
NMR and MS urinary metabolic phenotyping in kidney diseases is fit-for-purpose in the presence of a protease inhibitor†
Claire L.
Boulangé‡
 a,
Ilse M.
Rood
b,
Joram M.
Posma
c,
John C.
Lindon
ac,
Elaine
Holmes
ac,
Jack F. M.
Wetzels
b,
Jeroen K. J.
Deegens
b and
Manuja R.
Kaluarachchi
*a
a,
Ilse M.
Rood
b,
Joram M.
Posma
c,
John C.
Lindon
ac,
Elaine
Holmes
ac,
Jack F. M.
Wetzels
b,
Jeroen K. J.
Deegens
b and
Manuja R.
Kaluarachchi
*a
aMetabometrix Ltd, Sir Alexander Fleming Building, South Kensington, London SW7 2AZ, UK. E-mail: manuja.kaluarachchi@imperial.ac.uk
bDepartment of Nephrology, Radboud University Medical Center, Nijmegen, Geert Grooteplein Zuid 8, PO Box 9101, 6500 HB, Nijmegen, The Netherlands
cImperial College London, Division of Computational and Systems Medicine, Department of Surgery and Cancer, Faculty of Medicine, Sir Alexander Fleming Building, South Kensington, London SW7 2AZ, UK
First published on 9th January 2019
Abstract
Nephrotic syndrome with idiopathic membranous nephropathy as a major contributor, is characterized by proteinuria, hypoalbuminemia and oedema. Diagnosis is based on renal biopsy and the condition is treated using immunosuppressive drugs; however nephrotic syndrome treatment efficacy varies among patients. Multi-omic urine analyses can discover new markers of nephrotic syndrome that can be used to develop personalized treatments. For proteomics, a protease inhibitor (PI) is sometimes added at sample collection to conserve proteins but its impact on urine metabolic phenotyping needs to be evaluated. Urine from controls (n = 4) and idiopathic membranous nephropathy (iMN) patients (n = 6) were collected with and without PI addition and analysed using 1H NMR spectroscopy and UPLC-MS. PI-related data features were observed in the 1H NMR spectra but their removal followed by a median fold change normalisation, eliminated the PI contribution. PI-related metabolites in UPLC-MS data had limited effect on metabolic patterns specific to iMN. When using an appropriate data processing pipeline, PI-containing urine samples are appropriate for 1H NMR and MS metabolic profiling of patients with nephrotic syndrome.
Introduction
Proteinuria is an important manifestation of nephrotic syndrome (NS). In adults the most common causes of NS include (idiopathic) membranous nephropathy (iMN), (idiopathic) focal segmental glomerulosclerosis (iFSGS) and minimal change disease (MCD).1 Diagnosis is based on renal biopsy and treatment consists of prolonged aggressive therapy with (various) immunosuppressive drugs to induce remission and to reduce the risk of renal function deterioration.2 Not all patients respond to this therapy, and exposure to the therapeutics puts these patients at risk of complications. Therefore there is an urgent need for prognostic and/or diagnostic biomarkers that allow development of an individualized treatment strategy.Omic approaches (genomics, transcriptomics, proteomics and metabonomics) can be used to detect biomarkers and can help elucidate the pathological mechanisms related to disease. Recently we have used proteomic analyses to identify biomarkers in urinary microvesicles from patients with iMN, iFSGS and MCN. Lysosome membrane protein 2, (Limp 2) was identified as an upregulated biomarker in patients with iMN.3 However, the underlying pathophysiological mechanisms linked to an increase in the glomerular Limp 2 expression and the development of iMN remain to be understood. Ideally, combining different omic approaches will provide the community with a more complete profile of potential biomarkers of iMN.
In metabolic phenotyping, metabolite classes such as amino acids, organic acids and bases, fatty acids, bile acids, lipids and carbohydrates are routinely characterised. Changes in the level of these metabolites represents the ultimate and measurable response of biological systems to genetic differences, disease or environmental stimuli.4,5 By monitoring metabolite concentrations it is possible to obtain information for disease diagnosis, explain drug activity, and enable prognosis of outcomes providing an insight into the complex biochemical processes and their impact in human health and disease.6 To date, high resolution 1H NMR spectroscopy and ultra-performance liquid chromatography (UPLC) or gas chromatography (GC) coupled with mass spectrometry (MS) are the two main analytical platforms used to generate metabolic phenotyping data.7,8 The metabolic phenotype of urine from chronic kidney diseases (CKD) has been well characterised using MS and 1H NMR techniques and shows the value of applying such an approach for investigating CKD.9,10 A wide range of biomarkers has been reported in the context of CKD including proteins, peptides, amino acids, biogenic amines, dicarboxylic acids, lipids, nucleotides derivatives and phenolic compounds.11–13 Velenosi et al. showed an increase in gut-derived uremic toxins (4-cresol sulfate, hippurate, phenyl sulfate, pyrocatechol sulfate, 4-ethylphenyl sulfate, 4-cresyl glucuronide and equol-7-glucuronide) in plasma and tissues of CKD rats probably due to the dysfunction of the organic anion transporters (OAT) 1 and 3 in the kidney, which allow the renal clearance of uremic toxins.9,10 However, only limited results are available from metabolic phenotyping investigations of proteinuric samples from glomerular diseases.
In epidemiological projects studying kidney diseases, there is considerable interest in the use of urine for proteomic analysis as these samples potentially contain proteins secreted or shed directly from the kidney, bladder or prostate.14 For proteomics, it was initially recommended to add a protease inhibitor (PI) prior to freezing to prevent protein degradation caused by endogenous proteolytic enzymes. Consequently, some samples stored in biobanks contain PI. More recent studies have demonstrated that a PI can interfere with proteomic analysis. For instance, some of the PIs can form covalent bonds with proteins, modifying their isoelectric properties. The use of a PI in general and the specific type of PI to be employed is still a subject of debate in the renal and urinary proteomics community.15–17 With the increased importance of combining multi-omics techniques, it is important to know whether these archived samples containing a PI, are fit-for-purpose in metabolic phenotyping. In this study we have investigated the effect of PI addition on the 1H NMR and MS HILIC (ESI +/−) and RP (ESI +/−) metabolic profiles of urine from normal controls (NC) and from patients with NS due to iMN. It should be made clear that the aim of the study was not to identify metabolic biomarkers of the renal disease but to investigate whether addition of a PI affected the utility of a metabolic phenotyping analysis. Therefore, the current manuscript is concerned only with whether the use of a PI obscures information recovery and not with investigating the underlying biochemistry of the disease progress. That will be the subject of a subsequent publication. The analysis of this small sample set (NC; n = 4 and iMN; n = 6) indicated the sample class-independent impact of the PI presence on 1H NMR and MS metabolic profiles. Moreover, it has been shown that, after adequate processing of the 1H NMR and MS HILIC (ESI +/−) and RP (ESI +/−) urine metabolic profiles to eliminate the contribution of the PI, the data can be used to investigate the metabolic differences between NC and iMN.
Results
Sample details
The characteristics of patients with iMN are summarized in Table 1. Average urinary protein concentration was 13.2 (range 7.8–28.7) g/24 h. All patients with iMN received supportive therapy. None of the NC group used medication. Urine samples from NC (n = 4) and patients with iMN (n = 6) were divided into two aliquots with protease inhibitor added to one.| Clinical characteristics | Spot urine | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient ID | Age (years) | Sex (M/F) | Anti-PLA2R titera | Serum creatinine (μmol l−1) | Serum albumin (g dl−1) | Proteinuria (g/24 h) | Current medication | Creatinine (mmol l−1) | Proteinuria (g l−1) |
| a Serum anti-phospholipase 2 receptor (PLA2R), an antigen that has recently been associated as a cause for iMN.43 | |||||||||
| 1 | 74 | M | − | 236 | 0.9 | 28.7 | Gliclazide, enalapril, amlodipine, simvastatin, thyrax, furosemide, nifidipine | 6.5 | 12.9 |
| 2 | 72 | M | + | 67 | 2.4 | 7.8 | Furosemide, losartan | 5.9 | 3.6 |
| 3 | 48 | F | − | 58 | 1.2 | 14.1 | Furosemide, lisinopril, warfarin, simvastatin | 1.4 | 2.2 |
| 4 | 52 | M | +/− | 122 | 2.1 | 10.6 | Enalapril, furosemide, simvastatin, clopidogrel | 6.9 | 5.4 |
| 5 | 35 | M | + | 94 | 1.7 | 7.9 | Perindopril, candesartan, furosemide, dalteparin | 5.0 | 1.4 |
| 6 | 28 | M | + | 118 | 2.2 | 9.9 | Candesartan, spironolactone, calcium carbonate/cholecalciferol, insulin aspart (a short acting form of insulin) | 20.5 | 8.6 |
The protease inhibitor introduces contaminant peaks in the 1H NMR spectra
PI addition introduced exogenous signals superimposed onto the endogenous metabolite peaks in the 1H NMR metabolic profiles (Fig. S1A and B, ESI†). In particular, peaks from one excipient, identified as mannitol, obscures peaks from several endogenous species between 3.6 and 3.9 ppm (Fig. S1 and S2, ESI†). Two multiplets were also identified at 7.46 and 7.8 ppm which belong to an intermediate product of another PI constituent, i.e. 4-(2-aminonoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF) or a derivative of it. The NMR variables related to PI contaminants (at 2.05, 2.56, 3.11, 3.37, 3.67–3.89, 7.68, 7.80 ppm) were then excluded from the dataset prior to multivariate statistical analysis to prevent sample classification related to the PI presence.Presence of the PI does not affect the 1H NMR urine metabolic comparisons of controls and iMN patients
Following the removal of the PI contaminant peaks and application of median fold change (MFC) normalisation, principal component analysis (PCA) of the 1H NMR spectroscopic data revealed a good separation of the NC and iMN groups in the PC scores plot (Fig. 1A). An alternative normalisation method, total area normalisation, was explored and is presented in the ESI† (Fig. S4). There was no marked difference between sample aliquots with or without PI, indicating that the major source of variance in the data were not related to PI addition (Fig. 1A).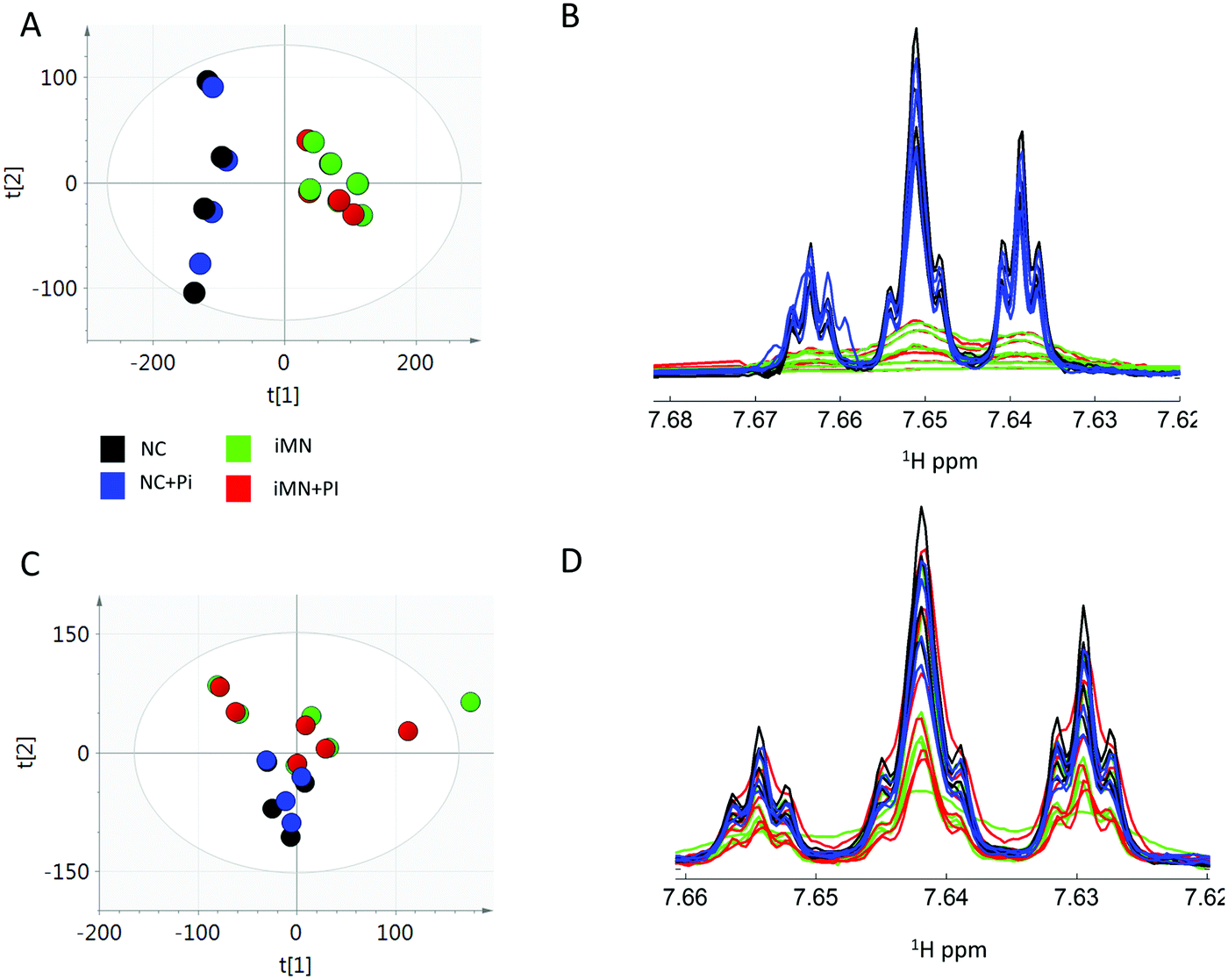 | ||
| Fig. 1 1H NMR spectroscopic metabolic profiles of control (NC) and nephrotic syndrome (iMN) patients with or without addition of a PI, before and after protein precipitation. PCA score plots of iMN, iMN + PI, NC and NC + PI of 1H NMR original (without protein precipitation) data showing a discrimination with NC/NC + PI and iMN/iMN + PI groups (A). Hippurate peak shapes in iMN (green), iMN + PI (red), NC (black) and NC + PI (blue) at 7.66 ppm highlighting a peak broadening in 1H NMR spectra of iMN/iMN + PI groups (B). PCA score plots of protein-precipitated data presenting clustering of NC, NC + PI, iMN and iMN + PI (C). Hippurate peaks shape at 7.66 ppm of protein precipitated data showing an increase in peak resolution in 1H NMR spectra of iMN/iMN + PI after protein precipitation (D). Additional 1H NMR spectral sections are presented in Fig. S5 (ESI†). | ||
The comparison of 1H NMR spectra of NC (blue and green) and iMN (black and red) groups highlighted an increase in the line width and reduced intensity of several spectral peaks (Fig. S5, ESI†) and particularly hippurate in iMN individuals compared to NC individuals (Fig. 1B). On the other hand, the line width of the spectra from the same samples in the presence or absence of PI were similar. The peak broadening phenomenon is characteristic of nonspecific binding of these small molecules with proteins, which depending on the relative mole fractions of the free and the bound forms, and the exchange rate between free and bound forms, results in a shortening of the NMR transverse relaxation time and an increase in peak width. This is in accordance with the fact that urinary protein concentration is significantly higher in iMN compared to NC individuals. The peaks were not found to be lower in intensity in the NC + PI/iMN + PI groups in comparison with the NC/iMN, groups which also suggests that urinary protein concentration, is unchanged with PI addition. Measurement of urinary proteins by a protein assay also showed no significant difference in samples with presence or absence of PI (Wilcoxon Mann Whitney (WMW) p-values > 0.05 when comparing NC versus NC + PI or iMN versus iMN + PI) and as expected proteinuria was higher in iMN patients compared to NC individuals (Fig. S6, ESI†). The difference in intensity of small molecule peaks between NC and iMN individuals could be caused by the increased interactions of highly bound molecules with proteins in iMN samples rather than being a reflection of direct biological changes, thus raising the false positive rate for biomarker discovery in proteinuric kidney diseases.
To fully remove the contribution of proteins in the endogenous metabolic profiles, the urine samples then underwent protein precipitation with methanol prior to NMR analysis. The protein quantification revealed that most proteins were removed from the urine of NC and iMN patients after protein precipitation (Fig. S6, ESI†). After removal of PI contaminant peaks and MFC-normalisation, the PCA scores plot showed no clear discrimination in the urine samples between presence and absence of PI (Fig. 1C). In addition, comparable line widths were found for hippurate in NC, NC + PI, iMN and iMN + PI samples indicating that a large proportion of this molecule is now free in solution (Fig. 1D). Importantly, the discrimination between NC and iMN samples in PCA score plots disappeared after protein precipitation suggesting that the largest source of variation between NC and iMN spectra are related to the protein content. In addition, a larger intra-group variability was observed in the PCA first component (T1, 15% of variance) that could also affect the discrimination between NC and iMN groups. This may have been introduced by the variability of the protein precipitation steps.
Orthogonal partial least squares discriminant analysis (OPLS-DA) was employed to investigate the effect of PI in urine metabolic profiles in the context of biomarker discovery for iMN in the original (without protein precipitation), and the protein–precipitated datasets. In both datasets, as expected, no difference was found between NC and NC + PI groups, nor between the iMN and iMN + PI groups in the OPLS-DA models (CV-ANOVA p-values > 0.05, Table S2, ESI†). A good discrimination was observed between NC and iMN in the original but not in the protein-precipitated datasets (CV-ANOVA p-value = 6 × 10−4, 0.17 respectively, Table S2, ESI†). These observations are in accordance with the PCA model. The OPLS loadings plot allowed the establishment of the spectral regions that influenced the discrimination between NC and iMN in the OPLS-DA models (Fig. 2A and B). The correlation patterns in most of the NMR variables of the protein-precipitated dataset were similar to those found in the original dataset (Fig. 2B) although the correlation coefficients were weaker. In addition, we also observe a moderate correlation of the trigonelline level (R2 = 0.65 at 4.46 ppm) with iMN only in the protein-precipitated dataset. Therefore, in the current study, the presence of proteins in the iMN patients has limited impact on the direction of association of biomarkers with iMN but can lower the resolution of specific peaks such as trigonelline, thus affecting the statistical comparison. Univariate analysis of the original NMR dataset confirmed the multivariate analysis results, i.e. that hippurate (7.83, 7.66 ppm, 7.56 ppm), creatinine (3.06, 4.08 ppm), phenylacetate (3.57 ppm), citrate (2.66 ppm) and unknown metabolites (at 1.23 (d) and 8.35 (d) ppm) significantly decreased in iMN patients. (WMW p-values and Benjamini–Hochberg adjusted p-values (pFDR) < 0.05). Given the low number of samples in both iMN and NC groups, we recognise that this study does not have enough statistical power to identify reliable markers of iMN, and indeed this is not the aim of the current investigation. These metabolites can only be considered as putative, unvalidated markers of iMN. None of the iMN-associated metabolites were significantly affected by PI addition (WMW p-values and pFDR >0.05, Table 2).
 | ||
| Fig. 2 OPLS-DA loadings plots of 1H NMR original (without protein precipitation) (A) and protein precipitated (B) metabolic profiles comparing NC and iMN. The pseudo-spectra represent the covariance between the NMR variables and the group discrimination. The positive peaks are more concentrated in the iMN group and the negative peaks are excreted in higher concentration in the NC group. NC: normal controls, iMN: idiopathic membranous nephropathy PI: protease inhibitor. The squared regression coefficients (R2) between the NMR variables and the logistic variable defining the groups are mapped and color-coded onto the pseudo-spectrum. The hot colours are allocated to highly correlated variables whereas the cold colours show variables with non-significant correlation 1: unknown, 2: citrate, 3: dimethylamine, 4: creatinine, 5: unknown, 6: glucose, 7: phenylacetate, 8: hippurate, 9: trigonelline. | ||
| 1H NMR analysis | NC vs. iMN | Effect of PI (NC/iMN − vs. NC + PI/iMN + PI) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Methods | Metabolites | 1H ppm | Multiplicity | MWM p-value | FDR | Fold changea | MWM p-value | FDR | Fold changeb |
| a Metabolite higher in iMN if fold change >1, and lower in iMN if fold change <1. b Metabolite higher in presence of PI if fold change >1 and lower with presence of PI if fold change <1. | |||||||||
| 1D standard experiment | UA1 | 1.23 | d | 0.0095 | 0.0304 | 0.72 | 0.3447 | 1 | 0.84 |
| Citrate | 2.668 | d | 0.0190 | 0.0304 | 0.43 | 0.9097 | 1 | 0.95 | |
| Dimethylamine | 2.722 | s | 0.0095 | 0.0304 | 0.36 | 0.6776 | 1 | 0.80 | |
| Creatinine | 3.063 | s | 0.0095 | 0.0304 | 0.38 | 0.6232 | 1 | 0.88 | |
| Phenylacetate | 3.57 | s | 0.0095 | 0.0304 | 0.67 | 0.5708 | 1 | 1.04 | |
| Hippurate | 7.563 | m | 0.0095 | 0.0304 | 0.14 | 0.6776 | 1 | 0.90 | |
| UA2 | 8.35 | d | 0.0095 | 0.0304 | 0.17 | 0.85 | 0.99 | 0.78 | |
| UPLC-MS analysis | NC vs. iMN | Effect of PI (NC/iMN − vs. NC + PI/iMN + PI) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Methods | Metabolites | [Adduct] (as detected) | m/z | Retention time (minutes) | MWM p-value | FDR | Fold change | MWM p-value | FDR | Fold change |
| HILIC UPLC MS (ESI +) | Hydroxyhexanoycarnitine | [M + H]+ | 276.162 | 7.81 | 0.0095 | 1 | 0.64 | 1.0000 | 1 | 0.67 |
| Asymmetric dimethylarginine | [M + 2Na − H]+ | 247.166 | 8.01 | 0.0190 | 1 | 1.37 | 0.0257 | 0.98 | 0.90 | |
| L-Carnitine | [M + H]+ | 162.581 | 7.20 | 0.0381 | 1 | 0.73 | 0.6776 | 0.99 | 0.90 | |
| UA 3 | [M + H]+ | 286.13 | 6.81 | 0.0381 | 1 | 0.99 | 0.7054 | 0.99 | 0.86 | |
| HILIC UPLC MS (ESI −) | α-N-Phenylacetyl-L-glutamine | [M − H]− | 145.096 | 5.14 | 0.0190 | 1 | 1.89 | 0.0312 | 0.83 | 1.74 |
| 4-Cresol sulfate | [M − H]− | 186.978 | 0.68 | 0.0381 | 1 | 1.01 | 0.0036 | 0.83 | 1.10 | |
| RP UPLC MS (ESI +) | UA 4 | Isotope | 537.166 | 10.13 | 0.0381 | 1 | 1.49 | 0.5205 | 0.92 | 0.74 |
| RP UPLC MS (ESI −) | 3,4-Dihydroxyphenylalanine | [M − H]− | 195.05 | 0.49 | 0.0095 | 0.26 | 1.86 | 0.6776 | 1 | 1.12 |
| Isocitrate | [M − H]− | 191.055 | 0.54 | 0.0095 | 0.26 | 2.83 | 0.1041 | 1 | 0.65 | |
The protease inhibitor introduces contaminant peaks in the UPLC-MS metabolic profile of urine
Two types of UPLC-MS analyses were carried out. These comprised reverse phase (RP) and hydrophilic interaction liquid chromatography (HILIC) with mass spectrometric detection using both positive and negative electrospray ionisation modes (ES + and ES −). Mass spectrometric features relating to the components of the PI were observed in the UPLC-MS metabolic data (Fig. S3, ESI†). By HILIC UPLC-MS (ESI +), it was possible to detect the ion from AEBSF at 204.05 m/z at a retention time of 3.54 min (Fig. S3A, ESI†). Another component, leupeptin hemisulfate was detected in both ionisation modes using HILIC UPLC-MS, detecting the positive ion 427.303 m/z at 4.64 min (Fig. S3B, ESI†) and negative ion 425.285 at 4.419 min (MS data not shown). In the RP UPLC-MS (ESI +) data, the AEBSF ion at 204.05 m/z was detected at a retention time of 3.90 min (MS data not shown). Moreover, the component mannitol that was observed strongly in the NMR spectra could also be detected in the HILIC-MS (ESI −) data at 181.073 m/z and retention time 4.793 min.Such exogenous peaks cause fewer problems for MS-based multivariate analysis than NMR features. This is because each of the undesired non-endogenous feature comprises only a single narrow entry in the data whereas for NMR spectroscopy any given substance (e.g. mannitol) can show several bands many of which can be complex multiplets of peaks.
Several UPLC-MS detectable biomarkers discriminate NC from iMN
HILIC UPLC-MS analyses (in positive and negative ionisation detection modes) was used to capture the signals of a large range of metabolites from small polar molecules. The PCA score plots (PC1 versus PC2) of the metabolic data indicated that differences between NC and iMN groups were the main source of variation in the HILIC UPLC-MS (ESI +) and HILIC UPLC-MS (ESI −) data.The pair-wise OPLS-DA models comparing NC vs. iMN were predictive for both HILIC UPLC-MS data sets (Table S2, ESI†). Univariate analysis was employed to investigate the data sets for features significantly (WMW p-value < 0.05) associated to iMN. Four metabolites (hydroxyhexanoycarnitine, asymmetric dimethylarginine, carnitine and one unidentified substance designated UA3) were discovered from the HILIC UPLC-MS (ESI +) and two metabolites (α-N-phenylacetyl-glutamine and 4-cresol sulfate) were found in the HILIC UPLC-MS (ESI −) data set (Table 2). The features and thus the metabolites they correspond to were not significant after adjusting the WMW p-values for the false discovery rate (pFDR > 0.05).
RP UPLC-MS (ESI +/−) analyses were also employed to capture the signals of a large range of metabolites from less polar molecules including lipids. The PCA score plots (PC1 versus PC2) of the metabolic data indicates that the main source of variation (PC1) in the RP UPLC-MS (ESI +) data is between samples with and without PI (Fig. 3), with the separation of the data based on NC and iMN groups explained by PC2. To remove the separation of the data based on presence or absence of PI, the features contributing to this natural separation (the features associated to the presence of the PI) were excluded. PCA of this new dataset (referred to from here on in as RP-UPLC MS (ESI + exc)) revealed that the separation in the data based on the presence of PI had been removed. The RP-UPLC-MS (ESI −) data displayed a natural separation based on NC and iMN groups (Fig. 3).
 | ||
| Fig. 3 Effect of PI addition on MS metabolic profiles of NC and iMN individuals. PCA score plots of NC, NC + PI, iMN and iMN + PI, in HILIC UPLC-MS (ESI +) (A), HILIC UPLC-MS (ESI −) (B), RP UPLC-MS (ESI +) (C) and RP UPLC-MS (ESI −) (D), colored by group. NC: normal controls, iMN: idiopathic membranous nephropathy, PI: protease inhibitor. | ||
The pair-wise OPLS-DA models comparing NC vs. iMN were not significant (CV-ANOVA p-values > 0.05) in the RP UPLC-MS (ESI +/+ exc/−) data. When the data from all samples were used, the OPLS-DA model comparing iMN/iMN + PI and NC/NC + PI was predictive (CV-ANOVA = 7.2 × 10−3, 1.66 × 10−4, 2.91 × 10−6) in RP UPLC-MS (ESI +, ESI + exc and ESI − respectively, Table S2, ESI†). This could be a result of increased statistical power when using the larger number of samples. It is possible, given the low number of samples, that subtle metabolic differences between NC and iMN are missed using multivariate analysis.18 As with the HILIC UPLC-MS data, univariate analysis was used to further investigate the features associated to iMN. Features significantly (WMW p-value < 0.05) associated to iMN, revealed one metabolite (an unidentified substance designated UA4) was discovered from the RP UPLC-MS (ESI +) and two metabolites (3,4-dihydroxyphenylalanine and isocitrate) in the RP UPLC-MS (ESI −) sets (Table 2). These metabolites were not significant after adjusting the WMW p-values for the false discovery rate (pFDR > 0.05).
For each metabolite discovered in the UPLC-MS datasets discriminating NC and iMN, the WMW p-value describing the association of the given metabolite to the presence of PI is also reported in Table 2. Asymmetric dimethylarginine was significantly lower in the presence of PI (WMW p-value = 0.026) while α-N-phenylacetyl-glutamine and 4-cresol sulphate significantly increased with PI (WMW p-value = 0.031 and 0.004 respectively, Table 2). These findings were based on the WMW p-value < 0.05, but did not survive after adjusting the WMW p-values for false discovery rate (pFDR > 0.05). The rest of the metabolites that correlate to iMN were not significantly affected by the presence of PI (WMW p-value < 0.05). These metabolites include 3,4-dihydroxyphenylalanine, hydroxyhexanoylcarnitine, isocitrate and carnitine.
Discussion
PI is often added to urine samples intended for proteomic analysis to inhibit protease activity and thus maintain the urinary protein pool.15 With the additional information offered by way of metabolic phenotyping, it is important to know whether these samples are fit for purpose. This study has explored the influence of PI on the metabolic profile of urine and shows that use of a PI introduces contaminant peaks which can be detected by 1H NMR spectroscopy and HILIC and RP UPLC-MS. By careful processing of the data, taking the spectral contaminants into account, it was possible to apply metabolic phenotyping to urine samples with added PI.In the 1H NMR spectra, PI contaminant peaks were found to cover those from endogenous metabolites triggering some difficulties in recovering the spectral information in these regions. However, the identification of endogenous metabolites is still possible because in general, molecules produce several peaks across the 1H NMR chemical shift range which are not all covered by the contaminant peaks. In addition, spectroscopic and mathematical techniques can be employed to help recover the obscured spectral data such as JRES spectroscopy or STORM (subset optimization by reference matching) and STOCSY-editing.19–22 The 1H NMR data processing is also essential for optimising the spectral information for metabolic phenotyping. Importantly, 1H NMR spectral alignment may improve the selection and the removal of the PI contaminants peaks although spectral artefacts are often introduced during this process.23,24 In this study, MFC and total area normalisation methods were compared (Supplementary information results, Fig. S4, ESI†), MFC proved to be more suitable to correct for dilution effects and this is in accordance with the literature.25 Application of the MFC normalisation is recommended where drug metabolites or excipients are present in high concentrations in urine samples.26,27 Once the contaminant peaks have been carefully excluded and the 1H NMR spectra appropriately processed, the data can be used for the metabolic comparison of control individuals and iMN patients although the sample numbers per class were low. Alternatively, protein precipitation of urine samples eliminates the contribution of proteins in endogenous profiles but may introduce extra intra-group variability and obscure the true biochemical difference between iMN and NC individuals. Several metabolites were identified as possible biomarkers of iMN, but since the sample numbers were so low in this study little significance can be attached to them and a properly statistically-powered study needs to be undertaken. PI-derived contaminants were also observed in HILIC UPLC-MS (ESI +/−) and RP UPLC-MS (ESI +/−) methods; nevertheless, non-supervised and supervised multivariate analyses highlighted no metabolic pattern specific to the PI presence in all datasets except in the RP UPLC-MS (ESI +) data. In RP UPLC-MS (ESI +) data, most of the features responsible for sample clustering were assigned to PI related compounds (AEBSF, leupeptin hemisulfate, trypsin inhibitor).
By comparing the features associated with iMN and those associated with PI presence, it was found that among the iMN-associated metabolites, two metabolites significantly increase with PI presence while one metabolite is reduced in the presence of the PI. This indicates that the PI probably suppresses or enhances the ionisation of several molecules. This comparison allows to flag potential false positive or negative markers that could be affected by the presence of PI and thus may be useful to identify robust and reliable biomarkers in metabolic profiling regardless of the presence of PI. To perform this analysis, analysis of some reference samples representative of each sample group with the presence or absence of PI was necessary. A large range of PI or other preservatives is commercially available with different protease inhibitors and additives whose function is generally to improve molecular stability15,28 (details of the PI product in Table S1, ESI†). Their utilisation might result in the introduction of other contaminant peaks and have different effects on the endogenous molecules.29 Therefore, if samples preserved in this manner are included in metabolic phenotyping studies, a similar approach to that described here should be carried out to evaluate the impact of any preservative on the 1H NMR and MS metabolic profiles.
A previous study has reported degradation of protein in samples stored with PI for three years.30 In the present study, it was found that although PI is added to stop protease activity, the protein pool is kept intact in the urine samples with PI. This may be due to the relative short storage (one year) of the samples in the presence of the PI. The utilisation of protease inhibitors or other preservatives in different biological matrices that contain a large amount of proteins, may modify the protein content and thus affect the 1H NMR spectroscopic and MS metabolic profiles.31–33 It is well known that proteins in biofluids can introduce large broad peaks in 1H NMR spectra that obscure peaks from small molecule metabolites and additionally they can lower the detectability of small molecules through protein binding.33,34 Therefore it is also advisable to estimate whether preservative addition increases the protein concentration in the samples.
Of note, drugs, preservatives and other biological additives often do not list the excipients and non-essential ingredients in the product composition. In this case, the mannitol which may be used as a binder/filler in the formulation of the PI tablet,35 was identified in high concentration in 1H NMR spectral urine profiles of NC + PI and iMN + PI patients although they are not mentioned in the Roche PI tablet description (Table S1, ESI†).
The main limitation of this study is the low sample size, which is a drawback when identifying metabolic difference between NC and iMN. The relatively low number of discriminating metabolites between NC and iMN patients could be due to a low number of samples in each group limiting the statistical strength of the analysis. In addition, the samples were not matched regarding to the conventional confounding factors (age, gender, ethnicity, infection screening and urine collection date). However, it is less of an issue here as the study aims to identify disruption of the PI on the urine metabotype (which is largely visible in the NMR and MS spectra) in the context of a kidney disease and not to evaluate the specific metabolic signature of the disease itself which is the subject of future studies.
A decision-making process for the metabolic analysis of urine samples both with and without a PI addition by 1H NMR spectroscopy, HILIC UPLC-MS and RP UPLC-MS is proposed in Fig. 4. This includes the following procedures:
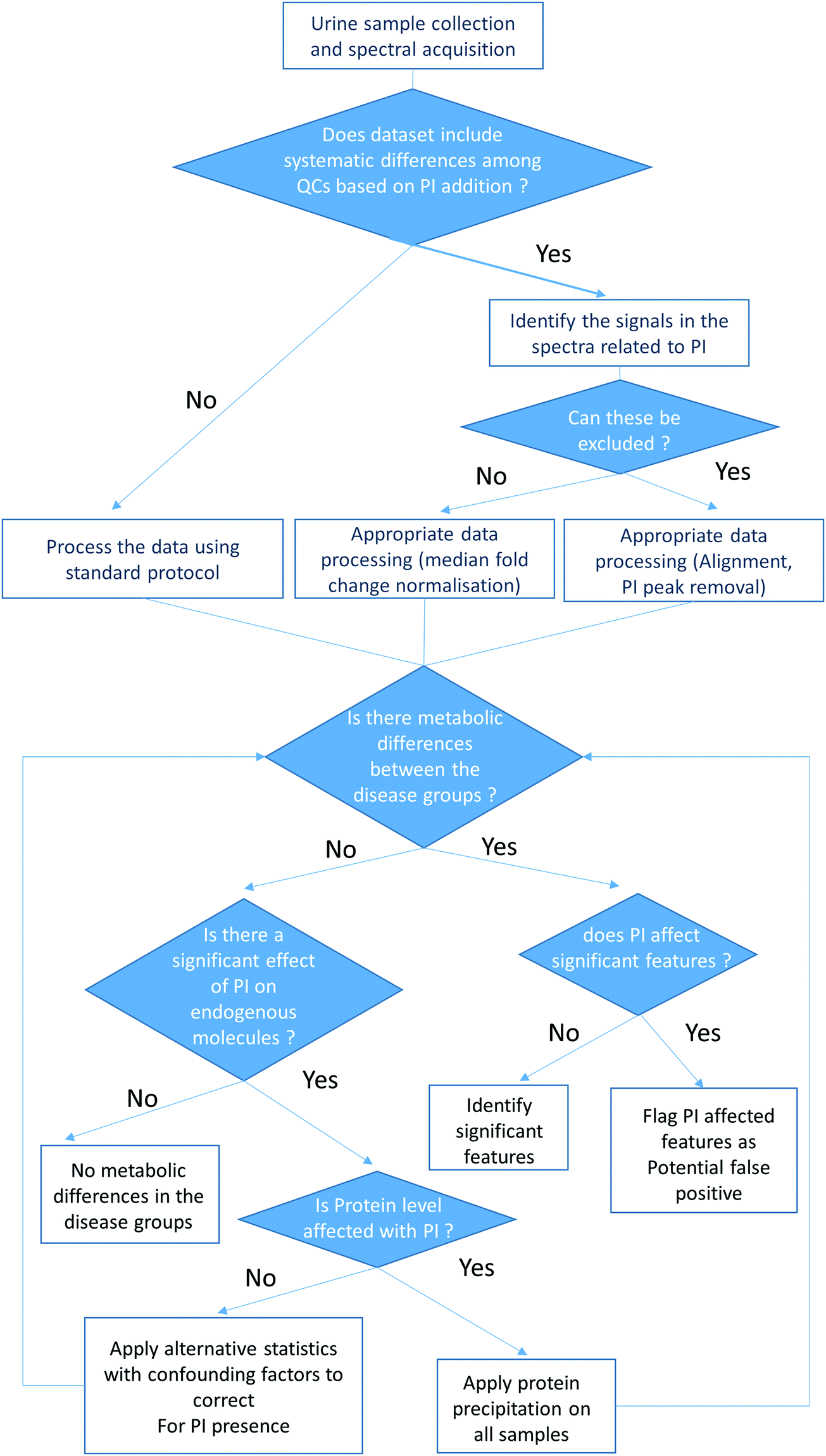 | ||
| Fig. 4 Flow chart of a proposed decision-making process for metabolic profiling of urine samples with presence and absence of a PI. | ||
– Preparation of quality control samples representative of each group of interest with the presence or absence of PI for analysis with the sample set.
– Measurement of urinary protein levels to evaluate whether PI addition affects the protein concentrations in the samples.
– Appropriate data processing (alignment and median fold change normalisation) and PI peak removal in the 1H NMR spectra.
– If PI significantly impacts the endogenous metabolites in the samples, alternative statistical procedures which take into account confounding factors can be applied to correct for the presence of PI.36
These procedures can help limit the identification of false positive and negative markers in urine samples containing preservatives and ultimately improve the biomarker discovery in renal disease.
Conclusion
This study demonstrated that it is possible to include proteinuric urine samples containing a PI for 1H NMR spectroscopic and HILIC UPLC-MS and RP UPLC-MS metabolic profiling. Careful data processing is required to eliminate the contribution of the PI on the 1H NMR dataset. In MS datasets, it is important to estimate the impact of PI on metabolite ionisation to flag potential false positive markers. We report a generalizable workflow for sample analysis where PI is part of the study design. The utilisation of urine samples for multi-omics analysis may prove useful in the identification of new biomarkers for renal diseases and could allow the development of an individualized treatment strategy.Experimental section
Urine collection and storage
This work forms of part of the Eurenomics EC-funded project. Full ethical approval was obtained by all the clinical collaborators within the Eurenomics project. All subjects also gave their informed consent. Once received, and prior to analysis, the samples were further anonymised by assigning a randomised study number. The samples were used only for study-specific objective as agreed with the sample holders prior to commencing the investigation. Urine samples from normal controls (NC) (n = 4) and patients with a NS due to biopsy-proven idiopathic membranous nephropathy (iMN) (n = 6) were collected under human participant research protocols approved by the Institutional Review Boards of Radboud University Medical Center, The Netherlands. NS was defined as proteinuria >3 g/24 h and a serum albumin <3.0 g dL−1. Urine samples were centrifuged at 3000g for 10 min at 4 °C. The supernatant was divided into two 15 ml aliquots. A protease inhibitor (Complete Mini, Roche, Indianapolis, IN, USA) was added to one 15 ml aliquot of each sample. The samples were stored for a year in 2 ml aliquots at −80 °C until further use. Protein content was measured in each sample by optical density at an absorbance of 600 nm (see Supplementary material and methods, ESI†). Proteins in the samples were also precipitated to remove the contribution of protein peaks in the 1H NMR spectra. To perform the protein precipitation, the samples were stored at −20 °C overnight with cold deuterated methanol. The supernatants were collected, dried down, and stored in −80 °C. Dried protein pellets were also re-suspended with water prior to protein measurement (see Supplementary material and methods, ESI†) and 1H NMR spectroscopic analysis.1H NMR spectroscopic analysis
Samples were thawed at room temperature and prepared according to a published protocol.37 Samples that underwent a protein precipitation were re-suspended with water prior to sample preparation (see Supplementary material and methods, ESI†). For each sample, a volume of 350 μl was combined with 350 μl of phosphate buffer (1.5 M KH2HPO4, pH = 7.4, 100% D2O, 1.9 mM sodium azide and 5 mM trimethylsilyl[2H4] propionic acid sodium salt (TSP)). The samples were then centrifuged for 5 min at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g at 4 °C and 600 μl of the supernatant was transferred into 5 mm outer diameter NMR tubes for analysis as published by Dona et al.371H NMR spectra were acquired at a temperature of 300 K, using a standard pulse sequence with water suppression (noesypr1d, (RD)–90°–t1–90°–tm–90°–acquisition) at 600.13 MHz, a relaxation delay (RD) of 4 s, a mixing time (tm) of 10 ms, a 90° pulse set at 16.3 μs, and 32 free induction decays (FIDs) using 64 K data-points. The FIDs were multiplied by an exponential factor to give a line broadening of 1 Hz and Fourier transformed to obtain the usual frequency spectrum (TOPSPIN 3.2 software, Bruker Biospin, Rheinstetten, Germany). The spectra were automatically phased, baseline corrected and calibrated using the TSP signal at δ 0 using a standard Bruker routine, then imported into MATLAB (version R2014a, the Math works, Natick MA, USA) and digitised to 32 K data-points. The water peak resonance (δ 4.7–5.05) was removed from each spectrum. The spectra were then aligned using an algorithm published by Veselkov et al.24 and the chemical shifts from any PI contaminants peaks were also excluded from each spectrum. The 1H NMR spectra were normalised with the median fold change (MFC) or the total area normalisation method38 prior to statistical analysis (further details on normalisation methods are given in ESI†).
000g at 4 °C and 600 μl of the supernatant was transferred into 5 mm outer diameter NMR tubes for analysis as published by Dona et al.371H NMR spectra were acquired at a temperature of 300 K, using a standard pulse sequence with water suppression (noesypr1d, (RD)–90°–t1–90°–tm–90°–acquisition) at 600.13 MHz, a relaxation delay (RD) of 4 s, a mixing time (tm) of 10 ms, a 90° pulse set at 16.3 μs, and 32 free induction decays (FIDs) using 64 K data-points. The FIDs were multiplied by an exponential factor to give a line broadening of 1 Hz and Fourier transformed to obtain the usual frequency spectrum (TOPSPIN 3.2 software, Bruker Biospin, Rheinstetten, Germany). The spectra were automatically phased, baseline corrected and calibrated using the TSP signal at δ 0 using a standard Bruker routine, then imported into MATLAB (version R2014a, the Math works, Natick MA, USA) and digitised to 32 K data-points. The water peak resonance (δ 4.7–5.05) was removed from each spectrum. The spectra were then aligned using an algorithm published by Veselkov et al.24 and the chemical shifts from any PI contaminants peaks were also excluded from each spectrum. The 1H NMR spectra were normalised with the median fold change (MFC) or the total area normalisation method38 prior to statistical analysis (further details on normalisation methods are given in ESI†).
Mass spectrometry analysis
LC-MS grade solvents were used throughout. A volume of 300 μl of urine was combined with 900 μl isopropanol (IPA). The sample:IPA mix was incubated for 2 hours at −20 °C, centrifuged at 2700g for 20 min and the supernatant aliquoted. Quality control (QC) samples were prepared by pooling 100 μl of each sample. The samples were randomised and maintained at 4 °C until prior to analysis. Separation of metabolites was performed using a Waters Acquity Ultra Performance LC system (Waters, Milford, MA, USA). Each sample was analysed using RP and HILIC UPLC methods as described in the literature.8,39The acquisition of ESI-MS data was performed on a Xevo G2 Q-TOF mass spectrometer (Waters, Milford, MA, USA) in positive and negative ESI modes. The two ionisation modes complement each other, as metabolites may preferentially ionise in either of the two modes according to their functional groups- which carry the charge of the molecule.
Capillary and cone voltages were set at 1.5 kV and 30 V respectively. The desolvation gas was set to 1000 L h−1 at a temperature of 600 °C; the cone gas was set to 50 L h−1 and the source temperature was set to 120 °C. For mass accuracy a lockspray interface was used with a leucine enkephalin (556.27741/554.2615 amu) solution at a concentration of 2000 ng ml−1, at a flow rate of 15 μl min−1 as the lock mass. The raw spectrometric data were processed using XCMS (version 1.50.0) in R (3.1.2, R Foundation for Statistical Computing, Vienna, Austria), using the centwave peak picking method to detect chromatographic peaks.40
Data analysis
1H NMR and UPLC-MS pre-processed data were analysed using a multivariate approach in SIMCA (SIMCA-P13 UMETRICS, Umea, Sweden). PCA was performed using the full resolution NMR (32 K data points) and MS data to assess sample clustering behaviour and intra-group variation. OPLS-DA was employed to maximise the metabolic differences between groups corresponding to NC, NC + PI, iMN and iMN + PI.23 The OPLS-DA model with a CV-ANOVA p-value < 0.05 was considered as predictive. Wilcoxon–Mann–Whitney (WMW) tests, adjusted using the Benjamini–Hochberg False Discovery Rate (pFDR), were also calculated for each feature. However, considering the low number of samples, the important features for group discrimination were identified if they had WMW p-values < 0.05 regardless of pFDR values.Metabolite identification
Metabolite assignments in NMR data were performed by matching the chemical shifts and J-coupling information with NMR spectral databases41 and literature. STOCSY (Statistical Total Correlation SpectroscopY)19 as well as 2-D 1H–13C and 1H–1H NMR experiments, were employed to help the identification of unknown metabolites.The metabolite identification in MS data was achieved by comparing the molecular weight and MS–MS fragmentation patterns of the features of interest with available literature and online databases such as METLIN,42 and the Human Metabolome Database (HMDB).41
Abbreviations
| NS | Nephrotic syndrome |
| PI | Protease inhibitor |
| NC | Normal control |
| iMN | (Idiopathic) membranous nephropathy |
| UPLC-MS | Ultra-performance liquid chromatography-mass spectrometry |
| NMR | Nuclear magnetic resonance |
| RP | Reverse phase |
| ESI | Electrospray ionisation |
| MVA | Multivariate analysis |
| iFSGS | (Idiopathic) focal segmental glomerulosclerosis |
| MCD | Minimal change disease |
| CKD | Chronic kidney disease |
| TSP | Trimethylsilyl[2H4]propionic acid |
| FID | Free induction decay |
| RD | Relaxation delay |
| MFC | Median fold change |
| QC | Quality control |
| PCA | Principal component analysis |
| OPLS | Orthogonal partial least squares |
| OPLS-DA | OPLS-discriminant analysis |
| AEBSF | 4-(2-Aminoethyl)benzenesulfonyl fluoride hydrochloride |
| STOCSY | Statistical total correlation spectroscopy |
| J-Res | J-Resolved |
| STORM | Subset optimization by reference matching. |
Conflicts of interest
All the authors declare no competing interests.Acknowledgements
The work is funded by the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 305608. I. M. Rood was supported by an AGIKO-grant from ZonMw-NWO (grant 92003587).References
- M. Haas, S. M. Meehan, T. G. Karrison and B. H. Spargo, Am. J. Kidney Dis., 1997, 30, 621–631 CrossRef CAS PubMed.
- V. Perkovic, R. Agarwal, P. Fioretto, B. R. Hemmelgarn, A. Levin, M. C. Thomas, C. Wanner, B. L. Kasiske, D. C. Wheeler, P. H. Groop and P. Conference, Kidney Int., 2016, 90, 1175–1183 CrossRef PubMed.
- I. M. Rood, M. L. Merchant, D. W. Wilkey, T. Zhang, V. Zabrouskov, J. van der Vlag, H. B. Dijkman, B. K. Willemsen, J. F. Wetzels, J. B. Klein and J. K. Deegens, Proteomics, 2015, 15, 3722–3730 CrossRef CAS PubMed.
- J. K. Nicholson, J. C. Lindon and E. Holmes, Xenobiotica, 1999, 29, 1181–1189 CrossRef CAS PubMed.
- C. L. Gavaghan, E. Holmes, E. Lenz, I. D. Wilson and J. K. Nicholson, FEBS Lett., 2000, 484, 169–174 CrossRef CAS PubMed.
- J. K. Nicholson, I. D. Wilson and J. C. Lindon, Pharmacogenomics, 2011, 12, 103–111 CrossRef CAS PubMed.
- J. C. Lindon, E. Holmes, M. E. Bollard, E. G. Stanley and J. K. Nicholson, Biomarkers, 2004, 9, 1–31 Search PubMed.
- M. R. Lewis, J. T. Pearce, K. Spagou, M. Green, A. C. Dona, A. H. Yuen, M. David, D. J. Berry, K. Chappell, V. Horneffer-van der Sluis, R. Shaw, S. Lovestone, P. Elliott, J. P. Shockcor, J. C. Lindon, O. Cloarec, Z. Takats, E. Holmes and J. K. Nicholson, Anal. Chem., 2016, 88, 9004–9013 CrossRef CAS PubMed.
- T. J. Velenosi, A. Hennop, D. A. Feere, A. Tieu, A. S. Kucey, P. Kyriacou, L. E. McCuaig, S. E. Nevison, M. A. Kerr and B. L. Urquhart, Sci. Rep., 2016, 6, 22526 CrossRef CAS PubMed.
- Z. H. Zhang, F. Wei, N. D. Vaziri, X. L. Cheng, X. Bai, R. C. Lin and Y. Y. Zhao, Sci. Rep., 2015, 5, 14472 CrossRef CAS PubMed.
- L. Chen, W. Su, H. Chen, D. Q. Chen, M. Wang, Y. Guo and Y. Y. Zhao, Adv. Clin. Chem., 2018, 85, 91–113 Search PubMed.
- Y. Y. Zhao and R. C. Lint, Adv. Clin. Chem., 2014, 65, 69–89 CAS.
- Y. Y. Zhao, N. D. Vaziri and R. C. Lin, Adv. Clin. Chem., 2015, 68, 153–175 Search PubMed.
- R. G. Fassett, S. K. Venuthurupalli, G. C. Gobe, J. S. Coombes, M. A. Cooper and W. E. Hoy, Kidney Int., 2011, 80, 806–821 CrossRef CAS PubMed.
- L. Musante, D. Tataruch, D. Gu, X. Liu, C. Forsblom, P. H. Groop and H. Holthofer, J. Diabetes Res., 2015, 2015, 289734 Search PubMed.
- P. O. Havanapan and V. Thongboonkerd, J. Proteome Res., 2009, 8, 3109–3117 CrossRef CAS PubMed.
- H. Zhou, P. S. Yuen, T. Pisitkun, P. A. Gonzales, H. Yasuda, J. W. Dear, P. Gross, M. A. Knepper and R. A. Star, Kidney Int., 2006, 69, 1471–1476 CrossRef CAS PubMed.
- E. Saccenti and M. E. Timmerman, J. Proteome Res., 2016, 15, 2379–2393 CrossRef CAS PubMed.
- O. Cloarec, M. E. Dumas, A. Craig, R. H. Barton, J. Trygg, J. Hudson, C. Blancher, D. Gauguier, J. C. Lindon, E. Holmes and J. Nicholson, Anal. Chem., 2005, 77, 1282–1289 CrossRef CAS PubMed.
- C. Ludwig and M. R. Viant, Phytochem. Anal., 2010, 21, 22–32 CrossRef CAS PubMed.
- C. J. Sands, M. Coen, A. D. Maher, T. M. Ebbels, E. Holmes, J. C. Lindon and J. K. Nicholson, Anal. Chem., 2009, 81, 6458–6466 CrossRef CAS PubMed.
- J. M. Posma, I. Garcia-Perez, M. De Iorio, J. C. Lindon, P. Elliott, E. Holmes, T. M. Ebbels and J. K. Nicholson, Anal. Chem., 2012, 84, 10694–10701 CrossRef CAS PubMed.
- L. Eriksson, P. L. Andersson, E. Johansson and M. Tysklind, Mol. Diversity, 2006, 10, 169–186 CrossRef CAS PubMed.
- K. A. Veselkov, J. C. Lindon, T. M. Ebbels, D. Crockford, V. V. Volynkin, E. Holmes, D. B. Davies and J. K. Nicholson, Anal. Chem., 2009, 81, 56–66 CrossRef CAS PubMed.
- J. Hochrein, H. U. Zacharias, F. Taruttis, C. Samol, J. C. Engelmann, R. Spang, P. J. Oefner and W. Gronwald, J. Proteome Res., 2015, 14, 3217–3228 CrossRef CAS PubMed.
- A. M. De Livera, D. A. Dias, D. De Souza, T. Rupasinghe, J. Pyke, D. Tull, U. Roessner, M. McConville and T. P. Speed, Anal. Chem., 2012, 84, 10768–10776 CrossRef CAS PubMed.
- T. De Meyer, D. Sinnaeve, B. Van Gasse, E. R. Rietzschel, M. L. De Buyzere, M. R. Langlois, S. Bekaert, J. C. Martins and W. Van Criekinge, Anal. Bioanal. Chem., 2010, 398, 1781–1790 CrossRef CAS PubMed.
- V. Thongboonkerd and P. Saetun, J. Proteome Res., 2007, 6, 4173–4181 CrossRef CAS PubMed.
- C. Lopez-Otin and C. M. Overall, Nat. Rev. Mol. Cell Biol., 2002, 3, 509–519 CrossRef CAS PubMed.
- M. P. Schuh, E. Nehus, Q. Ma, C. Haffner, M. Bennett, C. D. Krawczeski and P. Devarajan, Am. J. Kidney Dis., 2016, 67, 56–61 CrossRef CAS PubMed.
- D. Vuckovic, Anal. Bioanal. Chem., 2012, 403, 1523–1548 CrossRef CAS PubMed.
- J. C. Chatham and J. R. Forder, Biochim. Biophys. Acta, 1999, 1426, 177–184 CrossRef CAS.
- C. A. Daykin, P. J. Foxall, S. C. Connor, J. C. Lindon and J. K. Nicholson, Anal. Biochem., 2002, 304, 220–230 CrossRef CAS PubMed.
- J. K. Nicholson and K. P. Gartland, NMR Biomed., 1989, 2, 77–82 CrossRef CAS PubMed.
- H. L. Ohrem, E. Schornick, A. Kalivoda and R. Ognibene, Pharm. Dev. Technol., 2014, 19, 257–262 CrossRef CAS PubMed.
- A. C. Skelly, J. R. Dettori and E. D. Brodt, Evid. Based Spine Care J., 2012, 3, 9–12 Search PubMed.
- A. C. Dona, B. Jimenez, H. Schafer, E. Humpfer, M. Spraul, M. R. Lewis, J. T. Pearce, E. Holmes, J. C. Lindon and J. K. Nicholson, Anal. Chem., 2014, 86, 9887–9894 CrossRef CAS PubMed.
- F. Dieterle, A. Ross, G. Schlotterbeck and H. Senn, Anal. Chem., 2006, 78, 4281–4290 CrossRef CAS PubMed.
- S. M. G. Isaac and G. Astarita, Lipid Separation using UPLC with Charged Surface Hybrid Technology, accessed 15 August, 2016.
- N. G. Mahieu, X. Huang, Y. J. Chen and G. J. Patti, Anal. Chem., 2014, 86, 9583–9589 CrossRef CAS PubMed.
- D. S. Wishart, T. Jewison, A. C. Guo, M. Wilson, C. Knox, Y. Liu, Y. Djoumbou, R. Mandal, F. Aziat, E. Dong, S. Bouatra, I. Sinelnikov, D. Arndt, J. Xia, P. Liu, F. Yallou, T. Bjorndahl, R. Perez-Pineiro, R. Eisner, F. Allen, V. Neveu, R. Greiner and A. Scalbert, Nucleic Acids Res., 2013, 41, D801–D807 CrossRef CAS PubMed.
- C. A. Smith, G. O'Maille, E. J. Want, C. Qin, S. A. Trauger, T. R. Brandon, D. E. Custodio, R. Abagyan and G. Siuzdak, Ther. Drug Monit., 2005, 27, 747–751 CrossRef CAS PubMed.
- L. H. Beck, Jr., R. G. Bonegio, G. Lambeau, D. M. Beck, D. W. Powell, T. D. Cummins, J. B. Klein and D. J. Salant, N. Engl. J. Med., 2009, 361, 11–21 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8mo00190a |
| ‡ Present address: Phenome Centre, Singapore, Nanyang Technological University, Lee Kong Chian School of Medicine, Experimental Medicine Building, 59 Nanyang Drive, Singapore 636921. |
| This journal is © The Royal Society of Chemistry 2019 |