
Quantitative proteomics discloses monacolin K-induced alterations in triple-negative breast cancer cell proteomes and phosphoproteomes†
Federica
del Gaudio
abc,
Ida Chiara
Guerrera
d,
Raffaele
Riccio
a and
Maria Chiara
Monti
 *a
*a
aDepartment of Pharmacy, University of Salerno, Via Giovanni Paolo II, 132, 84084 Fisciano (SA), Italy. E-mail: mcmonti@unisa.it
bPhD Program in Drug Discovery and Development, Dipartimento di Farmacia, University of Salerno, Via Giovanni Paolo II 132, 84084 Fisciano (SA), Italy
cBiomolecular Mass Spectrometry and Proteomics, Bijvoet Center for Biomolecular Research and Utrecht Institute for Pharmaceutical Sciences, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands
dProteomics Platform 3P5-Necker, Université Paris Descartes – Structure Fédérative de Recherche Necker, INSERM US24/CNRS UMS3633, Paris 75014, France
First published on 3rd December 2019
Abstract
A positive prognosis of triple-negative breast cancer can be considered as one of the major challenges in clinical studies; accordingly, scientific research has the mission to find out novel chemotherapeutics to make it curable. In recent times, a good potential of dietary bioactive natural substances, called nutraceuticals, in suppressing cancer cell proliferation via gene expression regulation has been discovered: this effect and the lack of toxicity make nutraceuticals potentially effective agents against cancers. Monacolin K from red rice, a FDA-approved and well-tolerated compound generally employed to treat hypercholesterolemia, has been proved to have anti-proliferative and apoptotic effects in a wide panel of triple-negative breast cancers. Thus, an unbiased analysis of monacolin K-induced MDA-MB-231 cellular pathway alterations has been carried out by quantitative proteomics exploiting isobaric tags. Despite the positive modulation of some proteins already reported in the literature, an increased concentration of the tissue-type plasminogen activator PLAT has interestingly been found. This is a marker of good prognosis in mammary cancer, suggesting the anti-metastatic properties of this molecule as strongly associated with the alterations in the cytoskeleton organization and the consequent modulation of adhesion, motility and proteolysis. In accordance, some of the found monacolin K-induced phosphoproteome alterations have a tight connection to cell migration mechanisms. In this setting, the over-phosphorylation of Lamin A and of melanophilin induced by monacolin K has been very attractive. Moreover, monacolin K exerts its effect on the over-expression of the tissue inhibitor metalloproteinase-2 (TIMP-2), an endogenous metalloproteinase inhibitor. This protein modulates growth, migration and invasion of tumor cells and inhibits tumor angiogenesis.
Introduction
A favorable prognosis of breast cancer, the most frequent cancer in women, still remains a major challenge in clinical studies. Around 1–1.3 million breast cancers are annually diagnosed, and approximately 20% belong to one of the triple-negative breast cancer (TNBC) subgroups1 with an incidence ranging between 10% and 20% among the female population under 50.2,3TNBCs have a large variety of subtypes sharing a negative profile for ER (ER-), progesterone receptor (PR-) and human epidermal growth factor receptor (HER2-), the three markers exploited to classify breast cancers.1,3 High nuclear grade, accelerated tumor proliferation rate, high nuclear-cytoplasmatic ratio and increased mitotic activity are the most common histopathological features of triple-negative breast cancers.3–6 Based on their gene expression profiling, six different subgroups of triple-negative breast cancers have been distinguished but, due to this inter- and intra-heterogeneity, the detection of clinical biomarkers for early diagnosis is still a challenging issue. In parallel, it could be helpful to find molecular targets for directed therapies, leading to novel chemotherapeutics.
For many years, plant-based natural products have been the source for many successful drugs, as their peculiar chemical structures are usually optimized along evolution, making the biomedical potential of natural organisms widely recognized. Moreover, they could be helpful to inspect proteins involved in biological processes to contribute to the advancement in target discovery. The crucial point is their involvement in different cellular pathways influencing protein expression, their co-/post-translational modifications, their interactome and crosstalk. In particular, many studies found that nutraceuticals may also reverse, repress, or prevent the carcinogenesis process, mainly suppressing cell proliferation and blocking the mutagenic activity. Dietary bioactive substances may have a great impact on the regulation of gene expression, even at very low concentrations:7 their effects in combination with the lack of toxicity make them potentially efficient anti-cancer agents.8
In this scenario, we were intrigued by monacolin K (MNK), a polyketide pigment produced by the mold of the Monascus species (mostly M. purpureus, M. ruber and M. pilosus) upon the fermentation of red rice (Oryza sativa, L.). Among all the metabolites isolated from red yeast rice, monacolins have been identified to be responsible for the red yeast rice-related bioactivities on dyslipidaemias, the major therapeutic effect of this functional food. MNK has been disclosed to act as the drug lovastatin with the advantage of a drop in the severe side effects reported for the latter synthetic alternative. Its foremost use involves lowering the hematic concentration of cholesterol acting as an inhibitor of 3-hydroxy-3-methyl-glutaryl coenzyme A (HMG-CoA) reductase.9 From a chemical point of view, MNK is a polyketide, whose reactive centre is the γ-lactone ring which is present as a mixture of the closed ring and the β-hydroxy acid open form in equilibrium; the latter form is the active one for HMG-CoA reductase inhibition.10 Nowadays, recent scientific evidences link this molecule, and more generally statins, to anti-proliferative and apoptotic effects in a wide panel of cancers such as human colon HCT-116 and HT-29 cell lines, human adenocarcinoma A549 cell line, human prostate cancer PC-3 cell line and in vivo related model, hepatocarcinoma HepG2 cells, human intestinal Caco-2 and human breast adenocarcinoma MCF-7 as well as breast cancers and TNBCs.11 Besides, red yeast rice has been investigated for its anti-inflammatory effect,12 hypotensive and cardioprotective activities, anti-diabetic potential and osteo-protective effect. Since statins are FDA-approved, well tolerated and affordable, they provide the opportunity for an accelerated repurposing as cancer therapeutics. Particularly, their anti-proliferative activity on in vitro models of breast cancers has been evaluated: only lipophilic statins (fluvastatin, simvastatin and lovastatin) were able to pass the cell membrane and affect cellular proliferation;13 furthermore, their IC50s were sensitively lower for the triple-negative breast cancer models (as MDA-MB-231 cells) than for the non-triple-negative breast cancer models (MCF-7 cells and SKBr3 cells).14,15
Therefore, MNK has got a good potential to prevent or treat a subset of breast cancers such as TNBCs. However, the mechanism of MNK action as an anticancer in TNBCs is still unexplored. Often, small molecules, particularly natural compounds, have more than one cellular targets affecting cell response from more than one point of the same pathway and/or from several levels of different pathways, either involved in crosstalk or non-communicating directly.
In this work, we performed MS-based proteomics for a detailed and unbiased analysis of the role of MNK in the context of TNBC cell models. In particular, we investigated protein expression and phosphorylation affected by MNK in MDA-MB-231 cells, applying a quantitative proteomic approach exploiting isobaric tags (TMT™-10plex). Real-time assays on live cell systems were performed to identify the optimal MNK concentrations and times of stimulation. MDA-MB-231 cells were treated with MNK and the corresponding mixtures of proteins were digested and processed following a quantitative MS-based workflow to find out the changes in both proteomes and phosphoproteomes. Western blotting analysis was performed to validate proteomics data and to confirm MNK-based alterations in TNBC cells.
Results
Cell proliferation assay
Cell proliferation assays were performed to profile the cytotoxic activity in MDA-MB-231 cells in the presence or absence of MNK ranging from 0.5 μM to 200 μM. By using IncuCyte ZOOM™ Live-Cell analysis system, proliferation kinetics was followed in real time and cell viability parameters were continuously recorded (Fig. 1A). Thus, the concentration of MNK inhibiting 20% and 50% of cell proliferation was calculated for each of the evaluated seeding density, converging in values of IC20 = 0.51 ± 0.07 and IC50 = 4.02 ± 1.23 μM, respectively, in accordance with the literature (Fig. 1A).11 Moreover, MNK at 0.5 and 5 μM was tested over time, and the obtained curves clearly showed two most noteworthy time points after 6 hours and 24 hours from the seeding (Fig. 1B): the 6 hour-time-point was considered as the first time point in which both proliferation curves of the treated cells have a significant difference in comparison with the control curve; the 24 hour-time-point was pointed out because it is the first time point of the cell-growth plateau for the cells treated with MNK at its IC50 concentration (Fig. 1B). In addition, a third 30 minute-time-point was selected for control.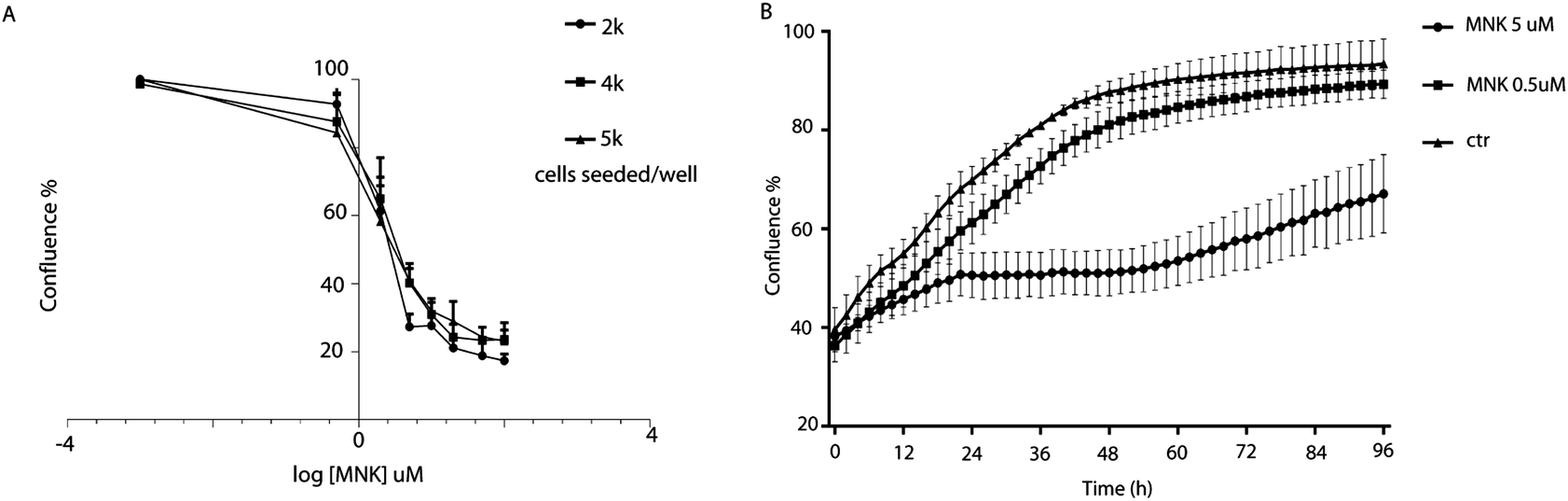 | ||
| Fig. 1 (A) Percentage of the MDA-MB-231 confluence among different MNK concentrations. Logarithmic curves were obtained based on the AUC representing the cumulative MNK effect on the growth over the time. The value of −3 on the x-axis is a hypothetical point in which the cells possess 100% confluence. (B) MDA-MB-231 proliferation curves at MNK concentrations of 0.5 and 5 μM, corresponding to IC20 and IC50, respectively, over time as graph output from the IncuCyte software. | ||
Drug treatment for proteomics
In order to perform proteomic and phosphoproteomic profiling, living cells were treated with two concentrations of the natural compound corresponding to its IC20 and IC50 ([MNK] = 0.5 μM and [MNK] = 5 μM, respectively). As a reference control, a treatment with 1% DMSO was performed. For each condition, three different stimulation time points were selected: a short time, ST = 30 minutes, an intermediate time, IT = 6 hour, and a long time, LT = 24 hours. After harvesting, the obtained nine samples (ST_CTRL; ST_MNK0.5; ST_MNK5; IT_CTRL; IT_MNK0.5; IT_MNK5; LT_CTRL; LT_MNK0.5; and LT_MNK5) were lysed and the aliquots containing the same content of proteins were digested. Fig. 2 shows the general workflow of the quantitative proteomic and phosphoproteomic experiments.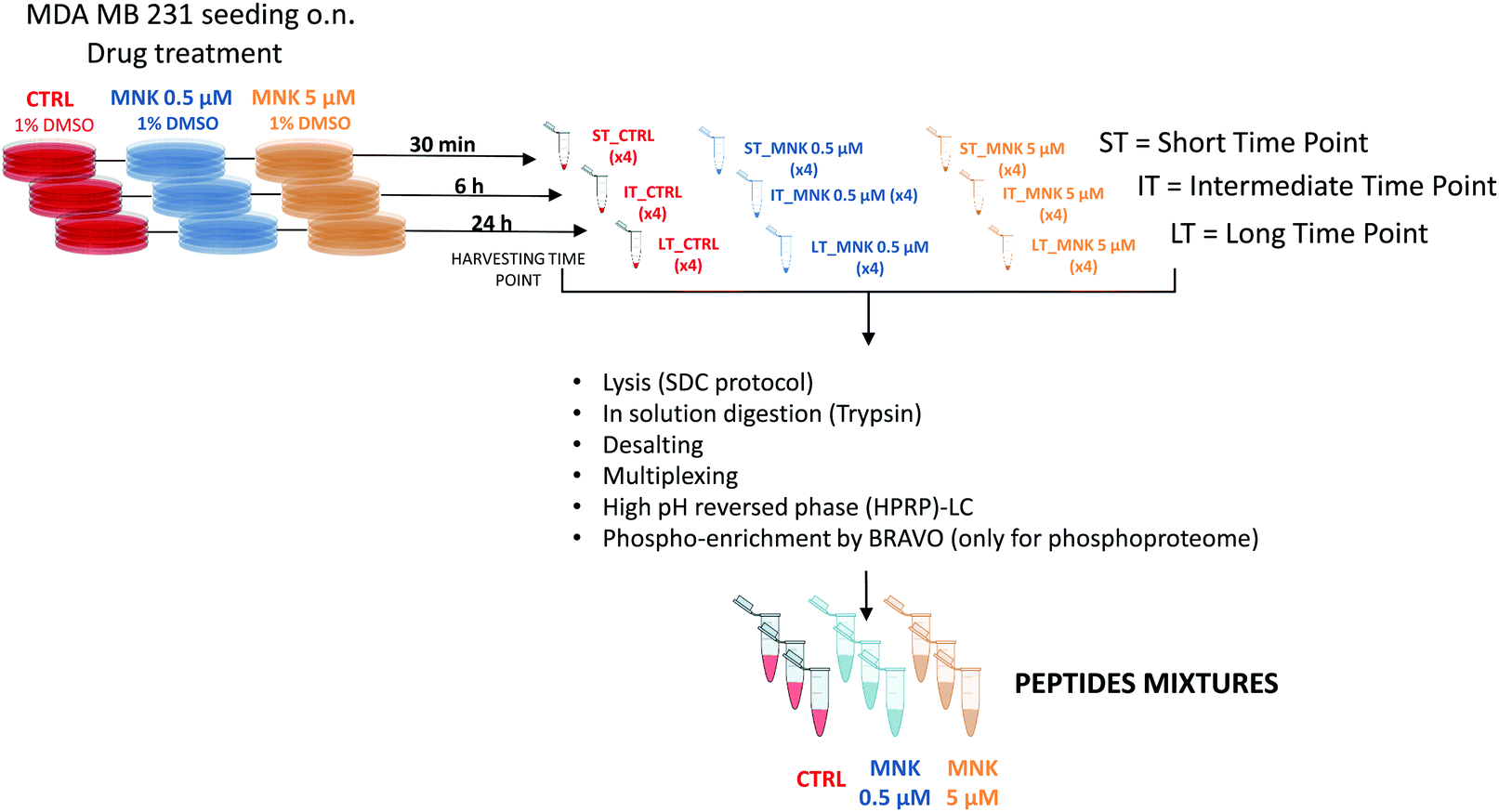 | ||
| Fig. 2 Proteomic and phosphoproteomic workflow: MDA-MB-231 cells were treated with two concentrations of MNK at its IC20 and IC50 ([MNK] = 0.5 μM and [MNK] = 5 μM, respectively). As a reference control, a treatment with the vehicle was performed. For each of the concentration-related conditions, three different harvesting time points (a short time, ST = 30 minutes, an intermediate time, IT = 6 hour, and a long time, LT = 24 hours) were performed. After the harvesting, the obtained nine samples of pelleted cells (ST_CTRL; ST_MNK0.5; ST_MNK5; IT_CTRL; IT_MNK0.5; IT_MNK5; LT_CTRL; LT_MNK0.5; and LT_MNK5) were lysed and digested by in-solution trypsinisation. All the obtained samples were labelled upon treatment with the TMT-10plex reagents. After the multiplexing, the obtained samples, one for each of the replicates, were purified and fractionated by HPRP-LC. Eight fractions for each replicate were obtained after the reunion step. For the phosphoproteome, aliquots of these 8 fractions were pooled again in 4 fractions for each replicate. A BRAVO Agilent system was used to run the phospho-enrichment of the peptides contained in these 4 fractions obtaining enrichment of phosphopeptides. All the fractions were then purified by Sep-Pak and prepared for the following mass spectrometric analysis. | ||
Quantitative proteomic and phosphoproteomic analysis
All the obtained samples were labelled upon treatment with the TMT-10plex reagents.16 The labelling efficiency was tested to be higher than 99%. After multiplexing, four samples were obtained, one for each of the biological replicates containing the nine conditions of stimulation by MNK. The four mixtures were purified and fractionated by high-pH reversed-phase (HPRP) liquid chromatography17 to access the deeper proteome. A total of 5408 proteins across samples were identified and reliably quantified.For each protein, the intensity was normalized to non-treated cells, used as the control (ESI,† Table S1). As expected, there is certain variation between the different replicates and, for this reason, two-way ANOVA statistical test on the identified proteins was performed setting two categorical classes for the stimulation: time of stimulation and MNK concentration. Thus, 10 proteins were quantified whose abundance was altered by the drug treatment with a p value <0.01. These proteins were clustered and visualized in a heat map after normalization by the z-score: they were mainly increased at the highest MNK dose (5 μM) and the longest time of exposure (24 hours as reported in Fig. 3A and 4A). Only lethal(3)malignant brain tumour-like protein 3 (L3MBTL3) was drastically increased already at MNK 0.5 μM concentration, suggesting a marked effect of the natural compound on L3MBTL3 expression (Fig. 3A and A). The same behavior was not revealed at an MNK concentration of 5 μM: anyway, in this condition several other proteins were overexpressed.
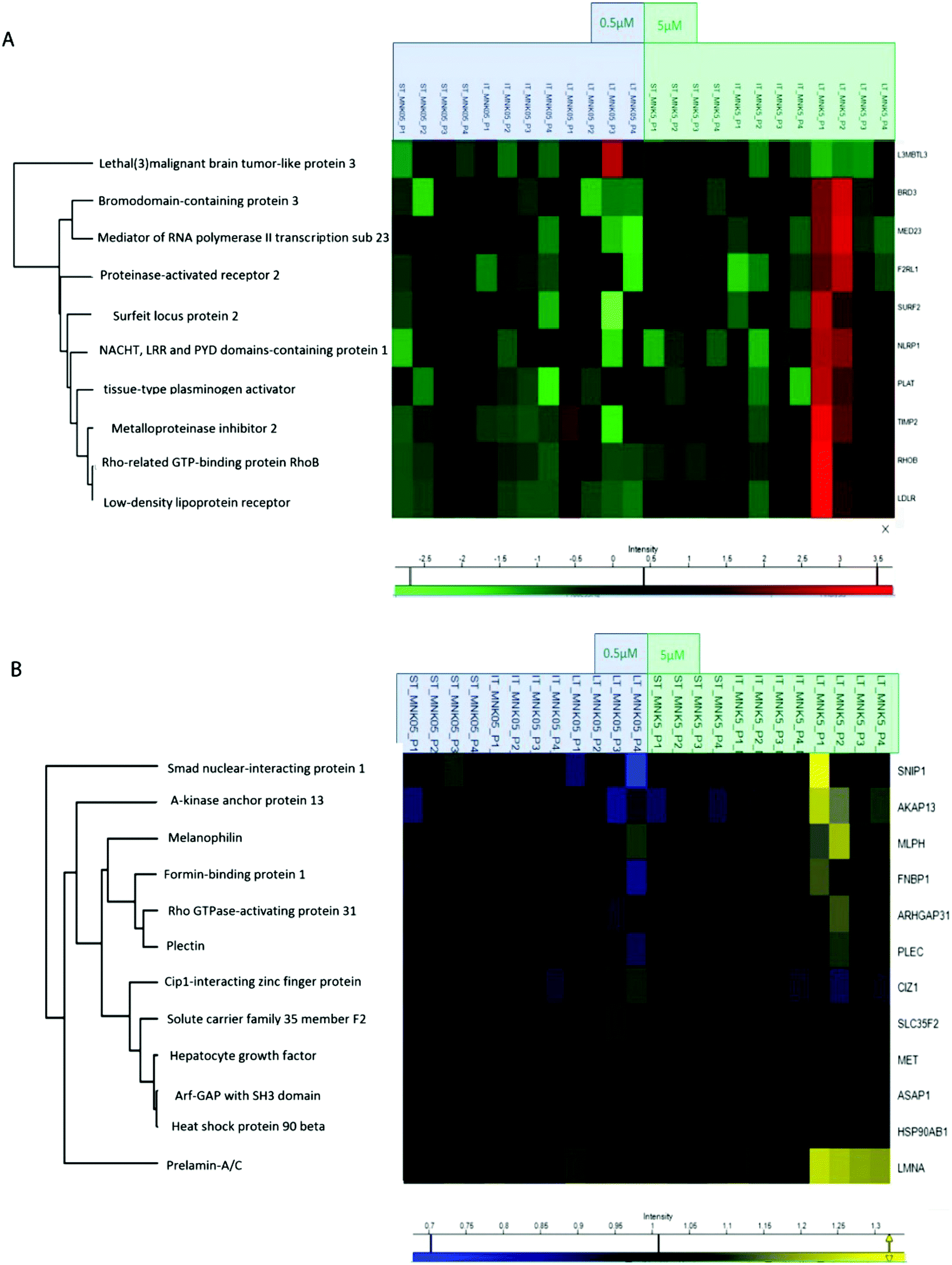 | ||
| Fig. 3 (A) Hierarchical clustering of the MNK-induced significative alterations of the MDA-MB-231 proteome. (B) Hierarchical clustering of the MNK-induced significative alterations of the MDA-MB-231 phosphoproteome. | ||
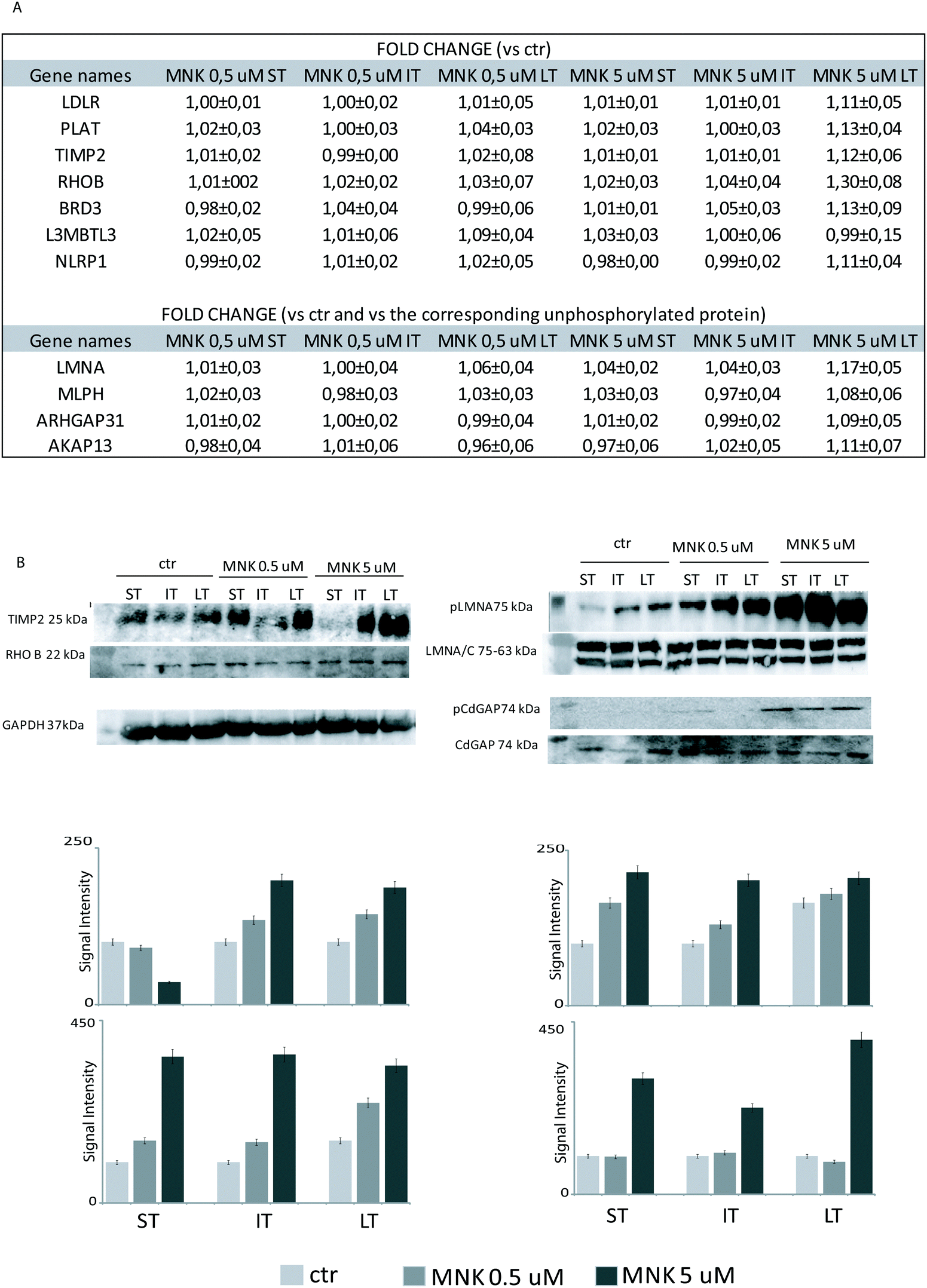 | ||
| Fig. 4 (A) The mean of the fold-changes measured on proteomes and phosphoproteomes of several proteins were reported for MNK-treated samples at different times in respect of the untreated samples. For phosphoproteins, the fold changes were reported as the ratios between the enrichment of each phosphoprotein and the corresponding one in its unphoshorylated form. The isoform ARHGAP31 was not identified and quantified in its unphosphorylated form, thus the fold changes of the phosphorylated one induced by MNK are reported in respect of the control without MNK. The error was conveniently calculated. (B) Western blot analysis on MDA-MB-231 cell lysates treated or not with MNK using antibodies against the tissue inhibitor of metalloproteinase 2 (TIMP2), the Rho-related GTP-binding protein (RhoB) and the phosphorylated form of Lamin A (pLMNA) and of Rho GTPase-activating protein 31 (ARHGAP31 or pCdGAP). For full proteome analysis on the left, GAPDH was used as a loading normalizer; for phosphoproteome analysis on the right, LMNA/C was used as a normalizer for pLMNA (against Lamin A and C at 70 and 65 kDa, respectively) and CdGAP, which, in turn, was used as a normalizer for the corresponding phosphorylated forms. | ||
Many of the overexpressed proteins are involved in cell growth, proliferation and/or migration. For some, particularly the low-density lipoprotein receptor (LDLR), the NACHT, LRR and PYD domain-containing protein 1 (NLRP1), the Rho-related GTP-binding protein (RHOB) and the tissue-type plasminogen activator (PLAT), there are some evidences in the literature linking MNK and/or other statin treatments of MDA-MB-231 and/or other cancers models with anti-proliferative/apoptotic/anti-migration effects.18–25
These results support the pertinence and the reliability of the reported experimental workflow and data analyses. Besides these, further unforeseen expression profiles were altered by MNK treatment of the MDA-MB-231.
Treating MDA-MB-231 with MNK at a concentration of 5 μM for 24 hours, the amount of tissue inhibitor of metalloproteinase 2 (TIMP2) was increased compared to naïve MDA-MB-231 cells. Analogously, MNK was responsible for the spread of the expression of the bromodomain-containing protein 3 (BRD3), the mediator of RNA polymerase II transcription subunit 23 (MED23) and the surfeit locus protein 2 (SURF2).
In order to obtain quantitative MS information for the phosphoproteome besides those for the proteome, phospho-enrichment of the peptides was performed using a BRAVO Agilent system, obtaining an enrichment of around 90% in phosphopeptides. All the fractions were then purified by Sep-Pak and prepared for the following mass spectrometric analysis. The phosphoproteomic analysis allowed the reliable quantification of 5331 phosphosites, precisely on 4824 serines, 488 threonines and 18 tyrosines (ESI,† Table S2). Then, two-way ANOVA statistical test on the identified proteins was performed as reported above (p value < 0.05). The proteins with enhanced phosphorylation were clustered and visualized in a heat map after normalization by the z-score (as reported in Fig. 3B and 4A).
Indeed, MNK treatment induces alterations in the phosphorylation level of 12 proteins. For instance, the Rho GTPase-activating protein 31 (ARHGAP31 also reported as CdGAP), the A-kinase anchor protein 13 (AKAP13), the Lamin A/C (LMNA) and the melanophilin (MLPH) phosphorylation states were increased upon MNK treatment of the MDA-MB-231 cells (Fig. 3B and 4A). Then, immunoblotting analyses were performed.
Immunoblotting analyses
Primary antibodies specific to the tissue inhibitor of metalloproteinase 2 (TIMP2), the Rho-related GTP-binding protein (RhoB), and against the phosphorylated form of Lamin A (pLMNA) and of Rho GTPase-activating protein 31 (pCdGAP) were used to carry out immunoblotting analyses for the validation of the full proteome analysis and the phosphoproteome one, respectively. For full proteome analysis, GAPDH was used as a loading normalizer; for phosphoproteome analysis, LMNA/C was used as a normalizer for pLMNA (against Lamin A and C at 70 and 65 kDa, respectively) and CdGAP was used as a normalizer for the corresponding phosphorylated forms. Indeed, to carefully evaluate the phosphorylation levels of a certain protein, the same protein has to be measured in its unphosphorylated form, under the same experimental conditions. As shown in Fig. 4B, both the over-expression of the tissue-inhibitor of metalloproteinase 2 and RhoB and the hyperphosphorylation of the Lamin A and Rho GTPase-activating protein 31 induced by MNK were verified and validated (Fig. 4B and Fig. S1, ESI†).Heat map visualization was obtained using the Perseus software. ST is short-time treatment of 30 minutes; IT is the intermediate-time treatment of 6 hours; and LT is long-time treatment of 24 hours with MNK at 0.5 μM concentration (MNK05) and at 5 μM concentration (MNK5). P1–P4 are the four biological replicates obtained for each MNK treatments. The color map is reported below together with the increase or decrease fold (for proteome from 3.5 to −2.5 and from 1.3 to 0.7 times in respect of the results obtained for the untreated samples which are not shown).
Discussion and conclusions
MNK, a natural statin abundantly extracted from red yeast rice, was selected for an isobaric-tags-based differential quantitative proteomic investigation on the proteome and the phosphoproteome of MDA-MB-231 model, since numerous scientific evidences of the anti-proliferative, apoptotic and anti-migration bioactivities have been found for lipophilic statins.11Regarding MNK-induced proteome alterations, the positive modulation of some of the reported proteins mediated by statins and/or MNK either in the MDA-MB cellular systems or in similar ones has already been deeply discussed in the literature. At times, some proteins over-expressed by MNK under our experimental conditions showed this trend due to statins’ action as HMG-CoA reductase inhibitors on cholesterol-related pathways. For instance, it is extensively reported that statins increased low-density lipoprotein receptor (LDLR) levels in cancer cells and that this effect is related to their action in reducing cardiovascular diseases.18 Indeed, since statins decrease the level of cholesterol, the intracellular cholesterol synthesis is blocked and, consequently, the expression of LDLR is increased. Furthermore, it has been reported that in the presence of pravastatin, LDLR gene expression in triple-negative breast cancer MDA-MB-231 cells is enhanced by almost 30 times.
Regarding the NACHT, LRR and PYD domain-containing protein 1 (NLRP1), it is reported that statins increased inflammasome gene and NLRP1 expression via regulating the sterol regulatory element-binding protein 1 (SREBP-1) pathway.19–22
The expression of the Rho-related GTP-binding protein RhoB has also been enhanced by MNK in the experiments carried out. It is reported that statins have anti-neoplastic effects by modulating prenylation of Ras and Rho proteins: in particular, Rho prenylation is important for Rho's interaction with upstream regulators and downstream effectors. It has been found that MNK increased RhoA and C solely in their unprenylated forms, but it increased both prenylated and unprenylated RhoB.23 This over expression affects cell adhesion and growth factor signaling pathways, playing a negative role in tumor genesis. Indeed, RhoB is considered as anti-proliferative and pro-apoptotic.24
Furthermore, an increased concentration of the tissue-type plasminogen activator PLAT, a marker of good prognosis in mammary cancers, has been found. A very similar result has been reported for breast cancer cells F3II treated with MNK.25 More in detail, it seems that MNK induced a significant increase in PLAT expression, suggesting the anti-metastatic properties of this molecule. This is strongly associated with the alterations in cytoskeleton organization and the consequent modulation of adhesion, motility and proteolysis.25 A similar behavior of MNK can be speculated also in MDA-MB-231 cells. A very interesting MNK effect has been exerted on the overexpression of the tissue inhibitor of metalloproteinase-2 (TIMP-2), an endogenous metalloproteinase inhibitor. This protein modulates the growth, migration and invasion of tumor cells and inhibits tumor angiogenesis. In a TNBC model, preliminary results showed that recombinant TIMP-2 protein inhibited tumor growth by 50%, causing the reduction of mammary tumor size in mouse models and revealing a therapeutic potential to decrease mammary carcinoma progression.26 Moreover, it has been reported that the ethanol extract of Ampelopsis japonica, a traditional oriental herb with anti-inflammatory and anticancer activities, increased the level of the same protein correlating this action with its high potential in suppressing migration and invasion in human MDA-MB-231 breast cancer cells.27
Another MNK key point action on TNBC cells regards the bromodomain-containing protein 3 (BRD3) overexpression. The contribution of bromo- and extra-terminal (BET) domain proteins to cancer progression has been largely reported, reinforcing their value as therapeutic targets notably for several cancers. In particular, it has been reported that BRD3 depletion is mostly associated with an upregulation of the major epithelial-to-mesenchymal transition (EMT) transcription factors, suggesting that this protein acts as a repressor of this pathway. Moreover, the EMT developmental program is usually activated by cancer cells to acquire a highly plastic phenotype that promotes invasion, metastasis, as well as chemoresistance and cancer stem cell generation.28 Thus, the MNK-induced enhancement of BRD3 concentration found in MDA-MB-231 cells correlates well with its action on downgrading invasion, metastasis, as well as chemo-resistance and cancer stem cell generation.
Finally, MNK increases the lethal(3)malignant brain tumor-like protein 3 (L3MBTL3) which is a poorly characterized member of the MBT (malignant brain tumor) family of methyl-lysine readers that act as chromatin-interacting transcriptional repressors.29,30 It is considered the universal modulator of Notch signaling: in particular, L3MBTL3 acts as a negative regulator of Notch target genes. Since breast cancers, like many other tumors, have been shown to over-express the Notch signaling pathway, it seems to be of interest to switch off this pathway.31
Regarding MNK-induced phosphoproteome alterations, increased phosphorylation of Rho GTPase-activating protein 31 (also called ARHGAP31) has been reported. Its phosphorylation reduces the GTPase-accelerating protein (GAP) activity. Indeed, the aberrant activity of Ras homologous (Rho) family of small GTPases is implicated in cancers and other human diseases.32–34 Similarly, an increase in phosphorylation of AKAP 13 protein has been measured after the cell treatment with MNK. The A-kinase anchor proteins (AKAPs) are a group of structurally diverse proteins that have the common function of binding to the regulatory subunit of protein kinase A (PKA). This subunit is associated with the guanine nucleotide exchange activation for the Rho/Rac family of small GTP-binding proteins, resulting in the conversion of the inactive GTPase to the active form capable of transducing signals. It interacts with RhoA, RhoB and RhoC.
One of the key features of triple-negative breast cancers is the enhanced capability of the cells to migrate, leading both to an amplified invasiveness of the tumor and to a wider risk for developing numerous metastatic loci.1,3,35,36 Mutations and/or alterations in protein expression and PTM levels involving cytoskeleton and/or cytoskeleton-associated proteins have been recognized as the molecular basis in tumor cell migration, invasion and metastasis.37 In this case, some of the MNK-induced phosphoproteome alterations have a tight connection to cell migration mechanisms as the overphosphorylation of Lamin A. It is well known that Lamin A is overexpressed in triple-negative breast cancer cells,38 but its phosphorylation has not been studied in detail. The nucleoskeletal protein prelamin A/C has been shown to contribute to cell sorting and survival in migration via constraining micropores in diverse human cells; indeed, the right amount of lamins is thus a requirement to ensure both successful 3D-migration and survival. Although insights into the phosphorylation levels of lamins during 3D-migration are scarce, the increased mobility of the phosphorylated protein has been speculated to be one mechanism promoting nuclear deformability, thereby facilitating migration. On the contrary, it is also reported that an increased phosphorylation of lamins generates softer nuclei, which are prone to fragmentation leading to cell death since they can easily be squeezed through tissue during migration.39,40
Cytoskeleton-related signaling pathways mediating cell migration and invasion involve both actin and microtubule networks. The scientific suggestions of the relationship between a distortion in microtubules dynamics and tumor process are clear, and they are fueling a novel interest in the role of microtubules-dependent networks in tumor metastatic processes.37–41 Taking into account these evidences, the hyper-phosphorylated level of melanophilin (MLPH) due to MNK treatments has been considered as an interesting result. In cell, MLPH is the most versatile component of a tripartite complex together with the Rab27a and myosin Va (MyoVa) subunits, and it also binds the actin and the microtubule's positive terminal upon ABD domain, by its C-terminal domain. In an appealing study, Oberhofer and co-workers elucidated the molecular basis for the switching of the MLPH between the actin and microtubule networks.42 Based on their findings, the ABD domain of the MLPH binds the actin upon electrostatic interactions, while the microtubule binding is regulated in a phosphorylation-dependent manner.42 As reviewed by Hall, different cell types – but even the same cell type in response to different external stimuli and environments – can change their modes of migration by switching between actin-based and microtubule-based mechanisms.41 Based on this evidence reported in the literature, it can be speculated that the hyper-phosphorylation of the MLPH induced by MNK could result in an altered migration capability of MDA-MB-231 cells.
Notably, it has been already reported in the literature that MNK itself has an impact on the migratory capabilities of MDA-MB231 cells by functional experiments in agreement with our findings over the proteome and phosphoproteome alteration. Indeed, Issat and Klawitter,7,32 using wound healing and scratch assay test, showed that the untreated cells migrated to the scratched area and almost refilled the gap within 72![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) hours, whereas MNK significantly reduced the migration and the gap refilling of 20% and 30% after 24 and 72 hours, respectively. Therefore, MNK may reduce the invasiveness of cancer cells. Moreover, it has been demonstrated by Yang17 that MNK, when used at a concentration range between 0.1 and 10 μM, dose-dependently inhibited proliferation of MDA-MB-231 cells. In respect of other cancer cell lines, MNK preferentially suppressed proliferation and induced apoptosis in triple-negative breast cancer cells.32 In conclusion, the TMT-based quantitative proteomic approach described herein has successfully disclosed novel MNK-induced alterations on the proteome and phosphoproteome of the MDA-MB-231 cells, profiling new hypothesis to explain the molecular basis of the anti-proliferative and anti-migration properties of this natural statin. A deep investigation on the role of MNK or other statins in other triple-negative breast cancer models may be valuable to corroborate these findings, depicting a more general mechanism of action.
hours, whereas MNK significantly reduced the migration and the gap refilling of 20% and 30% after 24 and 72 hours, respectively. Therefore, MNK may reduce the invasiveness of cancer cells. Moreover, it has been demonstrated by Yang17 that MNK, when used at a concentration range between 0.1 and 10 μM, dose-dependently inhibited proliferation of MDA-MB-231 cells. In respect of other cancer cell lines, MNK preferentially suppressed proliferation and induced apoptosis in triple-negative breast cancer cells.32 In conclusion, the TMT-based quantitative proteomic approach described herein has successfully disclosed novel MNK-induced alterations on the proteome and phosphoproteome of the MDA-MB-231 cells, profiling new hypothesis to explain the molecular basis of the anti-proliferative and anti-migration properties of this natural statin. A deep investigation on the role of MNK or other statins in other triple-negative breast cancer models may be valuable to corroborate these findings, depicting a more general mechanism of action.
Experimental
Cell proliferation assay
MDA-MB-231 cells were acquired from the American Type Culture Collection (ATCC) and grown in the laboratories of the Bijvoet Center for Biomolecular Research, Utrecht University, The Netherlands. Cells were maintained following the ATCC recommendations, seeded in a 96-multiwell plate at a density of 2000, 4000 or 5000 cells per well (100 μL of cellular suspension per well) and cultured overnight (5% CO2 atmosphere, 37 °C) in DMEM (Dulbecco's modified Eagle's medium) supplemented with 10% (v/v) fetal bovine serum, 100 U mL−1 penicillin, and 100 mg mL−1 streptomycin. After the cells have reached 20% of confluence, the medium was refreshed to treat cells with different concentrations of MNK (final concentrations in the well, 1% of DMSO: 200 μM, 100 μM, 50 μM, 20 μM, 10 μM, 5 μM, 2 μM, and 0.5 μM). Controls either without drug and DMSO or with 1% Triton-X100 were included. The 96-multiwell plate was imaged using an IncuCyte ZOOM live-cell imaging system equipped with a CCD camera (Basler scA1400-30 gm), a Sony ICX285 CCD sensor (1392 px × 1040 px, 12 bit) and a Nikon Plan Apo λ objective (magnification: ×4; numerical aperture of the optical system: 0.20; pixel size: 3.05 μm), and placed into an incubator (5% CO2 atmosphere, 37 °C). After 30 minutes of exposure, the acquisition of cell parameters was started automatically using the Incucyte ZOOM software and stopped after around 4 days, when the curve of naïve MDA-MB-231 cell proliferation reached a plateau. The collected kinetic parameters were elaborated using Graph Pad Prism 7. The results are expressed as the average of three replicates for each condition.Optimizing parameters for the proteomic experiments
Totally, 0.3 × 106 MDA-MB-231 cells were seeded and then treated with MNK at 0.5 μM or 5 μM (final concentration). A positive control using only the vehicle of the drug (DMSO 1% v/v) was prepared as well. The kinetic parameters of cell proliferation were acquired continuously for 3 days using an IncuCyte ZOOM system, and the kinetic curves were visualized. The results are expressed as the average of two replicates for each treatment condition.Drug treatments for proteomics
Totally, 5 × 106 MDA-MB-231 cells were seeded in each of the nine 150 mm Petri dishes needed for the treatments (DMEM was supplemented with 10% (v/v) fetal bovine serum, 100 U per mL penicillin, 100 mg per mL streptomycin; 5% CO2 atmosphere, 37 °C). After the cells have reached 20% confluence, DMEM was replaced with a fresh medium containing 1% DMSO (CTRL), 0.5 μM of MNK (1% DMSO) (MNK0.5 μM) or 5 μM MNK (1% DMSO) (MNK5 μM). Cells from each condition were harvested by trypsinization after 30 minutes (short time, ST), 6 hours (intermediate time, IT) or 24 hours (long time, LT) of treatment and collected by centrifugation obtaining nine pellets named: ST_CTRL, ST_MNK0.5μM, ST_MNK5μM, IT_CTRL, IT_MNK0.5μM, IT_MNK5μM, LT_CTRL, LT_MNK0.5μM and LT_MNK5μM. All the treatments were performed four times in replicate. The pellets were suspended in a lysis buffer (1% w/v sodium deoxycholate, 10 mM tris(2-carboxyethyl)phosphine hydrochloride, 100 mM tris, 40 mM chloroacetamide, 1 tablet of complete Mini protease inhibitors and a tablet of PhosphoStop phosphatase inhibitors, pH = 8.5) and cells were lysed by heating the suspension at 95 °C for 5 minutes and by sonication for 15 minutes at level 5 (30 s on and 30 s off) (Bioruptor ACD-200, Diagenode). The cell debris was pelleted by centrifugation (20000 rcf, 10 minutes), and the resulting protein mixture from each sample was quantified by the Bradford spectrophotometric assay to normalize the protein concentrations between treatments. Then 500 μg of proteins from each experimental condition was in-solution digested. Briefly, proteins were diluted in 100 mM AmBic (pH = 8) and then treated with 5 mM DTT (60 minutes, 37 °C) and 14 mM iodoacetamide (45 minutes, RT, darkness). Digestion was allowed to proceed overnight (37 °C) treating the samples with trypsin (1:50) and Lys-C (1:75), and then the sample was quenched with 2% FA. Samples were centrifuged (20000 rcf, 20 minutes), and the supernatant containing peptide mixtures was desalted by C-18 solid phase extraction (Sep-Pak). The C-18 columns were conditioned with 1 mL of ACN (3 times) and equilibrated with 1 mL of H2O + 0.6% AA (3 times) before loading the sample. After washing with 1 mL of H2O and 0.6% AA (3 times), peptides were eluted with 80% ACN + 0.6% AA and dried under vacuum.
TMT-labelling and sample preparation
For each of the nine experimental conditions, aliquots of peptide mixtures corresponding to 10 μg of proteins were pooled together to obtain an internal control (MIX) for each one of the 4 replicates (MIX_Pool1; MIX_Pool2; MIX_Pool3; and MIX_Pool4). The obtained ten samples for each replicate of the experiment were chemically labelled with isobaric tandem mass tags. To this purpose, protein mixtures were solubilized in 70 μL of 50 mM Na–HEPES at pH 8.5 instead of the TEAB recommended by the manufacturer in order to avoid the formation of unidentified and unwanted side reaction products during the labelling of the peptides with the TMT-10plex reagents (Thompson et al., 2003). Anhydrous ACN was added to each sample (10 μL, 100%) to reach a percentage guaranteeing the stability of the tandem mass tags during the chemical labeling. The samples were shaken for 5 minutes at 300 rpm, and 8 μL from each peptide mixtures was pooled together to have an internal control for each of the four replicates. Each of the obtained ten samples, containing peptide mixtures from 90 μg of proteins in 72 μL of 50 mM Na–HEPES + 8% ACN, was treated with one of the reagents from the TMT-10plex reagent kit. Isobaric tags had been previously slowly thawed at RT without any harsh thermal shock avoiding the formation of condensates under the lids of the storing vials (TMTs are sensible to the moisture affecting the labeling yield), solubilized at a concentration of 10 μg μL−1 (800 μg of each powdered isotopomer of the kit in 80 μL of 100% anhydrous ACN) and occasionally gently vortexed for 5 minutes at room temperature. One TMT-10plex kit was used to label all the peptide mixtures from the 4 replicates using 200 μg of each tag to treat 90 μg of peptides. Chemical labeling was allowed to proceed for 1 hour at 25 °C and 300 rpm, and then quenched by adding hydroxylamine at a final concentration of 5% (15 minutes, RT). Finally, the samples were acidified with 25% FA to reach pH 2. In order to estimate the labeling efficiency, 1 μL from each sample (around 1 μg of peptides) was checked by UPLC-MS/MS, and the obtained mass spectrometric data were submitted to Proteome Discoverer to allow the calculation of the fraction of modified peptides on the total.‡ For the purpose of controlling the multiplexing ratio, 1 μL of each sample (around 1 μg of peptides) was unified obtaining a single pooled sample for each replicate. Then 1% of the pools was checked by UPLC-MS/MS and analyzed using Proteome Discoverer to calculate the needed ratio adjustments by selecting the scaling mode on channels average comparing the scaled reporter ion abundances for each channel of the TMT-10plex. Based on the results of the ratio adjustments, all the labeled peptides from each treatment were pooled together obtaining one sample for each of the four replicates that was dried under vacuum and then solubilized in 40 μL of 0.1% FA. Purification of the samples was achieved by a desalting step using C-18 solid phase extraction (Sep-Pak). The C-18 columns were conditioned with 80 μL of MeOH (3 times) and equilibrated with 40 μL of 5% FA (3 times) before loading the sample. After washing with 40 μL of 5% FA (3 times), peptides were eluted in 40 μL 40% ACN + 55% H2O + 5% FA and then dried under vacuum. Aiming to simplify the complex sample of labeled peptide mixtures, an off-line high-pH reversed-phase (HPRP) liquid chromatography was required before the standard UPLC-MS analysis. Peptide mixtures (from a total of 900 μg of digested proteins) were solubilized in 20 μL of 10 mM ammonium formate (pH = 10 adjusted with NH4OH) and resolved on a Phenomenex Kynetex EVO C-18 column (5 μm particles, 100 A, 150 × 2.1 mm) using an Agilent 1200 binary pump equipped with a photodiode array detector. A 115 minute linear gradient from 0 to 40% of buffer B has been selected to fractionate the samples (0.200 mL min−1, buffer A: 10 mM ammonium formate (pH = 10 adjusted with NH4OH), buffer B: 90% ACN + 10% H2O (pH = 10 adjusted with NH4OH)). The fractions were concentrated in 8 samples by combining these fractions with equal elution time interval, and around 90% of them were combined again with the same criterion to obtain other 4 samples for each replicate besides the starting 8. All the fractions were cleaned up via a desalting step by C-18 solid phase extraction (Sep-Pak) as previously described and dried under vacuum. These latter 4 fractions, containing in total 800 μg of peptides, were submitted to a phospho-enrichment step using a AssayMAP Bravo (Agilent Technologies) platform equipped with Fe(III)–NTA cartridges (Agilent Technologies). Peptides in each of the 4 fractions (around 200 μg) were solubilized in 200 μL of 80% ACN + 20% H2O + 0.1% TFA and loaded into the Fe(III)–NTA cartridges previously primed with 100% ACN + 0.1% TFA and equilibrated with 80% ACN + 20% H2O + 0.1% TFA. After washing with 80% ACN + 20% H2O + 0.1% TFA, phosphorylated peptides were eluted with 25 μL 1% ammonia into a 96-multiwell plate containing 25 μL 10% FA and then dried down.Mass spectrometric analysis
All the LC-MS/MS experiments were carried out using an UHPLC 1290 system (Agilent) interfaced to a Q Exactive Plus Hybrid Quadrupole-Orbitrap mass spectrometer. Peptides were solubilized at 0.1 μg μL−1, and 10 μL was injected into the UHPLC system. A trapping step was carried out for 5 minutes with 0.1% FA (Dr Maisch Reprosil C18, 3 μm, 2 cm × 100 μm) before the chromatographic separation of the peptides using an Agilent Poroshell EC-C18 column (2.7 μm, 50 cm × 75 μm). For the full proteome analysis, peptides were resolved within 115 minutes pumping buffer A (0.1% FA) and buffer B (80% ACN + 0.1% FA) as follows: from 0% to 10% of buffer B in 1 minute, from 10% to 36% of buffer B in 95 minutes and from 36% to 100% of buffer B in 3 minutes. For the phosphoproteome analysis, peptides were resolved within 115 minutes pumping buffer A (0.1% FA) and buffer B (80% ACN + 0.1% FA) as follows: from 0% to 8% of buffer B in 1 minute, from 8% to 32% of buffer B in 95 minutes and from 32% to 100% of buffer B in 3 minutes. The flow was passively split at 300 nl minute−1. Data-dependent acquisition was selected as an operating mode for the mass spectrometer, and a MS2 method was used. The MS1 scans were acquired between 300 m/z and 1500 m/z as mass range and at a resolution set at 60000. Isolation width for the isolation of the ten most intense peaks having ion intensities above 500 counts was set to 2 m/z, and the selected precursor ions (enabling monoisotopic precursor selection and charge state rejection) were fragmented upon high-collision dissociation (for the HCD, 45% normalized collision energy at 30 ms activation time was selected). For the FTMS1, the automatic gain control (ACG) target was set at 1e6, while for FT MSn the value was fixed at 5e4; for both, 250 ms was set as the maximum ion time.
MS data analysis
Both raw mass spectrometric data from full proteome analysis or phosphoproteome analysis were processed using MaxQuant for the identification and quantification of the proteins or phosphoproteins, respectively. Andromeda was used as an engine for the search against the human SwissProt Database (UniProt Release 2017_10; UniProtKB/Swiss-Prot: 556006 entries). The specific parameters for the group were set as follows: carboamidomethylation of the cysteines as “fixed modifications”; acetylation at the N-terminal and the oxidation of the methionine as “variable modifications”. Moreover phosphorylations of serines, threonines and tyrosines were included in the analysis of phosphoproteomic mass spectrometric data. Parameters for the in silico digestion of protein sequences were set selecting Trypsin/P as specific proteolytic enzyme and allowing up to two missed cleavages. The “reporter ion MS2” option was selected for the quantification: all the TMT-10plex modifications both at N-terminal, at cysteines and at lysines of the peptides, were included as isobaric labels and a tolerance of 0.003 Da was selected for matching between mass-to-charge ratios of reporter ions in the spectra and the theoretical ones for those obtained upon in silico experiments. The “re-quantify” option was selected. Concerning the global parameters, the false discovery rate (FDR) for the identification both at the peptide spectrum match level and at the protein level (both determined by the target-decoy approach) was set at 1%. Both unique and razor peptides were used for the quantification of proteins. The “match between runs” option has been flagged besides the advanced identification tools. All the other parameters were retained as in the default MaxQuant settings. Tables (.txt) for either “proteinGroups” or “Phospho (STY) sites” were loaded in the starting matrix in the Perseus loading graphic user interface for proteome or phosphoproteome analysis, respectively. Concerning the full proteome, the “reporter intensity corrected” for each mass reporter ion of the TMT-10plex reagents was used within the 4 biological replicates. Protein groups were filtered out for reverse hits, contaminants and hits that are only identified by site. Experimental conditions were created based either on temporal variables (short time, ST = 30 minutes; intermediate time, IT = 6 hours; and long time, LT = 24 hours) or on MNK concentration-related variables (CTRL; MNK0.5μM; and MNK5μM). The obtained matrix was further reduced by excluding proteins with any missing value (100% of valid values) in each time- or concentration-related group of variables. The log2 function was used to transform all the intensities. Then, the main columns were normalized against its control condition (i.e. the intensities referring to ST_CTRL, IT_CTRL and LT_CTRL were used as denominators for each experimental condition). Finally, a two-way ANOVA was used to point out the significative differences, both time- and concentration-related, between intensities referring to the expression level of the same protein under the different experimental conditions; p-value < 0.01. MNK-induced alterations of the proteome among the time were clustered and visualized as a heat map of the relative z-scored ratios of the altered proteins.
Concerning the phosphoproteome, the same kind of workflow was followed using Perseus to visualize the heat map of the MNK-induced significative alterations in the phosphorylation levels of the proteins. The “reporter intensity corrected” both for single-, double- and triply-phosphorylated species have been loaded as main columns to build up the first generic matrix in Perseus; the global “localization probability” was loaded as well in this first matrix as a numerical column. Besides the contaminants and the reverse hits, also the phosphosites with a localization probability <0.75 were filtered out from the initial matrix. The obtained main data were rearranged with the function “expand site table” allowing the visualization of one row for each phosphorylation site of the peptides and of a single column referring to the “multiplicity” of the phosphorylation on a peptide. The obtained matrix was further reduced by excluding phosphosites with any missing value (100% of valid values) in each time- or concentration-related group of variables, and transformation and normalization of the intensities were carried out as reported above. Then, besides protein specific annotation, sequence-specific annotations (Kinase_Substrate_Dataset.gz and Phosphorylation_site_dataset.gz were downloaded from PhosphoSitePlus website) were added to analyze the modifications. Particularly, the “position within protein”, “linear motifs”, “known phosphorylation sites” and “known kinase-substrate relations” columns have been created. The following steps to obtain the heat map were done as previously described for the full proteome analysis.
Immunoblotting analysis
MDA-MB-231 cells were treated with MNK as previously described and the pellets from each drug treatment were lysed in PBS, pH 7.4, containing both protease and phosphatase inhibitor cocktails (from Sigma Aldrich) by mechanical lysis using a Dounce homogenizer and alternating cooling (on ice)–thawing (RT) cycles. After pelleting cellular debris, the protein contents in each sample were determined by the Bradford spectrophotometric assay and the concentrations were normalized between experimental conditions. Aliquots of the same MNK treatment of different biological replicates were pooled together, and 20 μg of proteins for each sample was treated with Laemmli buffer and heated for 5 minutes at 95 °C before to be loaded into a 12% polyacrylamide gel for the monodimensional electrophoresis. Immunoblotting analyses were repeated twice. Resolved proteins were transferred onto a nitrocellulose membrane, then blocked with 3% BSA in TBS-T (blocking with milk proteins has been avoided to prevent the possibility of high background due to the aspecific interactions between caseins and phosphorylation-recognizing antibody) and hybridized either with a monoclonal primary antibody against tissue inhibitor of the metalloproteinase 2 and the Rho-related GTP-binding protein (Anti TIMP2 and Anti RhoB from SantaCruz Antibodies, 1:500 and 1:1000 in TBS-T + 5% milk) or with a monoclonal primary antibody against the phosphorylated form of the Lamin A and Rho GTPase-activating protein 31 (anti p-LMNA and anti p-CdGAP from SantaCruz Antibodies, 1:2000 both in TBS-T + 5% milk). After recognition of the primary antibodies from the proper HPR-tagged secondary ones, chemiluminescence developed upon treatment of the membrane with a mixture of luminol and hydrogen peroxide was detected using a ImageQuant LAS4000 film imaging system. The densitometric analysis was elaborated using ImageJ for the quantification of the pixels and Excel for the visualization of the resulting histograms. Normalizing densitometric values were calculated based on the signals obtained by hybridizing the membrane with a monoclonal antibody against GAPDH (Invitrogen by Thermo Scientific, 1:2000 in TBS-T + 5% milk); LMNA/C (against Lamin A and C at 70 and 65 kDa, respectively) and CdGAP antibodies (from SantaCruz Antibodies, 1:2000 in TBS-T + 5% milk).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from Università degli Studi di Salerno (grant number ORSA151559) and by POR CAMPANIA FESR 2014/2020 Asse 1 – Obiettivo specifico 1.2 – Azione 1.2. Progetto: Campania OncoTerapie CUP: B61G18000470007. The authors thank Stamatia Rontogianni, Kelly Stecker and A. F. Maarten Altelaar from the Bijvoet Center for Biomolecular Research and the Netherlands Proteomics Centre, Utrecht University, The Netherlands for the MS experiments.Notes and references
- C. K. Anders, A. M. Deal, C. R. Miller, C. Khorram, H. Meng, E. C. Livasy, K. Fritchie, M. G. Ewend and C. M. Perou, Cancer, 2011, 117, 1602 CrossRef PubMed.
- S. Badve, D. J. Dabbs, S. J. Schnitt, F. L. T. Decker, V. Eusebi, S. B. Fox, S. Ichihara, J. Jacquemier, S. R. Lakhani, J. Palacios, E. A. Rakha, A. L. Richardson, F. C. Schmitt, P. H. Tan, G. M. Tse, B. Weigelt, I. O. Ellis and J. S. Reis-Filho, Mod. Pathol., 2011, 24, 157 CrossRef PubMed.
- Z. Elsawaf and H. P. Sinn, Breast Care, 2011, 6, 273 Search PubMed.
- R. Dent, M. Trudeau, K. I. Pritchard, W. M. Hanna, H. K. Kahn, C. A. Sawka, L. A. Lickley, E. Rawlinson, P. Sun and S. A. Narod, Clin. Cancer Res., 2007, 13, 4429 CrossRef PubMed.
- J. Xu, W. Yang, Q. Wang, Q. Zhang, X. Li, X. Lin, X. Liu and Y. Qin, Int. J. Clin. Exp. Pathol., 2014, 7, 7915 Search PubMed.
- B. D. Lehmann, J. A. Bauer, X. Chen, M. E. Sanders, A. B. Chakravarthy, Y. Shyr and J. A. Pietenpol, J. Clin. Invest., 2011, 121, 2750 CrossRef CAS PubMed.
- T. Issat, D. Nowis, M. Legat, M. Makowki, M. Klejman, J. Urbanski, J. Skierski, M. Koronkiewicz, T. Stoklosa, A. Brzezinska, J. Bil, J. Gietka, M. Jakobisiak and J. Golab, Int. J. Oncol., 2007, 30, 1413 CAS.
- E. Ranzato, S. Martinotti, C. M. Calabrese and G. Calabrese, J. Food Res., 2014, 4, 18 CrossRef.
- Q. Chang, X. X. Yan, S. Y. Gu, J. F. Liu and D. C. Liang, Proteins, 2008, 73, 254 CrossRef CAS PubMed.
- Y. Li, F. Zhang, Z. Wang and Z. Hu, J. Pharm. Biomed. Anal., 2004, 35, 1101 CrossRef CAS PubMed.
- H. Yao, G. He, S. Yan, C. Chen, L. Song, T. J. Rosol and T. J. Deng, Oncotarget, 2017, 8, 1913–1924, DOI:10.18632/oncotarget.12284.
- S. Nofech-Mozes, M. Trudeau, H. K. Kahn, R. Dent, E. Rawlinson, P. Sun, S. A. Narod and W. M. Hanna, Breast Cancer Res. Treat., 2009, 118, 131 CrossRef PubMed.
- A. O. Mueck, H. Seeger and D. Wallwiener, Menopause, 2003, 10, 332 CrossRef PubMed.
- M. J. Campbell, L. J. Esserman, Y. Zhou, M. Shoemaker, M. Lobo, E. Borman, F. Baehner, A. S. Kumar, K. Adduci, C. Marx, E. F. Petricoin, L. A. Liotta, M. Winters, S. Benz and C. C. Benz, Cancer Res., 2006, 66, 8707 CrossRef CAS PubMed.
- T. Yang, H. Yao, G. He, L. Song, N. Liu, Y. Wang, Y. Yang, E. T. Keller and X. Deng, J. Cancer, 2016, 7, 192 CrossRef CAS PubMed.
- A. Thompson, J. Schäfer and K. Kuhn, et al. , Anal. Chem., 2003, 75, 1895 CrossRef CAS PubMed.
- T. S. Batth, C. Francavilla and J. V. Olsen, J. Proteome Res., 2014, 13, 6176 CrossRef CAS PubMed.
- Y. Furuya, Y. Sekine, H. Kato, Y. Miyazawa, H. Koike and K. Suzuki, Prostate Int., 2016, 4, 56 CrossRef PubMed.
- S. Bleda, J. de Haro, I. Lopez de Maturana, C. Varela, A. Ferruelo and F. Acin, Ann. Vasc. Surg., 2016, 36, 260 CrossRef PubMed.
- S. Bleda, J. de Haro and F. Acin, Int. J. Cardiol., 2017, 247, 12 CrossRef PubMed.
- S. Yu, B. Dong, L. Tang and S. Zhou, Int. J. Cardiol., 2017, 247, 11 CrossRef PubMed.
- N. Lénárt, D. Brough and Á. Dénes, J. Cereb. Blood Flow Metab., 2016, 36, 1668 CrossRef PubMed.
- S. J. Turner, S. Zhuang, T. Zhang, G. R. Boss and R. B. Pilz, Biochem. Pharmacol., 2008, 75, 405 CrossRef CAS PubMed.
- E. Sahai and C. J. Marshall, Nat. Rev. Cancer, 2002, 2, 133 CrossRef PubMed.
- H. Farina, D. Bublik, D. F. Alonso and D. F. Gomez, Clin. Exp. Metastasis, 2002, 19, 551 CrossRef CAS PubMed.
- S. Kuskunović-Vlahovljak, N. Čamdžić, S. Radović, M. Dorić, M. Babić, E. Lazović Salčin and L. Džananović, J. Health Sci., 2017, 7, 158 Search PubMed.
- K. J. Nho, J. M. Chun, D. S. Kim and H. K. Kim, Mol. Med. Rep., 2015, 11, 3722 Search PubMed.
- G. P. Andrieu and G. V. Denis, Mol. Cancer Res., 2018, 16, 580 CrossRef CAS PubMed.
- R. Bonasio, E. Lecona and D. Reinberg, Semin. Cell Dev. Biol., 2010, 21, 221 CrossRef CAS PubMed.
- N. Nady, L. Krichevsky, N. Zhong, S. Duan, W. Tempel, M. F. Amaya, M. Ravichandran and C. H. Arrowsmith, J. Mol. Biol., 2012, 423, 702 CrossRef CAS PubMed.
- T. Xu, S. Park and B. Giaimo, et al. , EMBO J., 2017, 36, e201796525 CrossRef PubMed.
- J. Klawitter, T. Shokati, V. Moll and U. Christians, Breast Cancer Res., 2010, 12, R16 CrossRef PubMed.
- D. R. Cook, K. L. Rossman and C. J. Der, Oncogene, 2014, 33, 4021 CrossRef CAS PubMed.
- F. Paul, H. Zauber, L. von Berg, O. Rocks, O. Daumke and M. Selbach, Mol. Cell. Proteomics, 2017, 16, 73 CrossRef CAS PubMed.
- J. G. Donaldson and C. L. Jackson, Nat. Rev. Mol. Cell Biol., 2011, 12, 362 CrossRef CAS PubMed.
- K. Pogoda, A. Niwińska, M. Murawska and T. Pieńkowski, Med. Oncol., 2013, 30, 388 CrossRef PubMed.
- C. M. Fife, J. A. McCarroll and M. Kavallaris, Br. J. Pharmacol., 2014, 171, 5507 CrossRef CAS PubMed.
- S. R. Setijono, M. Park, G. Kim, Y. Kim, K. W. Cho and S. J. Song, Biochem. Biophys. Res. Commun., 2018, 496, 826 CrossRef CAS PubMed.
- E. Torvaldson, V. Kochin and J. E. Eriksson, Nucleus, 2015, 6, 166 CrossRef CAS PubMed.
- T. Harada, J. Swift, J. Irianto, J.-W. Shin, K. Spinler, A. Athirasala, R. Diegmiller, P. Dingal, I. Ivanovska and D. Discher, J. Cell Biol., 2014, 204, 669 CrossRef CAS PubMed.
- A. Hall, Cancer Metastasis Rev., 2009, 28, 5 CrossRef PubMed.
- A. Oberhofer, P. Spieler, Y. Rosenfeld, W. L. Stepp, A. Cleetus, A. N. Hume, F. Mueller-Planitz and Z. Ökten, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E4714 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9mo00140a |
| ‡ The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD013037. |
| This journal is © The Royal Society of Chemistry 2020 |