
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Across the spectrum: integrating multidimensional metal analytics for in situ metallomic imaging
Theodora J.
Stewart
 ab
ab
aKing's College London, Mass Spectrometry, London Metallomics Facility, 4th Floor Franklin-Wilkins Building, 150 Stamford St., London SE1 9NH, UK. E-mail: theodora.stewart@kcl.ac.uk
bKing's College London, School of Biomedical Engineering and Imaging Sciences, 4th Floor Lambeth Wing, St. Thomas’ Hospital, London SE1 7EH, UK
First published on 30th November 2018
Abstract
To know how much of a metal species is in a particular location within a biological context at any given time is essential for understanding the intricate roles of metals in biology and is the fundamental question upon which the field of metallomics was born. Simply put, seeing is powerful. With the combination of spectroscopy and microscopy, we can now see metals within complex biological matrices complemented by information about associated molecules and their structures. With the addition of mass spectrometry and particle beam based techniques, the field of view grows to cover greater sensitivities and spatial resolutions, addressing structural, functional and quantitative metallomic questions from the atomic level to whole body processes. In this perspective, I present a paradigm shift in the way we relate to and integrate current and developing metallomic analytics, highlighting both familiar and perhaps less well-known state of the art techniques for in situ metallomic imaging, specific biological applications, and their use in correlative studies. There is a genuine need to abandon scientific silos and, through the establishment of a metallomic scientific platform for further development of multidimensional analytics for in situ metallomic imaging, we have an incredible opportunity to enhance the field of metallomics and demonstrate how discovery research can be done more effectively.
 Theodora J. Stewart | Dr Theodora Stewart is the lead scientist and manager of the new London Metallomics Facility (LMF) based at King's College London, focusing on developing correlative bioimaging workflows to advance our understanding of the roles of metals in biology. Her research interests lie at the interface of biology, chemistry and physics, with a specific focus on developing analytical techniques and workflows to quantify temporal dynamics of intracellular metal species, their subcellular localisation, and identification of key biomolecules involved in their binding and transport with minimal disruption to the system of study through the use of both elemental mass spectrometry and synchrotron X-ray spectroscopy based bioimaging techniques. |
Introduction
I often hear the phrase “there is no silver bullet” when it comes to metal analytics. Have a conversation with any analytical chemist and the multitude of limitations each technique inherently possesses shall be quickly revealed. As disheartening as this may be, I cannot argue with the obvious analytical challenges and fully realize the necessity of utilizing a portfolio of complementary techniques. Yet, from my own personal experience and speaking to researchers coming from a wide range of scientific disciplines, it is also clear that we are not even fully aware of how many “bullets” exist.We all have, at the very least, an intuitive sense that the functions of essential metals in biology are complex, dynamic and critical for life. Nature possesses a whole arsenal of diverse approaches to support such processes. Therefore, to reveal Nature's secrets we must approach metallomic studies with an equally rich toolkit of integrated analytics across multiple dimensions of space and time, which are specific, sensitive and flexible enough to accommodate the wide range of concentrations, complex chemistry, and dynamics of metals in biology. This article is not merely another review of analytical techniques, of which there are plenty of informative works. In this perspective piece, I present a different way to relate to and integrate the analytics we use, placing a specific focus on current and emerging techniques for in situ metallomic imaging. Seeing is believing, and a picture is worth a thousand words. In the following pages, I take us on a journey through a network of multidimensional analytical tools, using the electromagnetic spectrum as a platform, moving from microwaves to gamma rays, then into the worlds of mass spectrometry and particle based imaging techniques to understand how the interaction between energy and living matter can illuminate so many different functional and structural aspects, which we require as scientists to comprehend the rich world of metals in biology.
Metallomics is powerful in that it presents an opportunity to shift our scientific thinking, approach and methodologies to more comprehensively understand our system of study, offering integration of existing research disciplines at a higher level. This same approach can be taken when relating to metallomic analytics. Ironically, the phase “metallomic analytics” is, in fact, an oxymoron. The word analysis originates from the ancient Greek word 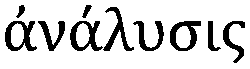 from ana- “up, throughout” and lysis “a loosening,” literally implying a breaking up, in seeming contradiction with metallomics, a bottom up synthesis approach to understanding metals in biology. How then can we use an approach that is inherently disruptive within the context of a field, which is inherently integrative? Rather than picking a few familiar techniques and operating within their analytical confines, what if we viewed our analytical possibilities in an integrated and comprehensive context? A systems analytical approach to systems biology questions, where just as in Nature, the whole is much more than the sum of its individual parts, demarcating a shift in the way we utilise analytical chemistry and physics from a series of separated individual tools to probe a living system towards an integrated network of analytics as powerful as the living biology it is used to study. The goal in integrating current techniques, whilst continuing further analytical developments directly informed by biological scientific needs, is to ultimately understand the role of a specific metal, at a specific location in time within the context of a cellular process and how those dynamic functions cascade to higher levels of function at the tissue, organ and organism levels. In the following sections, I discuss analytical techniques, which allow us to identify, quantify and directly probe the immediate surrounding chemical environment (oxidation state, local coordination geometry) of a metal, its localisation in both two and three dimensions at the cellular (Fig. 1), tissue (Fig. 2) and organ (Fig. 3) levels, and even assess temporal dynamics under certain conditions.
from ana- “up, throughout” and lysis “a loosening,” literally implying a breaking up, in seeming contradiction with metallomics, a bottom up synthesis approach to understanding metals in biology. How then can we use an approach that is inherently disruptive within the context of a field, which is inherently integrative? Rather than picking a few familiar techniques and operating within their analytical confines, what if we viewed our analytical possibilities in an integrated and comprehensive context? A systems analytical approach to systems biology questions, where just as in Nature, the whole is much more than the sum of its individual parts, demarcating a shift in the way we utilise analytical chemistry and physics from a series of separated individual tools to probe a living system towards an integrated network of analytics as powerful as the living biology it is used to study. The goal in integrating current techniques, whilst continuing further analytical developments directly informed by biological scientific needs, is to ultimately understand the role of a specific metal, at a specific location in time within the context of a cellular process and how those dynamic functions cascade to higher levels of function at the tissue, organ and organism levels. In the following sections, I discuss analytical techniques, which allow us to identify, quantify and directly probe the immediate surrounding chemical environment (oxidation state, local coordination geometry) of a metal, its localisation in both two and three dimensions at the cellular (Fig. 1), tissue (Fig. 2) and organ (Fig. 3) levels, and even assess temporal dynamics under certain conditions.
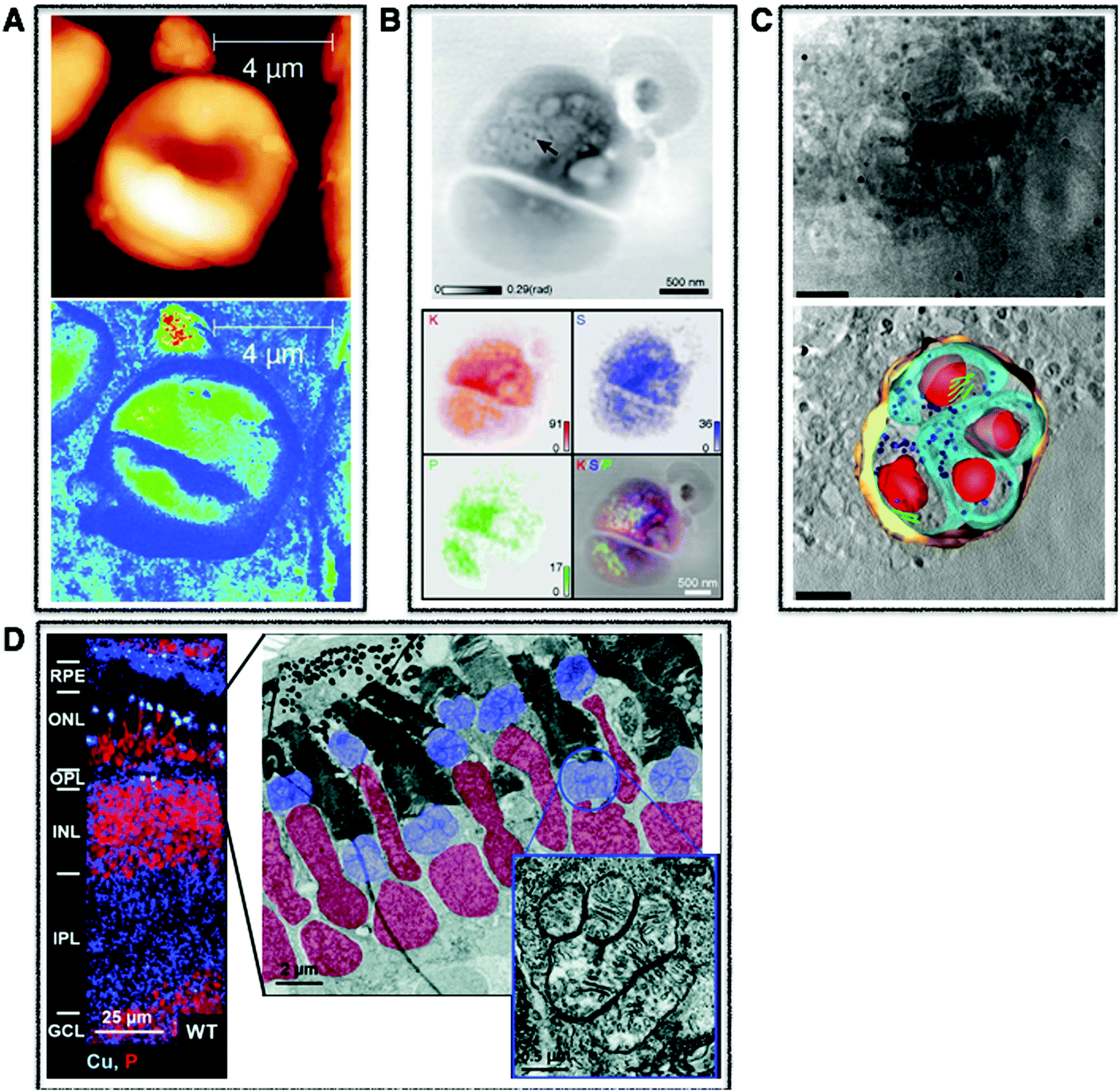 | ||
| Fig. 1 Cellular multimodal imaging. (A) AFM scan of a single red blood cell (upper) with parallel s-SNOM subcellular chemical contrast image (67 nm resolution) (lower). Adapted from Amrania et al.20 (B) Ptychographic image of frozen-hydrated Ostreococcus alga with arrow poiting to ribosome-like complexes (upper) and corresponding XRF maps of potassium (red), sulphur (blue) and phosphorus (green) with subsequent overlay (lower). Scale bars are 500 nm. Adapted from Deng et al.72 (C) Cryo-SXT image of a human fibroblast cell vacuole containing Toxoplasma gondii (upper) and the reconstructed volume showing the parasitophorous vacuole (yellow) with four parasites and their respective plasma membranes (cyan) rhoptries (green) and nuclei (red). Scale bars are 2.5 mm. Adapted from Harkiolaki et al.57 (D) NanoSIMS copper (blue) and phosphorous (red) overlay of wt zebrafish retina (left) and electron micrograph of similar region (right) with highlighted nuclei (red) and megamitochondria (blue) with inset corresponds to one megamitochondrion, indicating co-localisation of copper puncta and megamitochondria. Scale bars are 25 μm, 2 μm, and 0.5 μm, respectively. Adapted from Ackerman et al.135 | ||
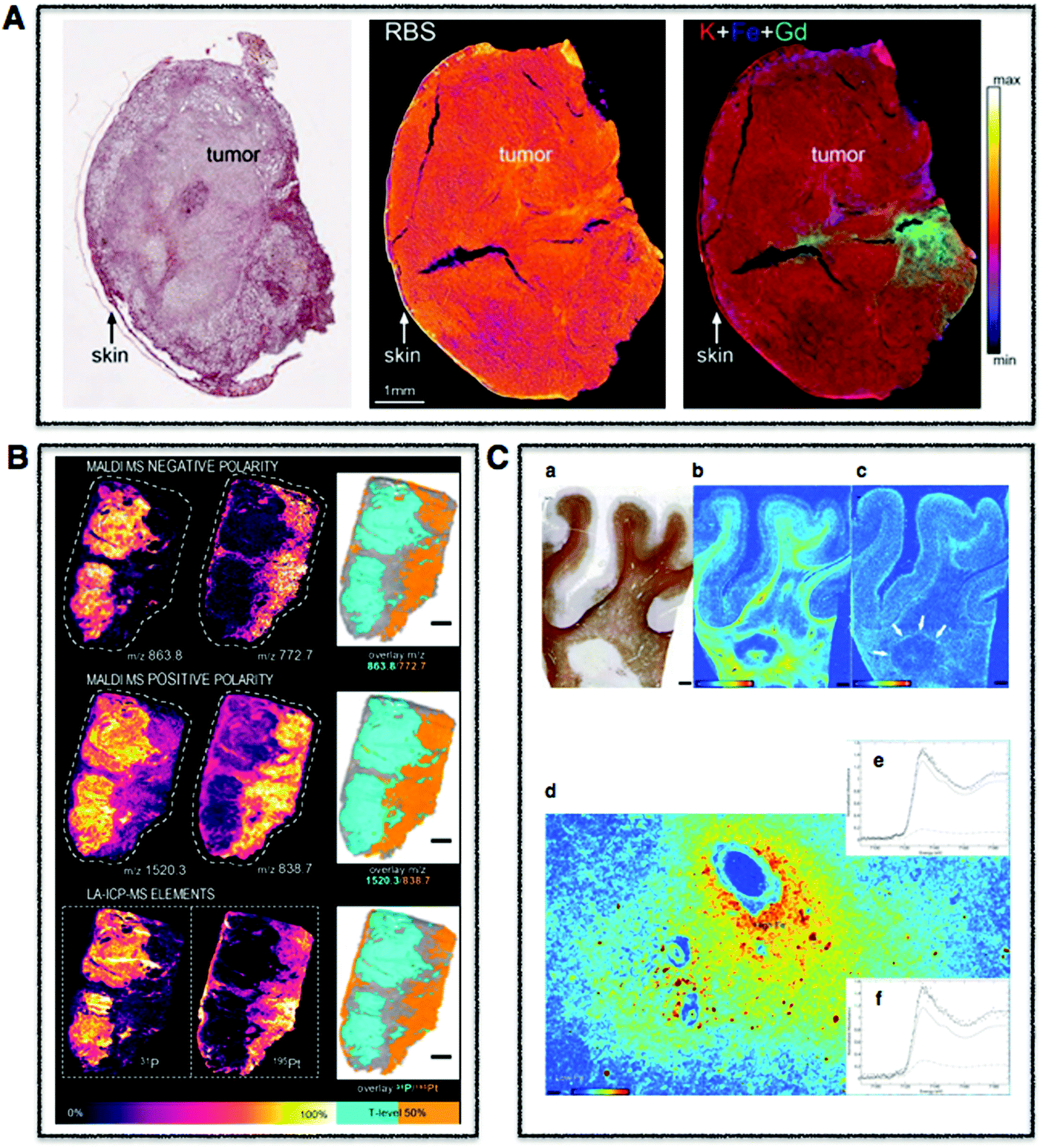 | ||
| Fig. 2 Tissue multimodal imaging. (A) Histological section of U87 human glioblastoma xenograft (left), RBS image of tissue section (middle), PIXE image of tissue section highlighting potassium (red), iron (blue), and gadolinium (green) corresponding to injected gadolinium NPs (right). Scale bar 1 mm. Adapted from Carmona et al.129 (B) Sections of human malignant pleural mesothelioma treated with cisplatin highlighting distribution of specifc phospholipids using MALDI-MSI in negative (m/z 772.7 and 863.8) (upper) and positive (838.7 and 1520.3) (middle) modes. LA-ICP-MS was used to map distribution of phosphorus and platinum. Scale bar 1 mm. Adapted from Holzlechner et al.136 (C) Histological section of apparently normal and periplaque white matter of multiple sclerosis lesion in human brain (a) and corresponding iron (b) and zinc (c) XRF maps. Scale bars 3 mm. Iron shown to accumulates perivascularly in astrocytes (d) and quantitative speciation of iron in concentrated regions (e) and in iron-poor regions (f). Scale bar 90 μm. Adapted from Popescu et al. under the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/).53 | ||
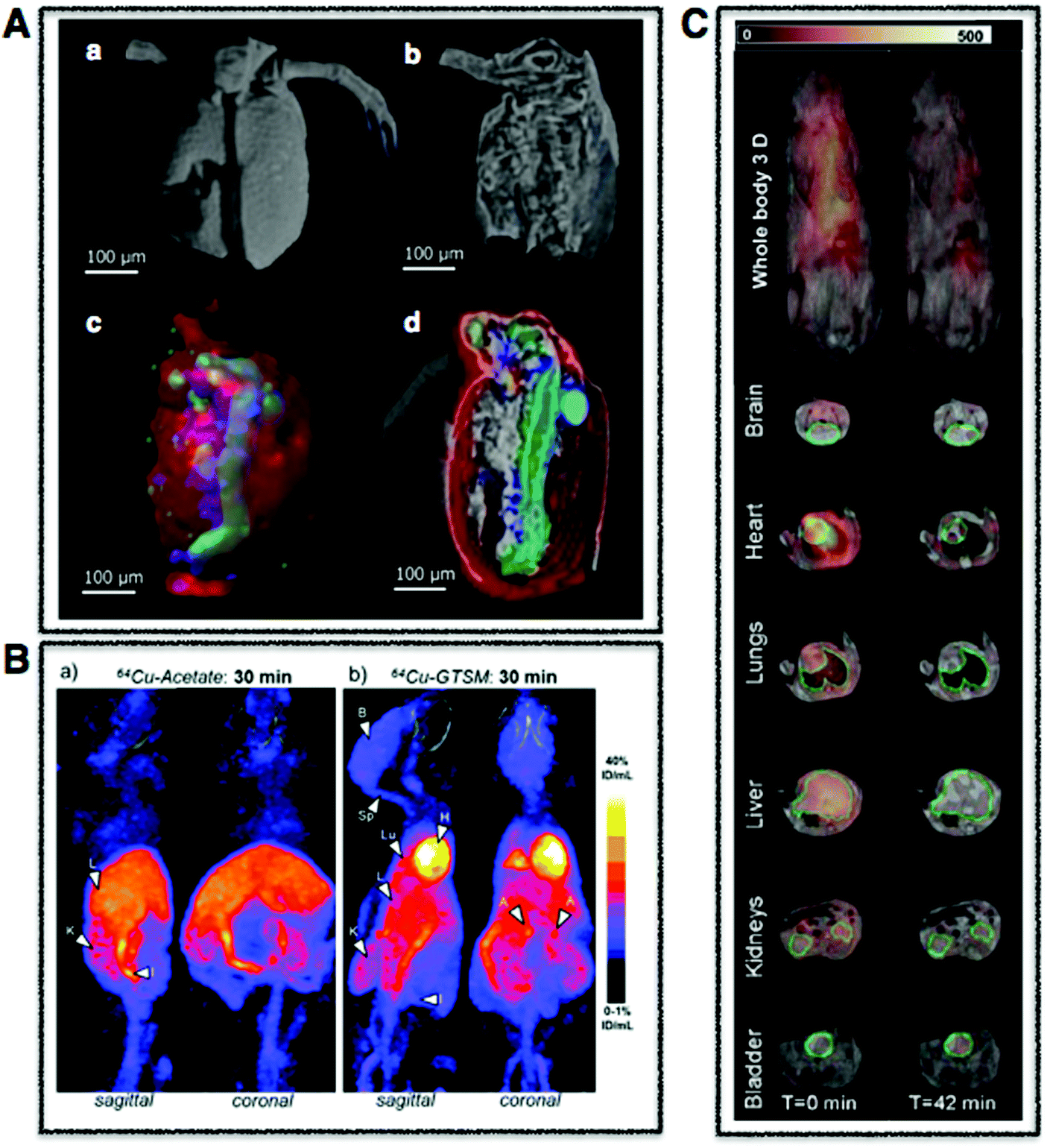 | ||
| Fig. 3 Whole body multimodal imaging. (A) μ-CT 3D renderings of C. dubia (a and b) and calcium (red), manganese (green), zinc (blue) isosurfaces using confocal μ-XRF (c and d). Scale bar is 100 μm. Adapted from Van Malderen et al.102 (B) PET/CT maximum intensity projections (MIPs) of 6–8-month-old wild-type mice at 30 min post-injection of Cu-acetate (a) and Cu-GTSM illustrating different uptake patterns. Adapted from Andreozzi et al.84 (C) Superimposed 3D EPRI and 3D proton MRI images in whole body and specific organs in cigarette exposed mice, where intensity distribution corresponds to EPR intensity of 3-CP nitroxide probe distribution at time 0 and 42 min. Adapted from Caia et al.137 | ||
Spectroscopy imaging
Each region of the electromagnetic spectrum possesses unique physical properties, which can be harnessed and utilised in analytical tools to gain information from the interaction of electromagnetic radiation and matter on questions pertaining to metal functions in biological processes (Fig. 4). When combined with optical, electron, or scanning probe microscopes, these spectroscopic signals are amplified to visualize structural and chemical information invisible to the naked eye. In the following section, I present several key spectroscopy imaging techniques for in situ metallomic studies ranging from whole body to subcellular imaging and highlight particular advances in both optical and X-ray microscopies that have been key in going beyond diffraction limitations to image at the nanoscale.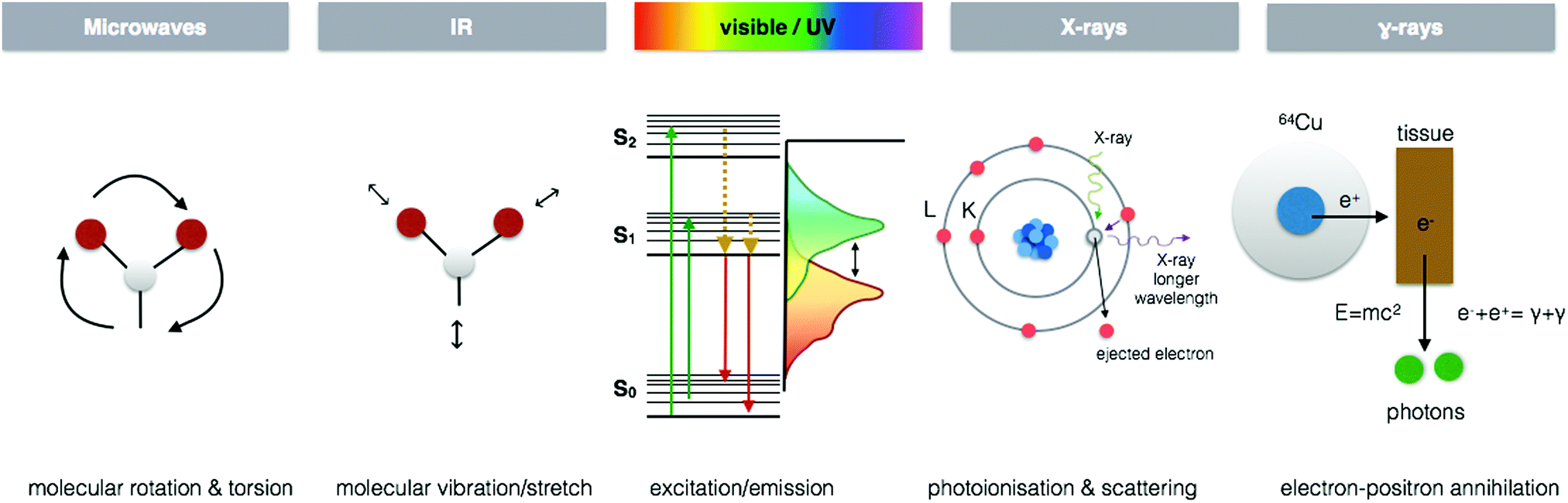 | ||
| Fig. 4 Energy–matter interactions of the electromagnetic spectrum. Spectroscopic imaging techniques arise from fundamental energy–matter interactions. EPRI exploits the molecular rotation and torsion caused by microwaves to detect paramagnetic species by measuring the magnetism of electrons and their change when bound to molecular structures. FTIR and Raman imaging take advantage of characteristic vibrations/stretching and even rotation associated with specific molecular species when interacting with infra-red for FTIR and typically extending to near infra-red and the visible region for Raman. CLSM and SRM utilise characteristic fluorophore excitation/emission profiles in the UV/vis region, exploiting the Stokes shift that occurs through loss of vibrational energy between absorption and emission to localise multiple fluorescent signals at the subcellular level. X-ray spectroscopic techniques benefit from element specific photoionisation energies and subsequent scattering of electrons to reveal detailed electronic structural information at the atomic scale. PET utilises gamma-ray emission that results from the annihilation of electrons in tissue from positron emitting isotopes. | ||
Electron paramagnetic resonance imaging (EPRI)
Electron paramagnetic resonance (EPR), also referred to as electron spin resonance (ESR), assesses the electronic structure of molecules through measuring the magnetism of unpaired electrons in an external magnetic field. Within the context of metallomics, EPR can be used to obtain detailed information on the coordination chemistry specifically of paramagnetic species (species with unpaired electrons), which include free radicals and some transition metal complexes in biological samples. For example, Mn2+ complexed by specific ligands at low temperature shows a significantly different spectrum relative to the free ion Mn2+.1 Although limited to paramagnetic and ferromagnetic species, conveniently these species are common intermediates in redox reactions found in both normal and aberrant metabolism, and transition metal complexes constitute the active centres of bio-macromolecules. EPR plays a particularly important role in elucidating mechanisms of metal redox chemistry.Traditionally, EPR has been used to characterize metal centres in both isolated and purified proteins and model compounds.2 However, EPR has probably gained its widest biological potential yet as EPR Imaging (EPRI) within the context of biomedical research.3 With EPRI, imaging of tissue O2 partial pressure (pO2)4 and reactive oxygen species (ROS) microenvironments is possible and ideal in the sense that it is both non-invasive and quantitative so that it can be carried out as an in vivo technique (Fig. 3C).5 By using a large range of commercially available paramagnetic probes, pO2 can be imaged and quantified, and by using a technique called spin-trapping, ROS can be imaged and followed over time.6 Unfortunately, EPRI does not provide anatomical information and suffers from poor spatial resolution (1 mm), relative to other imaging techniques. Even with this limited spatial resolution, EPRI has been used to map redox status in tumours, which can been used to predict resistance and reoccurrence to radiation therapies.4,7 When combined with other metal imaging modalities, EPRI can provide an important chemical context for tissue level metal redox processes involving unpaired electrons.
Magnetic resonance imaging (MRI)
The physical context is often critical for metallomic questions: which subcellular compartment or where in a tissue is the metal of interest? Developed as a tool to provide anatomic images, MRI has provided that contextual foundation during the course of its evolution over the past four decades. Paramagnetic metal ions and superparamagnetic iron oxide nanoparticles are often used as contrast agents, and greater refined metal-based complexes (i.e. Gd(III), Lu(III), Eu(III)) are now being used as reporter molecules for multimodal imaging approaches, combing the properties of MR contrast with fluorescence and possessing even targeting capabilities.8Typical resolutions for in vivo MRI fall around 1 mm3, however advances in both molecular and cellular MR imaging (MR-microscopy (MRM)) now allow for longitudinal monitoring of subcellular events whilst providing highly resolved anatomical contexts with spatial resolutions in the sub 100 μm range. MRM has been used to reveal characteristic pathologic features of both benign and malignant breast and lymph tissue,9 and as an in vitro tool to study myonuclei of individual muscle fibres down to 6 μm planar spatial resolution.10 Particularly relevant for metallomics, MRI chemical probes have also been synthesised for specific investigations of biologically important metal ions, such as potassium, magnesium, copper(I), copper(II), zinc, calcium and even toxic metals such as lead and cadmium.11 In the case of zinc, the Zn2+ sensor GdDOTA-diBPEN has been successfully used to image and quantify labile (non-protein bound) Zn2+ in the μM concentration range in mice within the context of zinc release in functional β-cells in the pancreas in vivo and, when administrated to type-1 diabetes mouse model, was sensitive to β-cell loss of function, detecting decreased release of Zn2+.12 The same probe has also been used to detect differences in healthy and tumorigenic prostate in mice, showing reduced concentrations in cancerous prostates, suggesting a possible diagnostic tool.13 With the help of metal specific MRI probes, an exciting era of research is emerging for in vivo deep tissue monitoring of metals.
Fourier Transform infrared (FTIR) imaging
Moving across the electromagnetic spectrum to the infrared region we come to Fourier Transform infrared spectroscopy (FTIR), one of several types of vibrational spectroscopies that uses differential absorbance, transmittance or reflectance of polychromatic infrared radiation to provide information on characteristic vibrational frequencies of chemical bonds, providing a chemical fingerprint sensitive to hetero-nuclear functional group vibrations as well as polar bonds. In fact, although not explicitly integrated into the names of other imaging techniques, FT has become an integral signal-processing tool in many of the techniques discussed below. With its ability to break down complex wave-like signals to extract meaningful spectra, like taking a cake (waveform) and extracting the recipe (the FT), no wonder physicist Lord Kelvin described Fourier's theorem as “an indispensible instrument in the treatment of nearly every recondite question in modern physics.”14 FTIR is particularly sensitive to changes in secondary structure of proteins, making it an important tool in the study of protein aggregation and misfolding, which is often observed in combination with altered distribution of metals. When used as a micro-spectroscopic technique, FTIR imaging is able to generate label-free, molecular-specific images to visualize the distribution of different components (i.e. protein, lipid, carbohydrate, nucleic acids) within tissue sections.15 In this sense, FTIR imaging can be considered as a complementary metabolomic tool to metallomic inquiries that allows for the in situ, non-destructive analysis of biological specimens.16,17To obtain biologically meaningful spatially resolved spectra in cells and tissues, the brilliance of a synchrotron source is often utilized to overcome challenges associated with small aggregates at the nanoscale level as well as to detect the often subtle changes in the FTIR spectra observed in vivo. By circularly accelerating electrons close to the speed of light to produce intense synchrotron light 10 billion times brighter than the sun, interactions at the finest atomic and molecular scales can be studied. Access to synchrotron facilities is free, but requires a peer-reviewed application process with calls typically every six months. Although access is limited, synchrotron radiation (SR) has proved to be an invaluable tool, measuring protein misfolding and aggregation in tissue in Alzheimer's, Parkinson's and Huntington's disease as well as amyotrophic lateral sclerosis and even scrapie.18 However, recent advances in scattering-type scanning near-field optical microscopy (s-SNOM), where near IR light is focused on a sample through the probe of an atomic force microscope (AFM), routinely allows for measurements at 10 nm and down to 1 nm.19 This principle has just been extended to cover the mid IR region, or the important “fingerprint” region, where a single red blood cell at 67 nm spatial resolution was recently imaged (Fig. 1A).20 When combined with techniques directly mapping metal distributions, FTIR can provide important information about the compositional environment of a metal at both the tissue21 and now cellular levels in two and three dimensions.22
Raman imaging
Raman spectroscopy also probes vibrational modes of molecules and is based on the similar principles underpinning FTIR vibrational spectroscopy, but differs in several fundamental ways by utilising monochromatic light (rather than polychromatic) to measure change in the polarizability (rather than dipole moment) of a molecule. Raman is sensitive to homo-nuclear molecular bonds and can distinguish between C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C
C and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds. Therefore, what is often “invisible” to FTIR is measured in Raman and vice versa. Raman has become a valuable tool for filling a gap through its ability to detect small structural changes and complementing other techniques like X-ray crystallography, which provide a larger structural picture.23 Although Raman is highly molecularly selective, it suffers from low sensitivity, often competing with the strong auto fluorescence generated from biological samples. However, specific types of Raman, primarily resonance Raman spectroscopy (RRS) and surface enhanced Raman spectroscopy (SERS), can be used, as signals are inherently enhanced.
C bonds. Therefore, what is often “invisible” to FTIR is measured in Raman and vice versa. Raman has become a valuable tool for filling a gap through its ability to detect small structural changes and complementing other techniques like X-ray crystallography, which provide a larger structural picture.23 Although Raman is highly molecularly selective, it suffers from low sensitivity, often competing with the strong auto fluorescence generated from biological samples. However, specific types of Raman, primarily resonance Raman spectroscopy (RRS) and surface enhanced Raman spectroscopy (SERS), can be used, as signals are inherently enhanced.
Just as in FTIR, Raman can be used as an imaging tool, providing morphological as well as both qualitative and quantitative biochemical information at subcellular resolution in living cells.24 In resonance raman spectroscopy (RRS), enhancement of specific chromophores are used, the most popular being π → π* transitions from conjugated π-bonds. Fortunately within the context of metallomics questions, these conjugated π systems are associated with critical cellular processes involving metals, as in the case of porphyrin derivatives, which constitute a main part of enzymatic cofactors. One such example is the enzyme cytochrome c, an iron containing heme protein essential in the respiratory system of eukaryotic organisms. Using RRS, the localisation of both the reduced (Fe2+) and oxidized (Fe3+) forms of cytochrome c and b in living murine cells were quantitatively determined, revealing that both cytochromes are co-distributed, but that cytochrome c is predominantly reduced whereas cytochrome b is predominantly oxidized.25 Obtaining this degree of chemical information in living cells at subcellular resolution presents exciting opportunities and a powerful context for specifically studying mitochondrial metal metabolism, which when disrupted, has been implied in a wide range of human diseases including red blood cell disorders, cardiomyopathy, skeletal myopathy, and neurodegeneration.26 Raman studies have also been used to characterize cancer tissues,27 which when combined with spatially resolved metal analyses have the potential to yield an even stronger identifying “fingerprint”. The reader is directed further to a recent detailed review of the different types of Raman spectroscopy and their state of the art applications in biology.28 Although advancements towards high-resolution spectroscopy for Raman have been made as illustrated with FTIR, the low sensitivity of Raman still requires larger laser power for tip-enhanced Raman, and therefore problematic for delicate biological samples. However, recent advances in spectroscopic stimulated Raman scattering (SRS) now allow for real-time metabolic imaging at the subcellular scale.29 Like in FTIR, Raman imaging provides a “molecular fingerprint” from which detailed information can be obtained on the local sites within larger macromolecules for metallomic studies.
Light microscopy
We often say that seeing is believing, and no other scientific technique underpins this statement more than light microscopy. There is an excitement and intuitive pull in moving beyond the inherent limitations of the human eye to see life at more subtle and powerful scales. Starting with the humble beginnings of light microscopy in 1665, marked by Hooke's manuscript “Micrographia” and pioneering work by van Leeuwenhoek, the inconvenient laws of physics have seemed to always present a barrier to witnessing the most fundamental processes of life at the subcellular scale. Even within the confines of that pesky 250 nm diffraction limit, significant advances in light microscopy have led from the evolution of transmission bright field to confocal laser scanning microscopy, which has revolutionised the field of biology in the past 20 years. With recent advancements in optics, electronics and mathematics we are finally able to overcome the diffraction limit, giving rise to super resolution microscopy, again shedding new light on cellular processes now at the molecular level. What sets light microscopy apart from techniques such as X-ray microscopy and mass spectrometry discussed later is the ability to non-destructively work with living samples, opening investigations into an additional dimension (4D) in time by capturing live cell dynamics.The use of fluorescent sensors to measure intracellular metal ions has been extensively reviewed.30 In brief, sensors can be broadly classified into three main types: molecular, genetically encoded, and hybrid. Some of the more commonly used sensors are molecular probes, which involve small-molecule fluorophores coupled to a metal chelator and must be externally delivered into the cell. However, these sensors often lack control over subcellular localisation and are unable to detect very low concentrations of free ions, which can be biologically relevant for certain metal ions like Zn2+ and Cu+. Genetically encoded probes are synthesised within the cell and can be used to overcome these limitations through the use of fluorescent proteins and a peptide or protein moiety for metal binding. When two fluorescent proteins are used, Förster resonance energy transfer (FRET) can be exploited creating a powerful ratiometric sensor where metal binding can either promote or disrupt FRET. Hybrid probes are simply a combination of small-molecule and genetically encoded probes, enabling protein/peptide tags to be used for localising small molecule sensors to specific subcellular compartments. It is important to note that “not all metals are created equal” and their wide range of concentrations and chemical properties present specific challenges for fluorescence based detection of certain intracellular metals like copper. Unlike Ca2+ found in relatively high concentrations within the cell, low intracellular Cu+ and Cu2+ concentrations are exquisitely maintained, balancing essential yet potent redox activity with a propensity to exacerbate oxidative stress. Although a handful copper sensors have been developed, far fewer are available relative to Ca2+ or Zn2+, as Cu+ can easily oxidise to Cu2+ and Cu0 in water and quench fluorescence due to its redox properties.
The case of zinc is exemplary in how visualisation and quantification of a metal in space and time can alter our understanding of the role of essential metals in biological processes. Zinc is recognised as an essential micronutrient required for cellular homeostasis. However, until recently it was thought to only serve structural and catalytic functions in proteins and enzymes. With the ability to quantify intracellular changes down to pM of free Zn2+ thanks to advances in zinc specific probes and sensors,33–35 along with their use to highlight the roles of zinc in processes spanning from modulating tyrosine phosphatase 1B activity36 to the quantitative mapping of zinc sparks released during fertilisation required for egg to embryo transition,37 its critical role in cell signalling has finally been acknowledged, opening the field of zinc biology to new exciting research questions.
X-Ray microscopy
Cellular processes involving metals are dictated by complex metal chemistry played out at the atomic and molecular scales. To glimpse into this nano world requires spectroscopic techniques utilising wavelengths powerful enough to excite and even ionise core electrons and at spatial scales corresponding to the Ångstrom distances found between interacting atoms. This level of detailed study requires X-rays, both scattering and refracting in their interaction with matter. By harnessing and taking advantage of these energy–matter interactions, X-ray spectroscopic techniques provide tools to probe local coordination geometry and chemical states of species as well as provide structural information at the subcellular level, thereby providing the unique advantage of determining metal speciation, a critical component in understanding the chemistry of metals in cellular processes. X-ray microscopy provides spatial resolutions down to tens of nm without the need for invasive approaches like labelling, dehydration, or chemical fixation, which can alter endogenous metal chemistry. The ability to obtain subcellular resolution is particularly important within the context of cellular metal chemistry, as metal homeostasis relies on tight regulation of metal species often through subcellular compartmentalisation. A few key examples include Fe–S clusters and heme synthesis products in the mitchochondria, zinc finger proteins required for gene transcription in the nucleus, and regulation of copper in the Golgi.40,41 Although the types of X-ray spectroscopies discussed are not limited to synchrotron radiation sources, in combination with optics allowing for sub-micron beam sizes, synchrotron radiation provides the high flux and brightness required for enhanced resolution and sensitivity required when imaging metals in biological samples at the subcellular level.X-ray microscopy can be broadly grouped into techniques providing elemental identification and speciation and those providing structural and morphological information. Localisation and quantification of elements in cells and tissues can be achieved using X-ray fluorescence (XRF), and when combined with X-ray absorption spectroscopy (XAS), additional speciation information is obtained on the chemical state and local coordination environment. To place such compositional information into a structural context, absorption or phase contrast tomography can be utilised to obtain information on cellular structure in both two and three dimensions, while X-ray diffraction techniques provide more detailed structural information and probe the local nanostructure and chemical composition of a sample. The most relevant X-ray microscopy methods for in situ metallomics along with examples are described below.
A wide range of metallomic questions have utilised both μ-XRF and nano-XRF, including questions regarding non-essential metal detoxification and effects to endogenous metals at the subcellular level, metal profiles in cellular processes and human disease, and subcellular localisation of metallo-based drugs, to highlight just a few examples. It was shown that silver and cobalt taken up in the unicellular algae Coccomyxa actinabiotis does not impact endogenous metal distributions, and that silver is taken up and seemingly sequestered in vacuoles, whereas cobalt is homogenously found directly outside the chloroplast.44 Intriguing work on the inheritance of trace metals during mitosis in NIH 3T3 mouse fibroblast cells showed the spatial dynamics of copper and zinc during metaphase, a 3-fold increase of zinc during interphase, and then resulting pairs of twin metal pools during cytokinesis, all indicating a more important physiological role of zinc than previously thought.45 Work regarding selenium-mediated arsenic hepatobiliary excretion in mammals used μ-XRF to co-localise arsenic and selenium in the liver, gall bladder and small intestine of hamsters.46 Metal dyshomeostasis is thought to be involved in many human diseases including neurodegenerative disease.47 Using μ-XRF it was shown that both iron and calcium increase in the brain of both control and Alzheimer's disease (AD) mice, and that copper, iron, zinc and calcium are all significantly higher in AD mice relative to controls indicating a possible role of metal dyshomeostasis in AD.48 Primary cultured neurons, derived from rat embryonic cortex (CTX) and two regions of the hippocampus, displayed characteristic spatial metal signatures (calcium, iron, zinc, copper, manganese), concluding that metal distribution in the brain is likely a result of not only extrinsic factors related to synaptic function, but also intrinsic factors controlling cellular metal content and distribution.49 Moving to the nano scale, nM concentration of an osmium based anti cancer drug were mapped in whole ovarian cancer cells with 50 nm spatial resolution, finally being able to resolve subcellular organelles and showing that osmium localises to the mitochondria not the nucleus, along with the mobilisation of calcium from the endoplasmic reticulum, signalling cell death.50 SXRF is the most sensitive of the X-ray based techniques, on par with that of mass spectrometry imaging based techniques (discussed later) and shown to be even more sensitive at the highest spatial resolutions.51 It is not surprising that to date, SXRF is considered to be the most reliable approach for quantitative and non-destructive imaging of elemental distributions at the sub-micron scale.
XAS mapping is used to determine speciation of metals in biological tissues and now possible at the cellular level with both scanning transmission X-ray microscopy (STXM) and the more recent introduction of hard X-ray nanoprobes. Determination of oxidation states and speciation “fingerprinting” through the spectral comparison with biologically relevant standards of metal complexes can be used to decipher the metal speciation in biological samples. Many of the SXRF examples previously highlighted were accompanied with XAS for metal speciation determination. For example, using XAS, the co-localisation of selenium and arsenic in liver was determined to be the seleno bis-(S-glutathionyl) arsinium ion, which was excreted via bile into the intestinal tract, thus supporting a previous hypothesis of hepatobiliar excretion.46 In the Alzheimer's Disease (AD) work, XANES showed evidence of copper redox properties in AD mouse brains,48 and accumulation and storage of ferritin in reactive astrocytes within smouldering and inactive plaques of Multiple Sclerosis (MS) patients were discovered using XANES (Fig. 2C).53 In work providing greater insight into the role of bromium in biological systems, it was shown that bromium is widely distributed across a range of mammalian tissues and fluids and that the major form is bromide, where it was seen in elevated concentrations in the sub-endothelial regions of arterioles in bovine ovaries.54 STXM is particularly unique in that it utilises X-rays extending into the soft-X-ray regime, allowing for speciation of elements such as carbon, nitrogen, and oxygen but also includes biologically important transition metals like iron and manganese. Because of this capability, certain transition metals can be speciated within the context of proteins, lipids, polysaccharides, and nucleic acids mapped down to 20 nm at certain synchrotron beam lines and can be done in 3D if proper optical configurations are present. The majority of work conducted has been within the context of biofilms and mineralizing bacteria.55 Utilising the principles of XAS as a microspectroscopic technique provides the invaluable opportunity for chemical mapping of metals down to the nanoscale, providing speciation and localisation critical for the mechanistic understanding of the role of metals in specific biological processes.
Although it does not provide metal-specific distribution information as in XRF and XAS, cryo-SXT has been used to visualise metal-based nanoparticle adsorption by cells58 and calcium-rich concentrates in algae,59 in addition to a whole range of processes including Plasmodium heme detoxification,60 bacterial activation of T lymphocytes,61 degranulation in mast cells,62 and chromatin reorganisation.63 In a correlative approach with cryo-SXT, immunogold markers, and cryo-fluorescence microscopy, endosomes and autophagosomes, normally negatively affected by sample preparation for electron microscopy, were imaged and the 3D ultrastructure of endocytic and autophagic structures, as well as clusters of omegasomes on the endoplasmic reticulum, which act as a platform for autophagosome biogenesis, were mapped, showing that these powerful approaches in combination can highlight cellular processes inaccessible using other imaging modalities,64 providing refined 3D subcellular morphological changes in response to experimental treatments, infection, and even disease in whole unstained cells (Fig. 1C).
One such CDI method that has risen to the forefront of bioimaging is ptychography.69,70 By using a coherent X-ray probe to scan and collect diffraction patterns from a series of partially overlapping regions in a sample, ptychography can provide detailed image reconstructions at near-wavelength lateral resolution and sub-nm depth resolution (Fig. 1B).71 Ptychography, although a relatively new technique, is currently being used in most synchrotron facilities with a wide range of biological applications in the literature. Both cellular structure and distribution of light elements (potassium, sulphur, phosphorus) in the frozen-hydrated green algae Ostreococcus sp., were recently imaged with high structural (<30 nm) and elemental composition (90 nm) resolution by combining ptychography and emitted fluorescence signals.72 To gain insights into the chemistry and biomineralisation function of magnetosomes (Fe3O4 nanoparticles) in magnetotactic bacteria (MTB), ptychography was used to image and speciate magnetosomes in Magnetovibrio blakemorei MV-1 cells, resulting in a proposed pathway of magnetosome biomineralisation.73 Within the context of nanomedicine, two oxidation states of individual iron oxide nanoparticles with a resolution of 16.5 nm internalized within individual HeLa cells were determined,74 and for the first time, ptychographic hard X-ray tomography was used to image 100 μm thick frozen biological tissue in 3D, allowing for the visualisation of myelinated axons and age-related pigmented cellular inclusions at 100 nm spatial resolution.75 Ptychography is still a relatively new approach but, if current results are any indication, the technique holds great promise for metallomic bioimaging applications going forward.
Mass spectrometry imaging
Moving from the multitude of analytical possibilities the electromagnetic spectrum offers for in situ metallomics, we come to the realm of mass spectrometry. Mass spectrometry imaging (MSI) can be conducted at both molecular and elemental levels and offers several major key capabilities to the metallomic analytical portfolio including spatially resolved molecular identification, isotopic sensitivity, and a wide dynamic range (five orders of magnitude) with the ability to quantify certain elemental isotopes down to parts per quadrillion (ppq) (1 × 10−15 g kg−1). In the following subsections, several MSI techniques specifically suited for in situ analysis of metals in biological samples are discussed. The combination of elemental and molecular mass spectrometry offers unprecedented opportunities for analytical biochemistry (Fig. 5).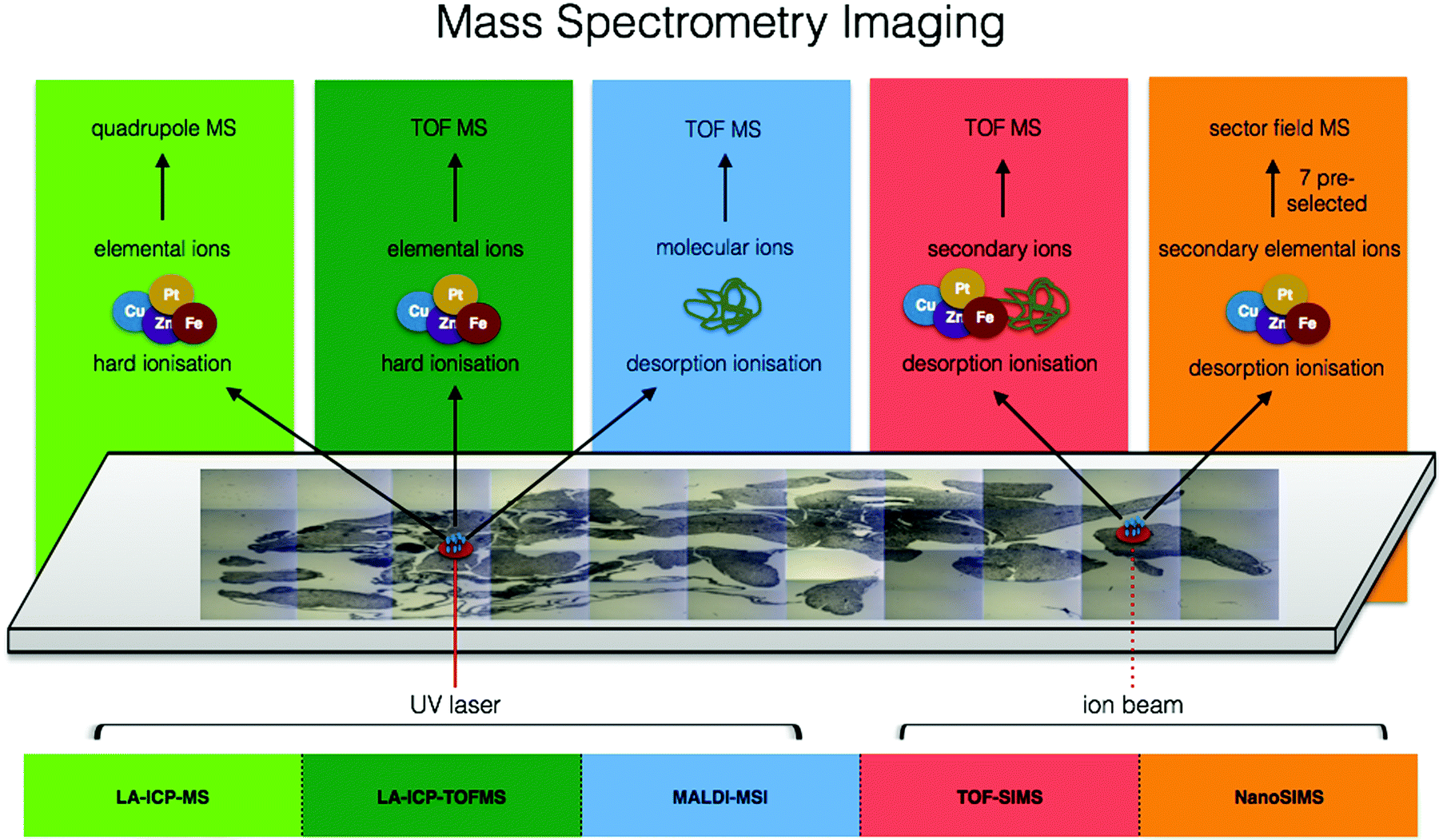 | ||
| Fig. 5 Comparison of ionisation sources, resulting elemental/molecular ions, and mass detectors among mass spectrometry imaging techniques. | ||
Laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS)
Of all the metallomic imaging approaches, ICP-MS based techniques have become a quantitative metal “go to” technique for good reason: ICP-MS provides some of the greatest sensitivities with detection limits of 10–100's of ppq for MS/MS systems and even down to single digit ppq for high-resolution magnetic sector field systems, boasts wide dynamic linear range of up to five orders of magnitude, and enables multi-element trace analysis of specific isotopes. These characteristics become critical for metallomic imaging, which requires the analysis of small amounts of biological material containing a wide range of elements in varying concentrations. Within a relatively short period of time, ICP-MS has replaced the previous workhorse of metal analysis, electro thermal atomic absorption spectroscopy (AAS).With front end coupling of laser ablation systems to ICP-MS, a focused laser beam is used to ablate solid sample material, with the resulting aerosol plume transferred into the ICP via a carrier gas. Through the reconstruction of ICP-MS signal counts from time to coordinate space, and with significant developments in the field of laser ablation over the past 10 years, fast generation of isotopically specific elemental distribution maps of biological samples down to spatial resolutions of one micron are opening applications to the cellular level, and sub-micron spatial resolution is now achievable utilising deconvolution methods.85 Recently reviewed by Van Malderen et al., new developments in cell ablation technology, faster washout speeds, and advances in post-acquisition image generation software for fast and powerful image rendering and analysis in both 2D and 3D86 are supporting an emerging surge of elemental bioimaging utilising LA-ICP-MS as a cornerstone technique.86 With additional developments in time-of-flight mass spectrometry (TOFMS), LA-ICP-TOFMS allows simultaneous determination of virtually the entire periodic table. Recent work coupling LA-ICP-TOFMS is incredibly promising for the field of metallomics, as it presents the first opportunity to capture the spatial distribution of the metallome in biological samples. Although ICP-TOFMS is less sensitive than ICP-MS by approximately a factor of 10, very recent advances in ablation cell technology, with introduction of the cobalt cell from Teledyne CETAC Technologies, has brought detection of heavier elements on par with most ICP-MS/MS. The overall result is extremely fast imaging capacity with near simultaneous measurements of the full elemental isotope mass range, supporting an emerging movement towards establishing LA-ICP-MS as a clinical diagnostic tool.
The use of LA-ICP-MS for bioimaging has reached the level of routine analysis in some specialized labs and has been thoroughly reviewed in the literature.87 Work is primarily dominated by endogenous mapping of metals in various types of organs (Fig. 2B)88–90 as well as determining penetration depth of specific metallodrugs,91,92 and can be done quantitatively using matrix matched standards for calibration. For quantification, a host of solid and liquid standard addition based methods including pressed pellets of reference materials, gelatin standards, metal spiked polymer film and tissue homogenates are thoroughly reviewed by Piekoszewski et al.93 Decisions on which calibration approach to utilise are taken at laboratory level. Therefore, there is a great need for leading research groups in the field of metallomic imaging to centralise expertise and develop standardised protocols that can be used by the growing metallomics community, key for extending the reach of metallomics and a clear goal for the newly established London Metallomics Facility, discussed at the end of this perspective.
Although tissue-scale analysis is becoming more routine, the increasing need for subcellular resolution, multiplexing capabilities for visualisation of biomarkers and even 3D metal distribution are driving the push past preconceived limitations of LA-ICP-MS with the help of several key researchers and commercial partners in the field. In relatively large fibroblast cells and macrophages, LA-ICP-MS has been used to track gold-labelled human regulatory macrophages in immune deficient mice,94 to image iodine as an elemental marker in differentiated fibroblast cells,95 the detection of Au NPs within fibroblast cells,96 and uptake of cadmium-based quantum dots in HeLa cells.97 Yet, many single cell analyses involve significantly smaller cells in the range of 10 μm. Recently, intracellular copper was quantified in the algae S. trochoidea by ablating whole individual cells to obtain information on distribution of copper uptake within the population as well as subcellular imaging using a spot size of 2 μm. Most importantly, this study cross-evaluated results with those obtained by SXRF, showing similar accumulation patterns and being the first to demonstrate the potential of LA-ICP-MS, not only for bioimaging at the subcellular level, but also its potential for high throughput single cell analysis.98 The increasing demand for establishing and imaging biomarkers within the context of human disease has led to multiplexing approaches in which metal lanthanide tagged antibodies are used to image specific proteins in tissue sections. Pioneering work illustrated this capability by imaging HER2 and 32 other proteins in human breast cancer tissue down to 1 μm spatial resolution,99,100 and this approach is now being explored in single cell imaging applications.101 Very recent software advances in post acquisition data processing and multi-modal data fusion have opened new possibilities for 3D LA-ICP-MS,102 demonstrated in a handful of publications looking at metals in murine brain with a spatial resolution of 80 μm,103 in mature wheat and rye grains with a spatial resolution of 20 μm,104 and most recently in Ceriodaphnia dubia with a spatial resolution of 5 μm using LA-ICP-TOFMS, correlated to microcomputed tomography (μ-CT).102 3D LA-ICP-MS has been time consuming, not only in data acquisition but also in data processing and image generation. However, as a result of recent combined hardware and software advances (HDIP software), 3D LA-ICP-MS is likely to play a greater role in the context of correlative 3D imaging.
Secondary ion mass spectrometry (SIMS)
Secondary ion mass spectrometry (SIMS) is by no means a new technique, being used for over half a century for surface analysis of inorganic materials. However, SIMS has undergone a revival with newly developed bioimaging applications, as it offers the highest lateral resolution of any MSI technique and is capable of imaging chemical species in tissues and single cells, due to its ability to achieve sub-micron spatial resolutions for both element and molecular imaging in the mass range from 1–2000 Da.105 In SIMS, a focused ion beam bombards a sample under vacuum, resulting in the ejection and ionisation of molecules from the sample surface. These ejected secondary ions are then analysed in a mass spectrometer. It is important to note that the majority of ejected molecules are in fact neutral and ionisation is not uniformly efficient for all species, illustrating one important caveat in SIMS with respect to metallomic applications involving transition metals that tend to ionise less easily relative to lighter elements. Despite these ionisation challenges, SIMS is still used to visualise metals in biological samples and adds the additional benefit of imaging lighter elements and molecular fragments to provide an informative molecular context for metals. SIMS can be run in both dynamic and static modes, where dynamic mode allows for depth profiling and excessive fragmentation leads to excellent elemental analysis, and where static mode minimizes surface erosion providing surface-sensitive molecular information.106 Although SIMS is not normally operated quantitatively with biological samples due to large variations in both sputtering efficiency (from heterogeneity of sample) and the ion yield of an element (from differences in inherent ionisation efficiencies), methods have been established to do so.93 Lighter elements are more readily ionised and thus SIMS is typically used to look at local intracellular concentrations of elements such as calcium, potassium, magnesium, and sodium.107Within the context of bioimaging, three main types of SIMS instruments have risen to the forefront: Fourier Transform ion cyclotron resonance (FTICR) SIMS, with its highly accurate mass-resolving power, time-of-flight SIMS (TOF-SIMS), chemically mapping samples at an order of magnitude better spatial resolution that FTICR-SIMS and with the additional benefit of depth profiling, and NanoSIMS, which offers sub-100 nm spatial resolution (Fig. 1D).108 A few examples include the use of NanoSIMS to visualize association of copper with cell components rich in proteins and phosphorus, highlighting the use of SIMS for intracellular tracking of essential trace elements in single cells.109 NanoSIMS showed the increase of ferritin in the coronal region of human AD plaques,110 whereas TOF-SIMS quantified cholesterol overload in the cerebral cortex of AD patients.111
Matrix assisted laser desorption/ionisation mass spectrometry imaging (MALDI-MSI)
Whereas SIMS is typically limited to a narrow mass range window of a few hundred Daltons, MALDI-MSI can expand this range and image molecules as large as 70 kDa in some instances,112 providing quantitative113 information on a heterogeneous molecular landscape for a wide range of analytes spanning proteins, peptides, and lipids to drugs and their metabolites.114 Despite MALDI-MSI being a label free approach, samples must be embedded in a chromophore-containing matrix to assist in the photo-volatilisation and ionisation process, which occurs as a result of a UV laser beam rastered across the sample surface under atmospheric pressure. The resulting ionised material is typically transferred to a TOF mass analyser, but higher mass resolving power can be obtained when coupled to an Orbitrap115 or FTICR mass analyser, allowing for protein identity assignment of similar average mass and with interspersed isotopomers revealing the presence of many protein adducts.116 With recent advances, spatial resolutions down to 1 μm are now possible, however 10–20 μm are more commonly used making this technique most appropriate for tissue level analyses.114Direct imaging of metal-containing compounds using MALDI-MSI is challenging due to poor ionisation efficiency and signal suppression from biological matrix effects.117 Therefore, MALDI-MSI is typically used in conjunction with LA-ICP-MS on sequential tissue sections and images are correlated in a “guilt by association” strategy (Fig. 2B). However, despite these analytical challenges, using an optimized sample preparation protocol, oxaliplatin and its metabolites were mapped in multicellular tumour spheroids for the first time, showing that most of the drug was detected in the outer area, and oxaliplatin complexed to methionine was at the core.118 When MALDI-MSI utilises high resolution mass spectrometry, the high mass resolving power of these techniques should allow for the identification of the molecular formulae including predictions of associated metals.1
Particle beam microscopy
We end our journey into the physical interactions of energy and matter with particle beam microscopy, where resulting physical phenomena from particle–matter interactions give rise to high-resolution images well beyond the diffraction limit of visible light, provide elemental information and chemical mapping, and fully quantitative elemental localisation (Fig. 6A, B). Taking advantage of the particle/wave dualism of the electron, its short wavelength (0.004 nm) allows for sub-nm spatial resolution of electron microscopy, providing unprecedented capabilities for obtaining subcellular structural context. When coupled to energy-dispersive (EDS/EDX) X-ray detectors and/or detectors for electron-energy loss spectroscopy (EELS), elemental identification and speciation can be incorporated. Particle induced X-ray emission (PIXE) provides increased sensitivity and fully quantitative elemental mapping, albeit at less refined spatial scales. Together these techniques fill important gaps in spatial resolution required for biological structural contexts as well as absolute quantitative mapping of metals down to the cellular level.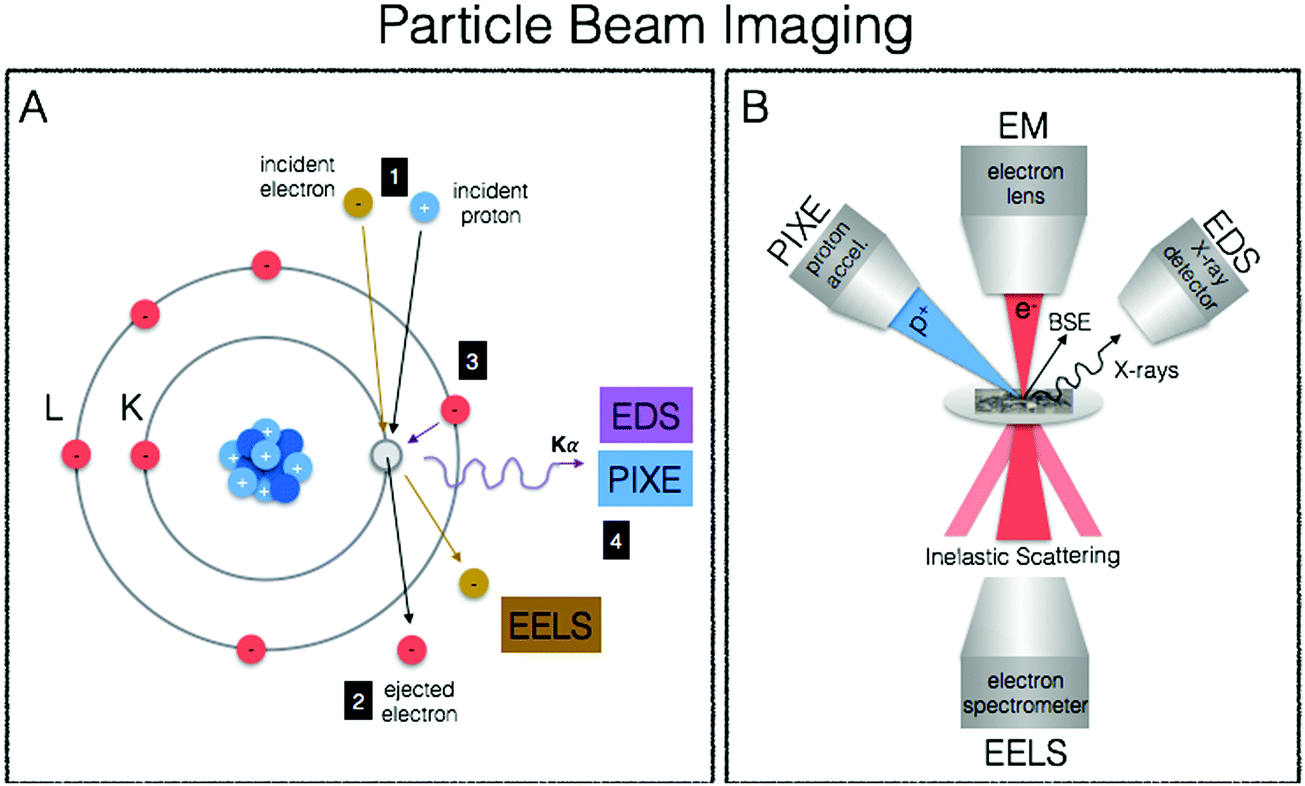 | ||
| Fig. 6 Principles of particle beam imaging techniques. (A) In both EM-EDS/EELS and PIXE a particle (electron or commonly proton, respectively) with sufficient energy is used to eject a core electron (1). In EM-EELS, the inelastically scattered incident electron is measured and the corresponding loss of energy provides element specific information such as coordination geometry and oxidation state (2). When an outer shell electron fills the inner core hole (3), element specific X-ray emission occurs and is measured in both EM-EDS and PIXE (4) to reveal elemental composition of a sample. (B) A schematic representation illustrating the principles of particle beam imaging techniques (BSE = backscattered electron). | ||
Electron microscopy (EM)
The best known particle-based imaging approach is no doubt electron microscopy (EM), operating on the same basic principles as light microscopy, but instead taking advantage of the short wavelength of electrons to achieve resolution many orders of magnitude better to reveal the finest details of internal structure (Fig. 1D), down to individual atoms in some cases. It is of no surprise then that EM has developed into an important cornerstone of biological imaging to address questions at the most refined spatial scales. Transmission EM (TEM), scanning transmission EM (STEM) and scanning EM (SEM) are used for analysis of biological material, major differences difference being that TEM/STEM sections are typically less than 100 nm, measure transmitted electrons, and offer the highest spatial resolution (down to 0.5 Å). In SEM, relatively thicker sections are used making sample preparation easier, and backscattered electrons are measured resulting in a loss of spatial resolution (0.4 nm) but increased depth of field. With advancements in cryo EM and developments in milling for serial block face imaging using focused ion beam (FIB), it is now possible to use cryo FIB-SEM for the direct and fast 3D imaging of large native frozen samples, including tissues.119TEM and SEM are not only used for generation of highly resolved ultrastructure, but when coupled EDS X-ray detectors and/or detectors for EELS, surface elemental composition can be obtained. When this analytical information is presented as a false colour overlay on traditional EM images, ions and biomolecules are directly visualised and elemental fingerprints of subcellular components are analysed in an approach coined as ColorEM.120 EDS and EELS are complementary in that EDS is particularly sensitive to heavier elements up to molybdenum, whereas EELS is best used for lighter elements ranging from carbon through the 3d transition metals. An additional benefit of EELS is the ability to image structure through carbon, nitrogen, and oxygen without counterstaining and speciation information of the coordination environment is possible. Although (S)TEM/SEM-EDS/EELS can provide the highest spatial resolutions, element detection is orders of magnitude less sensitive than compared to X-ray spectroscopic and mass spectrometric techniques due to intense Bremsstrahlung background, particularly for EDS detection, and applications in metallomic bioimaging have been recently reviewed.121
STEM-EELS is the preferred EM method for trace metal mapping in biological samples due to its increased sensitivity relative to EDS.122 Using STEM-EELS calcium concentrations in the endoplasmic reticulum and mitochondria were mapped in 100 nm thick sections of mouse cerebellar cortex, indicating the role of the endoplasmic reticulum in calcium regulation,123 and quantitative manganese mapping of rat brain cells showed that manganese particles primarily localised in mitochondria.124 This approach has also been extended to 3D with post acquisition computational methods, as illustrated in the generation of 3D maps of iron bound to ferritin in degenerating neurons of mice125 and new applications for the identification of markers and nanomaterials within the context of diagnostics and therapeutics have recently emerged.121
Particle-induced X-ray emission (PIXE)
Although a far cry from the spatial resolution capabilities of TEM/SEM-EDS/EELS, μ-PIXE is more sensitive and concentrations are fully quantitative without the need for calibration as particle beam interactions with electronic shells of the atoms have been extensively described.126 μ-PIXE is based on the same physical excitation phenomenon previously described for X-ray spectroscopy, with the difference being the source of excitation (protons in many cases). When carried out in combination with RBS (Rutherford Backscattering Spectrometry) to measure mass of sample, metal content can be spatially expressed in terms of weight of element per g of dry mass,127 with the powerful effect of being able to correct for changes in material density within a given sample. With spatial resolutions down to 1 μm and good sensitivity for essential trace elements (i.e. manganese, iron, copper, zinc, selenium), toxic heavy metals (mercury, lead) and pharmacological compounds (platinum), μ-PIXE provides a useful tool for metal distribution studies at the subcellular level.126For example, μ-PIXE showed that cobalt accumulated in the nucleus and peri-nuclear regions of skin human cells, causing a decrease in both magnesium and zinc and suggesting that cobalt may interfere in magnesium and zinc cellular homeostasis.128 Gadolinium-based nanoparticles along with essential elements (phosphorus, sulphur, chlorine, potassium, calcium, iron, copper, zinc) were quantitatively mapped in U87 human glioblastoma tumours with high spatial resolution using a combination of μ-PIXE and RBS, showing heterogeneous gadolinium NP distribution and effects to endogenous elemental distribution and concentrations (Fig. 2A).129
Multidimensional metallomics
At the heart of our metallomic investigations we ideally seek to quantify metal species in a particular location within a meaningful biological/functional context at a certain point in time. In reality, the ability to obtain this information in a single measurement is virtually impossible. We frequently face challenges of low concentrations in complex biological matrices often with the requirement for high spatial resolution and perhaps even measurement of temporal dynamics. Because of these circumstances, there is a clear need to use and continually develop new correlative workflows between analytical techniques that can provide complementary information to address questions of metal quantification, localisation, chemical and structural biological contexts, and temporal dynamics in metal- related biological processes. This statement is not new. Certainly we have read the growing number of calls in the literature for more correlative based approaches in science. However, even in reading state of the art reviews, one can get easily lost in a sea of acronyms, and despite reading countless articles, it feels quite a daunting task to have a comprehensive understanding of how such approaches can be combined and what each has specifically to offer.I have always envisioned a “go to” metallomic analytical table that finally presented the best selection of analytical tools for in situ metallomic studies. In this light, Table 1 presents a synthesis of techniques described in this perspective, meant to be an overview reference guide, which can be utilised in a multifaceted way. Rows display information on what each technique provides, grouped according to spectroscopy, spectrometry and particle beam approaches, each with corresponding literature citations of comprehensive reviews addressing specifics such as sample preparation and calibration. Columns allow for quick identification of techniques within the context of a specific requirement (i.e. quantification vs. speciation vs. localisation of metals). Additional colour coding allows for easy identification of techniques best suited for the level of study (i.e. subcellular, whole cell, tissue, organ). It should also be noted that often sophisticated and/or expensive instrumentation is required to conduct the analyses described, thus requiring use of core facilities and/or collaboration with specialised research groups as well as the pursuit of access calls at synchrotron facilities worldwide. Despite such approaches being far from fast bench-top solutions, the benefits of the required cross-disciplinary interactions speak for themselves in the results produced.
| a LOD for FTIR and Raman spectroscopy depends on concentration, sample thickness, and background interfering spectral features (i.e. S/N). b LOD dependent on the specificity and selectivity of the probe or sensor.30 For zinc, pM concentrations are detectable intracellularly.138 c A low primary ion beam current is used liberate ions, molecules and molecular clusters for analysis so that characterisation of organic macromolecules is possible, in contrast to NanoSIMS, which is better for quantitative analysis of elements due to a higher primary ion beam current and higher secondary ion yield but with a loss of organic compounds. d Resolution is 0.5 mm in animals and 0.5 cm in humans. e Typically performed on fixed or dehydrated samples due to H2O interferences, but live cell imaging at synchrotron facilities using microfluidic devices with D2O have been performed.139 f Scanning speeds can be decreased to less than 1 s if activity is high enough. |
|---|

|
Correlative workflows should provide complementary information, resulting in a greater understanding of a question at hand, which could not have been otherwise answered. This goal is different than using multiple techniques to confirm a specific observation (i.e. using μ-XRF and LA-ICP-MS to localise a metal at similar spatial resolutions). This statement is not meant to discredit the immense value and power of confirming an observation using multiple approaches, but rather to highlight the important distinction between corroboration and correlation of different information (i.e. speciation and localisation) to gain a richer understanding.
Recently, the linkage between bone crystallinity and ratios of Mg/Ca and Sr/Ca was explored in two rat models with chronic kidney disease (CKD) and diabetes mellitus (DM) by combining LA-ICP-MS, Raman spectroscopy and SEM. Higher Mg/Ca ratios, determined using LA-ICP-MS, correlated to a decrease in bone crystallinity, measured using Raman, and indicated calcium replacement by magnesium in deterioration of the hydroxyapatite (HAp) structure. To further investigate the change in crystallinity, SEM was used to measure HAp crystals, which decreased with increasing Mg/Ca and Sr/Ca. Interestingly, both crystallinity and Mg/Ca ratios for CKD rats were significantly different from controls and DM rats, highlighting that possible disease related alterations in elemental metabolism may influence ionic substitutions of bone calcium.130 Within the context of anti-cancer therapies, correlations between changes in protein secondary structure and platinum distribution were discovered, as well as between cell nuclear morphology and decrease of phosphorus, zinc, and magnesium through combining LA-ICP-MS and FTIR spectroscopy. Multivariate analyses of the results indicated different degrees of tumour viability and successfully discriminated dead tumour regions.131 Within the context of exploring the use of magnetic nanoparticles for diagnostic imaging, therapeutics and biotechnology, the uptake and interaction of Ag–magnetite and Au–magnetite composite nanoparticles in 3T3 fibroblast cells was studied. Cryo-SXT confirmed endocytotic uptake and stability of particles in endosomes and lysosomes, indicating their potential use as both optical and magnetic probes in living cells. SERS provided information on the local chemical environmental of the particles, indicating a change in surface composition and interaction of intracellular biomolecules specifically with the magnetite. Comparison of nanoparticle uptake and localisation with and without magnetite using LA-ICP-MS indicated that the magnetite is an important component for uptake.58 In another study, NanoSIMS allowed chemical imaging of macro and trace elements with subcellular resolution (element mapping). Calcium, magnesium, and phosphorus as well as the trace elements iron, copper, and zinc present at basal levels were detected in pyrenoids, contractile vacuoles, and granules. Some metals were even localized in small vesicles of about 200 nm size.132 In remarkable work, a conceptual model of the coccolith calcium pathway, essential for understanding coccolithophore calcification process within the global carbon cycle, was proposed based on results from a combination of techniques spanning light microscopy, X-ray spectroscopy and electron microscopy. Combining cryo-SXT, spatially and temporally resolved XANES, cryo-focused ion beam scanning electron microscopy (FIB-SEM), EDX and CLSM, a highly concentrated and previously unidentified pool of intracellular calcium was both imaged and speciated within the ultrastructural environment and showed that polyphosphates and other elements including magnesium, are co-localized with calcium.59 This work in particular highlights how a comprehensive picture of an important biological process can be ascertained when a wide range of analytical techniques are combined.
Conclusion & future directions
The field of metallomics takes a holistic approach to understand the metallobiochemistry of cells and organisms by utilising analytical tools to decipher the biological roles of metals in both space and time.133 Every cell utilizes a remarkably diverse range of metal ions (sodium, potassium, magnesium, calcium, iron, manganese, copper, zinc, and molybdenum) and non-metals (selenium, chloride, bromide and iodide) in addition to the standard elemental building blocks of biological molecules (carbon, oxygen, nitrogen, hydrogen, sulphur and phosphorus). The in situ metallomic imaging tools described in this perspective have the power to characterise these metallomes within the life span of individual cells and tissues with significant value for fundamental scientific questions relating to determination of cell fate and health, trace element metabolism, and for translational science in diagnosis, prognosis, and therapy. In fact, metallomes are as dynamic as the biological systems they exist within, opening another dimension of metallomics called meta-metallomics that addresses individual variation in the life trajectory as well as the influence of environmental factors such as nutrition, exposure and disease.134 There is currently an unmet recognized scientific need for establishing such profiles, and through the integration of state of the art analytical techniques described in this perspective, we can now generate elemental profiles with both spatial and temporal resolution within functional biological contexts at scales ranging from atomic to whole body.Not only is a more comprehensive utilisation of correlative techniques required to advance the field of metallomics, but, more significantly, a shift in our thinking and experimental approach in employing and applying these techniques. When I step back in life and summit that peak to get a full 360 degree view, magically everything's proper order and function becomes clear. Science is not isolated from life (quite the contrary), and this simplistic yet powerful clarity is blurred when our natural scientific tendency towards exclusivity dominates our thinking. From my many conversations with chemists, biologist, physicists, bioinformatisticians, engineers, and even clinicians from fields ranging from stem cell biology to global nutritional health to atmospheric pollution and environmental toxicology, one point is absolutely clear: there is a growing recognition that we cannot expect to solve our pressing scientific problems by summing up fragmented pieces of information obtained from the old “silo” approach. Across all disciplines, there is an overwhelming call in the literature for more integrative experimental approaches and analytical techniques to foster comprehensive cross-disciplinary research. To be able to put each analytical tool in its proper perspective and, rather than seeing individual disconnected methods, see a powerful and integrated network of connected approaches is the future trend, not only for the field of metallomics but science as a whole. What if we were to take a cue from Nature, where nothing operates under exclusivity and perfectly functions in an all-encompassing dynamic inclusivity. To fully understand the multitude of questions and be able to present real solutions for human health and disease along with our environmental impact, like Nature, we must also operate in a multiple and all-inclusive fashion in our experimental and analytical approach. The analytics must be placed into a larger context, and this perspective piece illustrates the multitude of synergistic capacities within a metallomics methodological approach, where with imagination, intuition and focused interaction between researchers from different fields, metallomics can grow to stand with the likes of genomics, proteomics and metabolomics.
To accomplish this goal, a scientific platform is required on which direct and productive interactions between researchers in all scientific fields, clinicians, and commercial partners are established. It requires us as scientists to transcend the present structure of education and research disciplines. With the recent creation of the London Metallomics Facility (LMF), finally comes the incredible opportunity to establish such a platform, which I am honoured to play a role in shaping. The vision of the LMF is not only to expand frontiers in correlative metal bioimaging through offering the most advanced-to-date portfolio of metallomics approaches to address the structural and functional roles of chemical elements in biology and medicine, but to also serve as a centralising hub to comprehensively integrate current analytical expertise in metallomics, foster direct interaction between leading research groups, and lead educational outreach and public engagement strategies. In this sense, metallomic analytics is more than about understanding the role of metals in biology. It presents a powerful opportunity to show in action how we can do science more effectively in a highly collaborative way.
Abbreviations
| pO2 | Oxygen partial pressure |
| μ-CT | Microcomputed tomography |
| AAS | Atomic absorption spectroscopy |
| AFM | Atomic force microscope |
| AS | Alzheimer's disease |
| CDI | Coherent diffractive imaging |
| CKD | Chronic kidney disease |
| CLSM | Confocal laser scanning microscopy |
| Cryo-SXT | Soft X-ray tomography |
| DM | Diabetes mellitus |
| EDS/EDX | Energy-dispersive X-ray spectroscopy |
| EELS | Electron-energy loss spectroscopy |
| EM | Electron microscopy |
| EPRI | Electron paramagnetic resonance imaging |
| ESR | Electron spin resonance |
| EXAFS | Extended X-ray absorption fine structure |
| FIB | Focused ion beam |
| FRET | Förster resonance energy transfer |
| FTICR | Fourier transform ion cyclotron resonance |
| FTIR | Fourier Transform infrared |
| HAp | Hydroxyapatite |
| LA-ICP-MS | Laser ablation inductively coupled plasma mass spectrometry |
| MALDI-MSI | Matrix assisted laser desorption/ionisation mass spectrometry imaging |
| MRI | Magnetic resonance imaging |
| MRM | Magnetic resonance microscopy |
| MSI | Mass spectrometry imaging |
| MS | Multiple sclerosis |
| PALM | Photo activated localisation microscopy |
| PET | Positron emission tomography |
| PIXE | Particle induced X-ray emission |
| RBS | Rutherford backscattering spectrometry |
| ROS | Reactive oxygen species |
| RRS | Resonance Raman spectroscopy |
| TEM | Transmission electron microscopy |
| TOFMS | Time-of-flight mass spectrometry |
| SEM | Scanning electron microscopy |
| SERS | Surface enhanced Raman spectroscopy |
| SIM | Structured illumination microscopy |
| SIMS | Secondary ion mass spectrometry |
| SMLM | Single molecule localisation microscopy |
| SPIM | Selective plane illumination microscopy |
| SR | Synchrotron radiation |
| SRM | Super resolution microscopy |
| SRS | Spectroscopic stimulated Raman scattering |
| s-SNOM | Scanning near-field optical microscopy (s-SNOM) |
| STED | STimulated emission depletion microscopy |
| STEM | Scanning transmission electron microscopy |
| STORM | Stochastic optical reconstruction microscopy |
| STXM | Scanning transmission X-ray microscopy |
| SXRF | Synchrotron X-ray fluorescence |
| XANES | X-ray absorption near edge structure |
| XAS | X-ray absorption spectroscopy |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
I would like to extend a sincere thank you to Wolfgang Maret for fruitful discussions and review of this manuscript, to William Hart for input on developing FTIR and Raman spectroscopic based imaging techniques, and to the London Metallomics Facility funded by the Wellcome trust (grant 202902/Z/16/Z).References
- W. Maret, J. A. Caruso, C. H. Contag, D. P. Giedroc, P.-L. Hagedoorn, A. Matusch, E. P. Skaar and R. B. Thompson, Trace Metals and Infectious diseases, 2015, pp. 1–61 Search PubMed.
- W. R. Hagen, Dalton Trans., 2006, 4415–4434 RSC.
- P. Danhier and B. Gallez, Contrast Media Mol. Imaging, 2015, 10, 266–281 CrossRef CAS.
- B. Epel, M. Kotecha and H. J. Halpern, J. Magn. Reson., 2017, 280, 149–157 CrossRef CAS.
- B. B. Williams and H. J. Halpern, Biomedical EPR, Part A: Free Radicals, Metals, Medicine, and Physiology, Springer, Boston, MA, New York, 2005, vol. 23, pp. 283–319 Search PubMed.
- N. Khan and H. Swartz, Mol. Cell. Biochem., 2002, 234–235, 341–357 CrossRef.
- S. Kishimoto, K.-I. Matsumoto, K. Saito, A. Enomoto, S. Matsumoto, J. B. Mitchell, N. Devasahayam and M. C. Krishna, Antioxid. Redox Signaling, 2018, 28, 1378–1393 CrossRef CAS.
- P. Caravan and Z. Zhang, The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, John Wiley & Sons, Ltd, Chichester, UK, 2013, vol. 221, pp. 311–342 Search PubMed.
- B. Z. Dashevsky, T. D'Alfonso, E. J. Sutton, A. Giambrone, E. Aronowitz, E. A. Morris, K. Juluru and D. J. Ballon, Sci. Rep., 2015, 5, 17435 CrossRef CAS PubMed.
- C. H. Lee, N. Bengtsson, S. M. Chrzanowski, J. J. Flint, G. A. Walter and S. J. Blackband, Sci. Rep., 2017, 7, 39496 CrossRef CAS.
- H. A. Arnon, N. D. Tirukoti and A. B. Shir, Isr. J. Chem., 2017, 57, 843–853 CrossRef.
- A. J. M. Lubag, L. M. De Leon-Rodriguez, S. C. Burgess and A. D. Sherry, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 18400–18405 CrossRef CAS PubMed.
- M. V. Clavijo Jordan, S.-T. Lo, S. Chen, C. Preihs, S. Chirayil, S. Zhang, P. Kapur, W.-H. Li, L. M. De Leon-Rodriguez, A. J. M. Lubag, N. M. Rofsky and A. D. Sherry, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E5464–E5471 CrossRef CAS.
- W. Thomson and T. Guthrie, Elements of Natural Philosophy, 1879 Search PubMed.
- M. Z. Kastyak-Ibrahim, M. J. Nasse, M. Rak, C. Hirschmugl, M. R. Del Bigio, B. C. Albensi and K. M. Gough, NeuroImage, 2012, 60, 376–383 CrossRef CAS.
- T. Nakamura, J. G. Kelly, J. Trevisan, L. J. Cooper, A. J. Bentley, P. L. Carmichael, A. D. Scott, M. Cotte, J. Susini, P. L. Martin-Hirsch, S. Kinoshita, N. J. Fullwood and F. L. Martin, Mol. Vision, 2010, 16, 359–368 CAS.
- M. J. Baker, J. Trevisan, P. Bassan, R. Bhargava, H. J. Butler, K. M. Dorling, P. R. Fielden, S. W. Fogarty, N. J. Fullwood, K. A. Heys, C. Hughes, P. Lasch, P. L. Martin-Hirsch, B. Obinaju, G. D. Sockalingum, J. Sulé-Suso, R. J. Strong, M. J. Walsh, B. R. Wood, P. Gardner and F. L. Martin, Nat. Protoc., 2014, 9, 1771–1791 CrossRef CAS.
- L. M. Miller, M. W. Bourassa and R. J. Smith, Biochim. Biophys. Acta, 2013, 1828, 2339–2346 CrossRef CAS.
- F. Keilmann and R. Hillenbrand, Philos. Trans. R. Soc. London, Ser. A, 2004, 362, 787–805 CrossRef CAS.
- H. Amrania, L. Drummond, R. C. Coombes, S. Shousha, L. Woodley-Barker, K. Weir, W. Hart, I. Carter and C. C. Phillips, Faraday Discuss., 2016, 187, 539–553 RSC.
- A. Balbekova, M. Bonta, S. Török, J. Ofner, B. Döme, A. Limbeck and B. Lendl, Anal. Methods, 2017, 9, 5464–5471 RSC.
- M. C. Martin, C. Dabat-Blondeau, M. Unger, J. Sedlmair, D. Y. Parkinson, H. A. Bechtel, B. Illman, J. M. Castro, M. Keiluweit, D. Buschke, B. Ogle, M. J. Nasse and C. J. Hirschmugl, Nat. Methods, 2013, 10, 861–864 CrossRef CAS.
- A. Torreggiani and A. Tinti, Metallomics, 2010, 2, 246–260 RSC.
- H. J. Butler, L. Ashton, B. Bird, G. Cinque, K. Curtis, J. Dorney, K. Esmonde-White, N. J. Fullwood, B. Gardner, P. L. Martin-Hirsch, M. J. Walsh, M. R. McAinsh, N. Stone and F. L. Martin, Nat. Protoc., 2016, 11, 664–687 CrossRef CAS.
- M. Kakita, M. Okuno and H.-O. Hamaguchi, J. Biophotonics, 2012, 6, 256–259 CrossRef.
- W. Xu, T. Barrientos and N. C. Andrews, Cell Metab., 2013, 17, 319–328 CrossRef CAS.
- Y. Zhou, C.-H. Liu, Y. Pu, G. Cheng, L. Zhou, J. Chen, K. Zhu and R. R. Alfano, in Optical Biopsy XIV: Toward Real-Time Spectroscopic Imaging and Diagnosis, ed. R. R. Alfano and S. G. Demos, International Society for Optics and Photonics, 2016, vol. 9703, p. 97031R Search PubMed.
- C. Krafft, M. Schmitt, I. W. Schie, D. C. May, C. Matthäus, T. Bocklitz and J. Popp, Angew. Chem., Int. Ed., 2017, 56, 4392–4430 CrossRef CAS.
- H. Lin, C. S. Liao, P. Wang, N. Kong and J.-X. Cheng, Light: Sci. Appl., 2018, 7, e17179 CrossRef.
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS.
- T. Fukada, S. Yamasaki, K. Nishida, M. Murakami and T. Hirano, J. Biol. Inorg. Chem., 2011, 16, 1123–1134 CrossRef CAS.
- W. Maret, Biometals, 2011, 24, 411–418 CrossRef CAS.
- A. Krezel and W. Maret, Arch. Biochem. Biophys., 2016, 611, 3–19 CrossRef CAS.
- H. Haase and W. Maret, Exp. Cell Res., 2003, 291, 289–298 CrossRef CAS.
- M. Wilson, C. Hogstrand and W. Maret, J. Biol. Chem., 2012, 287, 9322–9326 CrossRef CAS PubMed.
- E. Bellomo, A. Massarotti, C. Hogstrand and W. Maret, Metallomics, 2014, 6, 1229–1239 RSC.
- E. L. Que, R. Bleher, F. E. Duncan, B. Y. Kong, S. C. Gleber, S. Vogt, S. Chen, S. A. Garwin, A. R. Bayer, V. P. Dravid, T. K. Woodruff and T. V. O'Halloran, Nat. Chem., 2014, 7, 130–139 CrossRef.
- B.-C. Chen, W. R. Legant, K. Wang, L. Shao, D. E. Milkie, M. W. Davidson, C. Janetopoulos, X. S. Wu, J. A. Hammer, Z. Liu, B. P. English, Y. Mimori-Kiyosue, D. P. Romero, A. T. Ritter, J. Lippincott-Schwartz, L. Fritz-Laylin, R. D. Mullins, D. M. Mitchell, J. N. Bembenek, A.-C. Reymann, R. Böhme, S. W. Grill, J. T. Wang, G. Seydoux, U. S. Tulu, D. P. Kiehart and E. Betzig, Science, 2014, 346, 1257998 CrossRef.
- J. Vangindertael, R. Camacho, W. Sempels, H. Mizuno, P. Dedecker and K. P. F. Janssen, Methods Appl. Fluoresc., 2018, 6, 022003 CrossRef CAS.
- D. J. Hare, E. J. New, M. D. De Jonge and G. McColl, Chem. Soc. Rev., 2015, 44, 5941–5958 RSC.
- R. Polishchuk and S. Lutsenko, Histochem. Cell Biol., 2013, 140, 285–295 CrossRef CAS.
- B. De Samber, G. Silversmit, R. Evens, K. De Schamphelaere, C. Janssen, B. Masschaele, L. Van Hoorebeke, L. Balcaen, F. Vanhaecke, G. Falkenberg and L. Vincze, Anal. Bioanal. Chem., 2008, 390, 267–271 CrossRef CAS.
- R. Ortega, P. Cloetens, G. Devès, A. Carmona and S. Bohic, PLoS One, 2007, 2, e925 CrossRef.
- T. Leonardo, E. Farhi, A. M. Boisson, J. Vial, P. Cloetens, S. Bohic and C. Rivasseau, Metallomics, 2014, 6, 316–329 RSC.
- R. McRae, B. Lai and C. J. Fahrni, Metallomics, 2013, 5, 52–61 RSC.
- O. Ponomarenko, P. F. La Porte, S. P. Singh, G. Langan, D. E. B. Fleming, J. E. Spallholz, M. Alauddin, H. Ahsan, S. Ahmed, J. Gailer, G. N. George and I. J. Pickering, Metallomics, 2017, 9, 1585–1595 RSC.
- S. Pfaender and A. M. Grabrucker, Metallomics, 2014, 6, 960–977 RSC.
- H. Wang, M. Wang, B. Wang, M. Li, H. Chen, X. Yu, Y. Zhao, W. Feng and Z. Chai, Metallomics, 2012, 4, 289–296 RSC.
- R. A. Colvin, Q. Jin, B. Lai and L. Kiedrowski, PLoS One, 2016, 11, e0159582 CrossRef.
- C. Sanchez-Cano, I. Romero-Canelón, Y. Yang, I. J. Hands-Portman, S. Bohic, P. Cloetens and P. J. Sadler, Chem. – Eur. J., 2017, 23, 2512–2516 CrossRef CAS.
- K. M. Davies, D. J. Hare, S. Bohic, S. A. James, J. L. Billings, D. I. Finkelstein, P. A. Doble and K. L. Double, Anal. Chem., 2015, 87, 6639–6645 CrossRef CAS PubMed.
- S. A. James, D. J. Hare, N. L. Jenkins, M. D. De Jonge, A. I. Bush and G. McColl, Sci. Rep., 2016, 1–8 CAS.
- B. F. Popescu, J. M. Frischer, S. M. Webb, M. Tham, R. C. Adiele, C. A. Robinson, P. D. Fitz-Gibbon, S. D. Weigand, I. Metz, S. Nehzati, G. N. George, I. J. Pickering, W. Brück, S. Hametner, H. Lassmann, J. E. Parisi, G. Yong and C. F. Lucchinetti, Acta Neuropathol., 2017, 134, 45–64 CrossRef CAS.
- M. J. Ceko, K. Hummitzsch, N. Hatzirodos, W. Bonner, S. A. James, J. K. Kirby, R. J. Rodgers and H. H. Harris, Metallomics, 2015, 7, 756–765 RSC.
- M. Obst and G. Schmid, Methods in Molecular Biology, Humana Press, Totowa, NJ, 2013, vol. 1117, pp. 757–781 Search PubMed.
- W. G. Müller, J. B. Heymann, K. Nagashima, P. Guttmann, S. Werner, S. Rehbein, G. Schneider and J. G. McNally, J. Struct. Biol., 2012, 177, 179–192 CrossRef.
- M. Harkiolaki, M. C. Darrow, M. C. Spink, E. Kosior, K. Dent and E. Duke, Emerging Top. Life Sci., 2018, 2, 81–92 CrossRef.
- T. Büchner, D. Drescher, V. Merk, H. Traub, P. Guttmann, S. Werner, N. Jakubowski, G. Schneider and J. Kneipp, Analyst, 2016, 141, 5096–5106 RSC.
- S. Sviben, A. Gal, M. A. Hood, L. Bertinetti, Y. Politi, M. Bennet, P. Krishnamoorthy, A. Schertel, R. Wirth, A. Sorrentino, E. Pereiro, D. Faivre and A. Scheffel, Nat. Commun., 2016, 7, 11228 CrossRef CAS.
- S. Kapishnikov, A. Weiner, E. Shimoni, P. Guttmann, G. Schneider, N. Dahan-Pasternak, R. Dzikowski, L. Leiserowitz and M. Elbaum, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11188–11193 CrossRef CAS.
- A. Cruz-Adalia, G. Ramirez-Santiago, C. Calabia-Linares, M. Torres-Torresano, L. Feo, M. Galán-Díez, E. Fernández-Ruiz, E. Pereiro, P. Guttmann, M. Chiappi, G. Schneider, J. L. Carrascosa, F. J. Chichón, G. Martínez Del Hoyo, F. Sánchez-Madrid and E. Veiga, Cell Host Microbe, 2014, 15, 611–622 CrossRef CAS.
- H.-Y. Chen, D. M.-L. Chiang, Z.-J. Lin, C.-C. Hsieh, G.-C. Yin, I. C. Weng, P. Guttmann, S. Werner, K. Henzler, G. Schneider, L.-J. Lai and F.-T. Liu, Sci. Rep., 2016, 6, 34879 CrossRef CAS.
- M. A. Le Gros, E. J. Clowney, A. Magklara, A. Yen, E. Markenscoff-Papadimitriou, B. Colquitt, M. Myllys, M. Kellis, S. Lomvardas and C. A. Larabell, Cell Rep., 2016, 17, 2125–2136 CrossRef CAS.
- E. M. H. Duke, M. Razi, A. Weston, P. Guttmann, S. Werner, K. Henzler, G. Schneider, S. A. Tooze and L. M. Collinson, Ultramicroscopy, 2014, 143, 77–87 CrossRef CAS.
- J. Miao, P. Charalambous, J. Kirz and D. Sayre, Nature, 1999, 400, 342–344 CrossRef CAS.
- J. Miao, T. Ishikawa, I. K. Robinson and M. M. Murnane, Science, 2015, 348, 530–535 CrossRef CAS.
- A. Tripathi, J. Mohanty, S. H. Dietze, O. G. Shpyrko, E. Shipton, E. E. Fullerton, S. S. Kim and I. McNulty, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13393–13398 CrossRef CAS.
- D. A. Shapiro, Y.-S. Yu, T. Tyliszczak, J. Cabana, R. Celestre, W. Chao, K. Kaznatcheev, A. L. D. Kilcoyne, F. Maia, S. Marchesini, Y. S. Meng, T. Warwick, L. L. Yang and H. A. Padmore, Nat. Photonics, 2014, 8, 765 CrossRef CAS.
- F. Pfeiffer, Nat. Photonics, 2018, 12, 9–17 CrossRef CAS.
- A. M. Maiden, G. R. Morrison, B. Kaulich, A. Gianoncelli and J. M. Rodenburg, Nat. Commun., 2013, 4, 1669 CrossRef CAS.
- A. M. Maiden and J. M. Rodenburg, Ultramicroscopy, 2009, 109, 1256–1262 CrossRef CAS.
- J. Deng, D. J. Vine, S. Chen, Y. S. G. Nashed, Q. Jin, N. W. Phillips, T. Peterka, R. Ross, S. Vogt and C. J. Jacobsen, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2314–2319 CrossRef CAS.
- X. Zhu, A. P. Hitchcock, D. A. Bazylinski, P. Denes, J. Joseph, U. Lins, S. Marchesini, H.-W. Shiu, T. Tyliszczak and D. A. Shapiro, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E8219–E8227 CrossRef CAS.
- M. Gallagher-Jones, C. S. B. Dias, A. Pryor, K. Bouchmella, L. Zhao, Y. H. Lo, M. B. Cardoso, D. Shapiro, J. Rodriguez and J. Miao, Sci. Rep., 2017, 7, 4757 CrossRef.
- S. H. Shahmoradian, E. H. R. Tsai, A. Diaz, M. Guizar-Sicairos, J. Raabe, L. Spycher, M. Britschgi, A. Ruf, H. Stahlberg and M. Holler, Sci. Rep., 2017, 7, 6291 CrossRef CAS.
- T. R. DeGrado, B. J. Kemp, M. K. Pandey, H. Jiang, T. M. Gunderson, L. R. Linscheid, A. R. Woodwick, D. M. McConnell, J. G. Fletcher, G. B. Johnson, R. C. Petersen, D. S. Knopman and V. J. Lowe, Mol. Imaging, 2016, 15, 1–10 CrossRef CAS.
- G. Saar, C. M. Millo, L. P. Szajek, J. Bacon, P. Herscovitch and A. P. Koretsky, Mol. Imaging Biol., 2018, 20, 562–574 CrossRef CAS.
- M. Bruehlmeier, K. L. Leenders, P. Vontobel, C. Calonder, A. Antonini and A. Weindl, J. Nucl. Med., 2000, 41, 781–787 CAS.
- J. J. Bartnicka and P. Blower, J. Nucl. Med., 2018, 59, 1355–1359 CrossRef.
- R. Chakravarty, S. Chakraborty and A. Dash, Mol. Pharmaceutics, 2016, 13, 3601–3612 CrossRef CAS.
- F. Xie, Y. Xi, J. M. Pascual, O. Muzik and F. Peng, Metab. Brain Dis., 2017, 32, 717–726 CrossRef CAS.
- F. Peng, Ann. N. Y. Acad. Sci., 2014, 1314, 24–31 CrossRef CAS.
- J. B. Torres, E. M. Andreozzi, J. T. Dunn, M. Siddique, I. Szanda, D. R. Howlett, K. Sunassee and P. J. Blower, J. Nucl. Med., 2016, 57, 109–114 CrossRef CAS.
- E. M. Andreozzi, J. B. Torres, K. Sunassee, J. Dunn, S. Walker-Samuel, I. Szanda and P. J. Blower, Metallomics, 2017, 9, 1622–1633 RSC.
- S. J. M. Van Malderen, J. T. van Elteren and F. Vanhaecke, Anal. Chem., 2015, 87, 6125–6132 CrossRef CAS.
- S. J. M. Van Malderen, A. J. Managh, B. L. Sharp and F. Vanhaecke, J. Anal. At. Spectrom., 2016, 31, 423–439 RSC.
- D. Pozebon, G. L. Scheffler and V. L. Dressler, J. Anal. At. Spectrom., 2017, 32, 890–919 RSC.
- J. Sabine Becker, A. Matusch, C. Palm, D. Salber, K. A. Morton and J. Susanne Becker, Metallomics, 2010, 2, 104–111 RSC.
- S. G. Boaru, U. Merle, R. Uerlings, A. Zimmermann, C. Flechtenmacher, C. Willheim, E. Eder, P. Ferenci, W. Stremmel and R. Weiskirchen, J. Cell. Mol. Med., 2015, 19, 806–814 CrossRef CAS.
- E. Moreno-Gordaliza, C. Giesen, A. Lázaro, D. Esteban-Fernández, B. Humanes, B. Cañas, U. Panne, A. Tejedor, N. Jakubowski and M. M. Gómez-Gómez, Anal. Chem., 2011, 83, 7933–7940 CrossRef CAS.
- R. F. S. Lee, S. Theiner, A. Meibom, G. Koellensperger, B. K. Keppler and P. J. Dyson, Metallomics, 2017, 9, 365–381 RSC.
- S. Theiner, S. J. M. Van Malderen, T. Van Acker, A. Legin, B. K. Keppler, F. Vanhaecke and G. Koellensperger, Anal. Chem., 2017, 89, 12641–12645 CrossRef CAS.
- K. Jurowski, B. Buszewski and W. Piekoszewski, Talanta, 2015, 131, 273–285 CrossRef CAS PubMed.
- A. J. Managh, R. W. Hutchinson, P. Riquelme, C. Broichhausen, A. K. Wege, U. Ritter, N. Ahrens, G. E. Koehl, L. Walter, C. Florian, H. J. Schlitt, H. J. Reid, E. K. Geissler, B. L. Sharp and J. A. Hutchinson, J. Immunol., 2014, 193, 2600–2608 CrossRef CAS.
- C. Giesen, L. Waentig, T. Mairinger, D. Drescher, J. Kneipp, P. H. Roos, U. Panne and N. Jakubowski, J. Anal. At. Spectrom., 2011, 26, 2160–2167 RSC.
- D. Drescher, C. Giesen, H. Traub, U. Panne, J. Kneipp and N. Jakubowski, Anal. Chem., 2012, 84, 9684–9688 CrossRef CAS.
- J. Pisonero, D. Bouzas-Ramos, H. Traub, B. Cappella, C. Álvarez-Llamas, S. Richter, J. C. Mayo, J. M. Costa-Fernandez, N. Bordel and N. Jakubowski, J. Anal. At. Spectrom., 2018 10.1039/c8ja00096d.
- S. J. M. Van Malderen, E. Vergucht, M. De Rijcke, C. Janssen, L. Vincze and F. Vanhaecke, Anal. Chem., 2016, 88, 5783–5789 CrossRef CAS.
- C. Giesen, H. A. O. Wang, D. Schapiro, N. Zivanovic, A. Jacobs, B. Hattendorf, P. J. Schüffler, D. Grolimund, J. M. Buhmann, S. Brandt, Z. Varga, P. J. Wild, D. Günther and B. Bodenmiller, Nat. Methods, 2014, 11, 417–422 CrossRef CAS.
- H. A. O. Wang, D. Grolimund, C. Giesen, C. N. Borca, J. R. H. Shaw-Stewart, B. Bodenmiller and D. Günther, Anal. Chem., 2013, 85, 10107–10116 CrossRef CAS.
- L. Mueller, A. J. Herrmann, S. Techritz, U. Panne and N. Jakubowski, Anal. Bioanal. Chem., 2017, 409, 3667–3676 CrossRef CAS.
- S. J. M. Van Malderen, B. Laforce, T. Van Acker, C. Nys, M. De Rijcke, R. de Rycke, M. De Bruyne, M. N. Boone, K. De Schamphelaere, O. Borovinskaya, B. De Samber, L. Vincze and F. Vanhaecke, Anal. Chem., 2017, 89, 4161–4168 CrossRef CAS.
- B. Paul, D. J. Hare, D. P. Bishop, C. Paton, V. T. Nguyen, N. Cole, M. M. Niedwiecki, E. Andreozzi, A. Vais, J. L. Billings, L. Bray, A. I. Bush, G. McColl, B. R. Roberts, P. A. Adlard, D. I. Finkelstein, J. Hellstrom, J. M. Hergt, J. D. Woodhead and P. A. Doble, Chem. Sci., 2015, 6, 5383–5393 RSC.
- S. J. M. Van Malderen, B. Laforce, T. Van Acker, L. Vincze and F. Vanhaecke, J. Anal. At. Spectrom., 2017, 32, 289–298 RSC.
- B. Wu and J. S. Becker, Int. J. Mass Spectrom., 2011, 307, 112–122 CrossRef CAS.
- S. G. Boxer, M. L. Kraft and P. K. Weber, Annu. Rev. Biophys., 2009, 38, 53–74 CrossRef CAS.
- J. S. Becker, U. Breuer, H.-F. Hsieh, T. Osterholt, U. Kumtabtim, B. Wu, A. Matusch, J. A. Caruso and Z. Qin, Anal. Chem., 2010, 82, 9528–9533 CrossRef CAS.
- C. R. Anderton and L. J. Gamble, Microsc. Today, 2016, 24, 24–31 CrossRef CAS.
- V. I. Slaveykova, C. Guignard, T. Eybe, H.-N. Migeon and L. Hoffmann, Anal. Bioanal. Chem., 2008, 393, 583–589 CrossRef.
- C. Quintana, S. Bellefqih, J. Y. Laval, J. L. Guerquin-Kern, T. D. Wu, J. Avila, I. Ferrer, R. Arranz and C. Patiño, J. Struct. Biol., 2006, 153, 42–54 CrossRef CAS.
- A. N. Lazar, C. Bich, M. Panchal, N. Desbenoit, V. W. Petit, D. Touboul, L. Dauphinot, C. Marquer, O. Laprévote, A. Brunelle and C. Duyckaerts, Acta Neuropathol., 2012, 125, 133–144 CrossRef.
- A. van Remoortere, R. J. M. van Zeijl, N. van den Oever, J. Franck, R. Longuespée, M. Wisztorski, M. Salzet, A. M. Deelder, I. Fournier and L. A. McDonnell, J. Am. Soc. Mass Spectrom, 2010, 21, 1922–1929 CAS.
- C. W. Chumbley, M. L. Reyzer, J. L. Allen, G. A. Marriner, L. E. Via, C. E. Barry III and R. M. Caprioli, Anal. Chem., 2016, 88, 2392–2398 CrossRef CAS.
- M. Aichler and A. Walch, Lab. Invest., 2015, 95, 422–431 CrossRef CAS.
- E. Moreno-Gordaliza, D. Esteban-Fernández, A. Lázaro, B. Humanes, S. Aboulmagd, A. Tejedor, M. W. Linscheid and M. M. Gómez-Gómez, Talanta, 2017, 164, 16–26 CrossRef CAS.
- I. Piga, B. Heijs, S. Nicolardi, L. Giusti, L. Marselli, P. Marchetti, M. R. Mazzoni, A. Lucacchini and L. A. McDonnell, Int. J. Mass Spectrom., 2017, 1–7 Search PubMed.
- R. F. S. Lee, S. Theiner, A. Meibom, G. Koellensperger, B. K. Keppler and P. J. Dyson, Metallomics, 2017, 9, 365–381 RSC.
- X. Liu and A. B. Hummon, Sci. Rep., 2016, 6, 38507 CrossRef CAS.
- A. Schertel, N. Snaidero, H.-M. Han, T. Ruhwedel, M. Laue, M. Grabenbauer and W. Möbius, J. Struct. Biol., 2013, 184, 355–360 CrossRef CAS PubMed.
- N. M. Pirozzi, J. P. Hoogenboom and B. N. G. Giepmans, Histochem. Cell Biol., 2018, 150, 509–520 CrossRef CAS.
- V. L. Dressler, E. I. Müller and D. Pozebon, Adv. Exp. Med. Biol., 2018, 1055, 139–181 CrossRef.
- R. D. Leapman, Ultramicroscopy, 2017, 180, 180–187 CrossRef CAS.
- R. D. Leapman, J. A. Hunt, R. A. Buchanan and S. B. Andrews, Ultramicroscopy, 1993, 49, 225–234 CrossRef CAS.
- M. Morello, A. Canini, M. Grilli Caiola, A. Martorana, P. Mattioli, G. Bernardi and G. Sancesario, J. Trace Microprobe Tech., 2002, 20, 481–491 CrossRef CAS.
- P. Zhang, W. Land, S. Lee, J. Juliani, J. Lefman, S. R. Smith, D. Germain, M. Kessel, R. Leapman, T. A. Rouault and S. Subramaniam, J. Struct. Biol., 2005, 150, 144–153 CrossRef CAS.
- R. Ortega, G. Devès and A. Carmona, J. R. Soc., Interface, 2009, 6, S649–S658 CrossRef CAS.
- A. Carmona, G. Devès and R. Ortega, Anal. Bioanal. Chem., 2008, 390, 1585–1594 CrossRef CAS.
- R. Ortega, C. Bresson, A. Fraysse, C. Sandre, G. Devès, C. Gombert, M. Tabarant, P. Bleuet, H. Seznec, A. Simionovici, P. Moretto and C. Moulin, Toxicol. Lett., 2009, 188, 26–32 CrossRef CAS.
- A. Carmona, S. Roudeau, B. L'Homel, F. Pouzoulet, S. Bonnet-Boissinot, Y. Prezado and R. Ortega, Anal. Biochem., 2017, 523, 50–57 CrossRef CAS PubMed.
- Y.-K. Tanaka, N. Yajima, M. Okada, T. Matsumoto, Y. Higuchi, S. Miyazaki, H. Yamato and T. Hirata, Analyst, 2017, 142, 4265–4278 RSC.
- A. Balbekova, M. Bonta, S. Török, J. Ofner, B. Döme, A. Limbeck and B. Lendl, Anal. Methods, 2017, 9, 5464–5471 RSC.
- F. Penen, J. Malherbe, M.-P. Isaure, D. Dobritzsch, I. Bertalan, E. Gontier, P. Le Coustumer and D. Schaumlöffel, J. Trace Elem. Med. Biol., 2016, 37, 62–68 CrossRef CAS.
- S. Mounicou, J. Szpunar and R. Lobinski, Chem. Soc. Rev., 2009, 38, 1119–1138 RSC.
- W. Maret, Metallomics. Advances in Experimental Medicine and Biology, Springer International Publishing, Cham, 79th edn, 2018, vol. 1055, pp. 1–20 Search PubMed.
- C. M. Ackerman, P. K. Weber, T. Xiao, B. Thai, T. J. Kuo, E. Zhang, J. Pett-Ridge and C. J. Chang, Metallomics, 2018, 10, 474–485 RSC.
- M. Holzlechner, M. Bonta, H. Lohninger, A. Limbeck and M. Marchetti-Deschmann, Anal. Chem., 2018, 90, 8831–8837 CrossRef CAS.
- G. L. Caia, O. V. Efimova, M. Velayutham, M. A. El-Mahdy, T. M. Abdelghany, E. Kesselring, S. Petryakov, Z. Sun, A. Samouilov and J. L. Zweier, J. Magn. Reson., 2012, 216, 21–27 CrossRef CAS.
- W. Maret, Metallomics, 2015, 7, 202–211 RSC.
- L. Vaccari, G. Birada, G. Grenci, S. Pacor and L. Businaro, J. Phys.: Conf. Ser., 2012, 359, 012007 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |