
Mutant L-chain ferritins that cause neuroferritinopathy alter ferritin functionality and iron permeability

Abstract
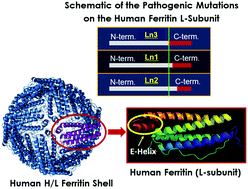
In mammals, the iron storage and detoxification protein ferritin is composed of two functionally and genetically distinct subunit types, H (heavy) and L (light). The two subunits co-assemble in various ratios, with a tissue specific distribution, to form shell-like protein structures of 24 subunits within which a mineralized iron core is stored. The H-subunits possess ferroxidase centers that catalyze the rapid oxidation of ferrous ions, whereas the L-subunit does not have such centers and is believed to play an important role in electron transfer reactions that occur during the uptake and release of iron. Pathogenic mutations on the L-chain lead to neuroferritinopathy, a neurodegenerative disease characterized by abnormal accumulation of ferritin inclusion bodies and iron in the central nervous system. Here, we have characterized the thermal stability, iron loading capacity, iron uptake, and iron release properties of ferritin heteropolymers carrying the three pathogenic L-ferritin mutants (L154fs, L167fs, and L148fs, which for simplicity we named Ln1, Ln2 and Ln3, respectively), and a non-pathogenic variant (L135P) bearing a single substitution on the 3-fold axes of L-subunits. The UV-Vis data show a similar iron loading capacity (ranging between 1800 to 2400 Fe(III)/shell) for all ferritin samples examined in this study, with Ln2 holding the least amount of iron (i.e. 1800 Fe(III)/shell). The three pathogenic L-ferritin mutants revealed higher rates of iron oxidation and iron release, suggesting that a few mutated L-chains on the heteropolymer have a significant effect on iron permeability through the ferritin shell. DSC thermograms showed a strong destabilization effect, the severity of which depends on the location of the frameshift mutations (i.e. wt heteropolymer ferritin ≅ homopolymer H-chain > L135P > Ln2 > Ln1 > Ln3). Variant L135P had only minor effects on the protein functionality and stability, suggesting that local melting of the 3-fold axes in this variant may not be responsible for neuroferritinopathy-like disorders. The data support the hypothesis that hereditary neuroferritinopathies are due to alterations of ferritin functionality and lower physical stability which correlate with the frameshifts introduced at the C-terminal sequence and explain the dominant transmission of the disorder.
 Please wait while we load your content...
Please wait while we load your content...