
Inhibition of arsenite methylation induces synergistic genotoxicity of arsenite and benzo(a)pyrene diol epoxide in SCC-7 cells†
Youxian
Li
,
Man
He
 ,
Beibei
Chen
* and
Bin
Hu
*
,
Beibei
Chen
* and
Bin
Hu
*
Key Laboratory of Analytical Chemistry for Biology and Medicine (Ministry of Education), Department of Chemistry, Wuhan University, Wuhan 430072, P. R. China. E-mail: binhu@whu.edu.cn; bbchen@whu.edu.cn; Fax: +86-27-68754067; Tel: +86-27-68752162
First published on 30th October 2018
Abstract
As is well-known, arsenite (As(III)) is a human carcinogen associated with many human cancers. As(III) can act as a co-carcinogen to induce DNA damage with other carcinogens. Benzo(a)pyrene diol epoxide (BPDE) is one of the most-studied environmental carcinogens, which exists ubiquitously in our daily life. The elucidation of the mechanism of As(III) as a co-carcinogen with BDPE in cells causing genotoxicity is beneficial for the evaluation of its bioeffect. In this study, a comprehensive analytical system is used for DNA damage evaluation, BPDE–DNA adduct detection, arsenic speciation and gene expression analysis. Based on the experimental results, it can be inferred that BPDE and As(III) synergistically cause genotoxicity, and the possible mechanism is that BPDE inhibits arsenic methylation, leading to cellular As(III) enrichment. As(III) inhibits nucleotide excision repair (NER) of the DNA adduct damage caused by BPDE. The synergistic effect of BPDE and As(III) causes DNA strand break damage, which further results in carcinogenesis.
Introduction
Arsenic is recognized as a human carcinogen that is widely spread in nature.1,2 There are many mechanisms of arsenic exposure leading to cancer. Excessive arsenic uptake can produce cell and gene toxicity via oxidative stress and inhibit DNA repair and signaling pathways;3–5 among them, arsenic as a co-carcinogen to inhibit DNA repair is considered as an important possible way of arsenic carcinogenesis.6 It is reported that arsenic does not directly react with DNA resulting in DNA damage. Arsenic is generally considered as a co-carcinogen affecting the teratogenic and carcinogenic properties of other reagents. For example, treating cells with arsenite (As(III)) and ultraviolet rays can reduce the removal of ultraviolet products in cells and increase the mutagenic rate of ultraviolet light.7Benzopyrene (BaP) is a kind of polyaromatic hydrocarbon (PAH) having the property of high carcinogenicity, and it is commonly found in food, cigarettes and burning environments. After uptake by cells, the cytochrome P450s in cells can catalyze BaP to benzo(a)pyrene diol epoxide (BPDE). After that, BPDE oxidizes DNA to form a BPDE–DNA adduct, especially the BPDE–G adduct (BPDE-N2-dG). Furthermore, BPDE-N2-dG serves as a “road block” to stop DNA replication, resulting in DNA damage. Fortunately, BPDE-N2-dG can be repaired by the spontaneous nucleotide excision repair (NER) process.8 However, cohort studies have shown that the synergistic reaction of BPDE and As(III) induces carcinogenesis.9–13 The content of BPDE–DNA adducts increases after As(III) and BPDE co-incubation in cells since the NER process is inhibited. However, the mechanism of the synergistic reaction of BPDE and arsenic is not fully understood. Therefore, it is of great significance to study the synergistic genotoxicity of BPDE and As(III).
High-performance liquid chromatography (HPLC), fluorescence and related techniques are commonly used in analyzing BPDE–DNA adducts.14–17 Quantitative analysis of BPDE–DNA adducts by using mass spectrometry (MS) itself is rare. The content of BPDE–DNA adducts in biomaterials is very low,18 and LOD of traditional MS detection is not satisfactory. The use of ultrasensitive high-resolution mass spectrometry (HR-MS) has effectively solved the problem.19 For DNA damage detection, the comet assay developed by Singh20 is always used. This method can quickly and intuitively detect DNA strand rupture damage, and it is recognized as a fast and sensitive method for DNA damage detection. Damaged DNA strands are released and migrate to an anode during electrophoresis, resulting in a comet-shaped image, and the DNA damage is quantified by the comet tail. The sensitivity of the comet assay is higher than that of a chromosome aberration assay. In addition, studies on the mechanism of As(III) as a co-carcinogen inhibiting the NER process to produce carcinogenicity have also focused on the gene expression analysis of NER-related genes, revealing corresponding enzyme physiological activity. However, the molecular mechanism of As(III) as a co-carcinogen inducing genotoxicity is still not clear due to the complexity of organisms.
A metallomic study to illustrate the transformation species of As(III) in cells is useful to reveal the metabolization pathway of As(III), which helps understand the biofunction of As(III). Inductively coupled plasma-mass spectrometry (ICP-MS)-based hyphenated techniques are the most widely used techniques for arsenic speciation.21–24 In our previous study,25 As(III) methylation metabolites in cells were detected via an ion pair reversed phase (IP-RP)-HPLC-ICP-MS equipped with the collision cell technique (CCT). It was found that at high concentrations of As(III) exposure, As(III) in cells cannot be methylated to a great extent. The methylation of As(III) is inhibited, which leads to the presence and enrichment of more toxic inorganic arsenic and its intermediate arsenic forms in the cells, possibly causing impairment of physiological activities and further leading to cell and gene toxicity of cells.26
In this study, a comprehensive analytical system was developed to study As(III) and BPDE synergistic effects on DNA damage in SCC-7 cells and the relation between As(III) methylation metabolism and genetic toxicity resulting from As(III). First, a comet assay was used to estimate the DNA strand break of the synergistic effect of BPDE and As(III). Then, HPLC-electrospray ionization-linear ion trap quadrupole (ESI-LTQ)-Orbitrap-MS analysis was performed for the determination of BPDE–DNA adducts. After that, intracellular As(III) methylation metabolites were studied by the IP-RP-HPLC-ICP-CCT-MS technology. A reverse transcription quantitative polymerase chain reaction (RT-qPCR) was used for gene expression analysis. At last, the relation between As(III) methylation and its genotoxicity as a co-carcinogen with BPDE was revealed.
Experimental
Instrumentation
Ultimate 3000 HPLC (Thermo Scientific™ Dionex) equipped with Acclaim Hypersil GOLD C18 (2.1 × 150 mm, 3 μM) (Thermo Scientific, USA) and Hybrid LTQ Orbitrap Elite Velos Pro high-resolution MS (Thermo Scientific, USA) were coupled for the detection of the BPDE–DNA adduct. Arsenic speciation analysis was carried out using an Ultimate 3000 series HPLC system (Thermo Scientific™ Dionex) equipped with a CAPCELL PAK C18 column (5 μm, 4.6 × 250 mm) (Shiseido, Japan) coupled to X series II ICP-MS equipped with CCT (Thermo Scientific, USA). A KQ 2200 ultrasonicator (Kunshan, China) was used to disrupt the cells, and the cell lysates were centrifuged by a legend micro 21R centrifuge (Thermo Scientific, Germany). A Mettler Toledo FE20 pH meter (Shanghai, China) was used to adjust the pH value. The mass balance (XS105) used in the experiment was purchased from Mettler Toledo (Shanghai, China).Reagents and standards
Sodium arsenite (As(III)) and sodium arsenate (As(V)) were purchased from Wako Pure Chemical Industries, Ltd (Japan). Monomethylarsenic disodium salt (MMA(V)) and dimethylarsenic acid (DMA(V)) were purchased from J&K Chemical Ltd (China) and Genebase Bioscience Co., Ltd (China), respectively. Stock solutions of all arsenic compounds (1000 mg L−1) were prepared from the corresponding standard compounds. All stock solutions were stored in the dark at 4 °C and diluted daily prior to use.BPDE was supplied by Toronto Research Chemicals (Ontario, Canada). To avoid hydrolysis of the epoxide, a stock solution of BPDE was always freshly prepared by dissolving BPDE in anhydrous dimethyl sulfoxide (DMSO) (>99.9% purity, Sigma-Aldrich) immediately before use. The final concentration of the solvent in the medium was kept at 0.05%.
Tetrabutylammonium hydroxide (TBAH) (Sigma-Aldrich), malonic acid (Sigma-Aldrich), ammonium hydroxide (Sinopharm, China) and methanol (Sinopharm, China) were used to prepare the mobile phase for the separation of arsenic species. The mobile phase was filtered through a 0.45 μm membrane before use. Methanol (MS grade) and triple deionized water were used to prepare the mobile phase for HPLC-ESI-LTQ-Orbitrap-MS analysis.
All reagents were at least of analytical reagent grade. High purity deionized water obtained from a Milli-Q system (18.2 MΩ cm, Millipore, France) was used throughout the experiments.
Cell culture
SCC-7 cells (squamous carcinoma cells) were provided by Prof. Xianzheng Zhang (Wuhan University, Wuhan, China). The cells were cultured in Dulbecco's modified Eagle's medium without sodium pyruvate (Hyclone, USA) supplemented with 10% fetal bovine serum (FBS, PAN) and 1% penicillin/streptomycin at 37 °C in a humidified atmosphere with 5% CO2. The cells were passaged when they reached 80% confluency.Comet assay
SCC-7 cells were seeded at a density of 1 × 105 cells per well in 24-well culture plates for 24 h. Then, the medium was replaced with a fresh FBS-free medium containing BPDE and/or various concentrations of As(III). After exposure for different durations, the cells were washed three times with cold phosphate buffer saline (PBS) and resuspended in PBS.The cells were immobilized in a 1.5 mL microcentrifuge tube containing 120 μL of 0.5% low melting point agarose and then applied to a cometslide, which was pretreated with 1% normal melting point agarose. Then, the cells were lysed with a lysis solution (2.5 mM NaCl, 100 mM EDTA and 10 mM Tris, adjust pH to 10.0) with 10% DMSO at 4 °C overnight and electrophoresed in NaOH buffer (300 mM NaOH, 1 mM EDTA, pH > 13) with 25 V and 300 mM for 20 min. Then, the slides were air-dried, stained with propidium iodide and imaged with an inverted fluorescence microscope (TE2000-U, Nikon, Japan). About 100 cells from each slide were scored using CometScore.
HPLC-ESI-LTQ-Orbitrap-MS analysis
SCC-7 cells were exposed to BPDE for various durations (0, 0.5, 1, 2 h) and then incubated with a fresh medium or medium containing As(III). After exposure for different durations, the cells were washed three times with cold PBS and centrifuged at 200 × g for 5 min. A Universal Genomic DNA Kit (CoWin Biosciences, China) was used for the extraction of genomic DNA. Two μL of the resulting DNA was used to generate single nucleosides by DNA Degradase Plus (Zymo Research, America) and then centrifuged three times in an Amicon filter at 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 × g to remove the enzyme. Also, 20 μL of the DNA digestion sample was injected into a HPLC-ESI-LTQ-Orbitrap-MS system for analysis.27,28 The conditions of HPLC-ESI-LTQ-Orbitrap-MS are listed in Table S1 (ESI†).
100 × g to remove the enzyme. Also, 20 μL of the DNA digestion sample was injected into a HPLC-ESI-LTQ-Orbitrap-MS system for analysis.27,28 The conditions of HPLC-ESI-LTQ-Orbitrap-MS are listed in Table S1 (ESI†).
HPLC-ICP-CCT-MS analysis
SCC-7 cells were exposed to As(III) or co-exposed to BPDE and As(III). After exposure for different durations, the cells were washed three times with cold PBS to remove the extracellular arsenic and transferred into Eppendorf tubes after being detached from the culture dishes by a cell scraper. The collected cells were diluted to 300 μL with PBS and lysed by ultrasonication on ice for 30 s 3 times. Then, the cell lysate was transferred into a 3 kDa NMWL Ultra-0.5 centrifugal filter device (Amicon, Millipore, France). After centrifugation at 21100 × g for 20 min to separate the inorganic and methylated arsenic species from the arsenic-bound proteins and other solid cellular structures, the centrifugation filtrate was collected for HPLC-ICP-CCT-MS analysis according to our previous study.25 The conditions for HPLC-ICP-CCT-MS are listed in Table S2 (ESI†).
RT-qPCR-based gene expression analysis
SCC-7 cells were exposed to 1 μM BPDE for 0.5 h or/and 2 μM As(III) for 24 h. RNA isolation was performed following the instruction manual (CoWin Biosciences, China). After the RNA concentrations were quantified and checked for purity (A260/A280 = 1.8–2.0) by NanoDrop™ 2000c (Thermo Scientific, USA), 1 μL RNA was reverse-transcribed to cDNA. For RT-qPCR, 1 ng cDNA was used, and 2× SYBR Green Master Mix (A) was bought from US Everbright Inc. (Suzhou, China).Seven target genes including AS3MT, MT1A, XPC, CSB, XPB, XPF and RAD50 as well as an internal reference gene GAPDH were detected by RT-qPCR. The thermal cycling program is 95 °C 30 s, (95 °C 30 s, 60 °C 30 s, 72 °C 30 s) 39×, 65 °C 5 s, 95 °C hold. The results were calculated by the 2−ΔΔCT (Livak) method and were expressed by the relative expression of the genes (fold increase), setting GAPDH as the reference gene. The primers used in RT-qPCR of each gene are shown in Table S3 (ESI†).
Results and discussion
DNA damage detected by comet assay
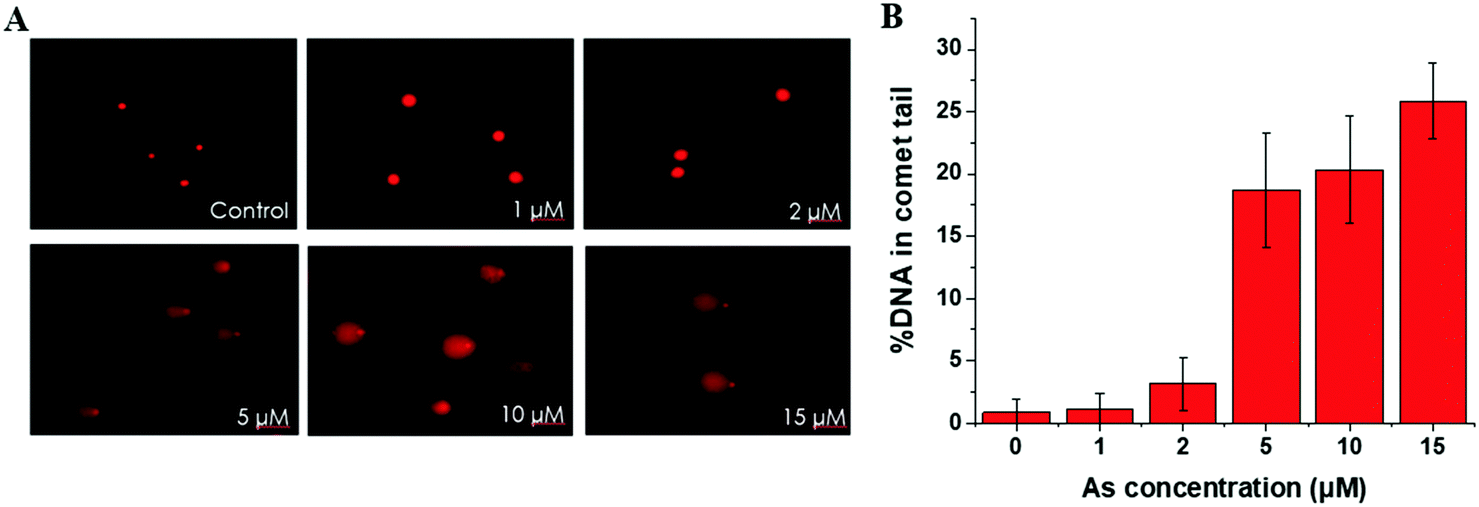 | ||
| Fig. 1 Arsenic-mediated DNA damage by comet assay. (A) Representative images of comet assay for analysis of SCC-7 cells treated with 0, 1, 2, 5, 10, 15 μM As(III) for 24 h (20× objective). (B) Quantitative analysis of comet assay images in SCC-7 cells with the same incubation conditions as described in (A). Error bars represent the standard deviation of triplicate experiments. | ||
This trend is coincident with our previously reported observation about the relation between As(III) exposure concentration and As(III) methylation.25 In the former results, when the As(III) exposure concentration was ≤2 μM, As(III) methylation was ongoing; when the concentration was ≥5 μM, As(III) methylation was inhibited. Therefore, we speculated that DNA damage may be associated with the degree of As(III) methylation. To confirm this hypothesis, we conducted an As(III) metabolism-related gene expression experiment. The results, shown in Table S4 (ESI†), indicated that a low concentration (2 μM) of As(III) caused an increase in the expression of AS3MT but had negligible influence on MT1A; a high concentration (10 μM) of As(III) largely inhibited the expressions of both AS3MT and MT1A. The inhibition of As(III) metabolism at a relatively high concentration of As(III) (≥5 μM) could be one of the reasons for cellular DNA damage.
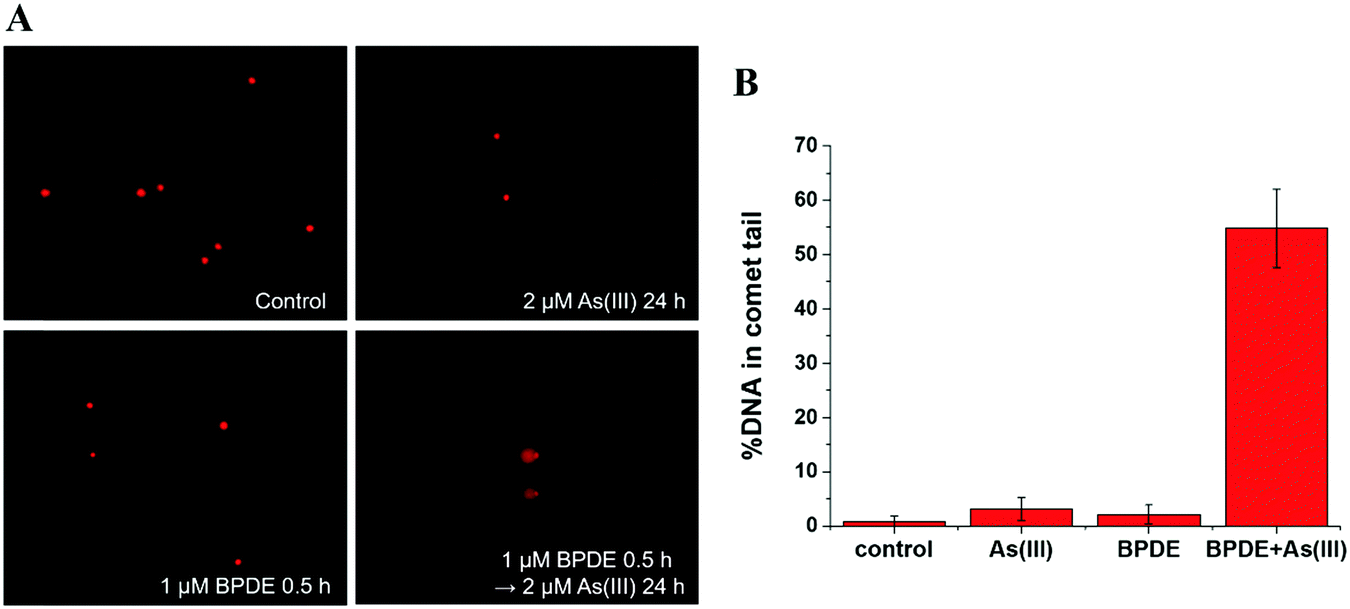 | ||
| Fig. 2 BPDE and arsenic-mediated DNA damage by comet assay. (A) Representative images of comet assay for analysis of SCC-7 cells treated with the following conditions: control: untreated cells; As(III): cells treated with As(III) (2 μM) alone for 24 h; BPDE: cells treated with BPDE (1 μM) alone for 0.5 h; BPDE + As(III): cells pretreated with BPDE (1 μM) for 0.5 h, followed by treatment with 2 μM As(III) for 24 h (10× objective). (B) Quantitative analysis of comet assay images in SCC-7 cells with the same incubation conditions as described in (A). Error bars represent the standard deviation of triplicate experiments. | ||
HPLC-ESI-LTQ-Orbitrap-MS analysis
HPLC-ESI-LTQ-Orbitrap-MS was used for the detection of A, T, C, G and DNA-BPDE adducts (BPDE-N2-dG). The relative concentration of BPDE-N2-dG is associated with DNA damage caused by BPDE. Typical extracted ion chromatograms of A, T, C, G and BPDE-N2-dG as well as the mass spectrum of BPDE-N2-dG are shown in Fig. S2 (ESI†).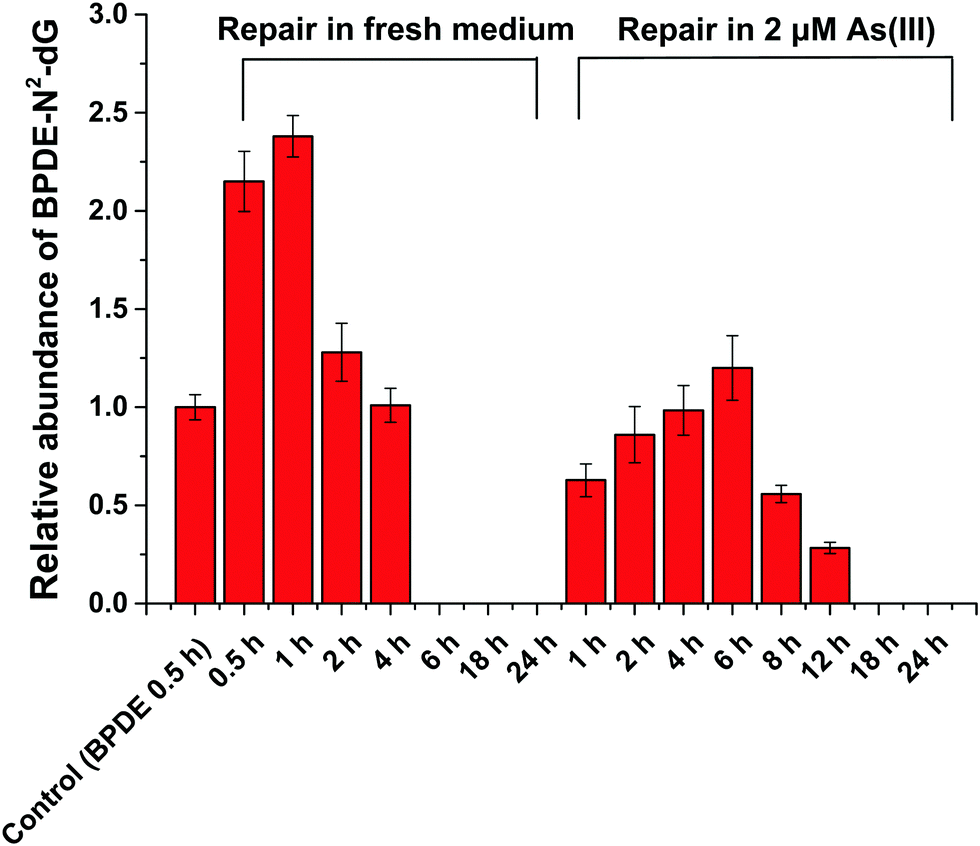 | ||
| Fig. 3 Effect of As(III) on the NER (BPDE-N2-dG repair) in SCC-7 cells. Semi-quantitative analysis of BPDE-N2-dG by HPLC-ESI-LTQ-Orbitrap-MS (set 1 μM BPDE incubated for 0.5 h as control) in SCC-7 cells treated with the following conditions: 1 μM BPDE incubated for 0.5 h and then in fresh medium for 0.5, 1, 2, 4, 6, 18 and 24 h, respectively; 1 μM BPDE incubated for 0.5 h and then in medium containing 2 μM As(III) for 1, 2, 4, 6, 12, 18 and 24 h, respectively. Error bars represent the standard deviation of triplicate experiments. | ||
HPLC-ICP-CCT-MS analysis
To reveal the methylation of As(III) in SCC-7 cells, the previously developed HPLC-ICP-MS method was used to compare the methylated metabolites in SCC-7 cells exposed to As(III) alone and co-exposed to BPDE and As(III); the results are displayed in Fig. 4A and B, respectively. From Fig. 4B, it can be found that As(III) was quickly metabolized to DMA(V) in cells undergoing an arsenic methylation pathway. The concentration of the final metabolite DMA(V) increased with the exposure time, whereas the concentrations of As(III), As(V) and MMA(V) in cells were very low. This phenomenon is in accordance with our previous result.25 When the cells were pre-incubated with 1 μM BPDE for 0.5 h, the intracellular arsenic species were quite different. As shown in Fig. 4A, As(III) was the dominant species and its concentration in cells increased with the exposure time; the methylation metabolite of DMA(V) increased very slightly, and other methylated metabolites were not detected. This indicates that the methylation of As(III) in BPDE-treated SCC-7 cells was greatly suppressed and accumulation of As(III) occurred in the cells. Therefore, it can be concluded that BPDE pre-treatment inhibited As(III) methylation and caused As(III) accumulation in cells.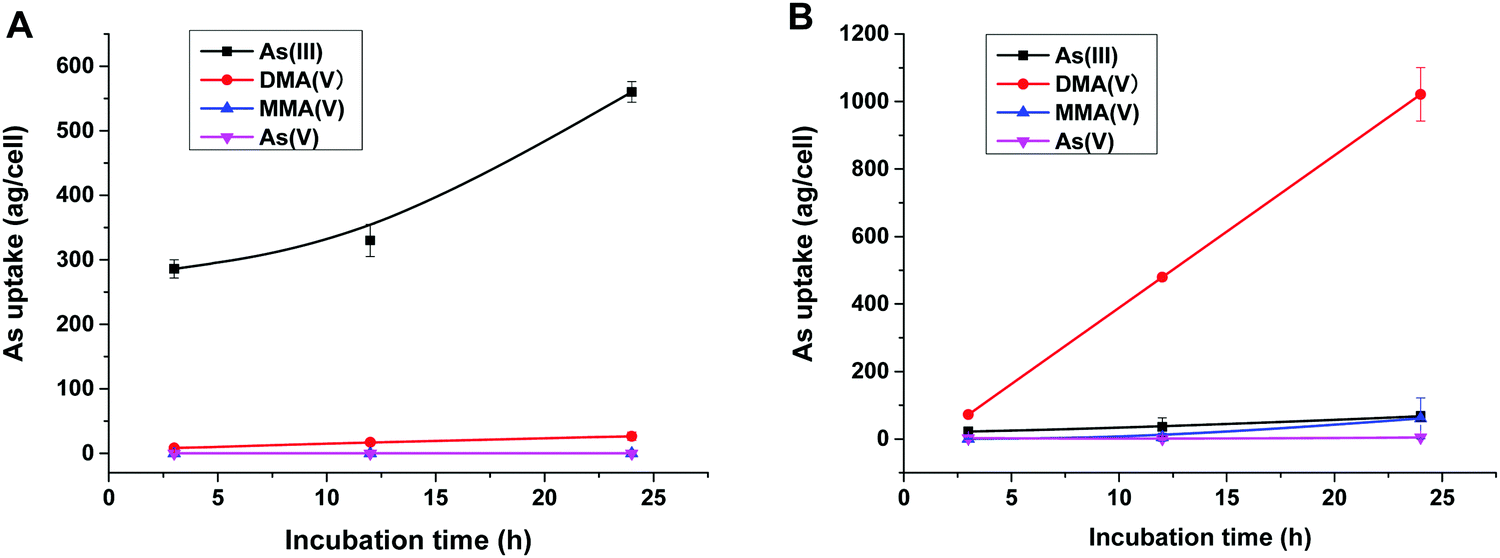 | ||
| Fig. 4 Concentrations of intracellular arsenic species in SCC-7 cells after exposure to (A) 1 μM BPDE for 0.5 h and then 2 μM As(III) for various times (3, 12, 24 h); (B) 2 μM As(III) for various times (3, 12, 24 h). Error bars represent the standard deviation of triplicate experiments. | ||
Gene expression
To further reveal the relationship between methylation of As(III) and genetic toxicity resulting from the co-exposure of As(III) and BPDE, real-time PCR was used to detect the expressions of seven related genes in SCC-7 cells. AS3MT (NCBI Gene ID: 57412) and MT1A (Gene ID: 4489) are closely related to As(III) methylation.31,32 The role of AS3MT is to catalyze the methyl transfer from SAM to As(III); it plays an important role in the methylation of arsenic. MT1A is a member of the Metallothionein family, which has a detoxification effect on heavy metals because it is rich in cysteine. XPC (Gene ID: 7508), CSB (Gene ID: 2074), XPB (Gene ID: 2071) and XPF (Gene ID: 2072) play vital roles in the NER process.33 The NER process includes two sub-pathways known as transcript-coupled repair (TRC-NER) and global genome repair (GGR-NER), of which the main difference is the first step for damage recognition. XPC and CSB were involved in the early recognition of genomic DNA damage of GGR-NER and TRC-NER, respectively. XPB and XPF are related to the steps of DNA unwinding and incisions, respectively, of the NER process. In addition, RAD50 (Gene ID: 10111), which is responsible for double DNA strand break repair, was also selected.As shown in Table 1, the expressions of AS3MT and MT1A are inhibited after BPDE and As(III) co-exposure compared to that observed for single exposure to As(III). This indicates that the metabolization of As(III) is suppressed by the pretreatment of BPDE, which is coincident with the results obtained by HPLC-ICP-MS. For the DNA double strand break repair related gene – RAD50, its expression is inhibited when cells are co-exposed to BPDE and As(III). These results agree well with the results of the comet assay, confirming that the synergistic reaction of As(III) and BPDE can lead to DNA double strand break. For the expression of NER-related genes, up-regulation of XPC, XPB and XPF was observed after incubating SCC-7 cells with only BPDE, demonstrating that GGR-NER was triggered. When the cells were exposed to As(III) alone, no clear effect on the expressions of XPC and CSB was observed, indicating that As(III) alone cannot directly cause DNA adduct damage and affect the NER process. However, after co-treatment with BPDE and As(III), the expressions of all four genes XPC, XPB, CSB and XPF were inhibited. It can be concluded that As(III) and BPDE synergistically inhibit the NER process. The results for gene expression analysis also reveal that the metabolization of As(III) is highly related to its carcinogenicity. BPDE suppressed the metabolization of As(III), causing an enrichment of As(III) in cells; the persistently existing As(III) inhibited NER of BPDE–DNA adducts, causing permanent DNA damage.
| Incubation conditiona | Gene expression fold change (set GAPDH as reference) | ||||||
|---|---|---|---|---|---|---|---|
| AS3MT | MT1A | XPC | XPB | CSB | XPF | RAD50 | |
| a As(III): 2 μM As(III) for 24 h; BPDE: 1 μM BPDE for 0.5 h; BPDE + As(III): 1 μM BPDE for 0.5 h, then 2 μM As(III) for 24 h. | |||||||
| As(III) | 1.59 ± 0.17 | 1.01 ± 0.23 | 1.24 ± 0.32 | 2.27 ± 0.51 | 1.19 ± 0.30 | 2.85 ± 0.66 | 1.88 ± 0.47 |
| BPDE | 1.29 ± 0.49 | 0.25 ± 0.05 | 5.12 ± 0.74 | 2.78 ± 0.63 | 1.60 ± 0.63 | 8.87 ± 0.62 | 2.66 ± 0.72 |
| BPDE + As(III) | 0.42 ± 0.21 | 0.23 ± 0.12 | 0.52 ± 0.19 | 0.65 ± 0.25 | 0.17 ± 0.03 | 0.74 ± 0.20 | 0.56 ± 0.21 |
Conclusion
In this study, a comprehensive analysis by integrating a comet assay, HPLC-ESI-LTQ-Orbitrap-MS, HPLC-ICP-MS and RT-qPCR was used to investigate the synergistic effect of BPDE and As(III) causing DNA damage in SCC-7 cells. The results showed that As(III) and BPDE synergistically potentiated the DNA strand break damage. The probable reason is that BPDE pre-exposure inhibited As(III) methylation, leading to accumulation of As(III), which suppressed the NER process of the BPDE-N2-dG adduct. The results of arsenic metabolization and NER process-related gene expression also corroborated this assumption. BPDE inhibits arsenic methylation by the inhibition of AS3MT and MT1A gene expressions; As(III) inhibits the repair of DNA damage caused by BPDE by inhibition of NER-related gene expression. Future research on the interaction between the metabolism and toxicity of As(III) will be continued in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (21575108, 21575107, 21375097, 21175102), the Science Fund for Creative Research Groups of NSFC (No. 20921062), and the National Basic Research Program of China (973 Program, 2013CB933900). We thank Professor Xiang Zhou from College of Chemistry and Molecular Sciences, Wuhan University for his assistant in HPLC-ESI-LTQ-Orbitrap-MS analysis, and Professor Zhixiong Xie from College of Life Science, Wuhan University for his assistant in RT-qPCR analysis.References
- R. S. Oremland and J. F. Stolz, The Ecology of Arsenic, Science, 2003, 34, 939–944 CrossRef PubMed.
- O. Neubauer, Arsenical Cancer: A Review, Br. J. Cancer, 1947, 1, 192 CrossRef CAS PubMed.
- H.-S. Yu, W.-T. Liao and C.-Y. Chai, Arsenic Carcinogenesis in the Skin, J. Biomed. Sci., 2006, 13, 657–666 CrossRef CAS.
- T. Schwerdtle, I. Walter, I. Mackiw and A. Hartwig, Induction of oxidative DNA damage by arsenite and its trivalent and pentavalent methylated metabolites in cultured human cells and isolated DNA, Carcinogenesis, 2003, 24, 967 CrossRef CAS.
- A. D. Kligerman and A. H. Tennant, Insights into the carcinogenic mode of action of arsenic, Toxicol. Appl. Pharmacol., 2007, 222, 281–288 CrossRef CAS PubMed.
- F. Faita, L. Cori, F. Bianchi and M. Andreassi, Arsenic-Induced Genotoxicity and Genetic Susceptibility to Arsenic-Related Pathologies, Int. J. Environ. Res. Public Health, 2013, 10, 1527 CrossRef CAS.
- Y. Sun, C. Kojima, C. Chignell, R. Mason and M. P. Waalkes, Arsenic transformation predisposes human skin keratinocytes to UV-induced DNA damage yet enhances their survival apparently by diminishing oxidant response, Toxicol. Appl. Pharmacol., 2011, 255, 242–250 CrossRef CAS PubMed.
- B. Hang, Formation and Repair of Tobacco Carcinogen-Derived Bulky DNA Adducts, J. Nucleic Acids, 2010, 2010, 709521 Search PubMed.
- S. Shen, J. Lee, W. R. Cullen, X. C. Le and M. Weinfeld, Arsenite and its mono- and dimethylated trivalent metabolites enhance the formation of benzo[a]pyrene diol epoxide-DNA adducts in Xeroderma pigmentosum complementation group A cells, Chem. Res. Toxicol., 2009, 22, 382–390 Search PubMed.
- S. Shen, J. Lee, M. Weinfeld and X. C. Le, Attenuation of DNA damage-induced p53 expression by arsenic: A possible mechanism for arsenic co-carcinogenesis, Mol. Carcinog., 2008, 47, 508–518 CrossRef CAS PubMed.
- J. Lee, X. Sun, M. Weinfeld and X. C. Le, Elevation of Cellular BPDE Uptake by Human Cells: A Possible Factor Contributing to Co-Carcinogenicity by Arsenite, Environ. Health Perspect., 2006, 114, 1832 CrossRef PubMed.
- T. Schwerdtle, I. Walter and A. Hartwig, Arsenite and its biomethylated metabolites interfere with the formation and repair of stable BPDE-induced DNA adducts in human cells and impair XPAzf and Fpg, DNA Repair, 2003, 2, 1449–1463 CrossRef CAS.
- H. C. Chiang and T. C. Tsou, Arsenite enhances the benzo[a]pyrene diol epoxide (BPDE)-induced mutagenesis with no marked effect on repair of BPDE–DNA adducts in human lung cells, Toxicol. In Vitro, 2009, 23, 897 CrossRef CAS.
- S. Pavanello and E. Clonfero, Determination of Anti-Bpde–DNA Adducts in Pah-Exposed Humans using the HPLC/Fluorescence Technique, Polycyclic Aromat. Compd., 1999, 17, 73–83 CrossRef CAS.
- F. Feng and H. Wang, Simultaneous analysis of four stereoisomers of anti-benzo a pyrene diol epoxide-deoxyguanosine adducts in short oligodeoxynucleotides using reversed-phase high-performance liquid chromatography, J. Chromatogr. A, 2007, 1162, 141–148 CrossRef CAS PubMed.
- F. Feng, J. F. Yin, M. Y. Song and H. L. Wang, Preparation, identification and analysis of stereoisomeric anti-benzo a pyrene diol epoxide-deoxyguanosine adducts using phenyl liquid chromatography with diode array, fluorescence and tandem mass spectrometry detection, J. Chromatogr. A, 2008, 1183, 119–128 CrossRef CAS.
- F. Feng, X. L. Wang, H. C. Yuan and H. L. Wang, Ultra-performance liquid chromatography-tandem mass spectrometry for rapid and highly sensitive analysis of stereoisomers of benzo a pyrene diol epoxide-DNA adducts, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 2104–2112 CrossRef CAS.
- B. H. Monien, F. Schumacher, K. Herrmann, H. Glatt, R. J. Turesky and C. Chesné, Simultaneous Detection of Multiple DNA Adducts in Human Lung Samples by Isotope-Dilution UPLC-MS/MS, Anal. Chem., 2015, 87, 641–648 CrossRef CAS.
- P. W. Villalta, J. B. Hochalter and S. S. Hecht, Ultrasensitive High-Resolution Mass Spectrometric Analysis of a DNA Adduct of the Carcinogen Benzo[a]pyrene in Human Lung, Anal. Chem., 2017, 89, 12735–12742 CrossRef CAS.
- N. P. Singh, M. T. McCoy, R. R. Tice and E. L. Schneider, A simple technique for quantitation of low levels of DNA damage in individual cells, Exp. Cell Res., 1988, 175, 184–191 CrossRef CAS.
- K. A. Francesconi and D. Kuehnelt, Determination of arsenic species: a critical review of methods and applications, 2000–2003, Analyst, 2004, 129, 373–395 RSC.
- X. C. Le, X. Lu and X.-F. Li, arsenic speciation, Anal. Chem., 2004, 27–33 Search PubMed.
- C. B'Hymer and J. A. Caruso, Arsenic and its speciation analysis using high-performance liquid chromatography and inductively coupled plasma mass spectrometry, J. Chromatogr. A, 2004, 1045, 1–13 CrossRef.
- M. Leermakers, W. Baeyens, M. D. Gieter, B. Smedts, C. Meert, H. C. D. Bisschop, R. Morabito and P. Quevauviller, Toxic arsenic compounds in environmental samples: Speciation and validation, Trends Anal. Chem., 2006, 25, 1–10 CrossRef CAS.
- Y. Li, B. Chen, M. He and B. Hu, Biomethylation metabolism study of arsenite in SCC-7 cells by reversed phase ion pair high performance liquid chromatography-inductively coupled plasma-mass spectrometry, Talanta, 2018, 188, 210–217 CrossRef CAS PubMed.
- E. Dopp, L. M. Hartmann, A. M. Florea, U. von Recklinghausen, R. Pieper, B. Shokouhi, A. W. Rettenmeier, A. V. Hirner and G. Obe, Uptake of inorganic and organic derivatives of arsenic associated with induced cytotoxic and genotoxic effects in Chinese hamster ovary (CHO) cells, Toxicol. Appl. Pharmacol., 2004, 201, 156–165 CrossRef CAS PubMed.
- C. Liu, Y. Wang, W. Yang, F. Wu, W. Zeng, Z. Chen, J. Huang, G. Zou, X. Zhang, S. Wang, X. Weng, Z. Wu, Y. Zhou and X. Zhou, Fluorogenic labeling and single-base resolution analysis of 5-formylcytosine in DNA, Chem. Sci., 2017, 8, 7443–7447 RSC.
- C. Liu, Y. Wang, X. Zhang, F. Wu, W. Yang, G. Zou, Q. Yao, J. Wang, Y. Chen, S. Wang and X. Zhou, Enrichment and fluorogenic labelling of 5-formyluracil in DNA, Chem. Sci., 2017, 8, 4505–4510 RSC.
- H.-P. Tran, A. S. Prakash, R. Barnard, B. Chiswell and J. C. Ng, Arsenic inhibits the repair of DNA damage induced by benzo(a)pyrene, Toxicol. Lett., 2002, 133, 59–67 CrossRef CAS.
- A. Luch, Nature and nurture-lessons from chemical carcinogenesis, Nat. Rev. Cancer, 2005, 5, 113–125 CrossRef CAS.
- D. S. Dheeman, C. Packianathan, J. K. Pillai and B. P. Rosen, Pathway of human AS3MT arsenic methylation, Chem. Res. Toxicol., 2014, 27, 1979–1989 Search PubMed.
- W. Qu and M. P. Waalkes, Metallothionein blocks oxidative DNA damage induced by acute inorganic arsenic exposure, Toxicol. Appl. Pharmacol., 2015, 282, 267–274 CrossRef CAS.
- N. Holcomb, M. Goswami, S. G. Han, T. Scott, J. D’Orazio, D. K. Orren, C. G. Gairola and I. Mellon, Inorganic arsenic inhibits the nucleotide excision repair pathway and reduces the expression of XPC, DNA Repair, 2017, 52, 70–80 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8mt00217g |
| This journal is © The Royal Society of Chemistry 2019 |