
Handling of nutrient copper in the bacterial envelope
Louisa J.
Stewart†
a,
Denis
Thaqi†
b,
Bostjan
Kobe
bc,
Alastair G.
McEwan
b,
Kevin J.
Waldron
 a and
Karrera Y.
Djoko
*d
a and
Karrera Y.
Djoko
*d
aInstitute for Cell and Molecular Biosciences, Newcastle University, Newcastle upon Tyne, NE24HH, UK
bSchool of Chemistry and Molecular Biosciences and Australian Infectious Diseases Research Centre, The University of Queensland, St Lucia, Queensland 4072, Australia
cInstitute for Molecular Bioscience, The University of Queensland, St Lucia, Queensland 4072, Australia
dDepartment of Biosciences, Durham University, Durham, DH13LE, UK. E-mail: karrera.djoko@durham.ac.uk; Tel: +44 191 3340809
First published on 10th October 2018
Abstract
In bacteria, copper (Cu) is often recognised for its potential toxicity and its antibacterial activity is now considered a key component of the mammalian innate immune system. Cu ions bound in weak sites can catalyse harmful redox reactions while Cu ions in strong but adventitious sites can disrupt protein or enzyme function. For these reasons, the outward transport of Cu from bacteria has received significant attention. Yet, Cu is also a bacterial nutrient, required as a cofactor by enzymes that catalyse electron transfer processes, for instance in aerobic and anaerobic respiration. To date, the inward flow of this metal ion as a nutrient and its insertion into target cuproenzymes remain poorly defined. Here we revisit the available evidence related to bacterial nutrient Cu trafficking and identify gaps in knowledge. Particularly intriguing is the evidence that bacterial cuproenzymes do not always require auxiliary metallochaperones to insert nutrient Cu into their active sites. This review outlines our effort to consolidate the available experimental data using an established energy-driven model for metalation.
Introduction: the challenge of handling nutrient copper
Approximately half of enzymes and a third of all proteins require metals to function – an often overlooked dimension of bacterial physiology and nutrition. Understanding the way in which the correct metal ion is inserted into enzymes and proteins still represents a major challenge in bioinorganic chemistry. In vitro, these biomolecules prefer the same relative order for metals that follows the Irving–Williams series. However, it is now understood that these universal preferences can, in general, be overcome in vivo because molecules within the crowded intracellular milieu buffer the available concentrations of metals in the inverse order.1 Tighter binding metals like copper (Cu) and zinc (Zn) are buffered at lower concentrations (and hence are less available), while weaker binding metals like manganese (Mn) are buffered at higher concentrations (and hence are more available).1 Once inside cells, metals flow down a thermodynamic gradient, i.e. from weaker (high energy) to tighter (low energy) sites in the buffer, via a series of stochastic, associative exchange reactions (Fig. 1a).2 A metalloprotein ultimately acquires the correct metal as long as its affinity for this metal is higher than the affinity of the buffer (i.e. the metal–protein complex is more stable than is the metal-buffer complex) (Fig. 1b).3,4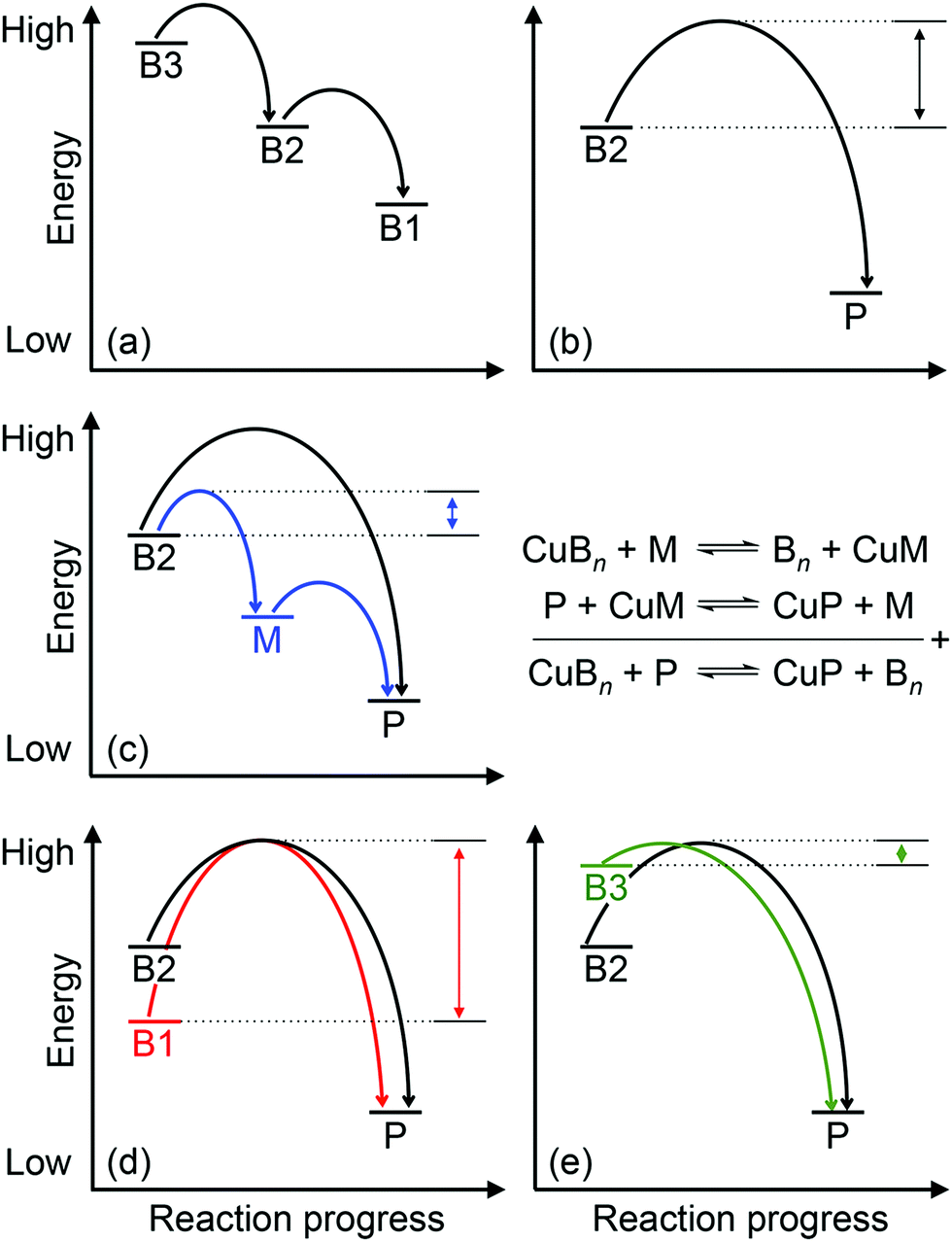 | ||
| Fig. 1 General energy-driven model for the insertion of Cu into cuproenzymes. The relative energy for each Cu-binding site, whether in the buffer (B1, B2, B3), cuproprotein (P), or metallochaperone (M) is shown. Curved arrows represent the forward transfer of Cu from one binding site to another while double-headed arrows represent the energy barrier that must be overcome. Several scenarios are depicted: (a) upon entry into cells, Cu fills the buffer by stepwise transfer from high energy or low affinity sites (denoted as B3) to low energy or high affinity sites (denoted as B1) in the buffer through stochastic exchange reactions. (b) Direct transfer of Cu from the buffer (in this example the mid-affinity or mid-energy site B2) to a cuproprotein (P). (c) Transfer of Cu from the mid-affinity buffer (B2) to a cuproprotein (P) via a metallochaperone (M). Equations representing these equilibria are shown on the right. (d) During conditions of Cu starvation, low affinity or high energy sites in the buffer (B3) start to empty, leaving only Cu that is bound in high affinity or low energy buffer sites (B1). Onward transfer of Cu from this low energy buffer to the cuproprotein (P) is shown with a high energy barrier. (e) During conditions of Cu stress, the excess Cu starts to fill the weaker sites in the buffer start (B3). Onward transfer from this high energy buffer to the cuproprotein (P) is shown, requiring a lower activation energy. | ||
Cu sits at the top of the Irving–Williams series and hence metalation of cuproenzymes is normally an endergonic or thermodynamically favourable process. By the same principle, Cu can also partition into stable sites in the wrong protein, leading to enzyme inactivation and bacterial poisoning. To minimise mis-metalation, cells employ metallochaperones that are thought to shuttle (or “chaperone”) the Cu ion from import pumps to target cuproproteins. Such pathways are well described for the eukaryotic cytosol and organelles.5–9 For prokaryotes, discussions of Cu homeostasis have revolved mainly around Cu tolerance,10–12i.e. removal of excess Cu from the cell under conditions of Cu surplus, when the cellular Cu buffer is “full”. By contrast, trafficking of nutrient copper, particularly when the buffer is “empty”, is less understood.
Cuproenzymes are thought to have evolved after the appearance of atmospheric O213 and so they are typically involved in reactions with oxygen and oxygen-containing species. In prokaryotes, Cu is a major nutrient for aerobic respiration (via haem-Cu oxidases in the electron transport chain), anaerobic respiration (via nitrous oxide reductases and Cu-containing nitrite reductases in the denitrification pathway), and removal of toxic reactive oxygen species (via Cu,Zn-superoxide dismutase). Intriguingly, the Cu-dependent enzymes in the aforementioned pathways are all localised to the bacterial envelope (i.e. in the periplasm of Gram-negative bacteria or on the surface of Gram-positive bacteria). Indeed, with the exception of plastocyanin and cytochrome oxidase in cyanobacteria, cuproproteins are not known to exist inside the bacterial cytoplasm. This apparent compartmentalisation of Cu to the extracytoplasmic space may represent a mechanism for balancing the physiological advantages of using nutrient Cu in catalysis while protecting against its potential toxicity. Indeed, Cu is generally considered to be more toxic in the cytoplasm and thus must be buffered at a lower availability (i.e. bound by higher affinity or lower energy sites in the buffer) relative to the extracytoplasmic space.
Metals in the bacterial envelope are readily exchangeable with the extracellular environment, for example via passive diffusion across outer membrane porins in Gram-negative organisms.14 Hence, fine control of metalation in this compartment may be more challenging than in the cytoplasm. This is considered particularly problematic for metalloproteins that are translocated via the Sec general secretory pathway and thus are folded (and metalated) in the extracytoplasmic space.15 By contrast, metalloproteins that are Tat substrates fold inside the cytoplasm and, at least in some cases, obtain their cognate metal prior to secretion.15 In the case of Cu, recent examination of the periplasmic multicopper oxidase CueO from Escherichia coli demonstrated that removal of the Tat signal sequence and expression of CueO in the cytoplasm led to isolation of only the apo-enzyme.16 In fact, all bacterial cuproproteins for which the steps of Cu insertion have been identified (detailed in this review) are thought to become metalated outside the cytoplasm, regardless of the translocation mechanism of the protein scaffold. One explanation is that the Cu affinities of these cuproenzymes are compatible with the buffered availability of Cu in the bacterial envelope but incompatible with that of the cytoplasm. In addition, the oxidation state of Cu in the buffer and the oxidation state preferred by the enzyme might further define extracytoplasmic metalation of cuproproteins.
What is the source of nutrient Cu for cuproenzymes in the bacterial envelope? In the simplest model, a buffered pool of Cu in the extracytoplasmic space acts as the Cu supplier. The molecular nature of this Cu buffer is presently unknown. It has been long assumed that thiols like glutathione (GSH) buffer Cu in the cytoplasm.17 There is also evidence that GSH is exported to the periplasm of Gram-negative organisms,18 and so it can presumably also buffer Cu in this compartment. However, the affinity of GSH for Cu is orders of magnitudes weaker when compared to those of bacterial Cu sensors in the cytoplasm19,20 or nutrient Cu metallochaperones in the periplasm.21 These relative values suggest that, despite its high intracellular concentrations, GSH would constitute a high energy buffer, filled only when an excess of Cu is available. By contrast, the identity of the low energy or high affinity buffer that contributes to normal Cu nutrition is unknown. Nevertheless, metalloproteomics examination of periplasmic extracts from Salmonella enterica sv. Typhimurium22 and Synechocystis23 indicated that periplasmic Cu is largely bound either to Cu metallochaperones or to unidentified low molecular weight proteins.
Regardless of the precise identity of the extracytoplasmic Cu buffer, it is presumably filled by Cu from the extracellular environment (Fig. 2). This exchange of Cu may occur via passive diffusion through porins14,24 or other unidentified mechanisms.25 Active uptake of Cu is also known to occur, for example via TonB-dependent receptors26 or via classical siderophores27 and Cu-binding metallophores (“chalkophores”) such as yersiniabactin and methanobactin.28–30 Once the buffer is filled by Cu, provided that the affinities of the cuproenzymes are higher than the affinity of the buffer, Cu will flow down the thermodynamic gradient and ultimately insert into target enzymes (Fig. 1b). Yet, there is now mounting evidence that Cu-exporting P-type ATPases embedded in the cytoplasmic membrane are involved in metalating extracytoplasmic cuproproteins.23,31,32 The implication is that nutrient Cu ions are trafficked through the cytoplasm en route to the extracytoplasmic targets and, if so, this must be a vital process for Cu homeostasis. When combined with the dearth of known cytoplasmic Cu importers, this apparently circuitous routing of Cu is one of the most puzzling aspects of nutrient Cu handling in bacteria.
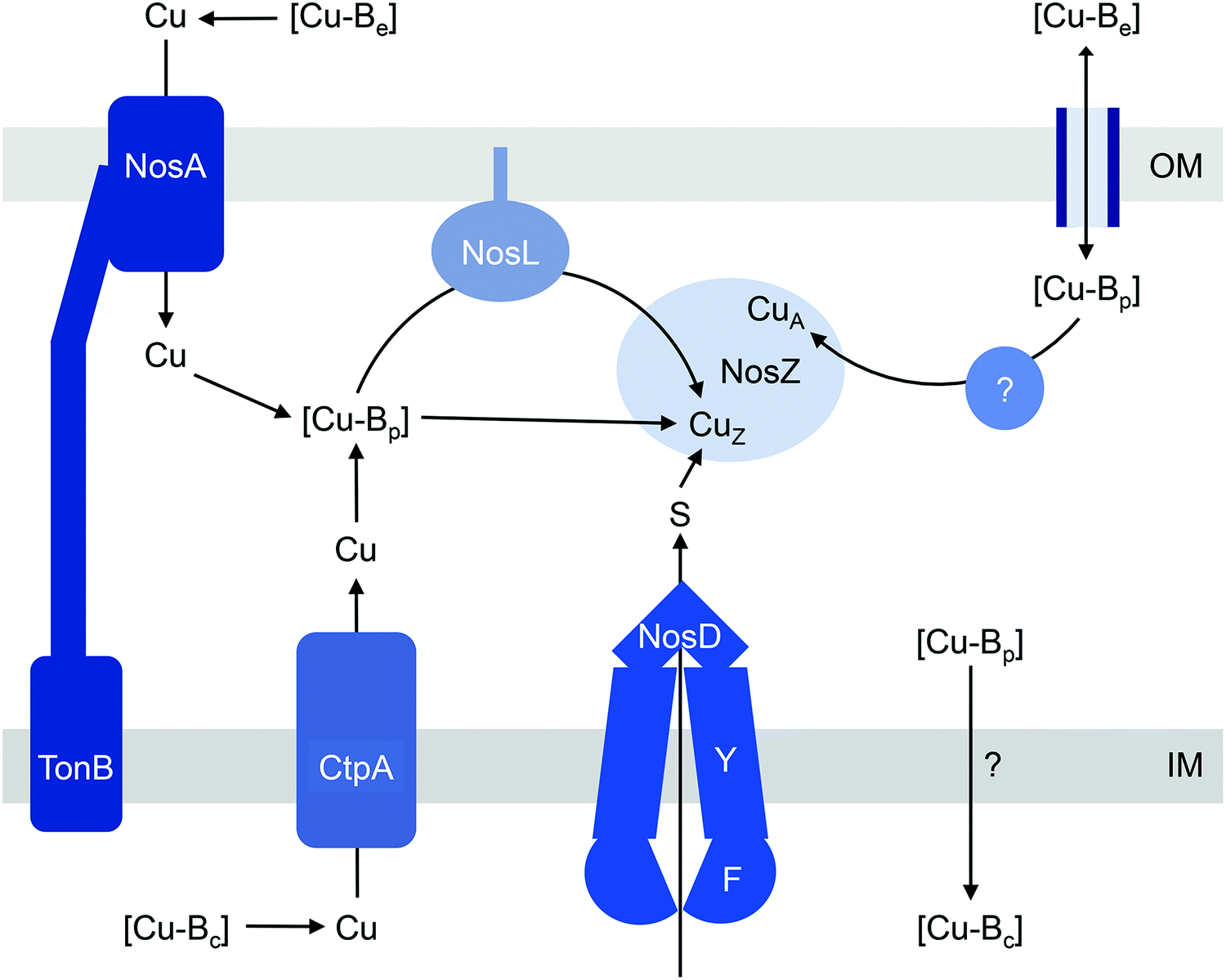 | ||
| Fig. 2 General model for the insertion of Cu into NosZ. [Cu-Be], [Cu-Bc], and [Cu-Bp] are buffered pools of Cu in the extracellular space, cytoplasm, and periplasm, respectively. In this model, [Cu-Bp] is filled by [Cu-Be] either via the TonB-dependent receptor NosA or possibly via direct exchange across outer membrane porins in NosA-deficient organisms. [Cu-Bp] is also filled by [Cu-Bc] via the P-type ATPase CtpA. How [Cu-Bc] is generated is unknown. The CuZ site in NosZ acquires Cu from [Cu-Bp] either directly or via the metallochaperone NosL, and this process is likely coupled with insertion of sulfur (S) by NosDFY. How the CuA site obtains Cu is unknown but this process likely resembles mechanisms for CuA assembly in haem-Cu oxidases. IM, inner membrane; OM, outer membrane. | ||
Our research groups have studied bacterial Cu tolerance for several years and have recently begun to investigate nutrient Cu handling, specifically in pathogenic Neisseria. This prompted us to revisit existing literature related to bacterial nutrient Cu trafficking and identify gaps in knowledge. We were particularly intrigued by the evidence that bacterial cuproenzymes do not always require auxiliary metallochaperones to insert nutrient Cu into their active sites. This review outlines our effort to consolidate the available experimental data by expanding an established energy-driven model for Cu trafficking.2 We focus on four major families of bacterial cuproenzymes: (1) nitrous reductases, (2) nitrite reductases, (3) Cu,Zn-superoxide dismutases, and (4) haem-Cu oxidases, and pay particular attention to the precise steps of Cu insertion. The genomic context and genetic distribution, structural features and properties of the Cu centres in the enzymes (and in the associated metallochaperones), as well as kinetic properties of these enzymes are already subjects of numerous excellent reviews and so will not be covered in detail.
Cu insertion into nitrous oxide reductases
Assembling a denitrification pathway is a Cu-expensive process since it involves at least one multicopper enzyme, namely nitrous oxide reductase (N2OR or NosZ), which catalyses the reduction of N2O to N2. NosZ homologues are classified as typical or atypical, distinguished by two key biochemical characteristics: (i) an additional haem c binding site is present near the C-terminus in atypical NosZ but is absent in typical NosZ; and (ii) translocation of atypical NosZ is Sec-dependent while that of typical NosZ is Tat-dependent.33 Both types of NosZ contain 6 Cu atoms per monomer (12 Cu per functional homodimer), arranged into one tetranuclear CuZ copper–sulfur (Cu4S2) cluster that binds and activates N2O during catalysis, and one binuclear mixed-valent CuA centre that acts as the site of electron entry.34–37Consistent with its high demand for Cu, NosZ activity in denitrifying organisms is greatly influenced by extracellular Cu levels.38–40 During conditions of Cu deficiency, NosZ activity decreases and N2O accumulates. This Cu-dependent regulation of NosZ occurs at the post-translational level, i.e. by modulating occupancy of the Cu centres. Growth in Cu-deficient conditions leads to production of NosZ in an inactive form. However, N2O reductase activity is restored by addition of exogenous Cu without the need for new protein synthesis.39 In bacteria possessing the typical NosZ, increases in extracellular Cu levels also induce the expression of nosZ. This requires at least one factor, the flavoprotein NosR, although the molecular details are yet to be elucidated.40,41 The nosR gene is not found in genomes encoding atypical NosZ,33 and whether Cu regulates nosZ transcription in these organisms is unknown.
The current models for CuZ and CuA biogenesis suggest that these Cu centres are assembled in the periplasm following secretion of the protein, for both the typical and atypical NosZ, regardless of the translocation mechanism (Fig. 2). Homologous expression of NosZ in the cytoplasm results in the production of neither the CuZ nor the CuA centre.42 Assembly of CuZin vivo requires NosDFY, an ABC-type transporter that may transport sulfur (Fig. 2)43 although this is yet to be confirmed experimentally. This requirement for NosDFY appears to be obligate and the genetic clustering of nosZ with nosDFY is absolutely conserved in all sequenced genomes that are currently available.33 N2O respiration is abolished if any of the nosDFY genes is mutated and this defect is not restored by addition of extracellular Cu.44,45 In addition, NosZ isolated from nosDFY-deficient strains contains only the CuA centre,46–48 indicating that NosDFY may not be required to assist CuA assembly.
Insertion of nutrient Cu into the CuZ cluster in vivo is likely facilitated by NosL, a small lipoprotein that is anchored to the outer membrane (Fig. 2). The soluble periplasmic domain of NosL binds one Cu(I) ion in vitro but its affinity has not been determined.49 The Cu ligands include one Cys and one Met, presumably from a conserved Cys-X-Met motif near the N-terminus.50,51 The third ligand, likely from a His residue, is yet to be identified, and no obvious candidate is found from analysis of amino acid sequences. Whether NosL delivers Cu(I) to NosDFY or directly to NosZ, whether metalation is coupled to sulfur insertion, and whether NosL assists in assembly of the CuA centre are yet to be established. None of the nos cluster genes appears to be essential for CuA assembly. Nevertheless, denitrifying organisms often possess additional Cu metallochaperones like Sco and PCuAC (described below), which may metalate the CuA sites in NosZ, but this remains to be elucidated.
Unlike NosDFY, NosL appears to be dispensable for CuZ assembly. Although fitness analyses of a mutant library suggest that nosL is essential for denitrification in Pseudomonas stutzeri,52 mutational inactivation of nosL in this organism does not yield an obvious defect in N2O reductase activity.53 Likewise, heterologous expression of NosZ in its active form in the periplasm of a nondenitrifying host does not necessitate the co-expression of NosL.46 Furthermore, nosL is absent from the nos gene cluster in many genomes and this absence does not correlate with the type of NosZ (typical or atypical) or the NosZ translocation mechanism.33
How does CuZ obtain nutrient Cu in the absence of NosL? There is a proposal that other Cu metallochaperones such as PCuAC (described below) can compensate, although this is yet to be tested experimentally. An alternative, and arguably simpler, hypothesis is that the CuZ site acquires Cu directly from the extracytoplasmic Cu buffer (Fig. 2). This reaction is thermodynamically favourable (“downhill” or exergonic) as long as the affinity of the CuZ scaffold for Cu is higher than the affinity of the buffer (i.e. the bound Cu ion in CuZ is lower in energy or more stable than is Cu in the extracytoplasmic buffer) (Fig. 1b). NosL may provide an “intermediate buffer” (with intermediate Cu affinities) that lowers the overall energy barrier for the transfer of Cu from the extracytoplasmic buffer to the CuZ scaffold, with Cu-NosL acting as a reaction intermediate (Fig. 1c). In this scenario, the absence of NosL would not affect the Cu occupancy of NosZ, provided that the buffered Cu availability is sufficiently high (i.e. Cu is bound by high energy or low affinity sites in the buffer) and the barrier for Cu transfer to NosZ is sufficiently low. NosL would become more important in Cu-deficient conditions, when the buffered Cu availability decreases (i.e. Cu is bound by low energy or high affinity sites in the buffer) and thus, presumably, the barrier for onward Cu transfer to NosZ increases (Fig. 1d).
Regardless of the precise role for NosL, the question remains: what is the source of the buffered Cu in the extracytoplasmic space? In the simplest model, this buffer is filled directly by Cu from the extracellular environment (Fig. 2). In some, but not all, denitrifying Gram-negative organisms, N2O respiration during conditions of Cu limitation requires NosA, a TonB-dependent receptor that may increase uptake of Cu into the periplasm (Fig. 2).26,46,54,55 Intriguingly, there is also evidence that the extracytoplasmic pool of Cu is filled by supply from the cytoplasm. NosZ activity in vivo was shown to depend on CtpA, a P-type ATPase that resembles known bacterial Cu-efflux transporters (Fig. 2).31 Mutation of ctpA leads to decreased NosZ activity but enzyme activity is restored by addition of Cu to the extracellular medium. This exogenous Cu presumably fills the extracytoplasmic Cu buffer, which in turn metalates NosZ (Fig. 2). If direct metalation of NosZ by the extracytoplasmic Cu buffer is possible in the ΔctpA mutant, why nutrient Cu must first be routed through the cytoplasm in the wild type organism appears a major conundrum.
Cu insertion into Cu-containing nitrite reductases
Cu-dependent nitrite reductase (Cu-NIR), usually called NirK, catalyses the reduction of NO2− to N2O. This enzyme contains a total of 2 Cu centres per monomer (6 Cu per functional homotrimer): one T1 “blue” Cu centre that acts as the site for electron entry and one T2 Cu centre that acts as the active site for NO2− binding and reduction.56 NirK is sometimes co-encoded in the genome with NirV, a protein of unknown function that does not appear to bind Cu.57,58 Only a minority of NirK homologues carry the Tat signal sequence, with most thought to be translocated via the Sec or other nonspecific secretory pathways.59T1 and T2 Cu centres are readily reconstituted by Cu salts in vitro and so insertion of Cu into NirK in vivo was previously assumed to require no accessory metallochaperones. However, a recent genetic screen identified that a soluble periplasmic Cu-binding protein, AccA, is required for metalating NirK (AniA) in pathogenic Neisseria (Fig. 3).60 AccA is a homologue of PCuAC, a metallochaperone that is thought to aid assembly of CuA and CuB centres in haem-Cu oxidases61–64 (described below). Like PCuAC, AccA binds one Cu(I) ion with a high apparent affinity,60 although precise quantification is still awaited. Conserved Met and His residues are likely involved in binding Cu(I). AccA also binds one additional Cu ion in the Cu(II) oxidation state.60 Several candidate ligands for Cu(II) are present in the His- and Met-rich C-terminus but their identities are yet to be determined. Programmes in our research groups are currently ongoing to determine which of the two bound Cu ions in AccA is loaded to which of the two Cu sites in AniA.
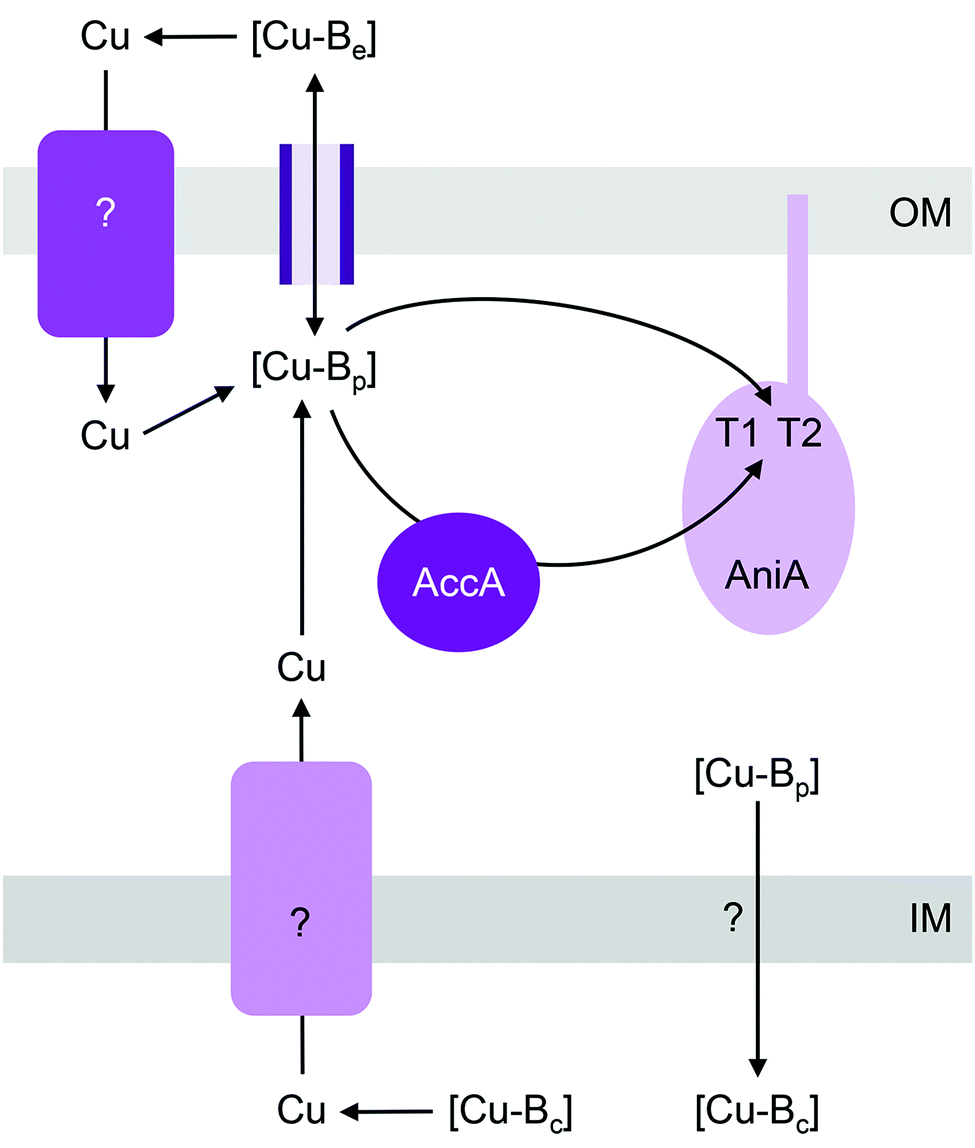 | ||
| Fig. 3 General model for the insertion of Cu into AniA (NirK). [Cu-Be], [Cu-Bc], and [Cu-Bp] are buffered pools of Cu in the extracellular space, cytoplasm, and periplasm, respectively. The T1 and T2 sites in AniA acquire Cu from [Cu-Bp] either directly or via the metallochaperone AccA (PCuAC). [Cu-Bp] is likely filled by [Cu-Be] via direct exchange across outer membrane porins. Whether an outer membrane importer or a cytoplasmic exporter is involved in filling [Cu-Bp] is yet to be determined. IM, inner membrane; OM, outer membrane. | ||
Mutants lacking accA generate wild type amounts of AniA but fail to reduce NO2−, suggesting that AniA is produced in the apo- or incorrectly metalated form. Consistent with this view, reduction of NO2− resumes, albeit only partially, upon addition of Cu salts into the extracellular media.60 Assuming that no other unidentified Cu trafficking pathway compensates for AccA, the observed recovery of AniA activity by exogenous Cu is consistent with the proposal that that this enzyme is metalated directly by a buffered Cu pool in the periplasm (Fig. 3). This reaction is energetically downhill as long as the affinity of AniA for Cu is higher than the affinity of the buffer (i.e. the bound Cu in AniA is more stable or less energetic) (Fig. 1b). As hypothesised earlier for NosL, the function of AccA may be to act as an intermediate buffer that lowers the overall energy barrier for Cu exchange and thus functionally “catalyses” the transfer of Cu from the buffer to the T1 and/or T2 sites of AniA (Fig. 1c). In the absence of AccA, provided that the buffered Cu availability is sufficiently high (i.e. Cu is bound by high energy or low affinity sites in the buffer), the barrier for onward Cu transfer decreases, and AniA becomes metalated (Fig. 1e).
There is evidence that deletion of the AccA homologue PCuAC in Bradyrhizobium japonicum also leads to transient accumulation of NO2−, implying a defect in NirK activity.65 Hence, although PCuAC primarily aids assembly of CuA centres (discussed below), the possibility that this metallochaperone inserts nutrient Cu into multiple cuproenzymes, including NirK, should not be disregarded. In the energy-driven model, the role of PCuAC is facilitative rather than obligatory (Fig. 1c). This model can rationalise why not all genomes that encode a NirK59 possess a PCuAC or AccA, and conversely, why the presence of pcuAC in denitrifying organisms does not exclusively correlate with the presence of nirK40 or even with CuA centres.66
Like pathogenic Neisseria, some NirK-containing organisms also possess the metallochaperone Sco.66,67 Together with PCuAC, Sco is thought to facilitate assembly of CuA and CuB centres in haem-Cu respiratory oxidases (described below). Whether Sco is required for inserting Cu into T1 and T2 sites of NirK is not known. Likewise, whether a Cu importer such as NosA from P. stutzeri or a Cu-exporting P-type ATPase such as CopA in pathogenic Neisseria is involved in metalating AniA or NirK is yet to be examined (Fig. 3).
Cu insertion into Cu,Zn-superoxide dismutase
The Cu,Zn-superoxide dismutase (SodC) is noted for its distribution among pathogenic bacteria68 and is often considered a virulence factor owing to its ability to detoxify the superoxide anion during phagocytosis.69–72 This enzyme contains one solvent-exposed T2 Cu centre (2 Cu per functional homodimer) in the active site. Like the other bacterial cuproenzymes discussed in this review, SodC is invariably localised to the extracytoplasmic space, either in its soluble (e.g. in the Gram-negative periplasm) or anchored form (e.g. on the surfaces of Gram-positive bacteria73 or on the outer membrane of some Gram-negative bacteria). It was previously assumed that SodC was secreted via the Sec pathway but it is now proposed that the hydrophobic signal sequence of SodC may interact with the Tat translocase.74The T2 centre in SodC assembles spontaneously in vitro without the need for assembly factors.75 Based on studies with Salmonella enterica sv. Typhimurium, metalation of SodC in vivo likely involves, but does not absolutely require, a soluble periplasmic metallochaperone named CueP (Fig. 4).32,76 Homologues of CueP are found in both Gram-positive and Gram-negative bacteria,77 but certainly not in all SodC producers (e.g. E. coli). Deletion of cueP impairs, but does not completely eliminate, the activities of the two SodC homologues in Salmonella, SodCI and SodCII.32,76 The reduction in enzyme activities correlates with decreased occupancy of the T2 Cu site, at least for SodCII.32 However, enzyme activity and/or Cu occupancy are restored in vivo by supplementing the culture medium with Cu salts or in vitro by addition of Cu into the cell-free extracts.32,76 Purified CueP binds one Cu(I) ion with high affinity using a combination of Cys and His ligands77,78 and it is indeed able to deliver this bound Cu to purified SodC in vitro.32
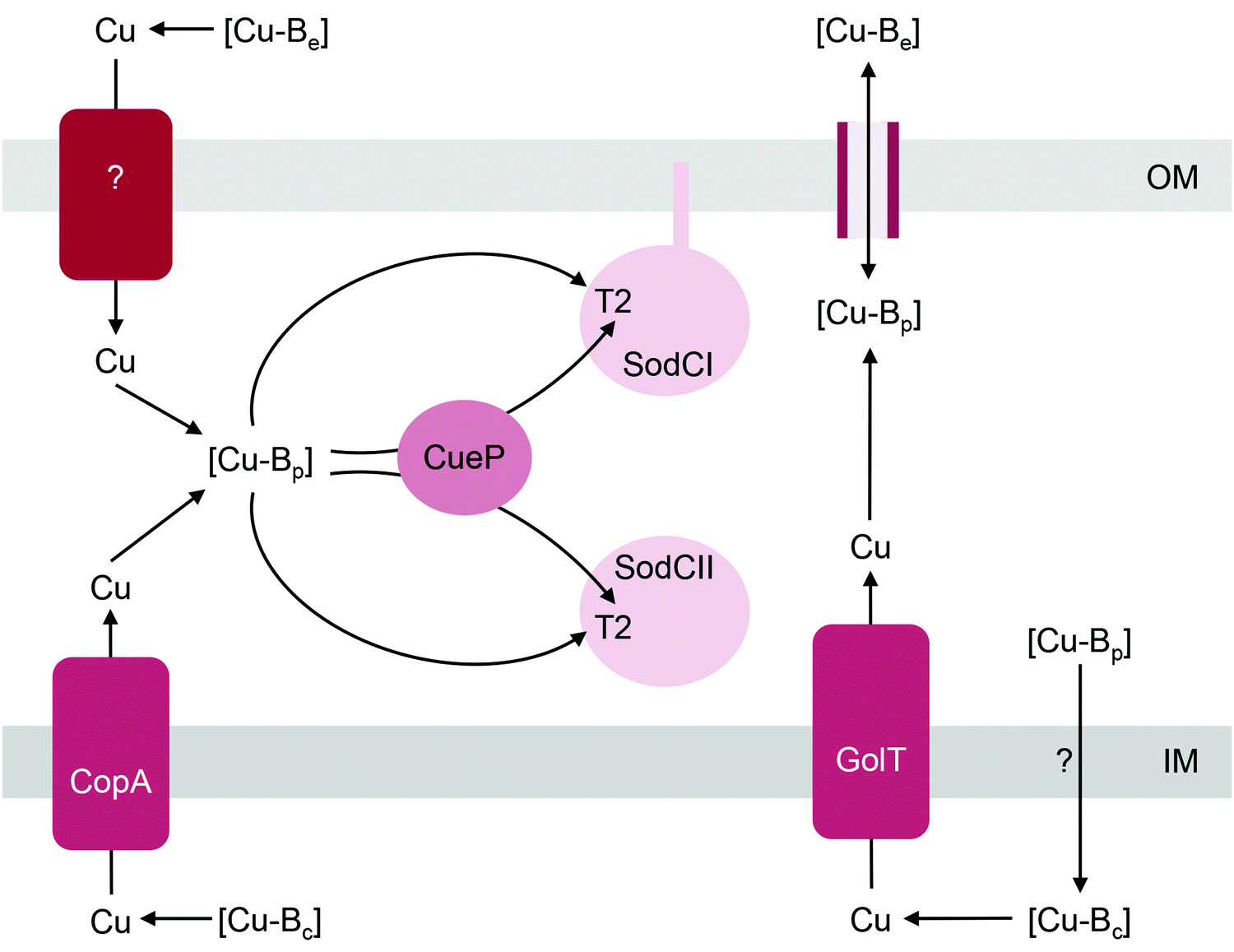 | ||
| Fig. 4 General model for the insertion of Cu into SodC. [Cu-Be], [Cu-Bc], and [Cu-Bp] are buffered pools of Cu in the extracellular space, cytoplasm, and periplasm, respectively. [Cu-Bp] is likely filled by [Cu-Be] via direct exchange across outer membrane porins. Whether an outer membrane importer is involved in this process is yet to be established. [Cu-Bp] is also filled by [Cu-Bc] via the P-type ATPases CopA or GolT. How [Cu-Bc] is generated is unknown. The T2 Cu site in SodC acquires Cu from [Cu-Bp] either directly or via the metallochaperone CueP. IM, inner membrane; OM, outer membrane. | ||
We noted that CueP is the first example of a bacterial Cu metallochaperone that participates in both Cu nutrition and Cu tolerance, and hence contributes fully to bacterial Cu homeostasis. Low basal amounts of CueP are produced during normal growth conditions but high amounts of this protein are generated during conditions of Cu surplus.22,79 Upregulation of cueP expression by Cu requires both the cytoplasmic Cu sensor CueR and CpxRA, which controls transcriptional responses to envelope stress.79 It has been proposed that CueP contributes to Cu tolerance by binding and sequestering excess Cu(I) in the periplasm. Consistent with this idea, CueP has been identified as a major Cu store in the Salmonella periplasm22 and the ΔcueP mutant is Cu-sensitive.77
How does CueP balance its seemingly dual role? The energy-driven model posits that as long as the T2 Cu site in SodC is more stable (i.e. higher in affinity or lower in energy) than is the periplasmic Cu buffer, SodC will acquire Cu directly from this buffer (Fig. 1b). Here, CueP acts as an intermediate buffer that lowers the energy barrier for Cu transfer, analogously to the other extracytoplasmic Cu metallochaperones described earlier (Fig. 1c). Consistent with this role as a functional catalyst (or Cu “insertase”), only low amounts of CueP need to be present. In the absence of CueP, the barrier for metalation of SodC is likely overcome by supplying excess extracellular Cu, which saturates the low energy (high affinity) sites and starts to fill the high energy (low affinity) sites in the periplasmic Cu buffer (Fig. 1e). This idea that SodC may acquire Cu directly from the periplasmic fluid has indeed been postulated previously.80
Even during conditions of Cu stress, i.e. when the low affinity and high energy sites in the buffer become full, SodC remains energetically downhill from the buffer, and so this enzyme will continue to be metalated correctly. However, adventitious protein sites may now also become downhill from the “full” buffer and subsequently mis-metalated by Cu (Fig. 5). Increasing the amounts of CueP under these conditions will generate alternative stable but, more importantly, specific sites for Cu binding. Thus, the excess Cu can “drain” from less stable (i.e. lower in affinity or more energetic) sites in the original buffer or in mismetalated proteins to the more stable (i.e. higher in affinity or less energetic) site in CueP (Fig. 5c). In this model, CueP essentially directs or regulates the flow of Cu down the thermodynamic gradient, both during conditions of normal Cu nutrition and conditions of Cu stress. This “intermediate buffering” function for Cu metallochaperones during Cu homeostasis has indeed been postulated previously81–83 although it has not been tested for the metallochaperones highlighted in this review. The challenge for bacteria is to control production of CueP such that it does not start to drain Cu from SodC as a consequence of mass action (e.g. see equation in Fig. 1). There is experimental evidence that correct amounts of CueP are indeed important. Expression of cueP from a CpxR-independent promoter leads to growth defects in the presence and absence of added Cu.79
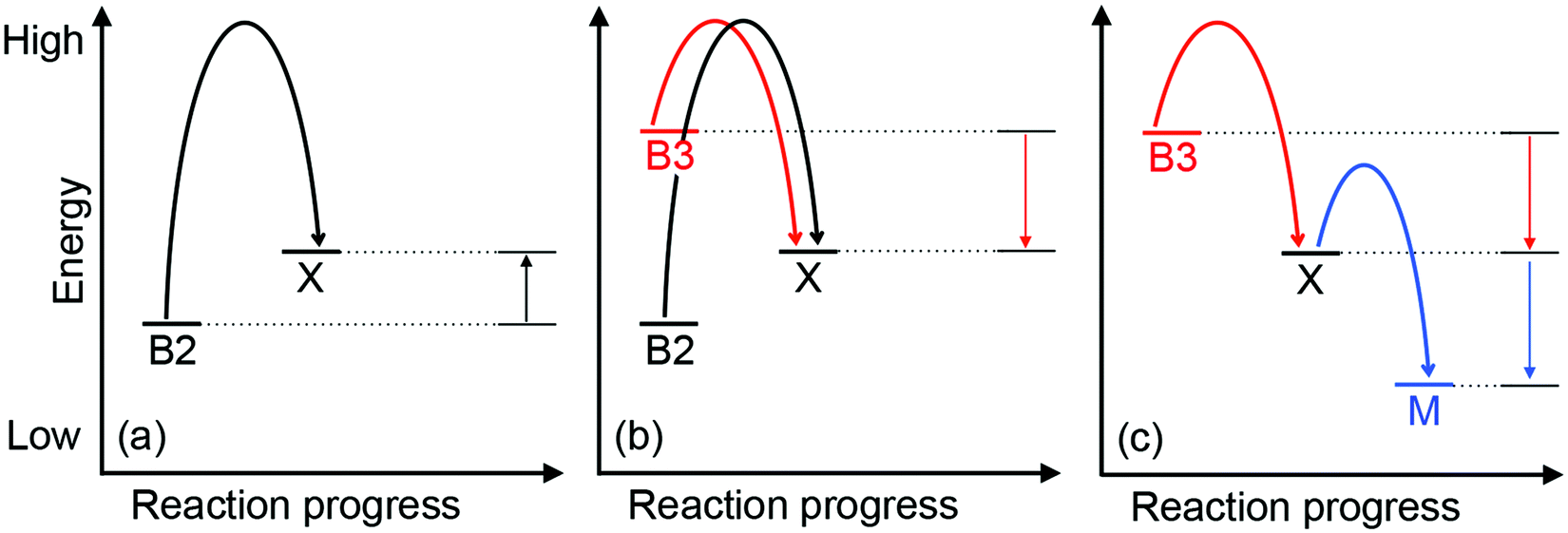 | ||
| Fig. 5 A general energy-driven model for the insertion of Cu into the wrong proteins (mismetalation). The relative energy for each Cu-binding site, whether in the buffer (B2, B3), Cu-binding metallochaperone (M), or a non-native adventitious protein (X) is shown. Curved arrows represent the forward transfer of Cu from one binding site to another. Several scenarios are depicted: (a) protein X, which is not a Cu-binding protein, binds Cu with an affinity that is weaker than that of the B2 buffer (i.e. the Cu-X complex is less stable or is more energetic than is the Cu-B2 complex). Hence, during normal Cu conditions, Cu transfer from buffer B2 to protein X is thermodynamically unfavourable (straight upward arrows), and X is not mismetalated by Cu. (b) During conditions of Cu stress, excess Cu enters cells and begins to fill low affinity or high energy sites in the buffer (B3). If this site is sufficiently high in energy, Cu will transfer out of the buffer into protein X, causing mismetalation. This transfer of Cu is now thermodynamically downhill and favourable (straight downward arrows). (c) Expression of a Cu-binding metallochaperone (M) during Cu stress conditions provides alternative, high-affinity or low energy but, more importantly, specific sites for Cu. Cu is thus transferred out of protein X and mismetalation is alleviated. | ||
Another mechanism to maintain SodC in its metalated form may involve control of the oxidation state of Cu. The ΔcueP mutant is Cu-sensitive only during anaerobic growth conditions.77 In the presence of O2, Cu(I) is removed from the buffer via oxidation to Cu(II) by the cuprous oxidase CueO,84,85 and thus additional buffering of Cu(I) by CueP may not be necessary. In addition, Cu in the resting form of SodC exists in the Cu(II) state. In vitro, this bound Cu(II) ion does not re-partition into apo-CueP.32 Thus, overexpression of CueP in vivo is unlikely to lead to de-metalation of SodC, at least under aerobic growth conditions, when SodC activity is essential.86 During anaerobic growth, when SodC is not required, extracytoplasmic reductants may reduce the Cu(II) ion in SodC and subsequent back-transfer of Cu(I) to CueP is plausible, although not yet demonstrated.
Metalation of SodC in vivo also depends, at least partly, on outward transport of Cu from the cytoplasm to the periplasm via either one of the two, functionally redundant, Cu efflux pumps in S. Typhimurium, CopA and GolT (Fig. 4). SodC isolated from mutant bacteria lacking both P-type ATPases contains only the Zn centre but readily acquires Cu upon addition of Cu(II) salts into cell-free extracts.32 This finding may further highlight the importance of the correct oxidation state for Cu. CopA and GolT transport Cu in the reduced Cu(I) form, and the relative affinities of the periplasmic domains of the P-type ATPases, the periplasmic buffer, CueP, and SodC for Cu(I) may be ordered such that metalation of SodC with Cu(I) is thermodynamically favourable. However, as already mentioned earlier, the T2 site in SodC is also competent to acquire Cu(II), at least in vitro.32In vivo, one possibility is that the buffered Cu(II) availabilities (or energies) in the periplasm are low and hence insertion of Cu(II) into SodC may be a thermodynamically uphill or unfavourable process. Measurements of the affinities of CueP and SodC each for Cu(I) and Cu(II), and comparisons with the buffered availabilities of Cu(I) and Cu(II) in the periplasm would be informative.
Cu insertion into haem-Cu respiratory oxidases
Haem-Cu respiratory oxidases are transmembrane, multi-subunit, multi-haem enzymes that catalyse the terminal step in the electron transport chain, namely the conversion of molecular oxygen to water. All members of this enzyme superfamily contain a mononuclear CuB centre that is spin-coupled to a haem. This is the site of O2 binding and reduction, and it is embedded deep within the transmembrane structures. Transfer of electrons from a cytochrome or quinol typically involves a relay of haem cofactors and, in some cytochrome c oxidases, a dinuclear CuA centre that is housed within a soluble periplasmic subunit. As anticipated from the complex enzyme architecture, assembly of haem-Cu oxidases likely requires a modular process that is synchronised both temporally and spatially, along with checkpoints that prevent folding of empty Cu sites into the mature but nonfunctional complex.87Of interest in this review are the precise steps of Cu insertion into CuB and CuA. These processes are most studied for mitochondrial cytochrome c oxidase (COX) in eukaryotes.88,89 Given the endosymbiotic bacterial origin of mitochondria, the mechanisms for metalation of mitochondrial COX and bacterial haem-Cu oxidases likely share some universal features. The bacterial metallochaperones involved in CuB and CuA assembly, namely Sco,64,90–94 PCuAC,61–63,65,95 or Cox11p,96–100 are, again, localised to the extracytoplasmic space (Fig. 6). These metallochaperones are structurally and functionally analogous to their eukaryotic counterparts (PCuAC acts as a functional Cox17 homologue). However, unlike the eukaryotic system, the precise contribution of each protein in the assembly of bacterial CuAvs. CuB centres and the sequence of Cu insertion events remain poorly defined and, bafflingly, appear to be organism-dependent.
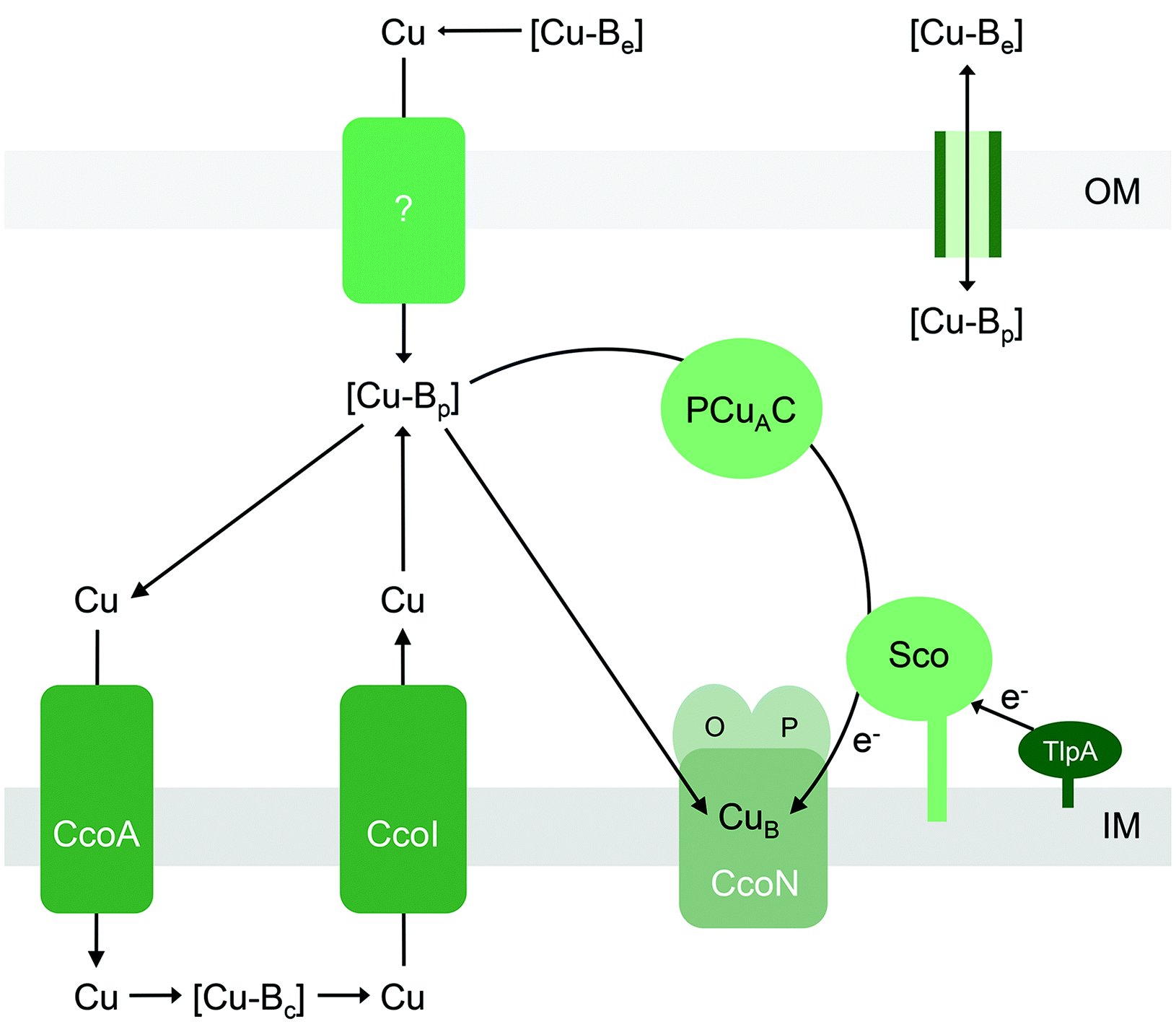 | ||
| Fig. 6 General model for the insertion of Cu into the CuB site into cytochrome cbb3 oxidase. Only the active site subunits CcoNOP are shown. This model may broadly apply to insertion of Cu into the CuA site in other haem-Cu oxidases. [Cu-Be], [Cu-Bc], and [Cu-Bp] are buffered pools of Cu in the extracellular space, cytoplasm, and periplasm, respectively. The CuB site in CcoN (and/or CuA site in other haem-Cu oxidases) may obtain nutrient Cu directly [Cu-Bp] or via the periplasmic Cu metallochaperones PCuAC and Sco. Based on studies on CuA assembly, Sco may also act as a thiol–disulfide reductase that maintains either the Cu ion or the CuB (or CuA) Cys ligands in their reduced forms. Upstream reductases such as TlpA may provide the reducing equivalents. Supply of Cu to [Cu-Bp] could occur by direct exchange across outer membrane porins or via an as yet unidentified importer. The MFS transporter CcoA supplies Cu to [Cu-Bc], with reduction from Cu2+ to Cu+ occurring either during transit or spontaneously in the reducing environment of the cytoplasm. Cu is routed back to the periplasm to fill [Cu-Bp] via the P-type ATPase CcoI. IM, inner membrane; OM, outer membrane. | ||
Part of the confusion can perhaps be ascribed to the varied genomic distributions of these metallochaperones. For instance, it is generally agreed that assembly of bacterial CuA centres in vivo involves both Sco and PCuAC (Fig. 6).21,63 There is indeed evidence that Sco and PCuAC form a transient complex in vitro and in vivo.63 However, the genes encoding these proteins are not always adjacent to each other in bacterial genomes.101 Moreover, sco and pcuAC are not always in close proximity with genes encoding CuA-containing oxidases. In some organisms, sco or pcuAC is instead associated with the nos cluster for nitrous reductase, nirK for nitrite reductase, putative operons for Cu homeostasis,65 other cuproenzyme genes, or genes with unknown functions.101 In addition, Sco and PCuAC homologues are present in bacteria that do not possess CuA (e.g. pathogenic Neisseria)66,90,102 and, in some organisms, multiple, functionally distinct homologues can exist.67
The current model for prokaryotes, which parallels that for eukaryotes, suggests that metalation of the CuA site is coupled to redox processes. The lipoprotein Sco contains a soluble, periplasmic thioredoxin-like domain and a conserved Cys–X–X–X–Cys motif, and thus it is not surprising that this protein displays thiol–disulfide reductase activity in vitro. Along with one additional His residue, the Cys thiols in Sco bind one Cu(I) ion with high affinity. This site also binds Cu(II) with an affinity that is higher than that for Cu(I).103 However, this Cu(II) ion is kinetically more inert than the bound Cu(I) ion and hence, exchange of Cu from Sco to its partners would occur only upon reduction to Cu(I).103 PCuAC displays a characteristic cupredoxin fold and binds one Cu(I) ion with high affinity using a combination of Met and His ligands (total of four) from a conserved HX6MX21HXM motif.62,65,95 Some homologues of PCuAC also bind Cu(II) in vitro but, in most cases, this binding is accompanied by reduction to Cu(I).21,60,62,95
Insertion of Cu into the CuA site in vitro does not require Sco, as long as the Cys ligands for CuA are present in their reduced forms.61In vivo, the oxygen-rich environment of the periplasm may promote oxidation of these Cys ligands. Under these aerobic conditions, in vitro experiments have shown that the CuA site is metalated only when both PCuAC and Sco are present, and only when Sco is provided in a reduced form.61 It is thus hypothesised that PCuAC acts as the Cu metallochaperone (or Cu donor) while Sco acts as a reductase that maintains either the Cu ion or the CuA cysteine ligands in the reduced forms (Fig. 6).64 Upstream reductases such as TlpA may provide the reducing power in vivo (Fig. 6).104,105
As mentioned earlier, not all bacterial haem-Cu oxidases contain a CuA centre. However, in contrast to the relative wealth of information available for CuA, current understanding of CuB assembly remains limited, mainly because the location of CuB deep within a transmembrane domain has largely precluded in vitro studies. Nevertheless, as discussed below, there is mounting in vivo evidence that Sco and PCuAC are also involved in CuB assembly, at least in some organisms. The mechanism may parallel that for CuA although the precise details still need investigation. In addition, the bacterial homologue of mitochondrial Cox11, Cox11p, has been implicated in forming bacterial CuB centres in vivo.98,100 Whether bacterial Cox11p coordinates its function with PCuAC and/or Sco is unknown.
Mutation of sco, pcuAC, or both, typically, but not always,60 leads to decreases, but not complete losses, in the activities of haem-Cu oxidases, regardless of whether the specific oxidase contains only CuB (e.g. cbb3 oxidase) or both CuB and CuA (e.g. cytochrome aa3 and ba3 oxidases).64,65,91,95,99,106,107 Deletion of sco typically produces the stronger phenotype.106,108 The defects in oxidase activities correlate with decreases in the amounts of mature subunits formed in vivo but these can be overcome, at least partially, by supplementing the extracellular medium with Cu salts.63–65,91,95,106–108
All of the abovementioned experimental data are again consistent with the model that the Cu sites in haem-Cu oxidases can acquire Cu directly from the extracytoplasmic Cu buffer (Fig. 6) as long as the energy of this buffer is sufficiently high (Fig. 1b). Although metalation with Cu in vitro is coupled to reduction (either of the Cu ion or of the CuA or CuB ligands), general extracytoplasmic reductases may provide this reducing power in vivo. This scenario is plausible for CuA because this centre is readily assembled in vitro in the presence of Cu salts and reductants.34,109 Provided that the thermodynamic gradients (i.e. buffered Cu availabilities, affinities of the metallochaperones, and affinities of the CuA scaffold) in vivo are appropriately setup for the exergonic Cu transfer from the buffer into the empty CuA sites, Cu will insert (Fig. 1b and c). For CuB, metalation may be combined with folding checkpoints to avoid accidental incorporation of the non-metalated sites in the mature complex. Indeed, potential roles in Cu insertion for other accessory components such as CcoG, CcoH, and CcoS (at least for the cbb3 oxidase) have been proposed, which may reflect their role in regulating such maturation checkpoints but mechanistic data are currently lacking.93,110,111
Like the other extracytoplasmic cuproenzymes described in this review, haem-Cu oxidases also appear to utilise nutrient Cu that has been routed via the cytoplasm, first via a major facilitator superfamily (MFS)-type transporter named CcoA that putatively imports Cu into the cytoplasm112,113 and subsequently via a Cu efflux pump (CcoI or CtpA)31,111,114,115 (Fig. 6). Deletion of each of these transporters leads to decreases in the activities of CuB and/or CuA-containing cytochrome oxidase activities, but these are, to some extent, alleviated by supplementation with Cu salts. This apparent routing of Cu through the intracytoplasmic compartment to metalate an extracytoplasmic cuproenzyme is one of the least understood aspects of nutrient Cu trafficking but, if it does occur, must represent a vital process in bacterial Cu homeostasis.
Outlook and perspectives: the need for systems approaches to examine nutrient Cu handling in bacteria
Among the six, first-row d-block transition metal ions that are considered as bacterial nutrients (Mn, Fe, Co, Ni, Cu, Zn), Cu is often highlighted for its potential toxicity. Cu ions bound in weak or high energy or unstable sites can catalyse harmful redox reactions, while Cu ions in strong, low energy or stable but non-native (adventitious) sites (mismetalation) can disrupt protein or enzyme function. While the outward transport of Cu as a bacterial poison has received significant attention from the metallomics community, inward flow of this metal ion as a bacterial nutrient remains less defined. Confounding this issue, known bacterial Cu importers and Cu-binding metallophores are still exceedingly rare and, as described in this review, while they are relatively more common, nutrient Cu metallochaperones are often functionally redundant.The apparent redundancy of Cu metallochaperones may be rationalised by the energy-driven model, in which target cuproenzymes obtain nutrient Cu directly from a buffered Cu pool via “downhill” or exergonic associative exchange reactions (Fig. 1b). It is our view that this model can universally rationalise all the available experimental evidence for the metalation of cuproenzymes in different bacterial organisms. In this model, the apo-metallochaperones can be considered as intermediate buffers or functional catalysts that lower the energy barrier for Cu transfer and regulate the flow of Cu down the thermodynamic gradient (Fig. 1c). The Cu-bound form of the metallochaperone thus represents a thermodynamic local minimum that limits “sideway” flows of Cu into adventitious sites (Fig. 1c). Hence, these metallochaperones are not obligate components for Cu homeostasis but are nonetheless able to provide alternative and more efficient routes for metalation during Cu nutrition, particularly when extracellular Cu is limiting, and for preventing (or correcting) mismetalation during Cu poisoning. This “intermediate buffering” function for Cu metallochaperones has indeed been proposed previously81–83 but how these metallochaperones lower the energy barrier for Cu transfer remains to be determined.
A key advantage of this model is that, in organisms where the metallochaperone is absent, there is no need to describe elaborate backup or compensatory mechanisms. Instead, the main considerations would be the oxidation state of Cu, as well as the relative amounts and Cu affinities of the target cuproenzymes, of the metallochaperones, and of the extracytoplasmic buffer. Differences in these properties may explain why periplasmic cuproproteins do not acquire Cu when expressed homologously in the cytoplasm. If the affinities of the cytoplasmic buffer for Cu are higher than the affinities of the cuproenzymes (i.e. bound Cu in the buffer is less energetic or more stable), transfer of Cu out of the buffer would be thermodynamically uphill or endergonic. Hence, knowledge of the relative tunings of extracytoplasmic buffer components compared to the cytoplasm becomes equally important.
The hypothesis that the cytoplasm supplies nutrient Cu to the extracytoplasmic space highlights a critical gap in knowledge. The extracytoplasmic space is largely contiguous with the extracellular environment. During conditions of environmental Cu deficiency, the Cu buffer could spontaneously drain, e.g. via diffusion through outer membrane porins, although this may be offset by outer membrane Cu uptake receptors or chalkophores, if they are present. By contrast, the cytoplasm is encapsulated within an impermeable lipid bilayer. While this appears to be a sensible solution for maintaining a stable supply of nutrient Cu, the sequence of events is unclear. How Cu fills the cytoplasmic buffer in the first place still needs investigation. Moreover, some Cu exporters (e.g. CopA and GolT from S. Typhimurium) operate under the control of cytoplasmic Cu sensors that activate transcription only when the cytoplasmic buffer is “full”. Under these conditions, buffered Cu availability outside the cytoplasm is presumably also elevated and indeed multiple extracytoplasmic components of Cu tolerance are usually produced.116 Why, then, is extracytoplasmic buffered Cu not used directly as the source of nutrient Cu? Is this related to the oxidation state of the metal? What is the contribution of Cu storage proteins like Csp, which can be present in the cytosol or the periplasm?117 Systems measurements of the buffered Cu availabilities, and comparisons between the cytoplasm and the periplasm, even if technically challenging, may prove illuminating. These can build on recent pioneering efforts by others in the metallomics community to decipher bacterial Cu (and metal) homeostasis.1,3,4,81,118,119
Conflicts of interest
We declare no conflict of interest.Acknowledgements
We thank Peter Chivers and Nigel Robinson (Department of Biosciences, Durham University) for enlightening discussions related to metal buffers. Louisa Stewart is supported by a Research Excellence Academy Studentship funded by Newcastle University. Denis Thaqi is supported by a Research Training Program Scholarship (Department of Education and Training, Commonwealth of Australia). Bostjan Kobe is supported by the National Health and Medical Research Council (NHMRC) Program Grant 1071659 and NHMRC Principal Research Fellowship (APP1110971). Kevin Waldron is supported by a Sir Henry Dale Fellowship (098375/Z/12/Z) co-funded by the Wellcome Trust and the Royal Society. Karrera Djoko is supported by the Royal Society Research Grant (RSG\R1\180044).References
- A. W. Foster, D. Osman and N. J. Robinson, Metal preferences and metallation, J. Biol. Chem., 2014, 289, 28095–28103 CrossRef CAS.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, T. Kozyreva, K. Zovo and P. Palumaa, Affinity gradients drive copper to cellular destinations, Nature, 2010, 465, 645–648 CrossRef CAS.
- D. Osman, A. W. Foster, J. Chen, K. Svedaite, J. W. Steed, E. Lurie-Luke, T. G. Huggins and N. J. Robinson, Fine control of metal concentrations is necessary for cells to discern zinc from cobalt, Nat. Commun., 2017, 8, 1884 CrossRef PubMed.
- A. W. Foster, R. Pernil, C. J. Patterson, A. J. P. Scott, L. O. Palsson, R. Pal, I. Cummins, P. T. Chivers, E. Pohl and N. J. Robinson, A tight tunable range for Ni(II) sensing and buffering in cells, Nat. Chem. Biol., 2017, 13, 409–414 CrossRef CAS PubMed.
- N. J. Robinson and D. R. Winge, Copper metallochaperones, Annu. Rev. Biochem., 2010, 79, 537–562 CrossRef CAS.
- A. D. Smith, B. L. Logeman and D. J. Thiele, Copper acquisition and utilization in fungi, Annu. Rev. Microbiol., 2017, 71, 597–623 CrossRef CAS.
- Z. N. Baker, P. A. Cobine and S. C. Leary, The mitochondrion: a central architect of copper homeostasis, Metallomics, 2017, 9, 1501–1512 RSC.
- S. Lutsenko, Copper trafficking to the secretory pathway, Metallomics, 2016, 8, 840–852 RSC.
- J. H. Kaplan and E. B. Maryon, How mammalian cells acquire copper: an essential but potentially toxic metal, Biophys. J., 2016, 110, 7–13 CrossRef CAS PubMed.
- J. M. Arguello, D. Raimunda and T. Padilla-Benavides, Mechanisms of copper homeostasis in bacteria, Front. Cell. Infect. Microbiol., 2013, 3, 73 Search PubMed.
- D. Osman and J. S. Cavet, Copper homeostasis in bacteria, Adv. Appl. Microbiol., 2008, 65, 217–247 CAS.
- E. Ladomersky and M. J. Petris, Copper tolerance and virulence in bacteria, Metallomics, 2015, 7, 957–964 RSC.
- H. Decker and N. Terwilliger, Cops and robbers: putative evolution of copper oxygen-binding proteins, J. Exp. Biol., 2000, 203, 1777–1782 CAS.
- J. F. Lutkenhaus, Role of a major outer membrane protein in Escherichia coli, J. Bacteriol., 1977, 131, 631–637 CAS.
- S. Tottey, K. J. Waldron, S. J. Firbank, B. Reale, C. Bessant, K. Sato, T. R. Cheek, J. Gray, M. J. Banfield, C. Dennison and N. J. Robinson, Protein-folding location can regulate manganese-binding versus copper- or zinc-binding, Nature, 2008, 455, 1138–1142 CrossRef CAS.
- P. Stolle, B. Hou and T. Bruser, The Tat substrate CueO is transported in an incomplete folding state, J. Biol. Chem., 2016, 291, 13520–13528 CrossRef CAS.
- J. H. Freedman, M. R. Ciriolo and J. Peisach, The role of glutathione in copper metabolism and toxicity, J. Biol. Chem., 1989, 264, 5598–5605 CAS.
- M. S. Pittman, H. C. Robinson and R. K. Poole, A bacterial glutathione transporter (Escherichia coli CydDC) exports reductant to the periplasm, J. Biol. Chem., 2005, 280, 32254–32261 CrossRef CAS.
- M. T. Morgan, L. A. H. Nguyen, H. L. Hancock and C. J. Fahrni, Glutathione limits aqua copper(I) to sub-femtomolar concentrations through cooperative assembly of a tetranuclear cluster, J. Biol. Chem., 2017, 292, 21558–21567 CrossRef CAS.
- Z. Xiao, J. Brose, S. Schimo, S. M. Ackland, S. La Fontaine and A. G. Wedd, Unification of the copper(I) binding affinities of the metallo-chaperones Atx1, Atox1, and related proteins: detection probes and affinity standards, J. Biol. Chem., 2011, 286, 11047–11055 CrossRef CAS.
- P. I. Trasnea, A. Andrei, D. Marckmann, M. Utz, B. Khalfaoui-Hassani, N. Selamoglu, F. Daldal and H. G. Koch, A copper relay system involving two periplasmic chaperones drives cbb3-type cytochrome c oxidase biogenesis in rhodobacter capsulatus, ACS Chem. Biol., 2018, 13, 1388–1397 CrossRef CAS.
- D. Osman, K. J. Waldron, H. Denton, C. M. Taylor, A. J. Grant, P. Mastroeni, N. J. Robinson and J. S. Cavet, Copper homeostasis in Salmonella is atypical and copper-CueP is a major periplasmic metal complex, J. Biol. Chem., 2010, 285, 25259–25268 CrossRef CAS.
- K. J. Waldron, S. J. Firbank, S. J. Dainty, M. Perez-Rama, S. Tottey and N. J. Robinson, Structure and metal loading of a soluble periplasm cuproprotein, J. Biol. Chem., 2010, 285, 32504–32511 CrossRef CAS.
- A. Speer, J. L. Rowland, M. Haeili, M. Niederweis and F. Wolschendorf, Porins increase copper susceptibility of Mycobacterium tuberculosis, J. Bacteriol., 2013, 195, 5133–5140 CrossRef CAS.
- X. Z. Li, H. Nikaido and K. E. Williams, Silver-resistant mutants of Escherichia coli display active efflux of Ag+ and are deficient in porins, J. Bacteriol., 1997, 179, 6127–6132 CrossRef CAS.
- H. S. Lee, A. H. Abdelal, M. A. Clark and J. L. Ingraham, Molecular characterization of nosA, a Pseudomonas stutzeri gene encoding an outer membrane protein required to make copper-containing N2O reductase, J. Bacteriol., 1991, 173, 5406–5413 CrossRef CAS.
- T. C. Johnstone and E. M. Nolan, Beyond iron: non-classical biological functions of bacterial siderophores, Dalton Trans., 2015, 44, 6320–6339 RSC.
- G. Ghssein, C. Brutesco, L. Ouerdane, C. Fojcik, A. Izaute, S. Wang, C. Hajjar, R. Lobinski, D. Lemaire, P. Richaud, R. Voulhoux, A. Espaillat, F. Cava, D. Pignol, E. Borezee-Durant and P. Arnoux, Biosynthesis of a broad-spectrum nicotianamine-like metallophore in Staphylococcus aureus, Science, 2016, 352, 1105–1109 CrossRef CAS.
- G. E. Kenney and A. C. Rosenzweig, Chalkophores, Annu. Rev. Biochem., 2018, 87, 645–676 CrossRef CAS.
- E. I. Koh, A. E. Robinson, N. Bandara, B. E. Rogers and J. P. Henderson, Copper import in Escherichia coli by the yersiniabactin metallophore system, Nat. Chem. Biol., 2017, 13, 1016–1021 CrossRef CAS.
- B. K. Hassani, C. Astier, W. Nitschke and S. Ouchane, CtpA, a copper-translocating P-type ATPase involved in the biogenesis of multiple copper-requiring enzymes, J. Biol. Chem., 2010, 285, 19330–19337 CrossRef CAS PubMed.
- D. Osman, C. J. Patterson, K. Bailey, K. Fisher, N. J. Robinson, S. E. Rigby and J. S. Cavet, The copper supply pathway to a Salmonella Cu,Zn-superoxide dismutase (SodCII) involves P(1B)-type ATPase copper efflux and periplasmic CueP, Mol. Microbiol., 2013, 87, 466–477 CrossRef CAS.
- R. A. Sanford, D. D. Wagner, Q. Wu, J. C. Chee-Sanford, S. H. Thomas, C. Cruz-Garcia, G. Rodriguez, A. Massol-Deya, K. K. Krishnani, K. M. Ritalahti, S. Nissen, K. T. Konstantinidis and F. E. Loffler, Unexpected nondenitrifier nitrous oxide reductase gene diversity and abundance in soils, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19709–19714 CrossRef CAS.
- J. A. Farrar, W. G. Zumft and A. J. Thomson, CuA and CuZ are variants of the electron transfer center in nitrous oxide reductase, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 9891–9896 CrossRef CAS.
- K. Brown, M. Tegoni, M. Prudencio, A. S. Pereira, S. Besson, J. J. Moura, I. Moura and C. Cambillau, A novel type of catalytic copper cluster in nitrous oxide reductase, Nat. Struct. Biol., 2000, 7, 191–195 CrossRef CAS PubMed.
- J. M. Charnock, A. Dreusch, H. Korner, F. Neese, J. Nelson, A. Kannt, H. Michel, C. D. Garner, P. M. Kroneck and W. G. Zumft, Structural investigations of the CuA centre of nitrous oxide reductase from Pseudomonas stutzeri by site-directed mutagenesis and X-ray absorption spectroscopy, Eur. J. Biochem., 2000, 267, 1368–1381 CrossRef CAS.
- T. Rasmussen, B. C. Berks, J. Sanders-Loehr, D. M. Dooley, W. G. Zumft and A. J. Thomson, The catalytic center in nitrous oxide reductase, CuZ, is a copper-sulfide cluster, Biochemistry, 2000, 39, 12753–12756 CrossRef CAS.
- H. Felgate, G. Giannopoulos, M. J. Sullivan, A. J. Gates, T. A. Clarke, E. Baggs, G. Rowley and D. J. Richardson, The impact of copper, nitrate and carbon status on the emission of nitrous oxide by two species of bacteria with biochemically distinct denitrification pathways, Environ. Microbiol., 2012, 14, 1788–1800 CrossRef CAS.
- T. Matsubara, K. Frunzke and W. G. Zumft, Modulation by copper of the products of nitrite respiration in Pseudomonas perfectomarinus, J. Bacteriol., 1982, 149, 816–823 CAS.
- M. J. Sullivan, A. J. Gates, C. Appia-Ayme, G. Rowley and D. J. Richardson, Copper control of bacterial nitrous oxide emission and its impact on vitamin B12-dependent metabolism, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19926–19931 CrossRef CAS.
- A. Black, P. C. Hsu, K. E. Hamonts, T. J. Clough and L. M. Condron, Influence of copper on expression of nirS, norB and nosZ and the transcription and activity of NIR, NOR and N2 or in the denitrifying soil bacteria Pseudomonas stutzeri, Microb. Biotechnol., 2016, 9, 381–388 CrossRef CAS.
- M. P. Heikkila, U. Honisch, P. Wunsch and W. G. Zumft, Role of the Tat ransport system in nitrous oxide reductase translocation and cytochrome cd1 biosynthesis in Pseudomonas stutzeri, J. Bacteriol., 2001, 183, 1663–1671 CrossRef CAS.
- W. G. Zumft and P. M. Kroneck, Respiratory transformation of nitrous oxide (N2O) to dinitrogen by bacteria and archaea, Adv. Microb. Physiol., 2007, 52, 107–227 CrossRef CAS.
- W. G. Zumft, A. Viebrock-Sambale and C. Braun, Nitrous oxide reductase from denitrifying Pseudomonas stutzeri. Genes for copper-processing and properties of the deduced products, including a new member of the family of ATP/GTP-binding proteins, Eur. J. Biochem., 1990, 192, 591–599 CrossRef CAS.
- N. Minagawa and W. G. Zumft, Cadmium-copper antagonism in the activation of periplasmic nitrous oxide reductase of copper-deficient cells from Pseudomonas stutzeri, Biol. Met., 1988, 1, 117–122 CrossRef CAS.
- P. Wunsch, M. Herb, H. Wieland, U. M. Schiek and W. G. Zumft, Requirements for Cu(A) and Cu–S center assembly of nitrous oxide reductase deduced from complete periplasmic enzyme maturation in the nondenitrifier Pseudomonas putida, J. Bacteriol., 2003, 185, 887–896 CrossRef CAS.
- J. Riester, W. G. Zumft and P. M. Kroneck, Nitrous oxide reductase from Pseudomonas stutzeri. Redox properties and spectroscopic characterization of different forms of the multicopper enzyme, Eur. J. Biochem., 1989, 178, 751–762 CrossRef CAS.
- A. Viebrock and W. G. Zumft, Molecular cloning, heterologous expression, and primary structure of the structural gene for the copper enzyme nitrous oxide reductase from denitrifying Pseudomonas stutzeri, J. Bacteriol., 1988, 170, 4658–4668 CrossRef CAS.
- M. A. McGuirl, J. A. Bollinger, N. Cosper, R. A. Scott and D. M. Dooley, Expression, purification, and characterization of NosL, a novel Cu(I) protein of the nitrous oxide reductase (nos) gene cluster, J. Biol. Inorg. Chem., 2001, 6, 189–195 CrossRef CAS.
- L. M. Taubner, M. A. McGuirl, D. M. Dooley and V. Copie, 1H, 13C, 15N backbone and sidechain resonance assignments of apo-NosL, a novel copper(I) binding protein from the nitrous oxide reductase gene cluster of achromobacter cycloclastes, J. Biomol. NMR, 2004, 29, 211–212 CrossRef CAS PubMed.
- L. M. Taubner, M. A. McGuirl, D. M. Dooley and V. Copie, Structural studies of apo-NosL, an accessory protein of the nitrous oxide reductase system: insights from structural homology with MerB, a mercury resistance protein, Biochemistry, 2006, 45, 12240–12252 CrossRef CAS PubMed.
- B. J. Vaccaro, M. P. Thorgersen, W. A. Lancaster, M. N. Price, K. M. Wetmore, F. L. Poole, 2nd, A. Deutschbauer, A. P. Arkin and M. W. Adams, Determining roles of accessory genes in denitrification by mutant fitness analyses, Appl. Environ. Microbiol., 2016, 82, 51–61 CrossRef CAS.
- A. Dreusch, J. Riester, P. M. Kroneck and W. G. Zumft, Mutation of the conserved Cys165 outside of the CuA domain destabilizes nitrous oxide reductase but maintains its catalytic activity. Evidence for disulfide bridges and a putative protein disulfide isomerase gene, Eur. J. Biochem., 1996, 237, 447–453 CrossRef CAS.
- K. Mokhele, Y. J. Tang, M. A. Clark and J. L. Ingraham, A Pseudomonas stutzeri outer membrane protein inserts copper into N2O reductase, J. Bacteriol., 1987, 169, 5721–5726 CrossRef CAS.
- H. S. Lee, R. E. Hancock and J. L. Ingraham, Properties of a Pseudomonas stutzeri outer membrane channel-forming protein (NosA) required for production of copper-containing N2O reductase, J. Bacteriol., 1989, 171, 2096–2100 CrossRef CAS.
- S. Horrell, D. Kekilli, R. W. Strange and M. A. Hough, Recent structural insights into the function of copper nitrite reductases, Metallomics, 2017, 9, 1470–1482 RSC.
- L. Philippot, Denitrifying genes in bacterial and archaeal genomes, Biochim. Biophys. Acta, 2002, 1577, 355–376 CrossRef CAS.
- R. Jain and J. P. Shapleigh, Characterization of nirV and a gene encoding a novel pseudoazurin in Rhodobacter sphaeroides 2.4.3, Microbiology, 2001, 147, 2505–2515 CrossRef CAS.
- D. Helen, H. Kim, B. Tytgat and W. Anne, Highly diverse nirK genes comprise two major clades that harbour ammonium-producing denitrifiers, BMC Genomics, 2016, 17, 155 CrossRef.
- F. E. Jen, K. Y. Djoko, S. J. Bent, C. J. Day, A. G. McEwan and M. P. Jennings, A genetic screen reveals a periplasmic copper chaperone required for nitrite reductase activity in pathogenic Neisseria, FASEB J., 2015, 29, 3828–3838 CrossRef CAS.
- L. A. Abriata, L. Banci, I. Bertini, S. Ciofi-Baffoni, P. Gkazonis, G. A. Spyroulias, A. J. Vila and S. Wang, Mechanism of Cu(A) assembly, Nat. Chem. Biol., 2008, 4, 599–601 CrossRef CAS.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, E. Katsari, N. Katsaros, K. Kubicek and S. Mangani, A copper(I) protein possibly involved in the assembly of CuA center of bacterial cytochrome c oxidase, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3994–3999 CrossRef CAS.
- P. I. Trasnea, M. Utz, B. Khalfaoui-Hassani, S. Lagies, F. Daldal and H. G. Koch, Cooperation between two periplasmic copper chaperones is required for full activity of the cbb3-type cytochrome c oxidase and copper homeostasis in Rhodobacter capsulatus, Mol. Microbiol., 2016, 100, 345–361 CrossRef CAS.
- A. K. Thompson, J. Gray, A. Liu and J. P. Hosler, The roles of Rhodobacter sphaeroides copper chaperones PCu(A)C and Sco (PrrC) in the assembly of the copper centers of the aa(3)-type and the cbb(3)-type cytochrome c oxidases, Biochim. Biophys. Acta, 2012, 1817, 955–964 CrossRef CAS.
- F. Serventi, Z. A. Youard, V. Murset, S. Huwiler, D. Buhler, M. Richter, R. Luchsinger, H. M. Fischer, R. Brogioli, M. Niederer and H. Hennecke, Copper starvation-inducible protein for cytochrome oxidase biogenesis in Bradyrhizobium japonicum, J. Biol. Chem., 2012, 287, 38812–38823 CrossRef CAS.
- K. L. Seib, M. P. Jennings and A. G. McEwan, A Sco homologue plays a role in defence against oxidative stress in pathogenic Neisseria, FEBS Lett., 2003, 546, 411–415 CrossRef CAS.
- L. Banci, I. Bertini, G. Cavallaro and A. Rosato, The functions of Sco proteins from genome-based analysis, J. Proteome Res., 2007, 6, 1568–1579 CrossRef CAS.
- J. S. Kroll, P. R. Langford, K. E. Wilks and A. D. Keil, Bacterial [Cu,Zn]-superoxide dismutase: phylogenetically distinct from the eukaryotic enzyme, and not so rare after all!, Microbiology, 1995, 141(Pt 9), 2271–2279 CrossRef CAS.
- K. L. Dunn, J. L. Farrant, P. R. Langford and J. S. Kroll, Bacterial [Cu,Zn]-cofactored superoxide dismutase protects opsonized, encapsulated Neisseria meningitidis from phagocytosis by human monocytes/macrophages, Infect. Immun., 2003, 71, 1604–1607 CrossRef CAS.
- K. E. Wilks, K. L. Dunn, J. L. Farrant, K. M. Reddin, A. R. Gorringe, P. R. Langford and J. S. Kroll, Periplasmic superoxide dismutase in meningococcal pathogenicity, Infect. Immun., 1998, 66, 213–217 CAS.
- A. Sansone, P. R. Watson, T. S. Wallis, P. R. Langford and J. S. Kroll, The role of two periplasmic copper- and zinc-cofactored superoxide dismutases in the virulence of Salmonella choleraesuis, Microbiology, 2002, 148, 719–726 CrossRef CAS.
- A. Battistoni, F. Pacello, S. Folcarelli, M. Ajello, G. Donnarumma, R. Greco, M. G. Ammendolia, D. Touati, G. Rotilio and P. Valenti, Increased expression of periplasmic Cu,Zn superoxide dismutase enhances survival of Escherichia coli invasive strains within nonphagocytic cells, Infect. Immun., 2000, 68, 30–37 CrossRef CAS.
- M. D'Orazio, S. Folcarelli, F. Mariani, V. Colizzi, G. Rotilio and A. Battistoni, Lipid modification of the Cu,Zn superoxide dismutase from Mycobacterium tuberculosis, Biochem. J., 2001, 359, 17–22 CrossRef.
- Q. Huang and T. Palmer, Signal peptide hydrophobicity modulates interaction with the twin-arginine translocase, mBio, 2017, 8, e00909-17 CrossRef.
- I. Bertini, S. Manganl and M. S. Viezzoli, in Advances in inorganic chemistry, ed. A. G. Sykes, Academic Press, 1998, vol. 45, pp. 127–250 Search PubMed.
- L. A. Fenlon and J. M. Slauch, Cytoplasmic copper detoxification in Salmonella can contribute to SodC metalation but is dispensable during systemic infection, J. Bacteriol., 2017, 199, e00437-17 CrossRef.
- L. B. Pontel and F. C. Soncini, Alternative periplasmic copper-resistance mechanisms in Gram negative bacteria, Mol. Microbiol., 2009, 73, 212–225 CrossRef CAS.
- B. Y. Yoon, Y. H. Kim, N. Kim, B. Y. Yun, J. S. Kim, J. H. Lee, H. S. Cho, K. Lee and N. C. Ha, Structure of the periplasmic copper-binding protein CueP from Salmonella enterica serovar Typhimurium, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1867–1875 CrossRef CAS.
- A. Pezza, L. B. Pontel, C. Lopez and F. C. Soncini, Compartment and signal-specific codependence in the transcriptional control of Salmonella periplasmic copper homeostasis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11573–11578 CrossRef CAS.
- A. S. Gort, D. M. Ferber and J. A. Imlay, The regulation and role of the periplasmic copper, zinc superoxide dismutase of Escherichia coli, Mol. Microbiol., 1999, 32, 179–191 CrossRef CAS.
- W. L. Pang, A. Kaur, A. V. Ratushny, A. Cvetkovic, S. Kumar, M. Pan, A. P. Arkin, J. D. Aitchison, M. W. Adams and N. S. Baliga, Metallochaperones regulate intracellular copper levels, PLoS Comput. Biol., 2013, 9, e1002880 CrossRef CAS.
- Y. Hatori and S. Lutsenko, An expanding range of functions for the copper chaperone/antioxidant protein Atox1, Antioxid. Redox Signaling, 2013, 19, 945–957 CrossRef CAS.
- D. L. Huffman and T. V. O'Halloran, Function, structure, and mechanism of intracellular copper trafficking proteins, Annu. Rev. Biochem., 2001, 70, 677–701 CrossRef CAS.
- K. Y. Djoko, L. X. Chong, A. G. Wedd and Z. Xiao, Reaction mechanisms of the multicopper oxidase CueO from Escherichia coli support its functional role as a cuprous oxidase, J. Am. Chem. Soc., 2010, 132, 2005–2015 CrossRef CAS.
- S. K. Singh, G. Grass, C. Rensing and W. R. Montfort, Cuprous oxidase activity of CueO from Escherichia coli, J. Bacteriol., 2004, 186, 7815–7817 CrossRef CAS.
- S. Ammendola, P. Pasquali, F. Pacello, G. Rotilio, M. Castor, S. J. Libby, N. Figueroa-Bossi, L. Bossi, F. C. Fang and A. Battistoni, Regulatory and structural differences in the Cu,Zn-superoxide dismutases of Salmonella enterica and their significance for virulence, J. Biol. Chem., 2008, 283, 13688–13699 CrossRef CAS.
- D. U. Mick, T. D. Fox and P. Rehling, Inventory control: cytochrome c oxidase assembly regulates mitochondrial translation, Nat. Rev. Mol. Cell Biol., 2011, 12, 14–20 CrossRef CAS.
- K. A. Jett and S. C. Leary, Building the CuA site of cytochrome c oxidase: a complicated, redox-dependent process driven by a surprisingly large complement of accessory proteins, J. Biol. Chem., 2018, 293, 4644–4652 CrossRef CAS.
- S. Schimo, I. Wittig, K. M. Pos and B. Ludwig, Cytochrome c oxidase biogenesis and metallochaperone interactions: steps in the assembly pathway of a bacterial complex, PLoS One, 2017, 12, e0170037 CrossRef.
- J. Buggy and C. E. Bauer, Cloning and characterization of senC, a gene involved in both aerobic respiration and photosynthesis gene expression in Rhodobacter capsulatus, J. Bacteriol., 1995, 177, 6958–6965 CrossRef CAS.
- D. L. Swem, L. R. Swem, A. Setterdahl and C. E. Bauer, Involvement of senC in assembly of cytochrome c oxidase in Rhodobacter capsulatus, J. Bacteriol., 2005, 187, 8081–8087 CrossRef CAS.
- E. Frangipani and D. Haas, Copper acquisition by the senC protein regulates aerobic respiration in Pseudomonas aeruginosa PA01, FEMS Microbiol. Lett., 2009, 298, 234–240 CrossRef CAS.
- E. Lohmeyer, S. Schroder, G. Pawlik, P. I. Trasnea, A. Peters, F. Daldal and H. G. Koch, The ScoI homologue SenC is a copper binding protein that interacts directly with the cbb(3)-type cytochrome oxidase in Rhodobacter capsulatus, Biochim. Biophys. Acta, 2012, 1817, 2005–2015 CrossRef CAS.
- A. G. McEwan, A. Lewin, S. L. Davy, R. Boetzel, A. Leech, D. Walker, T. Wood and G. R. Moore, PrrC from Rhodobacter sphaeroides, a homologue of eukaryotic Sco proteins, is a copper-binding protein and may have a thiol–disulfide oxidoreductase activity, FEBS Lett., 2002, 518, 10–16 CrossRef CAS.
- K. L. Blundell, M. A. Hough, E. Vijgenboom and J. A. Worrall, Structural and mechanistic insights into an extracytoplasmic copper trafficking pathway in Streptomyces lividans, Biochem. J., 2014, 459, 525–538 CrossRef CAS.
- P. Gurumoorthy and B. Ludwig, Deciphering protein–protein interactions during the biogenesis of cytochrome c oxidase from Paracoccus denitrificans, FEBS J., 2015, 282, 537–549 CrossRef CAS.
- D. Buhler, R. Rossmann, S. Landolt, S. Balsiger, H. M. Fischer and H. Hennecke, Disparate pathways for the biogenesis of cytochrome oxidases in Bradyrhizobium japonicum, J. Biol. Chem., 2010, 285, 15704–15713 CrossRef CAS.
- P. Greiner, A. Hannappel, C. Werner and B. Ludwig, Biogenesis of cytochrome c oxidase – in vitro approaches to study cofactor insertion into a bacterial subunit I, Biochim. Biophys. Acta, 2008, 1777, 904–911 CrossRef CAS.
- J. Bengtsson, C. von Wachenfeldt, L. Winstedt, P. Nygaard and L. Hederstedt, CtaG is required for formation of active cytochrome c oxidase in Bacillus subtilis, Microbiology, 2004, 150, 415–425 CrossRef CAS.
- L. Hiser, M. Di Valentin, A. G. Hamer and J. P. Hosler, Cox11p is required for stable formation of the Cu(B) and magnesium centers of cytochrome c oxidase, J. Biol. Chem., 2000, 275, 619–623 CrossRef CAS.
- F. Arnesano, L. Banci, I. Bertini and M. Martinelli, Ortholog search of proteins involved in copper delivery to cytochrome c oxidase and functional analysis of paralogs and gene neighbors by genomic context, J. Proteome Res., 2005, 4, 63–70 CrossRef CAS PubMed.
- K. R. Barth, V. M. Isabella and V. L. Clark, Biochemical and genomic analysis of the denitrification pathway within the genus Neisseria, Microbiology, 2009, 155, 4093–4103 CrossRef CAS.
- B. C. Hill and D. Andrews, Differential affinity of BsSco for Cu(II) and Cu(I) suggests a redox role in copper transfer to the Cu(A) center of cytochrome c oxidase, Biochim. Biophys. Acta, 2012, 1817, 948–954 CrossRef CAS.
- H. K. Abicht, M. A. Scharer, N. Quade, R. Ledermann, E. Mohorko, G. Capitani, H. Hennecke and R. Glockshuber, How periplasmic thioredoxin TlpA reduces bacterial copper chaperone ScoI and cytochrome oxidase subunit II (Coxb) prior to metallation, J. Biol. Chem., 2014, 289, 32431–32444 CrossRef CAS.
- E. Mohorko, H. K. Abicht, D. Buhler, R. Glockshuber, H. Hennecke and H. M. Fischer, Thioredoxin-like protein tlpa from Bradyrhizobium japonicum is a reductant for the copper metallochaperone ScoI, FEBS Lett., 2012, 586, 4094–4099 CrossRef CAS.
- E. Lohmeyer, S. Schroder, G. Pawlik, P. I. Trasnea, A. Peters, F. Daldal and H. G. Koch, The ScoI homologue senc is a copper binding protein that interacts directly with the cbb3-type cytochrome oxidase in Rhodobacter capsulatus, Biochim. Biophys. Acta, 2012, 1817, 2005–2015 CrossRef CAS.
- N. R. Mattatall, J. Jazairi and B. C. Hill, Characterization of YpmQ, an accessory protein required for the expression of cytochrome c oxidase in Bacillus subtilis, J. Biol. Chem., 2000, 275, 28802–28809 CrossRef CAS.
- A. K. Thompson, J. Gray, A. Liu and J. P. Hosler, The roles of rhodobacter sphaeroides copper chaperones PCu(A)C and Sco (PrrC) in the assembly of the copper centers of the aa3-type and the cbb3-type cytochrome c oxidases, Biochim. Biophys. Acta, 2012, 1817, 955–964 CrossRef CAS.
- J. A. Farrar, F. Neese, P. Lappalainen, P. M. H. Kroneck, M. Saraste, W. G. Zumft and A. J. Thomson, The electronic structure of CuA: a novel mixed-valence dinuclear copper electron-transfer center, J. Am. Chem. Soc., 1996, 118, 11501–11514 CrossRef CAS.
- H. G. Koch, C. Winterstein, A. S. Saribas, J. O. Alben and F. Daldal, Roles of the ccoGHIS gene products in the biogenesis of the cbb(3)-type cytochrome c oxidase, J. Mol. Biol., 2000, 297, 49–65 CrossRef CAS.
- C. Kulajta, J. O. Thumfart, S. Haid, F. Daldal and H. G. Koch, Multi-step assembly pathway of the cbb3-type cytochrome c oxidase complex, J. Mol. Biol., 2006, 355, 989–1004 CrossRef CAS.
- S. Ekici, H. Yang, H. G. Koch and F. Daldal, Novel transporter required for biogenesis of cbb3-type cytochrome c oxidase in Rhodobacter capsulatus, mBio, 2012, 3, e00293-11 CrossRef.
- B. Khalfaoui-Hassani, H. Wu, C. E. Blaby-Haas, Y. Zhang, F. Sandri, A. F. Verissimo, H. G. Koch and F. Daldal, Widespread distribution and functional specificity of the copper importer CcoA: distinct Cu uptake routes for bacterial cytochrome c oxidases, mBio, 2018, 9, e00065-18 CrossRef.
- M. Gonzalez-Guerrero, D. Raimunda, X. Cheng and J. M. Arguello, Distinct functional roles of homologous Cu+ efflux ATPases in Pseudomonas aeruginosa, Mol. Microbiol., 2010, 78, 1246–1258 CrossRef CAS.
- S. Ekici, S. Turkarslan, G. Pawlik, A. Dancis, N. S. Baliga, H. G. Koch and F. Daldal, Intracytoplasmic copper homeostasis controls cytochrome c oxidase production, mBio, 2014, 5, e01055 CrossRef.
- D. Raimunda, T. Padilla-Benavides, S. Vogt, S. Boutigny, K. N. Tomkinson, L. A. Finney and J. M. Arguello, Periplasmic response upon disruption of transmembrane Cu transport in Pseudomonas aeruginosa, Metallomics, 2013, 5, 144–151 RSC.
- C. Dennison, S. David and J. Lee, Bacterial copper storage proteins, J. Biol. Chem., 2018, 293, 4616–4627 CrossRef CAS.
- E. Pecou, A. Maass, D. Remenik, J. Briche and M. Gonzalez, A mathematical model for copper homeostasis in Enterococcus hirae, Math. Biosci., 2006, 203, 222–239 CrossRef CAS.
- J. H. Parmar, J. Quintana, D. Ramírez, R. Laubenbacher, J. M. Argüello and P. Mendes, An important role for periplasmic storage in Pseudomonas aeruginosa copper homeostasis revealed by a combined experimental and computational modeling study, Mol. Microbiol., 2018, 1–13 CAS.
Footnote |
| † These authors contributed equally to this work and the names are listed alphabetically. |
| This journal is © The Royal Society of Chemistry 2019 |