
Siderophore profiling of co-habitating soil bacteria by ultra-high resolution mass spectrometry†
Rene M.
Boiteau
 *ab,
Sarah J.
Fansler
c,
Yuliya
Farris
b,
Jared B.
Shaw
a,
David W.
Koppenaal
a,
Ljiljana
Pasa-Tolic
a and
Janet K.
Jansson
c
*ab,
Sarah J.
Fansler
c,
Yuliya
Farris
b,
Jared B.
Shaw
a,
David W.
Koppenaal
a,
Ljiljana
Pasa-Tolic
a and
Janet K.
Jansson
c
aEnvironmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, WA 99354, USA
bCollege of Earth, Ocean, Atmospheric Sciences, Oregon State University, Corvallis, OR 97330, USA. E-mail: rene.boiteau@oregonstate.edu
cBiological Sciences Division, Pacific Northwest National Laboratory, Richland, WA 99354, USA
First published on 14th November 2018
Abstract
The chemical structure of organic molecules profoundly impacts their interactions with metal ions and mineral phases in soils. Understanding the sources and cycling of metal-chelating compounds is therefore essential for predicting the bioavailability and transport of metals throughout terrestrial environments. Here we investigate the molecular speciation of organic molecules that solubilize trace metals in calcareous soils from Eastern Washington. Ultra-high performance Fourier transform ion cyclotron resonance mass spectrometry at 21 Tesla enabled fast and confident detection and identification of metal chelators that are produced by microbes that inhabit these soils based on screening for features that match diagnostic metal isotope patterns. We compared two approaches, one based on direct infusion using the incorporation of a rare isotope to validate true iron-binding features, and another based on separation with liquid chromatography and detection of isotopologues with coherent elution profiles. While the isotopic exchange method requires significantly shorter analysis time, nearly twice as many features were observed with liquid chromatography mass spectrometry (LCMS), mostly due to the reduction in ion suppression where major features limit the sensitivity of minor features. In addition, LCMS enabled the collection of higher quality fragmentation spectra and facilitated feature identification. Siderophores belonging to four major classes were identified, including ferrioxamines, pseudobactins, enterobactins, and arthrobactins. Each of these siderophores likely derives from a unique member of the microbial community, and each possesses different chemical characteristics and uptake pathways, likely contributing to fierce competition for iron within these soils. Our results provide insight into the metabolic pathways by which microbes that co-inhabit calcareous soils compete for this essential micronutrient.
 Rene M. Boiteau | Rene Boiteau is an assistant professor in the College of Earth, Ocean, and Atmospheric Sciences at Oregon State University and holds a joint appointment at the Pacific Northwest National Laboratory where he was previously a Linus Pauling Distinguished Postdoctoral Fellow. He and his group focus on understanding how microbes and plants impact the mobility and bioavailability of trace elements in marine and terrestrial environments. Much of this work relies on the development of analytical approaches to characterize biologically produced organic metal species and determine their kinetic properties using chromatography coupled with multimodal mass spectrometry. |
Significance to metallomicsWhile metals are essential micronutrients, the biochemical mechanisms that govern their bioavailability to microbes in alkaline soils with low iron solubility are poorly constrained due to the scarcity of these elements within a complex background of diverse organic carbon compounds. This study elucidates the siderophore profile of calcareous soil bacteria and demonstrates the diverse iron solubilizing strategies that coexist in these environments. These compounds have differential effects on the bioavailability of iron, and thus likely structure the competitive and complementary interactions that structure these soil ecosystems where free iron is a scarce resource. |
Introduction
One-third of global soils contain levels of calcium carbonate that buffer soil pH above 7, under which conditions soil iron becomes scarcely soluble and can become a limiting factor for plant and microbial growth.1 In these environments, iron is predominantly found as oxyhydroxide minerals, and concentrations of bioavailable free Fe(III) fall below the levels required for optimal growth of plants and microbes.2 Under such conditions, many plants, fungi, and bacteria can produce chelating agents termed siderophores that are secreted into their environment for the purpose of solubilizing iron and facilitating uptake through selective membrane receptors that import the iron-bound complex.3,4 The ability to produce and take up these compounds is thought to be an important factor that structures microbial ecosystems under conditions where iron scarcity exerts a selective ecological pressure.5While producing siderophores can increase iron uptake rates for the producers, the secretion of siderophores is costly from a nutrient and energy standpoint.6 As a result, there is a strong incentive for organisms to take up siderophores that they do not produce. Examples of siderophore ‘piracy’ are common among microbes and plants, as indicated by the possession of uptake pathways for siderophores that they cannot produce.7,8 These may arise either by mutations of transporters to take up exogenous siderophores, by horizontal gene transfer of the uptake pathway from siderophore producers, or by selective loss of biosynthesis genes.9,10
The prevalence of siderophore piracy induces a selective pressure for producers to invent new siderophores that cannot be taken up by competitors. This is reflected by the hundreds of unique siderophores that have been structurally elucidated to date.3,11 New siderophores can arise from divergent evolution of biosynthesis pathways, resulting in suites of structurally related siderophores with slight differences in chemical properties.12 For example, pseudomonas species produce dozens of structural variants of the siderophore pyoverdine that require different transporters to take up.13 At the same time, siderophore biosynthesis pathways provide elegant examples of convergent evolution, whereby different organisms independently evolved different pathways to produce siderophores with completely different structural components to fulfil the same end goal of iron uptake. Many siderophores, such as the tris-catechol molecule enterobactin, are produced via a series of ribosomal peptide synthetase enzymes,14,15 while others, such as ferrioxamines, are made via a non-ribosomal peptide synthetase independent pathway.16 Some organisms are capable of producing multiple siderophores, and the secretion of one or another dominates under different environmental conditions.17 This redundancy suggests that the diversity of siderophore chemical structures is in part tied to advantages within specific environmental niches that are not currently understood.
Despite the recognized importance for siderophore mediated iron acquisition strategies in shaping competition and symbioses within microbial populations, the essential details of which pathways and interactions are relevant in natural ecosystems where they predominantly function remain murky. Of the hundreds of structurally elucidated siderophores produced by microbes found in soils, few have been observed in nature. Several studies have developed indirect methods to estimate bulk siderophore abundances. Concentrations from soil extracts has been estimated by bioassays with indicator organisms, and estimates range from 0–300 μg per kg soil.18,19 However, the reliability of these assays is limited due to uncertainties regarding their specificity and difficulty of quantification.19 Other studies have used targeted chemical approaches to detect certain siderophores in soils. Schizokinen, a citrate based siderophore has been identified chemically in the soils of rice fields.20 In addition, ferrioxamines have been detected in forest podzol soils.21 Other work has quantified select fungal22 or plant23,24 derived chelators in soils. These studies provide an important but limited view of siderophore abundance and identity in soil systems.
More recently, high-resolution Fourier transform mass spectrometry has provided an untargeted window into the diversity of major metal binding species. The principal challenge of characterizing metal chelators in environmental systems is the complexity of the organic mixture and the trace concentrations of specific metal chelates. High-resolution mass spectrometry approaches are able to overcome these issues by resolving molecular features across a large mass range and providing accurate mass information that can be used to identify diagnostic isotope patterns for elements such as Fe. These approaches have been used to detect and identify some of the major chelators that naturally occur in aquatic and soil environments.5,25
Our previous studies on calcareous soils using these high resolution mass spectrometry based approaches have identified both plant and fungal chelators, but did not identify any bacterial compounds.25 One of the limitations of these untargeted methods is the reduced sensitivity compared to targeted analyses due to the broad mass ranges analyzed and the need to detect minor isotopologues in complex samples. In this study, we sought to identify the biosynthetic potential of soil bacteria within calcareous soils, with the goal of evaluating the diversity of siderophore metabolisms from coexisting microorganisms, and generating molecular targets for more sensitive detection of these compounds in soils. Soil samples were enriched with glucose, nitrogen, and phosphorous but not metals in order to induce microbial metabolisms for solubilizing iron from soil minerals. To characterize siderophores produced within these enrichment cultures, we used ultra-high resolution 21 Tesla (T) Fourier Transform Ion Cyclotron Resonance Mass Spectrometry (FT-ICR MS) to detect thousands of molecular features and identify those that contained iron. We employed and compare two approaches based on (1) direct infusion with validation by isotopic labeling (2) liquid chromatography (LC) separation with online FT-ICR MS and MS/MS. With these methods, we identified dozens of iron-bound features and structurally characterized compounds that belong to four major siderophore classes, highlighting the diversity of siderophores produced by co-habitating soil microbes.
Methods
Soil collection and enrichment
Soil was collected in October, 2017 from the upper 0–15 cm depth from the Washington State University Irrigated Agriculture Research and Extension Center near Prosser, WA and sieved through a 4 mm mesh sieve to remove large particles. The soils were stored at 4 °C. This soil is part of the Warden soil series, which consists of a calcareous coarse-silty, mixed, super active, mesic Xeric Haplocambid with a pH of 7.8 and DTPA extractable metals of 0.68 mg per kg Zn, 3.4 mg per kg Mn, 7.8 mg per kg Fe, 0.84 mg per kg Cu, and 0.17 mg per kg Ni (NAPT method S-6.10).Enrichment cultures were prepared by adding 100 mg of soil to 10 mL of filter sterilized media containing 1× M9 minimal salts with 0.2 mM CaCl2, 1 mM MgSO4, 0.4% glucose, and no added trace metals. 100 μL of the suspension was added to 25 mL of M9 minimal media in each of five 50 mL trace metal free polypropylene centrifuge tube (VWR), such that each tube received ∼1 mg of the homogenized soil. The five samples and a non-inoculated media blank were incubated in the dark at 30 °C on an orbital shaker set at 200 rpm for 48 h, growing to an optical density of 0.7 (measured as absorbance at 600 nm). The enrichment samples were centrifuged at 4500 rpm for 10 min at 4 °C and the supernatant was transferred to a new tube for mass spectrometry analysis. The pellet was frozen at −20 °C prior to DNA extraction for amplicon sequencing. DNA extraction and sequencing followed the Earth Microbiome Project protocol.26 DNA was extracted from 0.25 g of soil in triplicate using the PowerSoil DNA isolation kit (MoBio). Data quality control and processing was performed with Hundo (https://github.com/pnnl/hundo)27 and binned operational taxanomic unit results from the triplicate analyses were pooled together.
Analysis by direct infusion FT-ICR MS
For analysis by direct infusion, 6 mL of the enrichment supernatant was added to each of three 15 mL polypropylene centrifuge tubes. The tubes were amended with (1) nothing (2) 600 μL of 0.1 M FeCl3 (prepared from ferric chloride hexahydrate, Sigma-Aldrich) or (3) 100 μL of a 57Fe stock solution (prepared by dissolving 4 mg 57Fe oxide (Cambridge isotope laboratories) in 960 μL of concentrated HCl (Optima, Fisher Scientific) for 1 week). After incubating at room temperature for 1 h to allow for complexation, the residual iron and chloride were removed by solid phase extraction. C18 solid phase extraction columns (30 mg, 1 mL, Supelco) were primed with 1 mL MeOH and rinsed with 2 mL qH2O prior to extraction of the 6 mL culture media. The column was then rinsed with 2 mL qH2O and organic compounds along with chelated iron were eluted with 600 μL MeOH and collected in 2 mL polypropylene microcentrifuge tubes. Water (600 μL) was added to each tube and the samples were immediately analyzed by FT-ICR MS.The 21 T FT-ICR MS used for all analyses was designed and built in the Environmental Molecular Science Laboratory at the Pacific Northwest National Lab.28 Prior to analysis, the instrument was tuned using the Thermo Velos electrospray ionization (ESI) tune mix. Samples were introduced into the FT-ICR MS into a nano-ESI source using a syringe pump at a flow rate of 1 μL min−1 through a 360/50 etched silica emitter tip. Ions were detected over an m/z range of 220–1400 in positive mode using 3 second transients and averaging 100 scans (6 minutes per sample). Peaks from each mass spectrum with a signal to noise ratio above 7 were centroided and exported to R for alignment and isotope pattern screening using custom scripts.
Analysis by LC-FT-ICR MS
One of the advantages of the high magnetic field strength of the 21 T FT-ICR MS is the ability to collect high resolution spectra with 1–2 second scans, enabling the detection of molecules with online chromatography. We used reverse phase chromatography to separate organic species based on polarity.Enrichment supernatants were separated on a Dionex Ultimate 3000 bioinert high-pressure liquid chromatography (HPLC) system fitted with a poly-etheretherketone (PEEK) C18 column (2.1 × 100 mm, 3 μm particle size; Hamilton) and PEEK tubing and connectors. Samples were injected in a 50 μL volume and eluted with solvent A (5 mM aqueous ammonium formate) and solvent B (5 mM ammonium formate in mass spectrometry grade MeOH) with a 20 min gradient from 5 to 95% B, followed by isocratic elution at 95% B for 10 min at a flow rate of 0.2 mL min−1.
For determination of the siderophore mass, the flow from the LC was coupled to the 21 T FT-ICR MS using a heated ESI source set to a capillary voltage of 3500 V; sheath, auxiliary, and sweep gas flow rates of 12, 6, and 2 (arbitrary units); and ion transfer tube and vaporizer temperatures of 300 and 75 °C. Mass spectra were collected with 1–2 second transients. MS/MS fragmentation spectra were collected by collision induced dissociation (CID) of the major features using a collision energy of 40. Authentic standards of ferrioxamine E and enterobactin were purchased (Sigma Aldrich) and analyzed by LCMS at 5 μM concentration to validate the identity of these compounds. LCMS data was screened for iron isotope patterns using in-house scripts as described previously.5
Results
Direct infusion FT-ICR MS
We first identified the masses of iron chelating compounds by direct infusion FT-ICR MS by screening iron-spiked samples for features matching the iron isotope pattern, and then validating true features by demonstrating their binding to a rare iron isotope (57Fe, 2.12% natural abundance). This experiment enabled us to quickly screen the diversity of chelators in a complex sample and facilitated benchmarking the performance of 21 T FT-ICR MS for identifying rare isotopic patterns. Effective screening relies on sufficient resolving power to baseline separate the major carbon and iron isotopologues of organic molecules (the narrowest being M + 1 57Fe and 13C, Δm/z of 0.003). Resolving power (Δm at full width half maximum/m) decreases with increasing m/z (Fig. 1). At a scan rate of 3 seconds, the limit of resolving carbon and iron isotopologues (Δm = 2.9 mDa) by 2× the peak full width half maximum is >800 m/z. Higher resolution can be obtained with longer transient time and/or acquisition of mass spectra in absorption mode Fourier transform.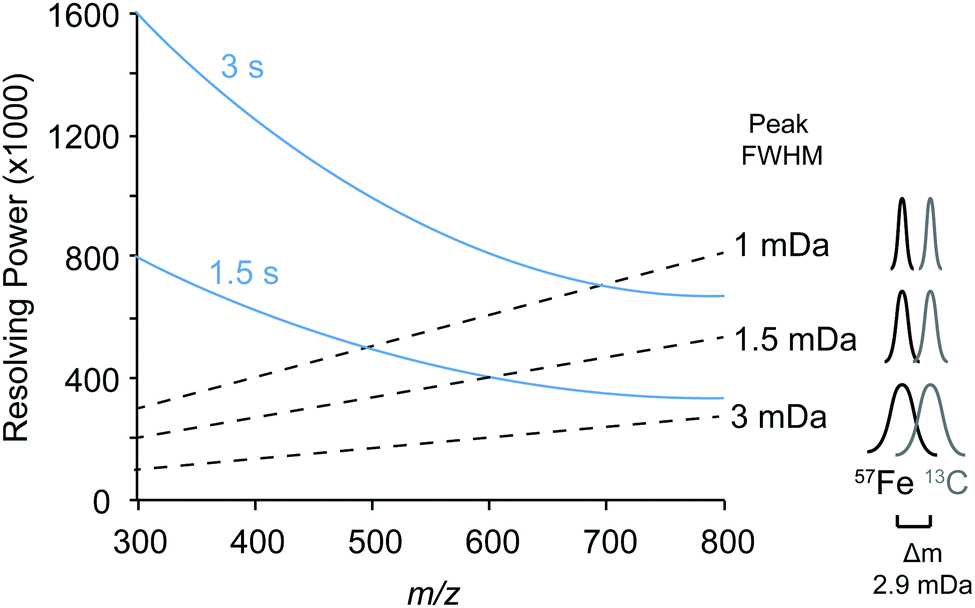 | ||
| Fig. 1 Resolving power of the 21 T FT-ICR MS (blue lines). Dashed lines indicate the mass peak full width half maxima (FWHM) peaks achieved across resolving powers and m/z values. FWHM values of less than 1.5 mDa are needed to resolve 57Fe and 13C isotopologues (shown in schematic on right). | ||
The Warden soil enrichment was spiked with either (1) no metal, (2) 56Fe, or (3) 57Fe and analyzed by direct infusion into the 21 T FT-ICR MS. Overall, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 102 peaks with a signal-to-noise ratio of >7 were detected across all analyses (Fig. 2). Features matching the iron isotope pattern were found by computationally screening for peak pairs with a mass difference of 1.9953 ± 0.001 m/z and a relative peak intensity of 0.0637 (54Fe/56Fe isotopologue) within a factor of 2. Of all possible peak pairs, 85 were identified that matched this criteria. For each of these candidates, the mass of the apo (no metal) and 57Fe forms were computed, and the intensity of these peaks, if present, were detected across data sets from all three treatments (Fig. 2). Fig. 2 shows the mass spectrum acquired for each of these conditions.
102 peaks with a signal-to-noise ratio of >7 were detected across all analyses (Fig. 2). Features matching the iron isotope pattern were found by computationally screening for peak pairs with a mass difference of 1.9953 ± 0.001 m/z and a relative peak intensity of 0.0637 (54Fe/56Fe isotopologue) within a factor of 2. Of all possible peak pairs, 85 were identified that matched this criteria. For each of these candidates, the mass of the apo (no metal) and 57Fe forms were computed, and the intensity of these peaks, if present, were detected across data sets from all three treatments (Fig. 2). Fig. 2 shows the mass spectrum acquired for each of these conditions.
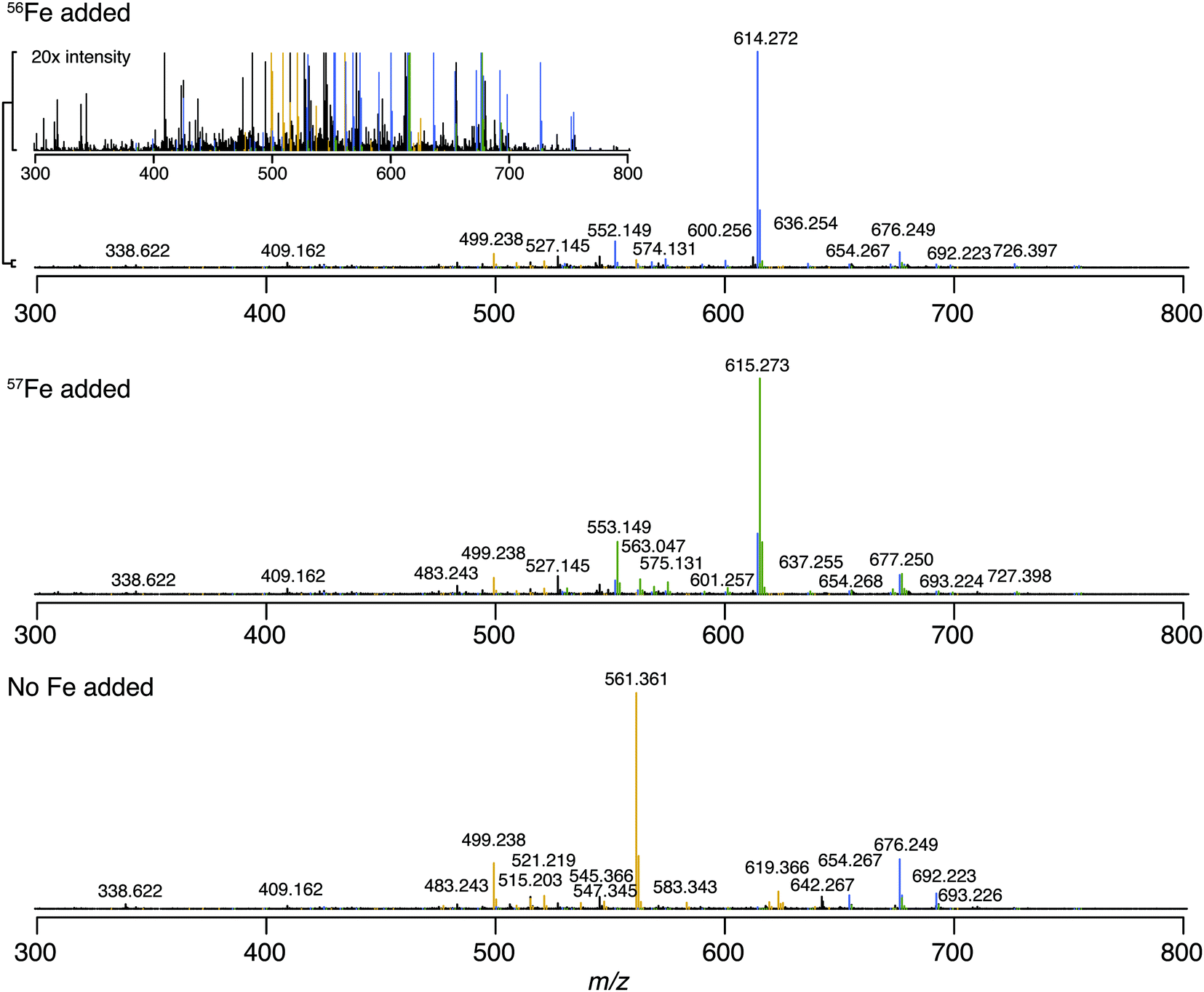 | ||
| Fig. 2 Direct infusion 21 T FT-ICR MS mass spectra of Warden soil enrichment supernatant amended with nothing (top), 56Fe (middle) or 57Fe (bottom). Peak colors correspond to unbound (yellow), 56Fe bound (blue) or 57Fe bound (green) organic complexes. | ||
We developed a quantitative selection criteria to distinguish true iron containing features from artifacts that coincidentally match the mass difference and relative abundance of 54Fe and 56Fe but do not contain iron. An enrichment factor was calculated for each of the 85 peak pairs based on peak intensities in the mass spectrum of the 57Fe addition sample:
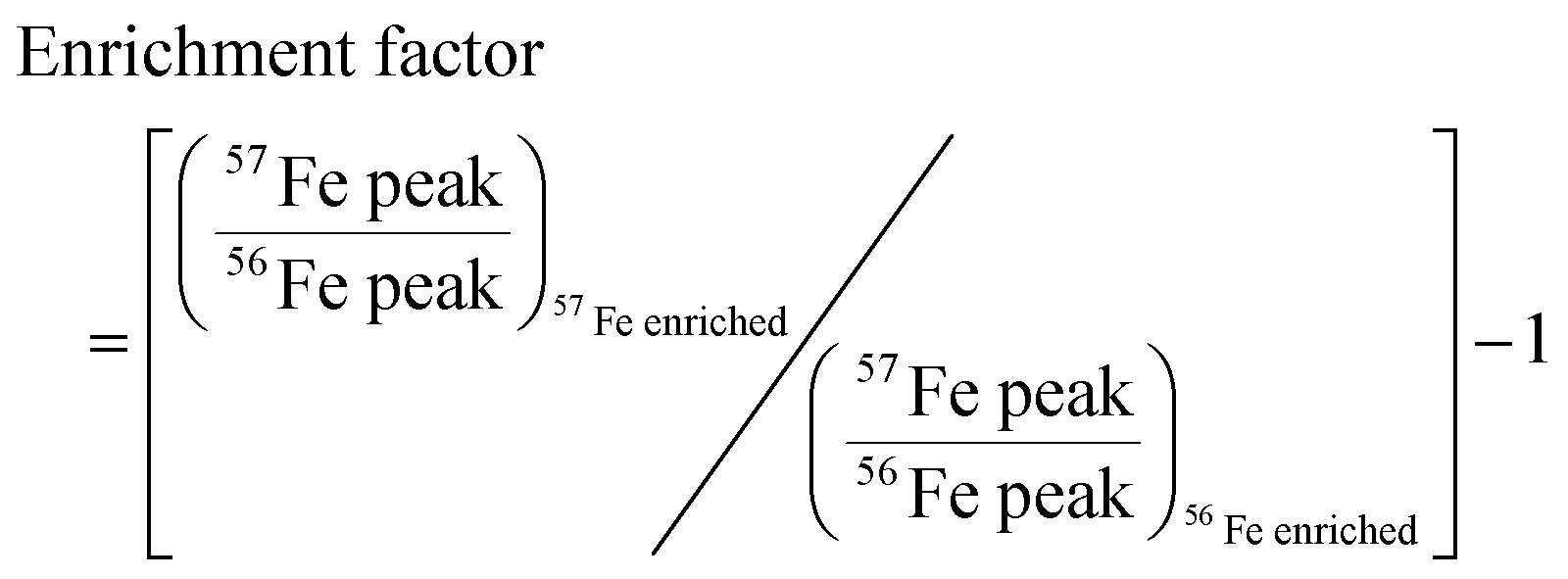
A positive enrichment factor is expected for true iron complexes due to the substitution of 56Fe for 57Fe. Of the 85 candidate peak pairs, 24 had enrichment factors >2 and were considered true iron-containing complexes (Fig. 3). The other peak pairs are likely coincidental matches to the isotope pattern due to the high peak density. After removing 13C isotopologues from the list, we were left with 17 features that each represent distinct iron containing species (Table S1, ESI†).
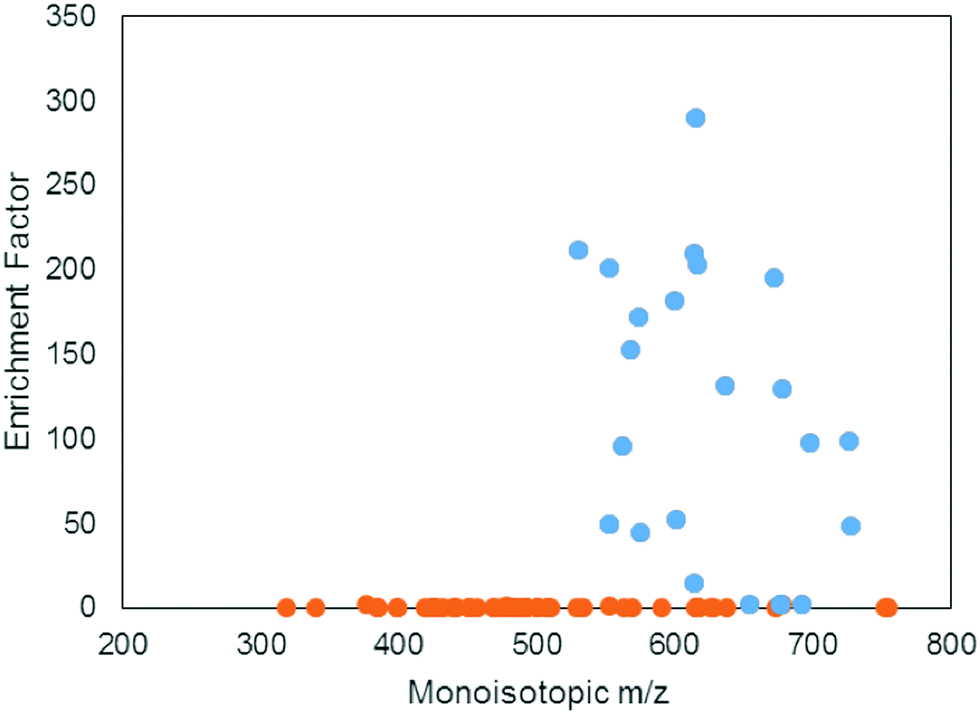 | ||
| Fig. 3 Benchmarking performance of the 21 T FT-ICR MS for identifying iron isotope pattern by direct infusion. Points above an enrichment factor of 2 were considered valid (blue points), while those below 2 were not (orange points). | ||
The mass range and intensity ratio of the validated peak pairs fell close to the true values, within a mass difference of ±0.0004 m/z and relative intensity of 0.04 to 0.07 with the exception of one intensity outlier at 0.097 (Fig. 4). In addition to these validated peaks, there were 22 of the 85 peaks where the 57Fe isotopologue was detected in the 57Fe enrichment sample, but the 56Fe isotopologue was not. For these compounds, which were mostly low abundance, it is possible that they are true Fe binding features, but that the 56Fe isotopologue fell below the current detection limit of the FT-ICR MS.
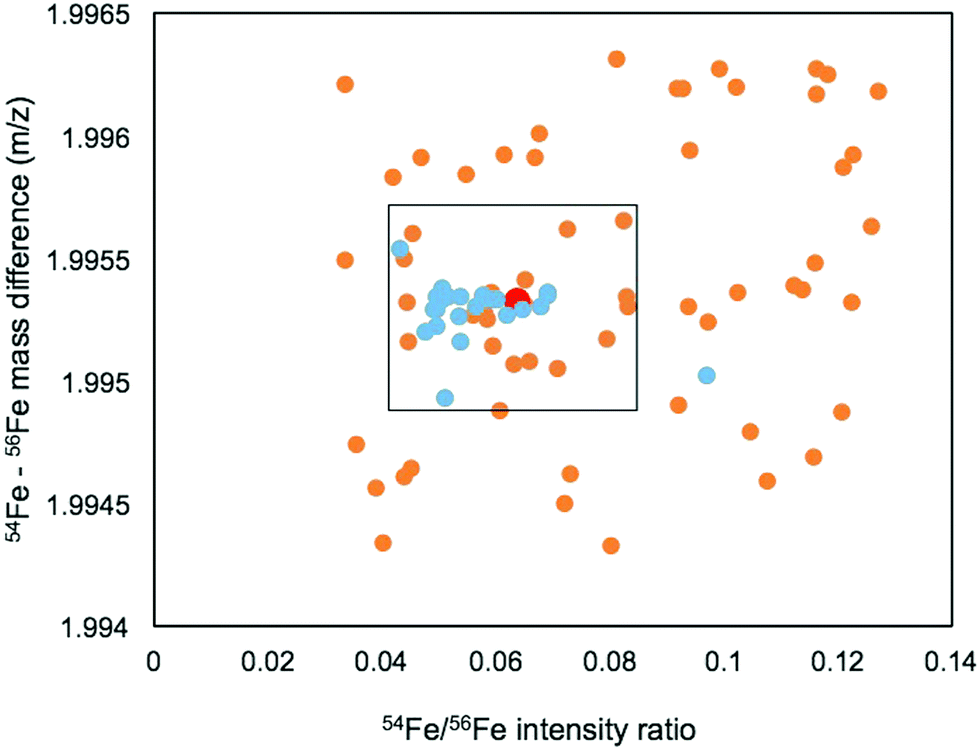 | ||
| Fig. 4 Mass difference and isotope ratio of peak pairs from direct infusion FT-ICR MS analysis that matched the iron isotopic pattern. Features validated by 57Fe addition are show in blue, while unvalidated features are plotted in orange. The true value is indicated in red, and the black box indicates the range of isotope ratio and mass difference criteria that were used to search for Fe features in subsequent LCMS analyses. | ||
LC-FT-ICR MS
Compounds were secondly identified by coupling liquid chromatography to the 21 T FT-ICR MS, taking advantage of the high acquisition speed at high resolving power (600000 at m/z 400 with a 1–2 second transient). Compounds associated with Fe were identified based on screening for features that match the Fe isotope pattern using the mass difference and ratio tolerance determined from the direct infusion data described in the previous section (Table S2, ESI†). The resulting mass list yielded a total of 46 distinct features that matched the iron isotope pattern after 13C isotopologues were removed from the feature list. This list included 12 of the 17 peaks detected by the direct infusion method. Of these monoisotopic features, 36 were singly charged and 11 were doubly charged. Tandem MS/MS fragmentation spectra were acquired for major features (Fig. 5, 6 and Table 1). A molecular network was constructed to facilitate compound identification, following the approach of Watrous et al. (2012).29 This was accomplished with custom scripts in R that calculate the cosine similarity scores between all possible pairings of MS/MS spectra (scores are the average of the dot product calculated based on peaks and intensity matrices, or based on neutral losses and intensity matrices, with a mass tolerance of 0.01 m/z). Networks were constructed with the R package igraph, with edges connecting nodes with a similarity score >0.15.
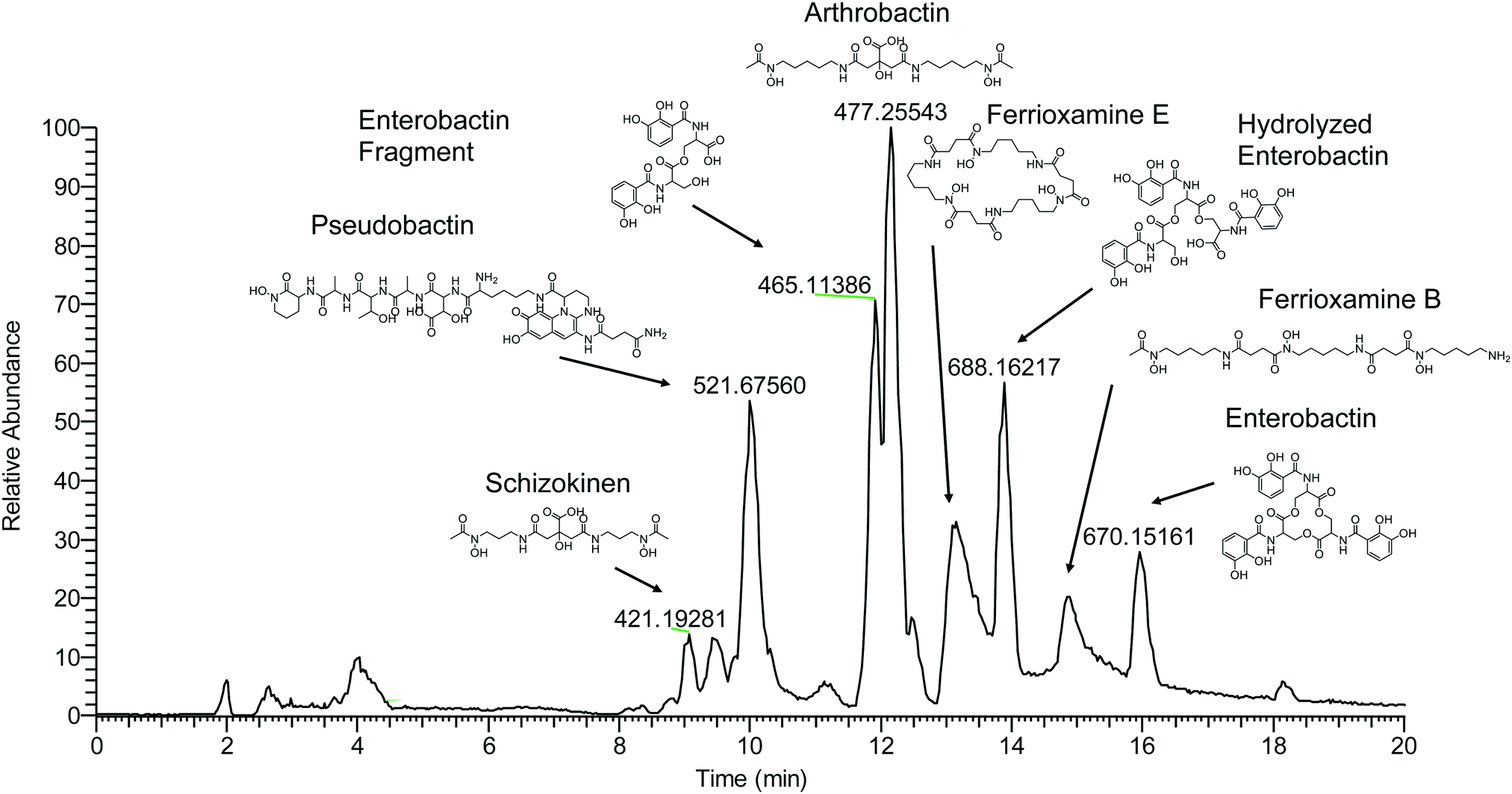 | ||
| Fig. 5 LCMS base peak chromatogram of soil enrichment with apo-masses (m/z) of major iron-binding species and identified compounds labeled. | ||
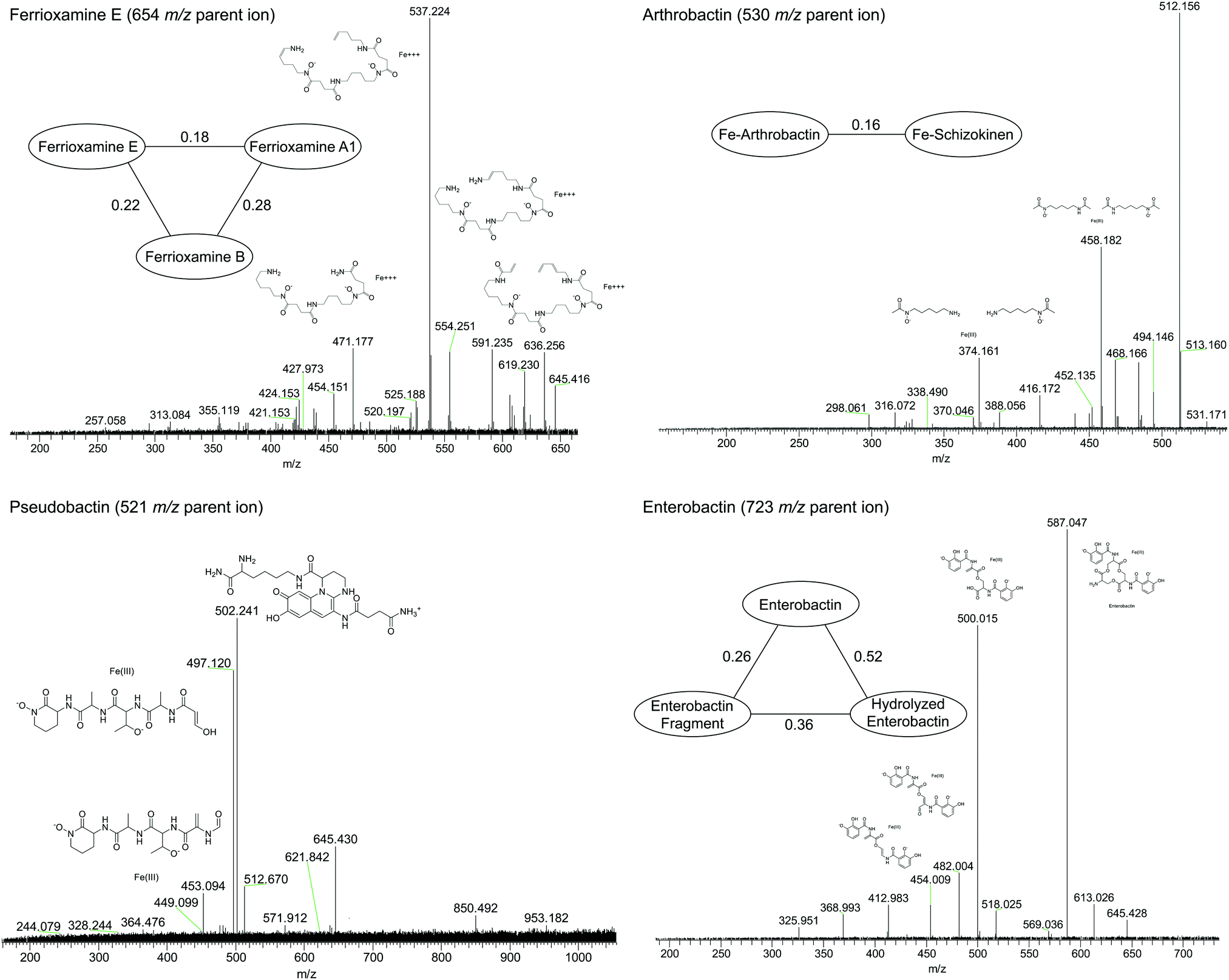 | ||
| Fig. 6 High-resolution tandem MS/MS fragmentation spectra of major iron complexes. MS/MS cosine similarity scores between related compounds are shown in edges connecting compound nodes. | ||
| Retention time (min) | Apo (m/z) | 54Fe (m/z) | 56Fe (m/z) | ID | Formula |
|---|---|---|---|---|---|
| a Validated by LCMS of authentic standard. | |||||
| Citrate di-hydroxamate | |||||
| 9.1 | 421.1935 | 472.1091 | 474.1043 | Schizokinen | C16H28N4O9 |
| 11.8 | 477.2556 | 528.1717 | 530.1670 | Arthrobactin | C20H36N4O9 |
| Catechol | |||||
| 16.1 | 670.1515 | 721.0677 | 723.0630 | Enterobactina | C30H27N3O15 |
| 13.9 | 688.1625 | 739.0782 | 741.0735 | Hydrolyzed enterobactin | C30H29N3O16 |
| Mixed catechol/hydroxamate | |||||
| 9.9 | 495.2198 | 520.6779 | 521.6755 [2+] | Pseudobactin (succinamide) | C42H60N12O16 |
| 8.7 | 495.7123 | 521.1699 | 522.1675 [2+] | Pseudobactin (succinic acid) | C42H59N11O17 |
| Tri-hydroxamate | |||||
| 13.4 | 547.3449 | 598.2612 | 600.2564 | Ferrioxamine A1 | C24H46O8N6 |
| 14.8 | 561.3606 | 612.2769 | 614.2721 | Ferrioxamine B | C25H48O8N6 |
| 13.2 | 601.3555 | 652.2717 | 654.2670 | Ferrioxamine Ea | C27H48O9N6 |
Fragmentation spectra and isotopic patterns for the metal-free apo form of major features were imported into Sirius, a machine learning fragmentation tree based platform for de novo identification of metabolites.30,31 The feature with an apo (iron-free) mass of m/z = 477.256 eluting at 11.8 min most closely matched arthrobactin, a citrate based siderophore with two hydroxamate moieties for iron chelation. Analysis of the Fe(III) complex fragmentation revealed similar neutral losses to those observed for losses within the citrate backbone of the structurally related siderophore synechobactin32 including neutral losses of H2O, CH2O2, CH2O3, C2O3, C4H2O4, and C6H4O5 (Fig. 6). Within the molecular network, another feature with m/z = 474.104 and retention time of 9.1 minutes closely matched the fragmentation pattern of arthrobactin and differed by C4H8. This molecule matched the molecular formula and fragmentation spectra expected from schizokinen, which differs from arthrobactin by shortening each acyl chain attached to the central citrate by two carbons.
A feature with apo-mass of m/z = 670.152 at 16.1 minutes was putatively identified as the tris-catechol siderophore enterobactin via Sirius. We were able to confirm this identity by analyzing an authentic standard and matching the retention time within 0.1 minutes and major features in the MS/MS fragmentation spectrum. The most abundant major fragments of enterobactin correspond to cleavages within the central ring that result in the loss of one catechol. From the network, several catechol degradation products were also identified. The most abundant was the hydrolyzed form of enterobactin (m/z = 688.162), which was detected as both the apo and iron bound form. We also detected a fragment of enterobactin (m/z = 465.113) corresponding to a second hydrolysis of the central ring resulting in the loss of one catechol. This fragment was only detected as the free form, suggesting that it has lower affinity for iron.
The 56Fe bound feature at m/z = 654.267 at 13.2 minutes was identified as the cyclic tri-hydroxamate siderophore ferrioxamine E and confirmed based on the exact mass and retention time of an authentic standard. The molecular network identified two similar features that differed by C2O and C3H2O, corresponding to the formula of the linear tri-hydroxamate siderophores ferrioxamine B and ferrioxamine A1. These molecules all possess diagnostic MS/MS fragments along the acyl backbone of the molecule.33
A series of doubly charged iron-binding compounds were also observed. The most abundant at 8.7 min had a monoisotopic iron bound m/z value of 521.675. Analysis of the fragmentation spectra of this molecule suggests that it is the succinic acid version of pseudobactin, a fluorescent catechol dihydroxamate siderophore in the pyoverdine class that is produced by certain Pseudomonas strains. Indeed, a pale bluish-green fluorescence was observed in the enrichment supernatant when exposed to ultraviolet light. The major fragments resulted from the cleavage along the peptide bond of the molecule and correspond to the florescent chain (m/z = 502) and iron binding peptide chain (m/z = 497). Another less abundant Fe-bound ion with monoisotopic mass of m/z = 522.168 exhibited a similar fragmentation pattern, likely corresponding to the succinamide form.
Numerous additional iron complexes were detected that did not have obvious similarities to the compounds that we putatively identified (Table S2, ESI†). These molecules may correspond to novel siderophores produced by the microbes that inhabit Warden soils, and their structural elucidation is the focus of ongoing work.
Bacterial 16S rRNA sequencing
Bacterial 16S rRNA PCR amplicon sequencing of the soil enrichment cultures revealed 26 operational taxanomic units. Four taxanomic orders accounted for 99.99% of all reads: Enterobacteriales (48%), Bacillales (29%), Pseudomonadales (22%) and finally Micrococcales (2%) (Fig. 7). These fast growing heterotrophic taxa were selectively enriched by the minimal media additives. Although they do not necessarily represent the dominant species in the native Warden soil, members of these orders are commonly among the most abundant and active taxa present in such environments.34,35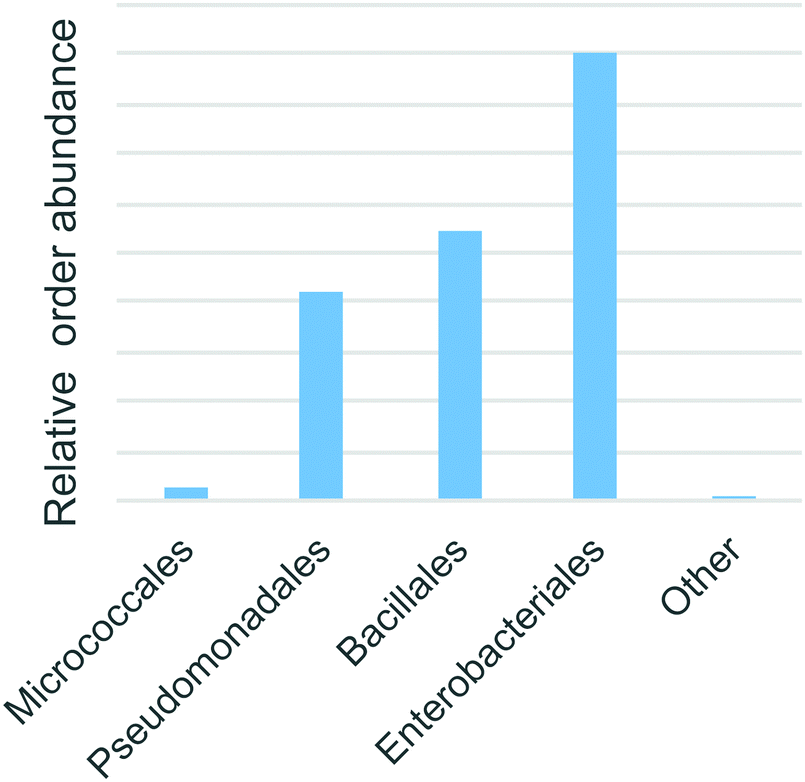 | ||
| Fig. 7 The abundance of predicted taxonomic annotations of enriched bacteria based on 16S rRNA amplicon sequences. | ||
Discussion
The two methods used in this study to characterize the metal-binding compounds each have their own unique advantages. Direct infusion FT-ICR MS is growing in popularity as a means of characterizing the diversity of organic species across environmental aquatic and soil samples due to its ability to sensitively detect molecular species in complex mixtures. Several studies have applied this approach towards the discovery of metal chelating agents from complex environmental and biological samples.36,37 The advantage of the direct infusion method is the simplicity of instrumentation (no chromatography is needed), fast analysis time (5–10 minutes per sample), and ease of data processing. Our results also demonstrate the value of isotopic validation for eliminating false positive matches to isotope patterns in complex samples.However, the benefit of chromatography with online FT-ICR MS detection is demonstrated by a significant improvement of the detection and identification of iron binding features. Over twice as many iron-bound features were detected. The primary reason for this improvement is the reduction of ion suppression due to the chromatographic separation and thus better signal from species that are less abundant or do not ionize well. This benefit is most clearly illustrated by the detection and identification of schizokinen using the LCMS approach, as the molecule was not detected by direct infusion. Furthermore, chromatography provides retention time information, which serves as an orthogonal identification criteria to MS and MS/MS, and enables confident structural identifications by comparison with authentic standards when available, as was the case for enterobactin and ferrioxamine E in this study.
Our results demonstrate that co-habitating microbes in calcareous grassland soils are capable of producing diverse suites of siderophores belonging to four major classes. The biosynthetic pathways for these different compound classes evolved independently to serve the convergent function of solubilizing and facilitating the uptake of micronutrient iron. The biosynthesis of enterobactin and pseudobactin, which are both produced by non-ribosomal peptide synthetase enzymes, can be reasonably attributed to the Enterobacteriales and Pseudomonadales within the enrichment cultures. Enterobactin is produced by many members of the Enterobacteriales class, including Escherichia, Staphylococcus, and Erwinia strains.15,38 However, it is worth noting that some isolated Bacillus species produce a structurally related compound, bacillibactin, using an analogous, and likely evolutionarily divergent, NRPS based pathway.39 Likewise, pyoverdine-like siderophores, while highly diverse in structure, are only known to be produced by Pseudomonadales including Pseudomonas and Azotobacter strains.13,17
It is less certain which taxa are responsible for producing the ferrioxamines or the citrate di-hydroxamate siderophores. Arthrobactin and schizokinen are most likely constructed by divergent nonribosomal peptide synthetase-independent biosynthesis pathways that modify a central citrate molecule with hydroxamate containing acyl groups.40 Schizokinen is known to be produced by several distinct bacterial lineages including the cyanobacteria Anabaena cylindrica,41 the Alphaproteobacteria Rhizobium leguminosarum,42 and Bacillus megaterium.43 Arthrobactin has been identified in cultures of the Arthrobacter pascens, belonging to the order Micrococcales.3 However, given the abundance of Bacillales in the soil enrichments, they are the expected producers of these citrate di-hydroxamate siderophores. Ferrioxamines are produced by a similarly wide variety of microbes including both Micrococcales16 and Enterobacteriacia.44 The diversity of the strains that produce these siderophores indicates significant horizontal gene transfer across the tree of life and makes it difficult to assign their biosynthesis to one particular taxa in our enrichments.
Our findings uncover some of the siderophores that likely play a role in the competition between microbes for scarce iron resources in soils. Previous studies have identified the co-habitation of bacterial strains in soils and aquatic systems that produce certain siderophores along with ‘cheater’ strains of the same genus that can take up the siderophores but have lost the ability to produce them.9,45 These interactions have important implications for the structure of resource/energy allocation by microbial communities. Similarly, here we demonstrate the co-habitation of bacteria that produce a variety of structurally unique siderophores within a single milligram of soil. Each of these siderophores has distinct transporters required for their uptake and utilization, meaning that they differentially impact iron availability to members of the microbial community. Microbes commonly possess transporters for siderophores that are only known to be produced by distantly related microbes. For example, some Enterobacteriales and Pseudomonadales strains possess transporters for ferrioxamines, even though no strains within these orders has been show to produce these compounds.44,46 Our result provides evidence that these organisms likely interact with distantly related siderophore producers within calcareous soil environments, and suggest that intense competition for iron in these conditions drives the diversification of siderophore biosynthesis strategies (and likely siderophore uptake). Many plant growth promoting microbes belong to bacterial orders found within our microbial enrichment. In some cases siderophore biosynthesis has been shown to improve growth of iron-starved crops.47 It is possible that some of the molecular strategies that we have identified may provide a direct or indirect benefit to crops growing in calcareous soils. Future work will focus on developing sensitive targeted analyses for quantifying these siderophore targets in soils and studying the environmental conditions that promote the dominance of certain pathways over others.
Conclusions
The advent of high resolution mass spectrometry has recently enabled untargeted approaches for detecting and identifying iron–organic species in complex samples by taking advantage of diagnostic isotope patterns imparted on the mass spectra of these molecules. This study demonstrates that co-habitating microbes in calcareous soils are capable of producing at least four major classes of siderophores: enterobactin, ferrioxamines, pseudobactins, and citrate di-hydroxamate siderophores. Our findings align well with the understanding that competition for iron under alkaline pH environmental conditions leads to the diversification of pathways for iron acquisition. Such molecular strategies of iron uptake likely contribute to the competitive success of microbes in these environments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Joseph Brown for bioinformatics assistance and Robert Starke for advice regarding microbial enrichment cultures. This research was supported by the U.S. Department of Energy Office of Biological and Environmental Research (BER) and is a contribution of the Scientific Focus Area ‘Phenotypic response of the soil microbiome to environmental perturbations’ (70880). R. Boiteau was funded by the Linus Pauling Postdoctoral Fellowship LDRD 204495 from the Pacific Northwest National Lab. This work was conducted at the Environmental Molecular Sciences Laboratory, a U.S. Department of Energy user facility, under project 49644. PNNL is operated for the DOE by Battelle Memorial Institute under Contract DE-AC05-76RLO1830.References
- M. L. Guerinot and Y. Yi, Plant Physiol., 1994, 104, 815–820 CrossRef CAS.
- C. Colombo, G. Palumbo, J.-Z. He, R. Pinton and S. Cesco, J. Soils Sediments, 2014, 14, 538–548 CrossRef CAS.
- R. C. Hider and X. Kong, Nat. Prod. Rep., 2010, 27, 637–657 RSC.
- M. Sandy and A. Butler, Chem. Rev., 2009, 109, 4580–4595 CrossRef CAS.
- R. M. Boiteau, D. R. Mende, N. J. Hawco, M. R. McIlvin, J. N. Fitzsimmons, M. A. Saito, P. N. Sedwick, E. F. DeLong and D. J. Repeta, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14237–14242 CrossRef CAS.
- C. Völker and A. Tagliabue, Mar. Chem., 2015, 173, 67–77 CrossRef.
- J. M. Harrington, O. W. Duckworth and K. Haselwandter, Biometals, 2015, 28, 461–472 CrossRef CAS.
- W. Lee, M. van Baalen and V. A. A. Jansen, Ecol. Lett., 2012, 15, 119–125 CrossRef.
- O. X. Cordero, L. A. Ventouras, E. F. DeLong and M. F. Polz, Proc. Natl. Acad. Sci. U. S. A., 2012, 1–6 CAS.
- M. Di Lorenzo, M. Stork, M. E. Tolmasky, L. A. Actis, D. Farrell, T. J. Welch, L. M. Crosa, A. M. Wertheimer, Q. Chen, P. Salinas, L. Waldbeser and J. H. Crosa, J. Bacteriol., 2003, 185, 5822–5830 CrossRef CAS.
- G. Winkelmann and H. Drechsel, Microbial Siderophores, 2008, vol. 7–12 Search PubMed.
- M. A. Fischbach, C. T. Walsh and J. Clardy, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4601–4608 CrossRef CAS.
- S. L. Hartney, S. Mazurier, M. K. Girard, S. Mehnaz, E. W. Davis, H. Gross, P. Lemanceau and J. E. Loper, J. Bacteriol., 2013, 195, 765–776 CrossRef CAS.
- M. P. Kem and A. Butler, Biometals, 2015, 28, 445–459 CrossRef CAS.
- K. N. Raymond, E. a Dertz and S. S. Kim, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3584–3588 CrossRef CAS.
- F. Barona-Gómez, U. Wong, A. E. Giannakopulos, P. J. Derrick and G. L. Challis, J. Am. Chem. Soc., 2004, 126, 16282–16283 CrossRef.
- O. Baars, X. Zhang, F. M. M. Morel and M. R. Seyedsayamdost, Appl. Environ. Microbiol., 2015, 81, 27–39 Search PubMed.
- P. E. Powell, G. R. Cline, C. P. P. Reid and P. J. Szaniszlo, Nature, 1980, 287, 833–834 CrossRef CAS.
- J. E. Loper and J. S. Buyer, Mol. Plant-Microbe Interact., 1991, 4, 5–13 CrossRef CAS.
- H. A. Akers, Appl. Environ. Microbiol., 1983, 45, 1704–1706 CAS.
- E. Ahmed and S. J. M. Holmström, Geochim. Cosmochim. Acta, 2014, 131, 184–195 CrossRef CAS.
- S. J. M. Holmström, U. S. Lundström, P. A. W. van Hees and R. D. Finlay, Biogeochemistry, 2004, 71, 247–258 CrossRef.
- Y. Schindlegger, E. Oburger, B. Gruber, W. D. C. Schenkeveld, S. M. Kraemer, M. Puschenreiter, G. Koellensperger and S. Hann, Electrophoresis, 2014, 35, 1375–1385 CrossRef CAS.
- Y. Schindlegger, E. Oburger, M. Puschenreiter, G. Stingeder, G. Koellensperger and S. Hann, J. Anal. At. Spectrom., 2015, 30, 1345–1355 RSC.
- R. M. Boiteau, J. B. Shaw, L. Pasa-Tolic, D. W. Koppenaal and J. K. Jansson, Soil Biol. Biochem., 2018, 120, 283–291 CrossRef CAS.
- J. G. Caporaso, C. L. Lauber, W. A. Walters, D. Berg-Lyons, J. Huntley, N. Fierer, S. M. Owens, J. Betley, L. Fraser, M. Bauer, N. Gormley, J. A. Gilbert, G. Smith and R. Knight, ISME J., 2012, 6, 1621–1624 CrossRef CAS.
- J. Brown, N. Zavoshy and C. Brislawn, pnnl/hundov1.2.1 (Version v1.2.1). Zenodo, 2018, DOI:10.5281/zenodo.1429364.
- J. B. Shaw, T. Y. Lin, F. E. Leach, A. V. Tolmachev, N. Tolić, E. W. Robinson, D. W. Koppenaal and L. Paša-Tolić, J. Am. Soc. Mass Spectrom., 2016, 27, 1929–1936 CrossRef CAS.
- J. Watrous, P. Roach, T. Alexandrov, B. S. Heath, J. Y. Yang, R. D. Kersten, M. van der Voort, K. Pogliano, H. Gross, J. M. Raaijmakers, B. S. Moore, J. Laskin, N. Bandeira and P. C. Dorrestein, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E1743–E1752 CrossRef CAS PubMed.
- S. Böcker, M. C. Letzel, Z. Lipták and A. Pervukhin, Bioinformatics, 2009, 25, 218–224 CrossRef.
- K. Dührkop, H. Shen, M. Meusel, J. Rousu and S. Böcker, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12580–12585 CrossRef.
- R. M. R. M. Boiteau and D. J. D. J. Repeta, Metallomics, 2015, 7, 877–884 RSC.
- E. Mawji, M. Gledhill, P. J. Worsfold and E. P. Achterberg, Rapid Commun. Mass Spectrom., 2008, 2195–2202 CrossRef.
- L. R. Thompson, J. G. Sanders, D. McDonald, A. Amir, J. Ladau, K. J. Locey, R. J. Prill, A. Tripathi, S. M. Gibbons, G. Ackermann, J. A. Navas-Molina, S. Janssen, E. Kopylova, Y. Vázquez-Baeza, A. González, J. T. Morton, S. Mirarab, Z. Z. Xu, L. Jiang, M. F. Haroon, J. Kanbar, Q. Zhu, S. J. Song, T. Kosciolek, N. A. Bokulich, J. Lefler, C. J. Brislawn, G. Humphrey, S. M. Owens, J. Hampton-Marcell, D. Berg-Lyons, V. McKenzie, N. Fierer, J. A. Fuhrman, A. Clauset, R. L. Stevens, A. Shade, K. S. Pollard, K. D. Goodwin, J. K. Jansson, J. A. Gilbert and R. Knight, et al. , Nature, 2017, 551, 457–463 CrossRef CAS.
- R. A. White, E. M. Bottos, T. Roy Chowdhury, J. D. Zucker, C. J. Brislawn, C. D. Nicora, S. J. Fansler, K. R. Glaesemann, K. Glass and J. K. Jansson, mSystems, 2016, 1, e00045 CrossRef.
- H. Waska, A. Koschinsky, M. J. Ruiz Chancho and T. Dittmar, Mar. Chem., 2015, 173, 78–92 CrossRef CAS.
- L. R. Walker, M. M. Tfaily, J. B. Shaw, N. J. Hess, L. Paša-Tolić and D. W. Koppenaal, Metallomics, 2017, 9, 82–92 RSC.
- C. T. Bull, C. A. Ishimaru and J. E. Loper, Appl. Environ. Microbiol., 1994, 60, 662–669 CAS.
- E. A. Dertz, J. Xu, A. Stintzi and K. N. Raymond, J. Am. Chem. Soc., 2006, 128, 22–23 CrossRef CAS.
- D. Oves-Costales, N. Kadi and G. L. Challis, Chem. Commun., 2009, 6530–6541 RSC.
- M. Deicke, J. F. Mohr, J.-P. Bellenger and T. Wichard, Analyst, 2014, 139, 6096–6099 RSC.
- E. P. Storey, R. Boghozian, J. L. Little, D. W. Lowman and R. Chakraborty, Biometals, 2006, 19, 637–649 CrossRef CAS PubMed.
- K. B. Mullis, J. R. Pollack and J. B. Neilands, Biochemistry, 1971, 10, 4894–4898 CrossRef CAS.
- I. Berner, S. Konetschny-Rapp, G. Jung and G. Winkelmann, Biometals, 1988, 111, 51–56 Search PubMed.
- E. Butaite, M. Baumgartner, S. Wyder and R. Kümmerli, Nat. Commun., 2017, 8, 1–12 CrossRef CAS.
- M. A. Llamas, M. Sparrius, R. Kloet, C. R. Jiménez, C. Vandenbroucke-Grauls and W. Bitter, J. Bacteriol., 2006, 188, 1882–1891 CrossRef CAS.
- W. Radzki, F. J. Gutierrez Mañero, E. Algar, J. A. Lucas García, A. García-Villaraco and B. Ramos Solano, Antonie van Leeuwenhoek, 2013, 104, 321–330 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8mt00252e |
| This journal is © The Royal Society of Chemistry 2019 |