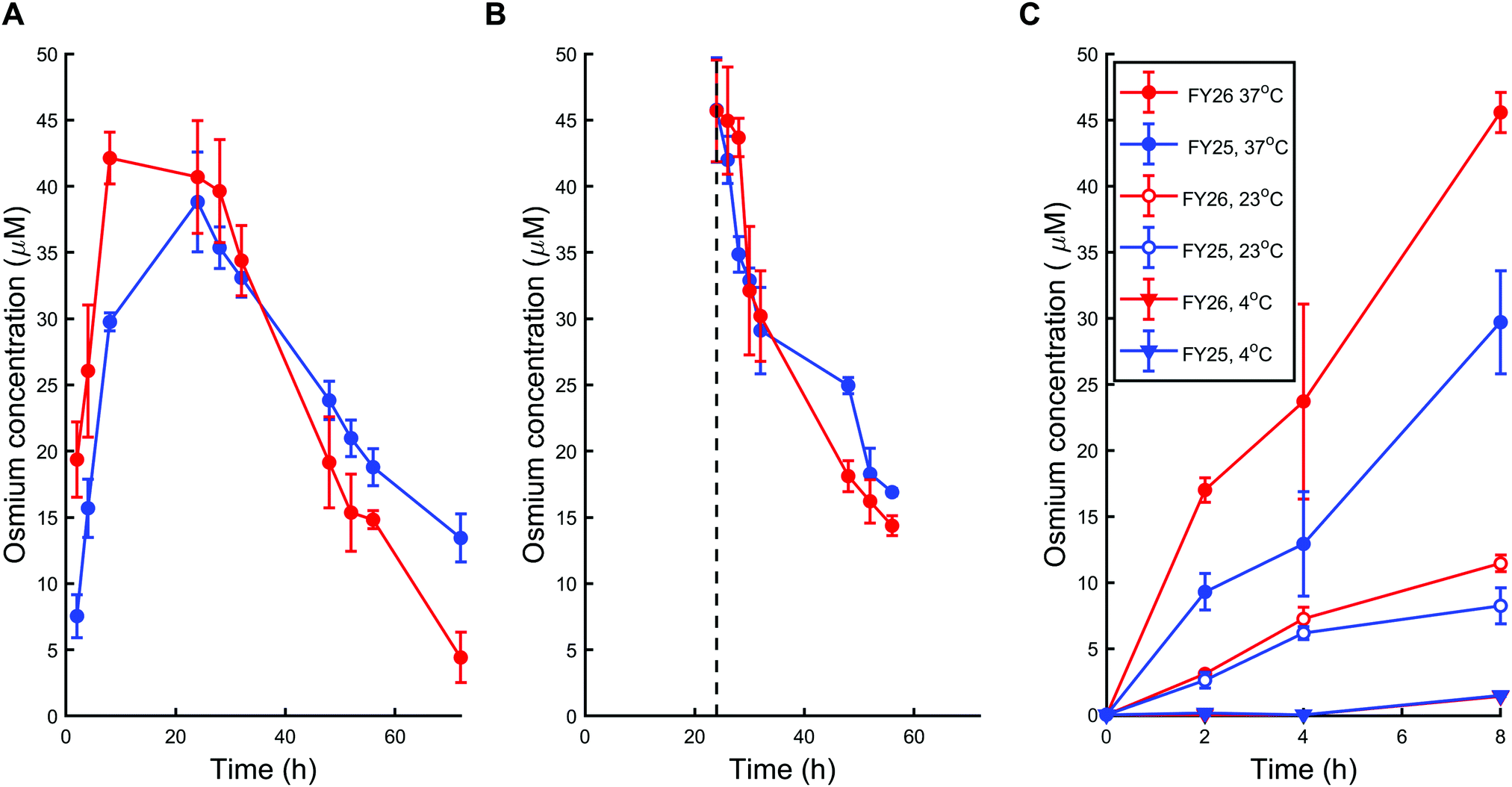 | ||
| Fig. 2 Cellular transport of FY25 (1.5 μM, blue) and FY26 (0.15 μM, red) in A2780 human ovarian cancer cells. (A) Intracellular osmium accumulation during exposure to chlorido complex FY25 or iodido complex FY26 over 72 h. (B) Intracellular osmium concentration after a 24 h-exposure to FY25 or FY26 followed by drug removal by replacement of the medium with drug-free medium. (C) Temperature-dependent intracellular osmium accumulation during exposure to FY25 or FY26 over 8 h at 4, 23 and 37 °C. Datapoints for FY25 and FY26 exposure at 4 °C are superimposed. | ||
To further investigate the cellular transport of FY25 and FY26, the decrease in intracellular accumulation of Os was determined after exposing the cells to these complexes for 24 h and then replacing the culture medium with fresh complex-free culture medium (so called ‘recovery time’ in complex-free medium). During this experiment, cells were washed with PBS before recovery time to ensure that the Os measured was indeed in the intracellular space and not on the outer cellular surface of the monolayer nor in the leftover medium in the well. After such careful removal of either complex from the extracellular medium, the intracellular concentration of Os decreased over the duration of the experiment, with similar kinetics for either FY25 or FY26 exposure (Fig. 2B).
To assess whether energy-dependent mechanisms are involved in the cellular transport of the complexes, ovarian cells were exposed to FY25 or FY26 at 4 °C, 23 °C or 37 °C over 8 h. For all of these time points, intracellular accumulation of Os increased with temperature. After 8 h of exposure to either FY25 or FY26, the Os intracellular concentration decreased by 3.6 and 4-fold respectively at 23 °C, and by 20.3 and 31.1 fold respectively at 4 °C, compared to those determined at 37 °C (Fig. 2C).
Mathematical modeling of FY25 and FY26 cellular pharmacokinetics
The designed models of the cellular pharmacokinetics (PK) of FY25 and FY26 aimed to investigate quantitatively the cell uptake and efflux of each complex together with the activation of FY26 into FY25 (Fig. S1 in ESI†). The first accumulation study without recovery time showed that Os intracellular concentration decreased after 24 h of exposure to either complex (Fig. 2). The hypothesis that FY26 and FY25 concentrations in the culture medium decreased over time was ruled out since (i) the complexes are stable in culture medium11 and (ii) the extracellular volume (10 mL) was much greater than the total intracellular volume of all cells (ca. 4 μL, calculated as 4 million cells × 1 pL each) so the quantity of Os accumulating in the cells was negligible with respect to extracellular concentrations of complexes. Thus, FY26 or FY25 were assumed not to disappear spontaneously from the extracellular compartment. The Os concentration versus time profiles which first increased and then decreased towards zero values could not be accounted for using a simple model incorporating linear uptake and efflux terms with constant reaction rates which allowed only for increasing profiles of Os intracellular concentration ultimately reaching a non-zero steady state level. Thus, two mathematical models were designed to investigate the intracellular mechanisms that might operate during exposure to complexes and affect their cellular transport. Such mechanisms could enable cancer cells to evade damage caused by the metal complex, and so, their molecular understanding might be important in further preclinical studies. Two hypotheses were investigated: the presence of either (i) enhanced efflux or (ii) reduced uptake, both as a result of exposure to the organo-osmium complexes (Fig. 3). The “enhanced efflux” model assumes that FY25 activates a Nuclear Factor (NF) that in turn enhances the transcription of efflux transporters, hence increasing efflux of the complex from the cells. Such assumptions rely on experimental studies demonstrating that the expression of the ATP-Binding Cassette transporters Abcb1 and Abcg2 is mediated by nuclear factors NFkB and Nrf2, respectively.21 In contrast, the “reduced uptake” model assumes that the decrease in intracellular Os accumulation results from the activation of an unspecified chemical species that increases the degradation of influx transporters leading to a decrease in cellular uptake of the complex. For both models, complex-induced activation leading to modified transport was assumed to occur when the intracellular concentration of the complex reaches a critical activation threshold.21 The models for FY25 exposure account only for the uptake and efflux of the complex, and the activation threshold depends on the intracellular concentration of FY25. For modeling FY26 exposure, an additional step corresponding to the transformation of FY26 into FY25 in the intracellular compartment was included. The transport of both FY25 and FY26 was included, and the activation threshold was assumed to depend on the sum of FY25 and FY26 intracellular concentrations. Equations for the models and their associated parameters are presented in the ESI,† (Tables S1–S3).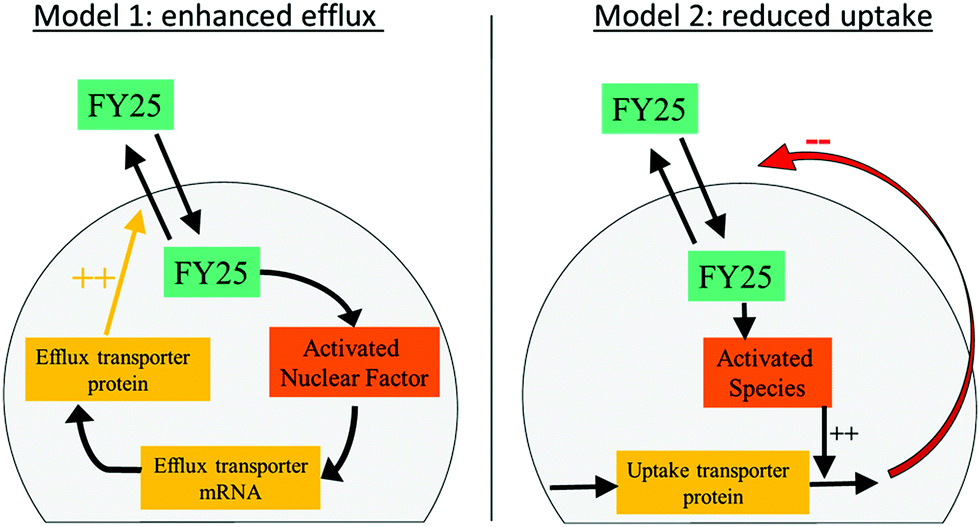 | ||
| Fig. 3 Mathematical models of FY25 cellular pharmacokinetics considering either an enhanced efflux or a reduced uptake of the complex as a result of its administration to A2780 cancer cells. | ||
Model fit to data provides molecular insights into cellular PK of complexes
Parameters of either the “enhanced efflux” or the “reduced uptake” models were fitted to the two available datasets for Os accumulation experiments, with and without recovery time (Fig. 4 and 5). The “enhanced efflux” model was not able to reproduce both datasets at the same time, the best-fit model achieving a poor fit for the experiment without recovery time and a good fit for the experiment with recovery time. That result suggested that this model did not correspond to a reasonable molecular mechanism applicable to these experiments (Fig. 4). In contrast, the “reduced uptake” model achieved a very good fit to the data (Fig. 5). The model-to-data distance evaluated by the Sum of Squared Residuals (SSR) was equal to 2826 for the “enhanced efflux” model and 291 for the “reduced uptake” model confirming the superiority of the second model (Table 1).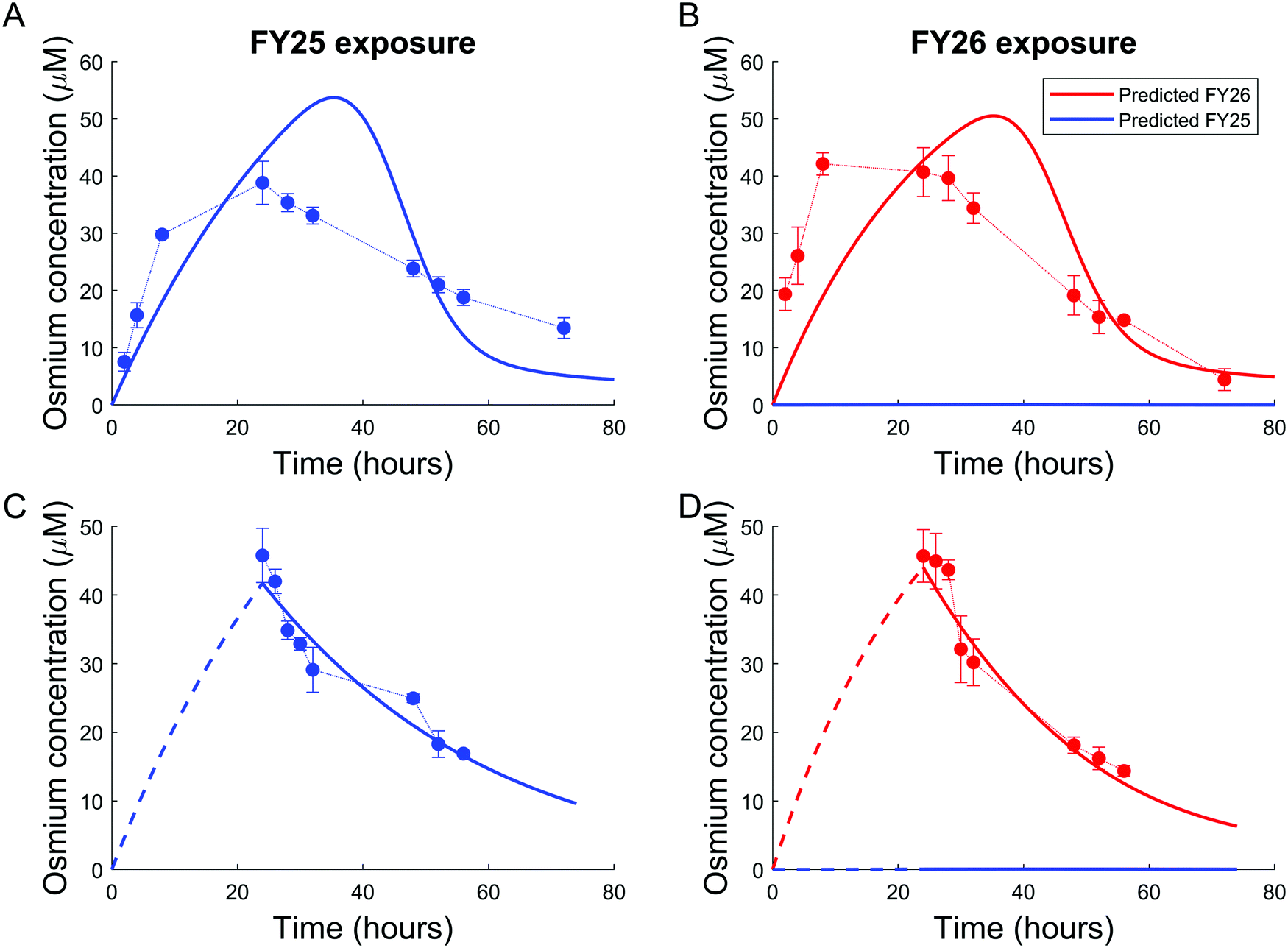 | ||
| Fig. 4 Poor data fit for the “enhanced efflux” model. No parameter set could be found to fit all datasets simultaneously. (A and B) experimental intracellular osmium accumulation (dots) and model-predicted intracellular concentrations of FY25 (blue solid lines) and FY26 (red solid lines) in A2780 cancer cells treated with equipotent concentrations (1× IC50) of FY25 (A) or FY26 (B). (C and D) Intracellular osmium accumulation (dots) and model-predicted intracellular concentrations of FY25 (blue solid lines) and FY26 (red solid lines) after 24 h exposure to FY25 (C) or FY26 (D), followed by 48 h recovery time in complex-free culture medium. | ||
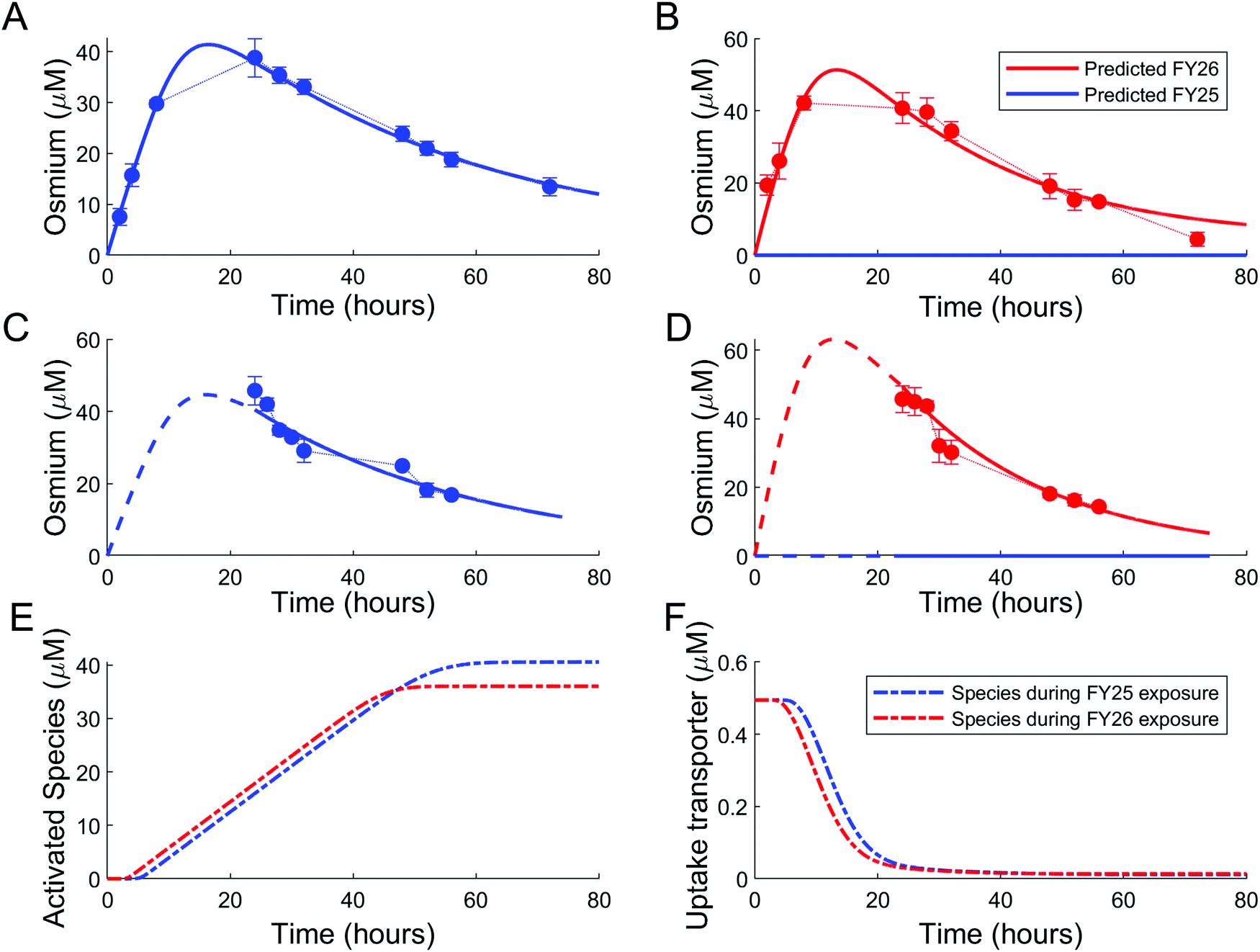 | ||
| Fig. 5 Good data fit for the “reduced uptake” model: (A and B). Intracellular osmium accumulation (dots) and model-predicted concentrations of FY25 (blue solid lines) and FY26 (red solid lines) in A2780 cancer cells treated with equipotent concentrations (1× IC50) of FY25 (A) or FY26 (B) for 72 h. (C and D): Intracellular osmium accumulation (dots) and model-predicted concentrations of FY25 (blue solid lines) and FY26 (red solid lines) after 24 h exposure to equipotent concentrations of FY25 (C) or FY26 (D), followed by 48 h recovery time in complex-free medium. (E and F) Intracellular concentrations of the complex-activated species (E) and of the uptake transporter (F) during exposure to either FY25 (blue) or FY26 (red) without recovery time (see Fig. 3). | ||
| Complex | Estimate | 95% Confidence interval | Units |
|---|---|---|---|
| FY25 active uptake | 6.03 | [5.3, 6.8] | μM−1 h−1 |
| FY25 active efflux | 108.3 | [103.5, 114.8] | h−1 |
| FY26 active uptake | 0.0265 | [0.0254, 0.0274] | μM−1 h−1 |
| FY26 active efflux | 0.0407 | [0.039, 0.0425] | h−1 |
| FY26 to FY25 transformation | 0 | [0, +∞] | h−1 |
| Activation threshold | 20.8 | [16.5, 30] | μM |
| Activation hill power | 14 | [2.2, +∞] | — |
| Activation rate constant | 0.86 | [0.74, 1.18] | h−1 |
To further investigate the validity of the “reduced uptake” model, parameter practical identifiability was investigated (see Methods and ESI,† Fig. S6). All model parameters were identifiable except two parameters, which proved the reliability of the model. The first parameter which was not identifiable corresponded to the rate constant for conversion of FY26 into FY25, whose optimal value was estimated as zero. However, this was actually an artefact arising from the datasets used in the estimation procedure as setting this parameter to zero made the estimation of transport parameters for FY25 and FY26 independent. Next, the second parameter which was not identifiable was the Hill power, which set the steepness of the Hill kinetics assumed for compound-induced uptake decrease. This parameter was estimated to the highest investigated value, that is 14, which corresponded to the highest steepness. However, any value of this parameter above 10 achieved the optimal value of the likelihood, hence the best fit to the data (Fig. 6).
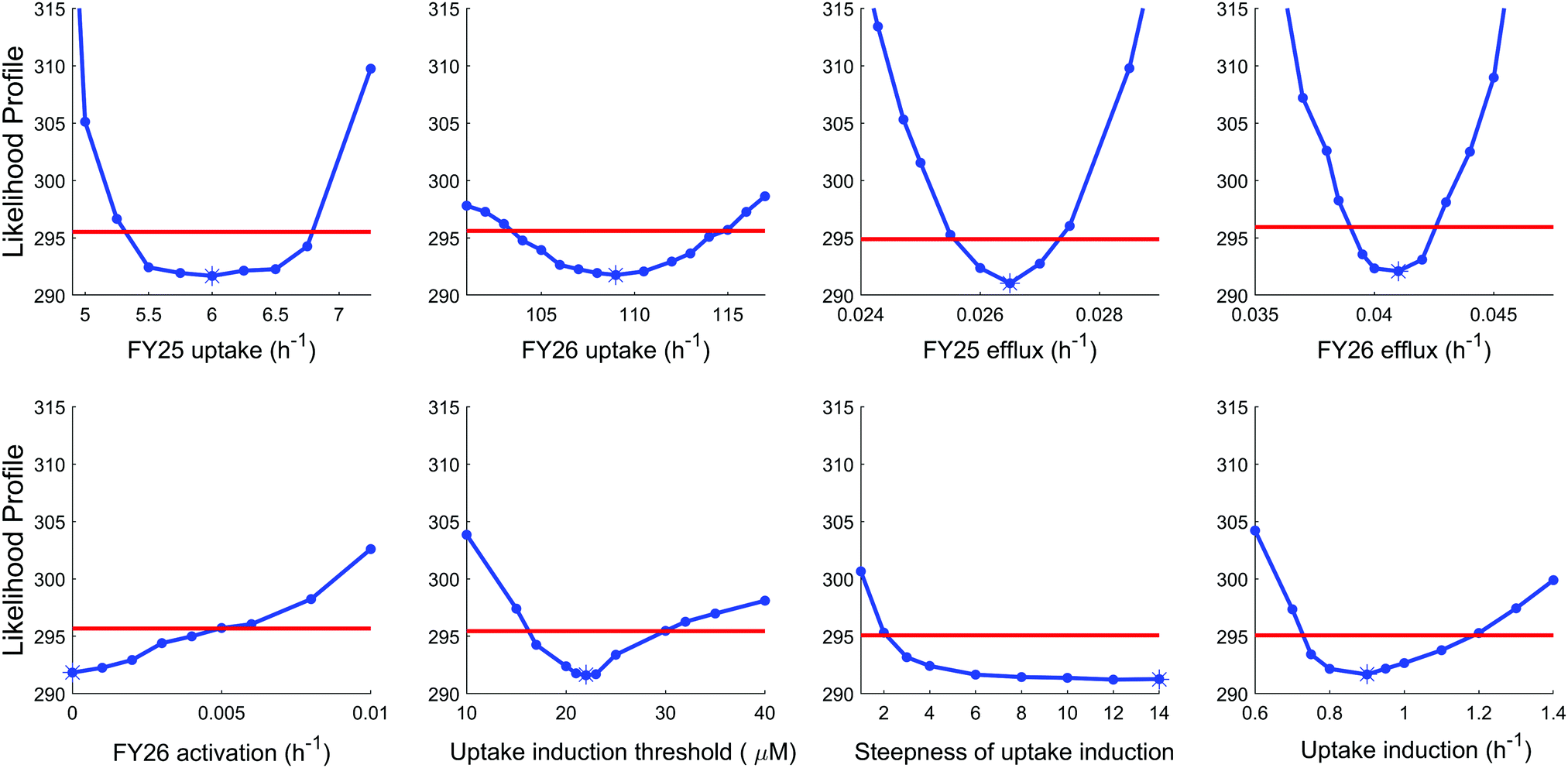 | ||
| Fig. 6 Likelihood profiles for parameters of the “reduced uptake” model. Stars represent the optimal parameter value. Red lines are the 95% confidence threshold value. If the likelihood profile crosses the threshold value twice (i.e. when increasing and decreasing parameter value starting from optimal value), this proves parameter identifiability. The points at which the likelihood profile crosses the threshold are the ranges of the parameter 95% confidence interval. | ||
In the “reduced uptake” model, the cellular transport rate for FY26 was estimated to be faster than that of FY25, by 1.5-fold for efflux and 17.7-fold for uptake (Table 1). This appears to correlate with the lower cytotoxicity of FY25 compared to that of FY26,22 and structurally related ruthenium complexes,23 thus providing a partial validation of the model. The activation threshold corresponding to the intracellular osmium concentration above which alteration in the rate of complex transport was triggered was estimated as 20.8 μM (95% confidence interval [16.5, 30]), and was reached 4 to 5 h after the start of FY25 or FY26 exposure. After that time, the unknown species was then activated in the mathematical model and enhanced the degradation of uptake transporters with similar kinetics for FY25 and FY26 exposure (Fig. 5E and F).
Next, we investigated whether these mathematical results could depend on the choice of kinetics used in the mathematical modelling to represent the PK of the complexes. Linear kinetics of FY25 and FY26 transport and of FY26 activation were changed to Michaelis–Menten terms. We observed that Michaelis–Menten kinetics did not enable the “enhanced efflux” model to fit the experimental data (SSR = 2650), but still allowed for a very good fit of the “reduced uptake” model (SSR = 291), thus indicating that the superiority of the second model did not depend on the choice of kinetics. Altogether, these results advocated decreased uptake of the complexes rather than increased efflux as an explanation for the overall decrease in osmium accumulation after 24 h of FY25 or FY26 exposure.
Temperature dependence of cellular transport parameters
We further investigated the temperature-dependence of Os accumulation during the first 8 h of exposure. Intracellular accumulation of Os in ovarian cancer cells exposed to either FY25 or FY26 decreased as a function of temperature (Fig. 7). The model developed above was used to analyse the temperature dependence of transport parameters. For the sake of simplicity and in the absence of detailed data, the influence of the complexes on their own cellular uptake was neglected as the analysis above predicted that the transport of the compounds was altered only after 4 to 5 hours of exposure. FY25 and FY26 uptake parameters were unchanged for the experiment performed at 37 °C and were estimated from the osmium accumulation datasets for experiments at 23 °C and 4 °C. All other model parameters were assumed not to be affected by temperature. The model calibrated from the datasets of Fig. 4 and 5 achieved a good fit to this new dataset obtained at 37 °C (Fig. 7). Furthermore, the model predicted that rate constants for FY25 uptake at 23 °C and 4 °C were equal to 36% and 3.1%, respectively, of those at 37 °C. Similarly, the rate constants of FY26 uptake at 23 °C and 4 °C were equal to 29% and 2.8%, respectively, of those at 37 °C (Fig. 7).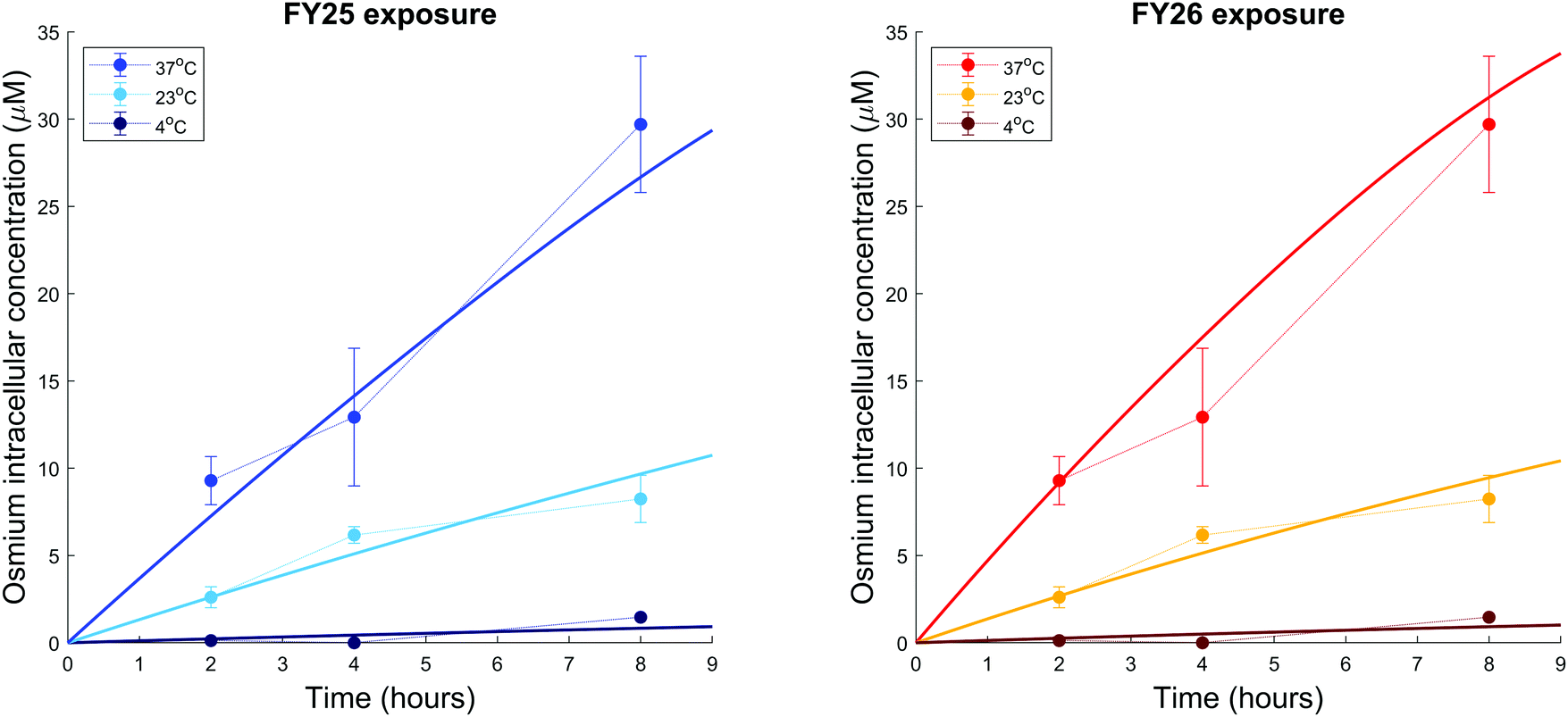 | ||
| Fig. 7 Temperature-dependence of osmium accumulation in A2780 cancer cells treated with equipotent concentrations of FY25 or FY26. Solid lines represent the best-fit of the model for FY25 (left) and FY26 (right) exposure at the stipulated temperatures. | ||
Discussion
Knowledge of the kinetics of drug uptake, efflux and accumulation in cancer cells is important for optimisation not only of drug design, but also of treatment regimens, including the selection of combination therapies. In general, our understanding of the uptake and efflux mechanisms for metallodrugs is poor. The most widely used drug in cancer chemotherapy is cisplatin and even now, 40 years after its introduction into clinical use, its transport mechanisms have not been fully elucidated.Here we have studied the time- and temperature-dependent accumulation of a promising iodido organo-osmium anticancer drug candidate [(η6-p-cymene)Os(Ph-azopyridine-NMe2)I]+ (FY26) by A2780 human ovarian cancer cells. This complex is inert and a pro-drug which is activated inside cancer cells to form the more reactive chlorido complex [(η6-p-cymene)Os(Ph-azopyridine-NMe2)Cl]+ (FY25) as a major metabolite. Other species that are likely to be formed include the hydroxido complex, the glutathione adduct, and oxidised glutathione sulfenate adduct.14 The ability of chloride and hydroxide to bind strongly to Os(II) even in the presence of a large molar excess of glutathione is notable and contrasts with the behaviour of Pt(II) in cisplatin.
We have compared the time-dependent accumulation of the chlorido complex FY25 and iodido complex FY26 in human ovarian cancer cells and constructed models which account for extent and time dependence of the influx and efflux of the complexes and the intracellular conversion of FY26 into FY25. The marked decrease in the accumulation of Os after 24 h of exposure to the complexes is well described by mathematical modelling which reveals that this can be explained by complex-induced decrease in osmium uptake, but not by enhanced osmium efflux (Fig. 4 and 5). The validity of the “reduced uptake” model was further strengthened by its accurate prediction of the 17× faster uptake of FY26 at 37 °C and 1.4× faster efflux of FY26 as compared to those of FY25, as demonstrated in previous experiments which were not used for model design nor calibration. Drug-induced uptake reduction could constitute a defence mechanism to slow down the accumulation of antiproliferative drugs into cancer cells, thus leading to a decreased cytotoxicity.
This combined experimental and modelling result suggests that specific active transporters are involved in the uptake of these organo-osmium complexes. Indeed, there are reported examples of transport proteins involved in the uptake and efflux of metallodrugs. Early on it was thought that the main route of cellular accumulation of cisplatin, present in extracellular media as the neutral molecule cis-[PtCl2(NH3)2], relied only on passive diffusion. However, more recent studies have suggested that active transport, including specific membrane proteins, might be also involved. These include the copper transporters Ctr1 and Ctr2,24,25 although the relevance of these to platinum treatment is currently in doubt.26 Alternatively, the p-type copper transporters ATP7A and ATP7B have also been linked to cisplatin resistance. However, the mechanism by which copper transporters might transport cisplatin (or other platinum drugs) is unclear. These proteins tend to be rich in sulfur ligands (methionine and cysteine side chains), and the strong trans effect of sulfur can readily lead to trans-ligand displacement from Pt(II). Organic cation transporters and multidrug resistance transporters (e.g. MDR1, p-glycoprotein) may also play a role in platinum transport and resistance.26 In comparison, the arsenic drug, arsenic trioxide, is transported into cells via aquaglyceroporin membrane transporters, perhaps understandable due to its structure in solution, [As(OH)3], which is a glycerol mimic.27
We can expect that the transporters used by metallodrugs will depend intimately on the structures of the complexes, including the metal and its oxidation state, the types and number of bound ligands, and the coordination geometry. Previously, we studied the A2780 cellular uptake and accumulation of iminopyridine complexes of Ru(II) arene iminopyridine complexes [Ru(η6-p-cymene)(N,N-dimethyl-N′-[(E)-pyridine-2-ylmethylidene]benzene-1,4-diamine)X]+ where X = Cl or I. Remarkably, whereas the chlorido complex was taken up largely by active transport, passive transport dominated for the iodido complex. Cellular efflux of Ru was slow, and was partially inhibited by the efflux pump inhibitor verapamil, which suggested a role for P-glycoprotein in the efflux of the drug.23 Furthermore, for the diamino arene Ru(II) complex [Ru(biphenyl)(ethylenediamine)Cl]+ which is cross-resistant with Adriamycin, drug resistance in A2780 cells was almost completely reversed by verapamil, suggesting that this protein plays a major role in the efflux of the ruthenium anticancer complex.28
Conclusions
Most metallodrugs are pro-drugs so understanding their transport and activation becomes important for understanding their mechanisms of action. Experimental work on the mechanisms of action of metallodrugs is challenging since it requires determination of not only the metal and its oxidation state, but also the nature of the bound ligands, both monodentate and chelated. Here we are fortunate that osmium is not a biologically essential metal, and thus its concentrations can be readily quantified using ICP-MS at pharmacologically-relevant doses. Although both cisplatin and the organo-osmium complexes studied here are activated by exchange of monodentate ligands, the mechanisms of exchange are quite different. In the case of the azopyridine complexes, it appears to involve attack on the azo bond of the chelated ligand by intracellular glutathione (GSH).14,15 Moreover the organo-osmium complexes appear to localise in the cytoplasm of cancer cells,29 whereas cisplatin exerts its effect in the nucleus.This combined experimental and mathematical study allowed us to identify a probable effect of FY25 and FY26 on their own cellular uptake leading to decreased intracellular accumulation. Future work will involve investigations of possible specific membrane transporter proteins for FY26. This complex induces redox changes in cancer cells,10 especially the production of reactive oxygen species (ROS). Whether this leads to protein damage and loss of function of a transporter, or whether attack on mRNA could be involved, requires further investigation. ROS-induced attack on intracellular proteins may be related to the apparent block of osmium uptake after an intracellular threshold osmium concentration has been reached. Further work is required to investigate whether this is due to degradation of a specific transporter protein or another mechanism. Such considerations are important for the preclinical development of FY26 and will affect the choice of treatable cancers and the choice of therapeutic combinations with other drugs.
Author contributions
I. R. C, P. J. S. and A. B. designed and coordinated the experiments. A. B. and F. B. carried out the mathematical modelling. I. R. C, C. S. C, J. P. C. C. and J. I. S. carried out the cell experiments. J. P. C. C. carried out the ICP-MS measurements. All authors contributed to the interpretation of the data and the writing of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Wellcome Trust (Grant No. 107691/Z/15), EPSRC (grants EPSRC EP/F034210/1; EP/P030572/1) and CRUK/EPSRC (grant number C53561/A19933) for financial support. AB is funded by the Plan Cancer of the French National Cancer Institute (INCa), through the ATIP-Avenir program.References
- L. Kelland, The resurgence of platinum-based cancer chemotherapy, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- B. van Zyl, D. Tang and N. A. Bowden, Biomarkers of platinum resistance in ovarian cancer: what can we use to improve treatment, Endocr. – Relat. Cancer, 2018, 25, 303–318 Search PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Third row transition metals for the treatment of cancer, Philos. Trans. R. Soc., A, 2015, 373, 20140185 CrossRef PubMed.
- M. Hanif, M. V. Babak and C. G. Hartinger, Development of anticancer agents: wizardry with osmium, Drug Discovery Today, 2014, 19, 1640–1648 CrossRef CAS PubMed.
- W. Su, Y. Li and P. Li, Design of Ru-arene complexes for antitumor drugs, Mini-Rev. Med. Chem., 2018, 18, 184–193 CAS.
- S. M. Meier-Menches, C. Gerner, W. Berger, C. G. Hartinger and B. K. Keppler, Structure-activity relationships for ruthenium and osmium anticancer agents – towards clinical development, Chem. Soc. Rev., 2018, 47, 909–928 RSC.
- J. P. C. Coverdale, I. Romero-Canelon, C. Sanchez-Cano, G. J. Clarkson, A. Habtemariam, M. Wills and P. J. Sadler, Asymmetric transfer hydrogenation by synthetic catalysts in cancer cells, Nat. Chem., 2018, 10, 347–354 CrossRef CAS PubMed.
- L. Feng, Y. Geisselbrecht, S. Blanck, A. Wilbuer, G. E. Atilla-Gokcumen, P. Filippakopoulos, K. Kraling, M. A. Celik, K. Harms, J. Maksimoska, R. Marmorstein, G. Frenking, S. Knapp, L. O. Essen and E. Meggers, Structurally sophisticated octahedral metal complexes as highly selective protein kinase inhibitors, J. Am. Chem. Soc., 2011, 133, 5976–5986 CrossRef CAS PubMed.
- S. D. Shnyder, Y. Fu, A. Habtemariam, S. H. van Rijt, P. A. Cooper, P. M. Loadman and P. J. Sadler, Anti-colorectal cancer activity of an organometallic osmium arene azopyridine complex, MedChemComm, 2011, 2, 666–668 RSC.
- J. M. Hearn, I. Romero-Canelon, A. F. Munro, Y. Fu, A. M. Pizarro, M. J. Garnett, U. McDermott, N. O. Carragher and P. J. Sadler, Potent organo-osmium compound shifts metabolism in epithelial ovarian cancer cells, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3800–3805 CrossRef PubMed.
- Y. Fu, A. Habtemariam, A. M. Pizarro, S. H. van Rijt, D. J. Healey, P. A. Cooper, S. D. Shnyder, G. J. Clarkson and P. J. Sadler, Organometallic osmium arene complexes with potent cancer cell cytotoxicity, J. Med. Chem., 2010, 53, 8192–8196 CrossRef CAS PubMed.
- J. P. C. Coverdale, H. E. Bridgewater, J. I. Song, N. A. Smith, N. P. E. Barry, I. Bagley, P. J. Sadler and I. Romero-Canelon, In vivo selectivity and localization of reaction oxygen species (ROS) induction by osmium anticancer complexes that circumvent platinum resistance, J. Med. Chem., 2018, 61, 9246–9255 CrossRef CAS PubMed.
- I. Romero-Canelon, M. Mos and P. J. Sadler, Enhancement of selectivity of an organometallic anticancer agent by redox modulation, J. Med. Chem., 2015, 58, 7874–7880 CrossRef CAS PubMed.
- R. J. Needham, C. Sanchez-Cano, X. Zhang, I. Romero-Canelon, A. Habtemariam, M. S. Cooper, L. Meszaros, G. J. Clarkson, P. J. Blower and P. J. Sadler, In-cell activation of organo–osmium(II) anticancer complexes, Angew. Chem., Int. Ed., 2017, 56, 1017–1020 CrossRef CAS PubMed.
- X. Zhang, E. Sicilia, P. J. Sadler and C. S. Cano, in preparation.
- J. M. Gallo, Pharmacokinetic/pharmacodynamic-driven drug development, Mt. Sinai J. Med., 2010, 77, 381–388 CrossRef PubMed.
- C. Venzago, M. Popp, J. Kovac and A. Kunkel, Pharmacopeial requirements for elemental impurities: a novel approach to the trace determination of osmium by oxidative pressure vessel sample digestion and measurement using inductively coupled plasma mass spectrometry (ICP-MS) after complexation and stabilisation, J. Anal. At. Spectrom., 2013, 28, 1125–1129 RSC.
- S. Dulong, A. Ballesta, A. Okyar and F. Levi, Identification of circadian determinants of cancer chronotherapy through in vitro chronopharmacology and mathematical modeling, Mol. Cancer Ther., 2015, 14, 2154–2164 CrossRef CAS PubMed.
- A. Ballesta, Q. Zhou, X. Zhang, H. Lv and J. M. Gallo, Multiscale design of cell-type-specific pharmacokinetic/pharmacodynamic models for personalized medicine: application to temozolomide in brain tumors, CPT: Pharmacometrics Syst. Pharmacol., 2014, 3, e112 CAS.
- A. Raue, C. Kreutz, T. Maiwald, J. Bachmann, M. Schilling, U. Klingmuller and J. Timmer, Structural and practical identifiability analysis of partially observed dynamical models by exploiting the profile likelihood, Bioinformatics, 2009, 25, 1923–1929 CrossRef CAS PubMed.
- A. Ballesta, J. Clairambault, S. Dulong and F. Lévi, Theoretical optimization of irinotecan-based anticancer strategies in case of drug-induced efflux, Appl. Math. Lett., 2011, 24, 1251–1256 CrossRef.
- S. H. van Rijt, I. Romero-Canelón, Y. Fu, S. D. Shnyder and P. J. Sadler, Potent organometallic osmium compounds induce mitochondria-mediated apoptosis and S-phase cell cycle arrest in A549 non-small cell lung cancer cells, Metallomics, 2014, 6, 1014–1022 RSC.
- I. Romero-Canelón, A. M. Pizarro, A. Habtemariam and P. J. Sadler, Contrasting cellular uptake pathways for chlorido and iodido iminopyridine ruthenium arene anticancer complexes, Metallomics, 2012, 4, 1271–1279 RSC.
- S. Ishida, J. Lee, D. J. Thiele and I. Herskowitz, Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14298–14302 CrossRef CAS PubMed.
- X. Lin, T. Okuda, A. Holzer and S. B. Howell, The copper transporter CTR1 regulates cisplatin uptake in Saccharomyces cerevisiae, Mol. Pharmacol., 2002, 62, 1154 CrossRef CAS PubMed.
- I. A. Riddell and S. J. Lippard, Cisplatin and oxaliplatin: our current understanding of their actions, Met. Ions Life Sci., 2018, 18, 1–42 CAS.
- R. Mukhopadhyay, H. Bhattacharjee and B. P. Rosen, Aquaglyceroporins: generalized metalloid channels, Biochim. Biophys. Acta, 2014, 1840, 1583–1591 CrossRef CAS PubMed.
- R. E. Aird, J. Cummings, A. A. Ritchie, M. Muir, R. E. Morris, H. Chen, P. J. Sadler and D. I. Jodrell, In vitro and in vivo activity and cross resistance profiles of novel ruthenium(II) organometallic arene complexes in human ovarian cancer, Br. J. Cancer, 2002, 86, 1652–1657 CrossRef CAS PubMed.
- C. Sanchez-Cano, I. Romero-Canelón, Y. Yang, I. J. Hands-Portman, S. Bohic, P. Cloetens and P. J. Sadler, Synchrotron X-ray fluorescence nanoprobe reveals target sites for organo-osmium complex in human ovarian cancer cells, Chem. – Eur. J., 2017, 23, 2512–2516 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9mt00173e |
| This journal is © The Royal Society of Chemistry 2019 |