
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Designing conjugated porous polymers for visible light-driven photocatalytic chemical transformations
Jeehye
Byun
bc and
Kai A. I.
Zhang
 *ab
*ab
aDepartmemt of Materials Science, Fudan University, Shanghai 200433, P. R. China. E-mail: kai_zhang@fudan.edu.cn
bMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: kai.zhang@mpip-mainz.mpg.de
cWater Cycle Research Center, Korea Institute of Science and Technology (KIST), Hwarangno 14-gil 5, Seongbuk-gu, 02792 Seoul, Republic of Korea
First published on 11th September 2019
Abstract
Conjugated porous polymers (CPPs) have recently emerged as a new class of visible light-active, organic and heterogeneous photocatalysts for visible light-mediated photoredox reactions. The CPPs have been established as a potential alternative to resolve critical drawbacks of traditional molecular and homogeneous photocatalysts due to their structural durability, non-toxicity, low cost due to the absence of noble metals, and high designability. Tremendous attempts have been made toward the design and synthesis of CPPs for a variety of visible light-promoted photocatalytic chemical transformations. Nevertheless, the concomitant design protocols of CPPs have not been well structured so far. Herein, in this review, we aim to summarize the recent developments in controlling the structural, photophysical and electronic properties of CPPs, and thereby extract the underlying design principles. According to the principle of the photocatalytic process, key parameters for the molecular design of CPPs were described in three sections: (1) light absorbance by energy band gap, (2) charge separation and transport, and (3) electron transfer to the target substrate. The macroscopic features, i.e. morphology, porosity and chemical functionality, and processibility of CPPs were also presented for the enhancement of their photocatalytic activity.
 Jeehye Byun | Jeehye Byun attained her PhD from Korea Advanced Institute of Science and Technology (KAIST) in 2016, and worked as a postdoctoral fellow in the group of Dr Kai Zhang at Max Planck Institute for Polymer Research supported by the Alexander von Humboldt Foundation. She is currently a senior researcher in Korea Institute of Science and Technology (KIST). Her research interests are designing porous materials for environmental applications, i.e. CO2 conversion and water treatment. |
 Kai A. I. Zhang | Kai Zhang did his PhD at the University of Cologne, Germany and partly at the University of Strathclyde, Glasgow, UK. After working as a postdoctoral researcher with Prof. Markus Antonietti at the Max Planck Institute of Colloids and Interfaces, Potsdam, Germany, he joined Max Planck Institute for Polymer Research in 2013 as a research group leader in the department of Physical Chemistry of Polymers under Prof. Katharina Landfester. In 2019, he joined as full professor the Department of Materials Science, Fudan University, Shanghai, China. His research interests include the development of organic porous materials, macromolecular photocatalysts and their applications in organic photoredox reactions, fine chemistry, photobiocatalysis etc. |
1. Introduction
Sunlight is an enormous power reservoir which daily supplies our planet with an immense amount of energy.1 For using solar energy as the most available renewable energy source, nature has shown its ability to utilize sunlight to drive chemical processes, i.e. photosynthesis. Inspired by nature processes, scientists have made remarkable efforts toward the development of efficient photocatalysts to facilitate chemical reactions with assistance from solar energy.2,3 Among the intensely investigated systems, homogeneous molecular photocatalysts have led the earlier discoveries of photocatalysis, where the established examples are transition metal complexes4–6 and organic dyes.7 Despite the performance, several drawbacks are still associated with homogeneous photocatalysts, to name but a few, toxicity of metals, high cost, and stability issues due to the photo-bleaching effect. Furthermore, the accompanying purification step of the homogeneous system is also a nettlesome obstacle that hinders their large-scale field applications.Metal-free and heterogeneous photocatalysts have thus drawn attention in recent years for resolving the disadvantages of homogeneous photocatalytic systems. Carbon nitrides, a state-of-the-art example of metal-free photocatalysts, have been deeply studied for visible light-driven photoredox reactions such as water splitting,8–13 CO2 reduction,14–16 bioimaging,17,18 humidity sensing,19 and homogeneous catalysis.20 π-Conjugated polymers are another emerging class as an alternative to the traditional homogeneous photocatalysts as they have versatile photophysical and electronic properties that can be controlled by the diverse combination of building blocks. Moreover, by virtue of structural designability, the conjugated polymers can be further endowed with morphological traits and specific chemical functionalities, facilitating a wider range of photocatalytic reactions with enhanced efficiency. Conjugated porous polymers (CPPs), in this regard, which combine promising features with controllable porosity and surficial properties, have arisen as an ideal candidate for visible-light-driven photocatalysis.21 In 2013, CPPs were first demonstrated for visible light-induced chemical transformations, in which a series of poly-benzothiadiazoles could catalyze singlet oxygen (1O2) generation under blue light illumination to convert α-terpinene into ascaridole.22 Since then, a considerable number of photoredox reactions have been conducted with CPPs as heterogeneous photocatalysts. Recent examples include selective oxidation of sulfides,23 free radical polymerization,24 dehalogenation of haloketones,25 oxidative coupling of amines,26 hydrogen27–31 and oxygen evolution,32,33 reduction of 4-nitrophenol,34 metal-free Stille-type C–C coupling,35 [2+2] cycloaddition,36 and enantioselective α-alkylation37etc.
A variety of synthetic protocols are well established to prepare CPPs,38 and the typical chemical routes are as follows; (i) metal-assisted cross coupling reactions (e.g. Suzuki-,24 Sonogashira-,22,39 Heck-,40 Yamamoto-41, Glaser-,42 Negishi-,43 Kumada coupling reaction,44 and oxidative polymerization45,46), (ii) acid-34,47–49 and base-catalyzed polymerization,27,50 and (iii) thermo-driven polymerization.9,51,52 Such a diversity of chemical reactions with various building blocks produces a number of CPPs for visible light-promoted photocatalytic applications.53,54 The π-conjugated skeleton and the corresponding properties are to be controlled by a suitable type of reaction and chemical composition, depending on the targeted applications.
Despite the performance of CPPs shown in recent studies, the associated design principles of CPPs for photocatalysis have not been well defined, raising questions such as (i) how the molecular composition and geometry affect the photocatalytic activity of the CPPs, (ii) how the functionality and morphology of the CPPs can improve the catalytic efficiency, and (iii) how the CPPs can be utilized in a large-scale system. There is therefore a demand to reveal the structure–property relationship of the CPPs for photocatalytic application. Here, carbon nitrides, which belong to their own class with a number of comprehensive reviews,55–58 will not be much covered. This review is to summarize the latest investigations upon how the molecular structures and physicochemical properties of CPPs correlate with their photocatalytic behavior. In addition, a design strategy to scale up and process CPPs for potential field applications is also discussed. We believe that this review can provide a general design concept to fine-tune the structure and property of CPPs for targeted photocatalytic chemical transformations under visible light.
2. Design of CPPs for photocatalysis
The photocatalytic performance of CPPs is originated from strong light-harvesting ability arising from the extended conjugation and the corresponding energy band gap. A few early examples of organic polymer photocatalysts were linear chains with delocalized π-systems,59,60 followed by recent studies of linear polymers with considerable photocatalytic performance.61–65 By integrating morphological and surficial properties in three dimensional structures, additional strengths such as high stability, reusability and selectivity could be generated as add-on advantages, resulting into recent research activities on CPPs. In the following sections, our main focus will be firstly on the molecular design of CPPs according to the principle of photocatalysis with semiconductive materials. Then, macroscopic controls in morphology, porosity, and functionality of CPPs, and large-scale process design of CPPs will be presented in turn.2.1. Structure design of CPPs on a molecular level
In principle, the main polymeric backbone of CPPs is sp2 hybridized and the pz orbitals are oriented perpendicular to the polymer chain, leading to π-delocalization and the concomitant energy band gap (Eg) by a HOMO (highest occupied molecular orbital)–LUMO (lowest unoccupied molecular orbital) gap.66 The molecular energy level of CPPs can be empirically determined by cyclic voltammetry or by ultraviolet photoelectron spectroscopy in vacuo.67 The use of computational calculations by density functional theory (DFT) has become useful to predict the energy level of CPPs.68 The first step of heterogeneous photocatalysis by CPPs is light absorbance and the generation of charge carriers within the structure. Upon light irradiation, the semiconductive CPP absorbs photons with energy greater or equal to its band gap energy, generating electron–hole pairs. The fate of the photogenerated electron and hole could be either (volume/surface) recombination or migration to the surface for electron/energy transfer to substrates. At the surface of CPPs, the migrated electron can be donated to an electron acceptor as a reduction process, and the hole can be combined with an electron from an electron donor as an oxidation process. Therefore, key parameters of CPPs for efficient photocatalysis can be considered as the following three steps (Fig. 1a); (1) light absorption in the visible light range with optimal energy band gap, (2) efficient dissociation of photogenerated charge carriers and their transport in CPPs, and (3) electron or energy transfer from the CPP to the target substrates associated between the band positions of the CPP and the redox potentials of the substrates. Fig. 1b summarizes the molecular design strategy of CPPs classified into three parts based on the photocatalytic reaction processes, which are described in the following sections.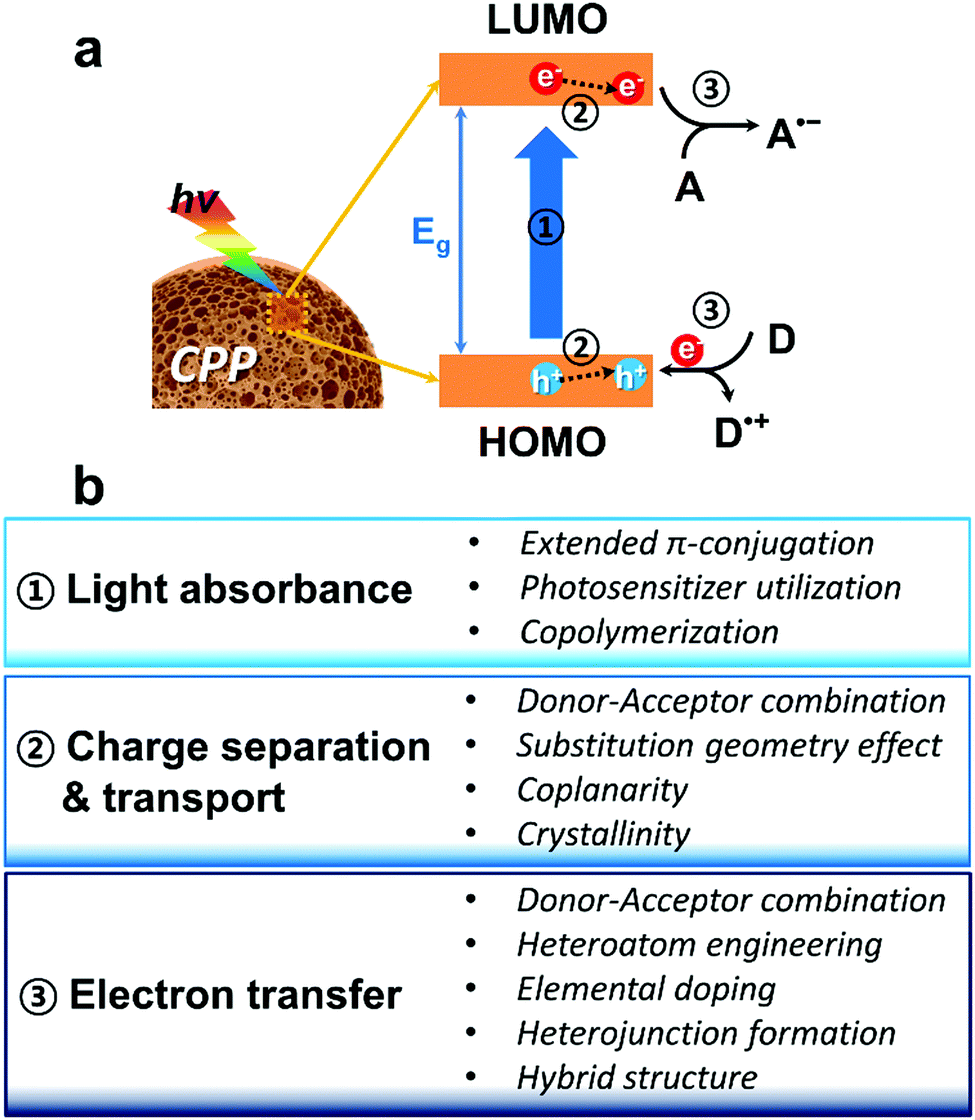 | ||
| Fig. 1 (a) Photocatalytic reaction process using CPPs as photocatalysts; (1) light absorbance by the energy band gap, (2) charge separation and transport inside the CPP, and (3) electron transfer between the CPP and substrates. HOMO and LUMO indicate the highest occupied molecular orbital and lowest unoccupied molecular orbital of CPP, respectively. D and A represent electron donor and electron acceptor, respectively. (b) Molecular design strategy of CPPs based on the photocatalytic reaction processes. | ||
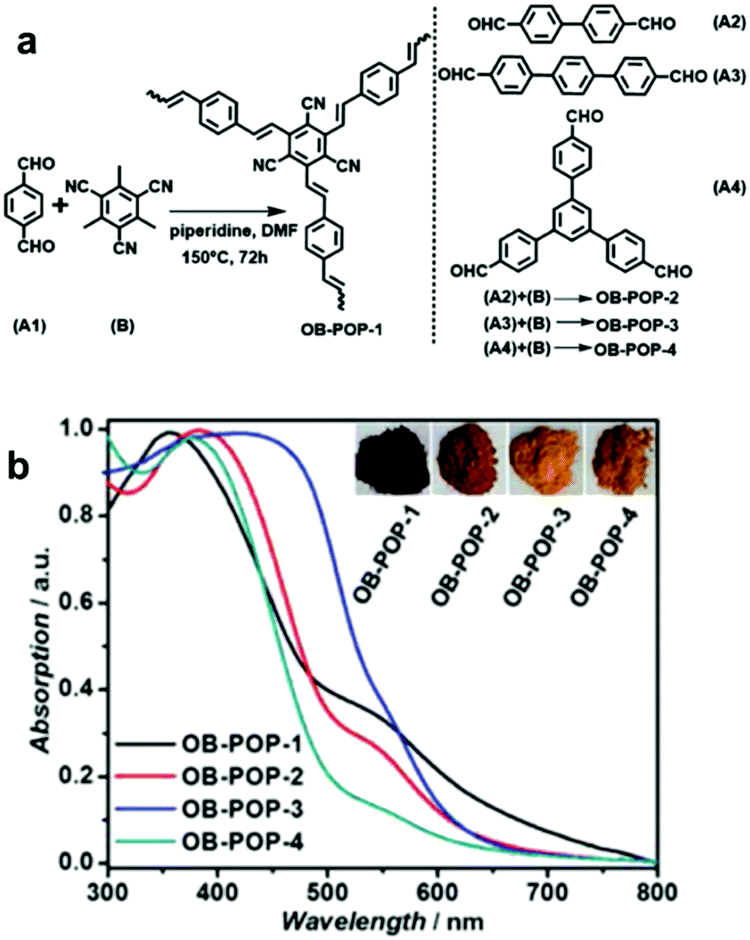 | ||
| Fig. 2 Enhanced light absorption using longer phenylene linkers in CPPs. (a) Synthetic process of olefin-bridged porous organic polymers (OB-POPs) and (b) their UV/vis diffuse reflectance spectra with photographs of the resulting polymers (inset). Reproduced with permission.27 Copyright 2017 Wiley-VCH. | ||
Incorporation of photosensitizer molecules within CPPs could be a simple approach to increase the number of conjugation units for light harvesting with the lower energy band gap. Bottom-up approaches of making polymeric structures with known photosensitizing units were recently reported. Dye molecules such as Rose Bengal,72 BODIPY73–76 and Eosin Y,77 metal complexes,78,79 metalloporphyrins,80,81 and metallophthalocyanine82 were merged into the CPP skeleton to generate porous materials with photophysical attributes. Cooper et al. integrated a Rose Bengal dye into conjugated microporous polymers via Pd-catalyzed Sonogashira–Hagihara polycondensation, showing a broad light absorption in the range from 350 nm to 700 nm (Fig. 3a).72 The obtained polymer RB-CMP1 showed high porosity (>830 m2 g−1) and reactivity for heterogeneous photocatalytic aza-Henry reaction. Similarly, a BODIPY chromophore was employed to generate polymer photocatalyst CMPBDP by Liras et al.,74 showing an intense light absorption at around 550 nm. The utilization of BODIPY into the polymer further enabled complexation with ruthenium, forming a hybrid structure for the improved catalytic efficiency.75 The incorporation of metallophthalocyanine photosensitizer in CPPs allowed light absorption in the long-wavelength visible to far-red regions (Fig. 3b).82 The attained polymer MPc-CMP could efficiently generate singlet oxygen (1O2) at 700 nm (M = Co, Ni, Cu, Zn), where the zinc- and copper-coordinated structures showed the highest efficiency among the four MPc-CMPs. Meanwhile, the binding motif of monomers and their stoichiometry determine the amount of photosensitizers loaded on CPPs. Lin et al. showed that the homocoupling of [Ru(bpy)3]2+ maximized the loading of chromophores into the polymer up to ∼90%,79 comparable to the cross-coupled non-conjugated structures with 2.2% of Ru photosensitizer (Fig. 3c).83 The higher loading of the photoactive unit resulted in enhanced visible light absorption and electron transfer. The Ru(bpy)32+-homocoupled polymer was almost nonporous with small surface area, however, it showed superior photocatalytic efficiency for organic transformations, i.e. aza-Henry reaction, oxidative coupling of amines, and reductive dehalogenation reaction, owing to efficient excited state migration through Ru chromophores.
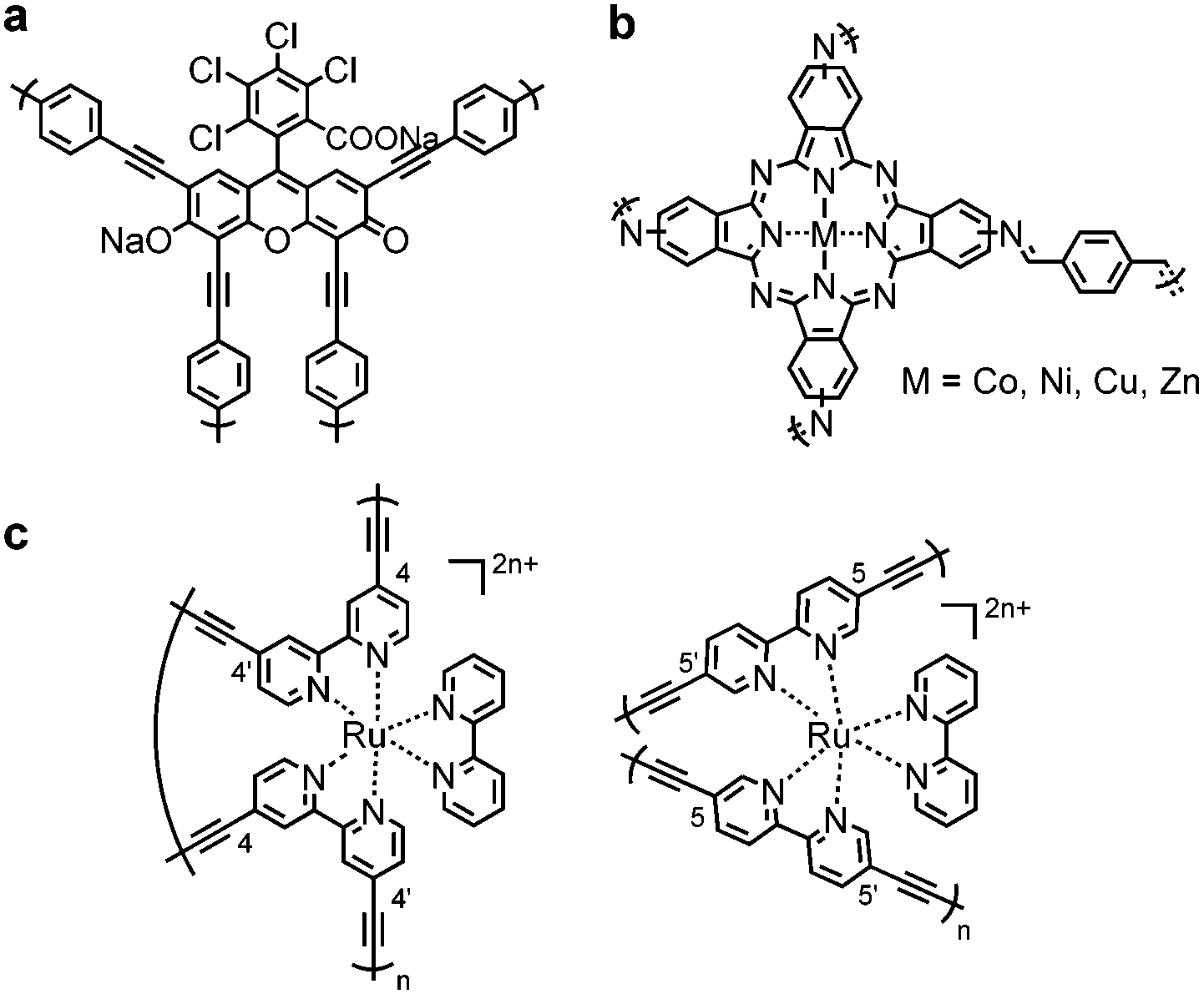 | ||
| Fig. 3 Incorporation of photosensitizers in CPPs for light harvesting. (a) Conjugated microporous polymer with Rose Bengal Dye (RB-CMP1) and (b) metallophthalocyanine (MPc-CMP, M = Co, Ni, Cu, Zn). (c) Ru(bpy)32+-homocoupled polymers containing high Ru(bpy)32+ loading up to 90%. | ||
Copolymerization of two or more monomers having different energy levels is the dominating synthetic route to control the photophysical properties of CPPs. The energy band gap of CPPs can be adjusted by the molecular hybridization between the monomers for intramolecular π electron interaction, resulting in the reduced band gap of CPPs. A representative example of the copolymerized CPPs for the band gap control was made by Cooper et al.28 A series of pyrene-based conjugated microporous polymers (CP-CMPn, n = 1 to 15) was prepared by statistical copolymerization between benzene and pyrene. The tunable band gaps by increasing pyrene composition were found to be altered from 2.95 to 1.94 eV along with the photoluminescent peak from 445 to 588 nm, respectively (Fig. 4). Time-dependent density functional theory (TD-DFT) revealed that the narrower band position of the pyrene-containing system than that of the benzene-containing system contributed to the optical gap shifts of pyrene-dominant structures to higher wavelengths. When applied for photocatalytic H2 evolution, CP-CMP10 with the optical band gap of 2.33 eV displayed the highest amount of H2 produced among the structures, clearly showing that the electronic structure and optical property should be well aligned for the enhanced photocatalytic activity.
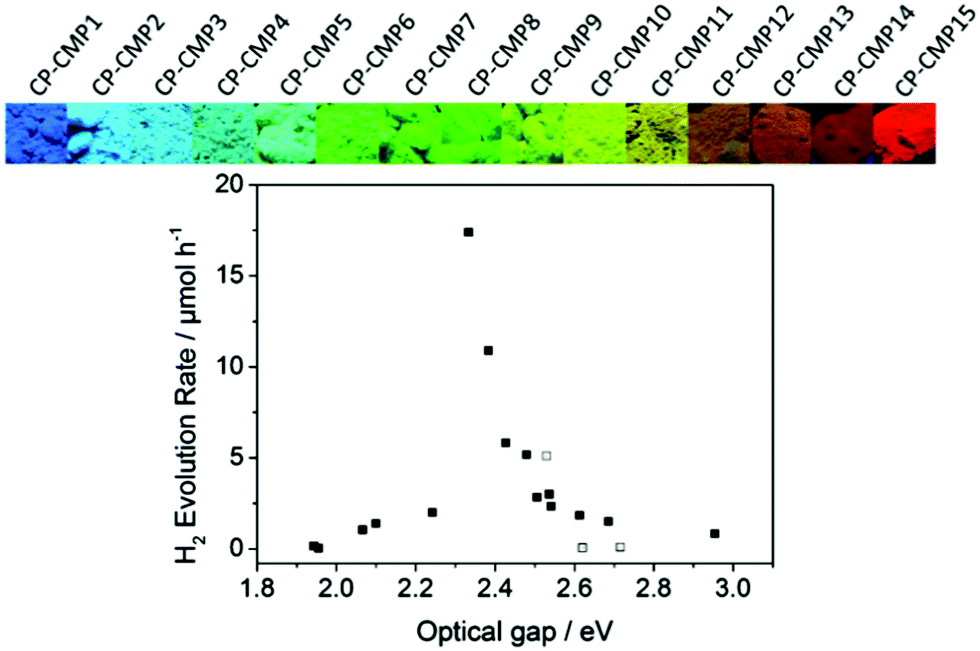 | ||
| Fig. 4 Statistical copolymerization of comonomers for band gap engineering of CPP. Pyrene-based conjugated microporous polymers and their photocatalytic H2 production efficiencies. Reproduced with permission.28 Copyright 2015 American Chemical Society. | ||
Li et al. reported on the design of molecular donor–acceptor heterostructures with covalent triazine frameworks (Fig. 5).86 Within the polymer network, benzothiadiazole and thiophene moieties were selectively incorporated as electron-withdrawing and electron-donating units by sequential copolymerization, respectively. The authors showed enhanced charge-carrier separation within the heterostructures, and therefore the photocatalytic H2 production could be efficiently facilitated, showing 4–6 times higher H2 evolution rate (6.6 mmol g−1 h−1) than the single component structures. Likewise, Zhang et al. showed the copolymerization of benzene with thiophene as a strong donor and benzothiadiazole as a strong acceptor to demonstrate that the D–A compositions in CPPs could improve the charge separation and mobility.87 In particular, the structure made out of benzene and benzothiadiazole (BBT) showed the most intense signal, indicating that the photogenerated charge separation was efficient when with a weak donor and strong acceptor combination. The BBT structure thereby gave the highest photocatalytic efficiency for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond activation of styrene to generate benzaldehyde as the main product. Similarly, three CPPs were synthesized with a triphenyltriazine core and three linkers of thiophene, benzene, and benzothiadiazole for photocatalytic reduction of CO2.88 Electrochemical impedance and photocurrent measurements confirmed that the CPP with the benzothiadiazole linker (CPs-BT) exhibited the minimum Nyquist radius and an improved photocurrent response, indicating higher charge separation and mobility. The CPs-BT thus gave a stable photoconversion of CO2 to CO with high selectivity, leading to a high quantum yield of 1.75% at 405 nm. Bojdys et al. recently showed a library of sulfur and nitrogen containing porous polymers, where the thiophene-based monomers played a role as an electron donor and the triazine-containing units as an electron acceptor.89 The authors demonstrated the optimal band gaps of the polymers for proton reduction and the oxidation of the sacrificial donor lay at 2.1–2.3 eV formed by the D–A interaction. More importantly, when the donor and the acceptor were spaced with a phenyl linker, the push–pull effect by D–A units was rather weakened to yield less charge mobility within the polymers. The polymer consisting of benzotrithiophene and triazine exhibited the superior H2 evolution rate of 3158 μmol h−1 g−1 with 4.5% of apparent quantum efficiency at 420 nm due to the D–A induced charge transfer.
C bond activation of styrene to generate benzaldehyde as the main product. Similarly, three CPPs were synthesized with a triphenyltriazine core and three linkers of thiophene, benzene, and benzothiadiazole for photocatalytic reduction of CO2.88 Electrochemical impedance and photocurrent measurements confirmed that the CPP with the benzothiadiazole linker (CPs-BT) exhibited the minimum Nyquist radius and an improved photocurrent response, indicating higher charge separation and mobility. The CPs-BT thus gave a stable photoconversion of CO2 to CO with high selectivity, leading to a high quantum yield of 1.75% at 405 nm. Bojdys et al. recently showed a library of sulfur and nitrogen containing porous polymers, where the thiophene-based monomers played a role as an electron donor and the triazine-containing units as an electron acceptor.89 The authors demonstrated the optimal band gaps of the polymers for proton reduction and the oxidation of the sacrificial donor lay at 2.1–2.3 eV formed by the D–A interaction. More importantly, when the donor and the acceptor were spaced with a phenyl linker, the push–pull effect by D–A units was rather weakened to yield less charge mobility within the polymers. The polymer consisting of benzotrithiophene and triazine exhibited the superior H2 evolution rate of 3158 μmol h−1 g−1 with 4.5% of apparent quantum efficiency at 420 nm due to the D–A induced charge transfer.
 | ||
| Fig. 5 Enhanced charge separation within a covalent triazine framework by creating an intramolecular heterostructure via sequential copolymerization strategy. Reproduced with permission.86 Copyright 2018 Wiley-VCH. | ||
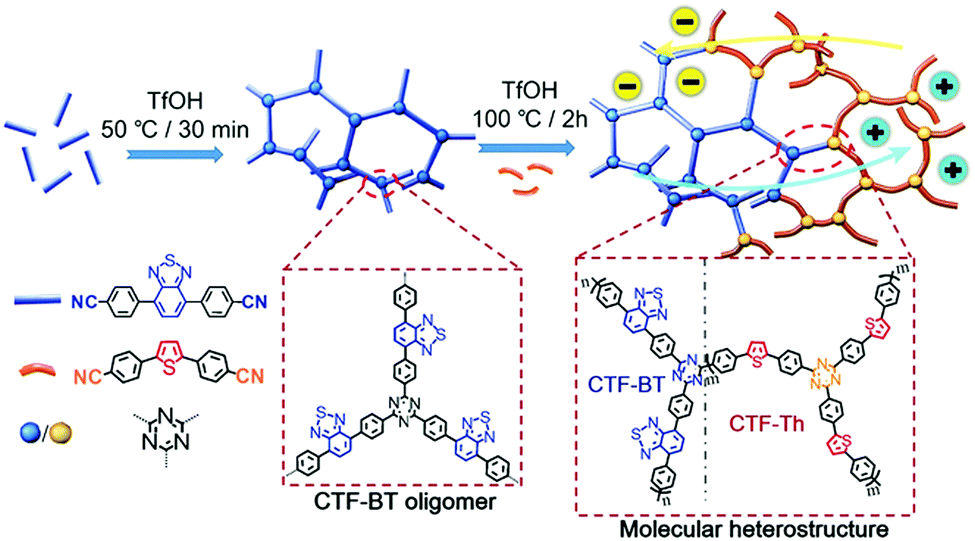
Multiple domain D–A compositions in a single CPP led to enhanced photoinduced charge separation via an intramolecular energy transfer. Zhang et al. reported an asymmetric covalent triazine framework (asy-CTF) consisting of four different molecular D–A domains in comparison to symmetric ones (Fig. 6a and b).90 Each domain showed different electron densities on the HOMO/LUMO levels (Fig. 6c), allowing the energy transfer cascade. Time-resolved photoluminescence (TRPL) spectra revealed that asy-CTF (1.27 ns) showed shorter fluorescence lifetime compared to the symmetric CTF-Th (1.66 ns). When in the selected time trace as shown in Fig. 6d and e, asy-CTF showed much higher and broader energy emission peak than CTF-Th, suggesting multiple overlapping emissive bands which red-shift to lower energy by successive excitation energy transfer. Compared to the symmetric CTFs, asy-CTF exhibited a suppressed energy band gap of 2.3 eV and a higher photocurrent, indicating the improved light absorbance and charge mobility, respectively. Owing to the upshift of HOMO/LUMO level, asy-CTF could have a higher reductive overpotential toward an oxidant molecule (Ered = −0.78 V vs. SCE) than the symmetric CTFs, leading to the highest photocatalytic efficiency for synthesizing benzophosphole oxide from secondary phosphine oxide and alkyne in the presence of N-ethoxy-2-methylpyridinium tetrafluoroborate as the oxidant.
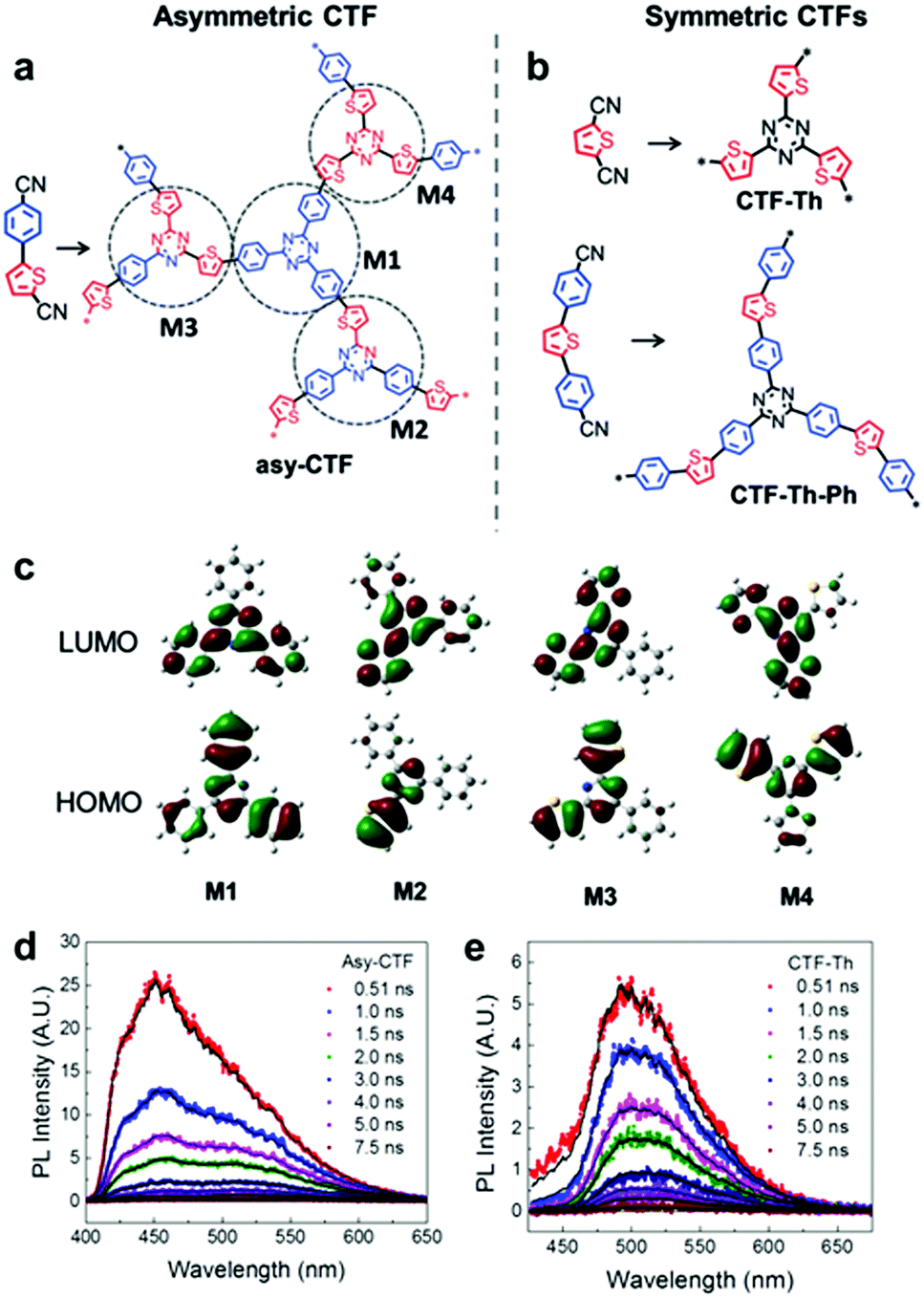 | ||
| Fig. 6 Multidomain D–A compositions in CPP for energy transfer cascade. (a) Representative structure of an asymmetric covalent triazine framework (asy-CTF) containing four different molecular D–A domains (M1–M4), (b) its symmetric counterparts CTF-Th and CTF-Th-Ph with single D–A domains, and (c) the calculated electron densities over HOMO/LUMO levels of the four D–A domains in asy-CTF. TRPL spectra of (d) asy-CTF and (e) CTF-Th at an excitation wavelength of 400 nm measured in the selected time trace of 0.51–7.5 ns. Adapted with permission.90 Copyright 2018 Wiley-VCH. | ||
The photogenerated electrons and holes migrate through the π-conjugated CPP chains and take part in photocatalytic reactions, and thus the longer lifetime of electron–hole pairs by the efficient charge transport can improve the photocatalytic activity of CPPs. Zhang et al. suggested the geometry design principle of benzoxadiazole-based CPPs by merely changing the substitution positions on the central phenyl group with divalent benzoxadiazole linker (Fig. 7).26 When the benzoxadiazole linker was connected to the 1,3,5-position on the phenyl unit, the CPP showed a large increase in the EPR (electron paramagnetic resonance) spectrum under light, indicating the longer living of charge carriers. On the other hand, the CPPs with 1,2,4- and 1,2,4,5-substitutions led to a marginal response in the EPR spectra, likely due to the lower charge mobility. The difference in structural configurations also resulted in the band position change in CPPs, and the CPP with 1,3,5-substitution gave the highest photocatalytic activity in the oxidative coupling of amines with the proper band alignment and the longer living electron–hole pair. The authors further showed a series of poly(benzothiadiazole)s according to the variation of substitution sites on the phenyl core with benzothiadiazole linker.31 The polymer with 1,4-substitution (B-BT-1,4) exhibited the narrowest band gap of 2.17 eV and enhanced photocurrent owing to the extended π-conjugation. The linear type B-BT-1,4 yielded a higher photocatalytic H2 production rate compared to the three-dimensional structures with 1,3,5-, 1,2,4-, and 1,2,4,5-substitution of the benzothiadiazole unit on the phenyl core, supporting the good charge transfer ability of B-BT-1,4. In the same vein, Zhang et al. mentioned that the meso-positions of the phenyl core were unfavorable for π-delocalization compared to the para-positions.27
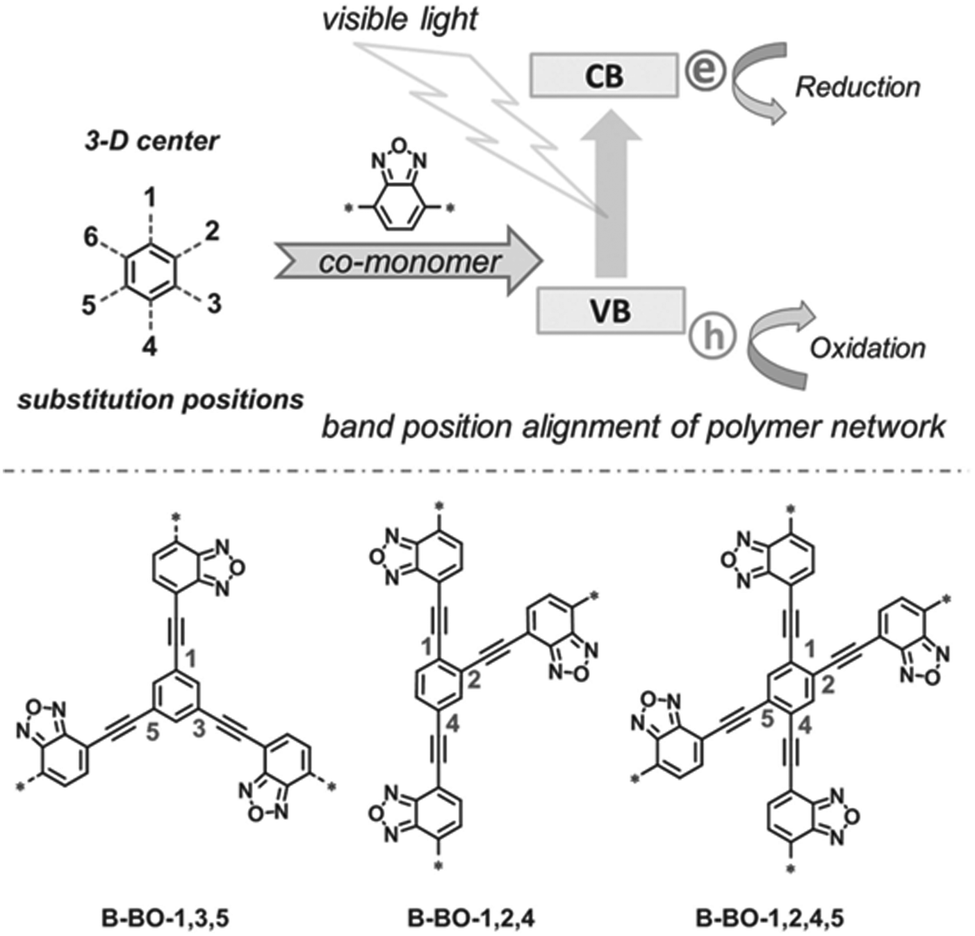 | ||
| Fig. 7 Substitution geometry effect for charge transport in CPPs. Structural design of poly(benzoxadiazole)s by changing the substitution position on the central phenyl ring with benzoxadiazole linkers. Adapted with permission.26 Copyright 2015 Wiley-VCH. | ||
The effect of structure conformation on charge transport was further investigated by using a variety of comonomers on CPPs. Müllen et al. employed different sizes of phenylene derivatives as building blocks in poly(azomethine) networks.47 As the size of the monomer increased (i.e. from benzene to anthracene), the optical band gap of the resulting polymers became narrower due to the extended π-conjugation as shown in Section 2.1.1. When the anthracene monomer was connected by the 9,10-position, however, the network linkage created a high torsion angle (∼30°), interrupting the transport of charge carriers and thereby lowering the photocatalytic activity for H2 production. With the connection to the 2,6-position on anthracene, the polymer network showed a planar structure with the torsion angle less than 2°, and the H2 evolution rate was far increased, demonstrating a clear deviation of planarity in the connecting struts to affect the photoactivity of CPPs. Similarly, Cai et al. synthesized CPPs with perylene diimide and bipyridyl linker for photocatalytic H2 evolution and dye degradation, where the coplanar structure exhibited better charge transport.91 A series of porous conjugated polymers was designed by copolymerizing electron-rich chromophores with bipyridyl comonomers.92 The connection between the bipyridyl unit and DBD (4,8-di(thiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene) as a strong electron-donor provided a coplanar structure of the CPP, leading to a favorable charge transport for photocatalytic H2 evolution. A clear example for the effect of a planar structure in CPP-based photocatalysis has been unveiled by Lotsch et al.49 Two-dimensional azine-based covalent organic frameworks (Nx-COFs) were produced using a series of triphenylarylaldehydes to vary the number of nitrogen atoms (x = 0 to 3) on the central aryl ring (Fig. 8). The increase of nitrogen content on the COF directly corresponded to the higher H2 production rate. The more nitrogen content on the central aryl ring resulted in less dihedral angle to improve crystallinity and stack two-dimensional layers of COF, leading to the enhanced charge mobility and light harvesting.
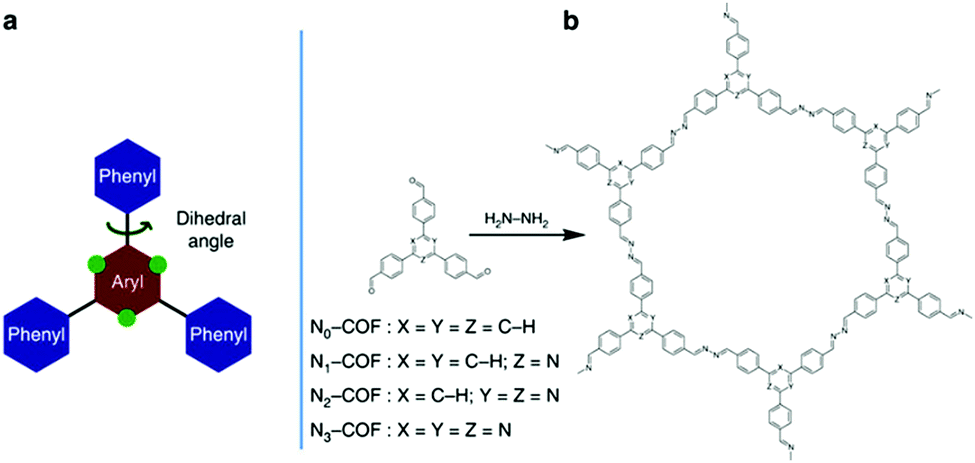 | ||
| Fig. 8 Planarity control in CPPs for charge mobility. (a) Schematic illustration of the triphenylarene core and (b) synthetic procedure for a tunable azine covalent organic framework. Adapted with permission.49 Copyright 2015 Macmillan Publishers Limited. | ||
A series of two-dimensional planar CPPs was lately reported for enhanced charge transport. Xu et al. have achieved overall water splitting using a 1,3-diyne-linked CPP, generating a stoichiometric amount of H2 and O2 under visible light.33 The ultrathin layer of CPPs allowed the excitons to directly migrate to the polymer surface, which minimized charge recombination to yield the high quantum efficiency of 10% at 420 nm. Besides, layer-structured covalent triazine frameworks (CTF) were proven to show higher photocatalytic activities via the improved charge mobility and the suppressed charge recombination. Jin and Tan et al. presented the CTFs constructed by the polycondensation reaction between aldehyde and amidine monomers under mild conditions, and the resulting CTF-HUSTs showed extremely high efficiency for photocatalytic H2 evolution.30 The same authors were further able to control the crystallinity of CTFs by decreasing the nucleation rate during the polycondensation process. Instead of directly using the aldehyde monomer, the authors chose alcohol monomer to be in situ oxidized to the aldehyde monomer for slowing down the initial nucleation of CTF (Fig. 9a). By the stepwise increase of the reaction temperature, CTF-HUST exhibited good crystallinity, leading to the improved charge transport in the framework (Fig. 9b). Indeed, the crystalline CTF-HUST-C1 gave 3.5 times higher H2 production rate than the layered CTF-HUST-1, showing 5100 μmol h−1 g−1 and 1460 μmol h−1 g−1, respectively. The crystalline CTFs and their photocatalytic behavior were also demonstrated by Thomas et al.93Via a two-step synthetic process combining the acid-catalyzed trimerization and ionothermal method, CTFs could show the enhanced crystallinity even in much shorter reaction time (max. 30 min) than the ionothermal synthesis (40 h). The H2 evolution efficiency of CTFs was increased with respect to the longer time of ionothermal reaction up to 10 min, likely due to higher crystallinity and polymerization degree of CTFs. When heated longer than 10 min, the H2 production rate of CTFs started to decrease owing to the partial carbonization of the structures. Very recently, Cooper et al. revealed that sulfone-containing covalent organic frameworks with high crystallinity outperformed the semicrystalline and the amorphous conjugated polymer for photocatalytic H2 evolution.94 The crystallinity of the polymer layers facilitated the π-delocalization through π–π stacking as well as the enhanced charge carrier transport within the polymer, leading to 9 times higher H2 production rate than the amorphous counterpart.
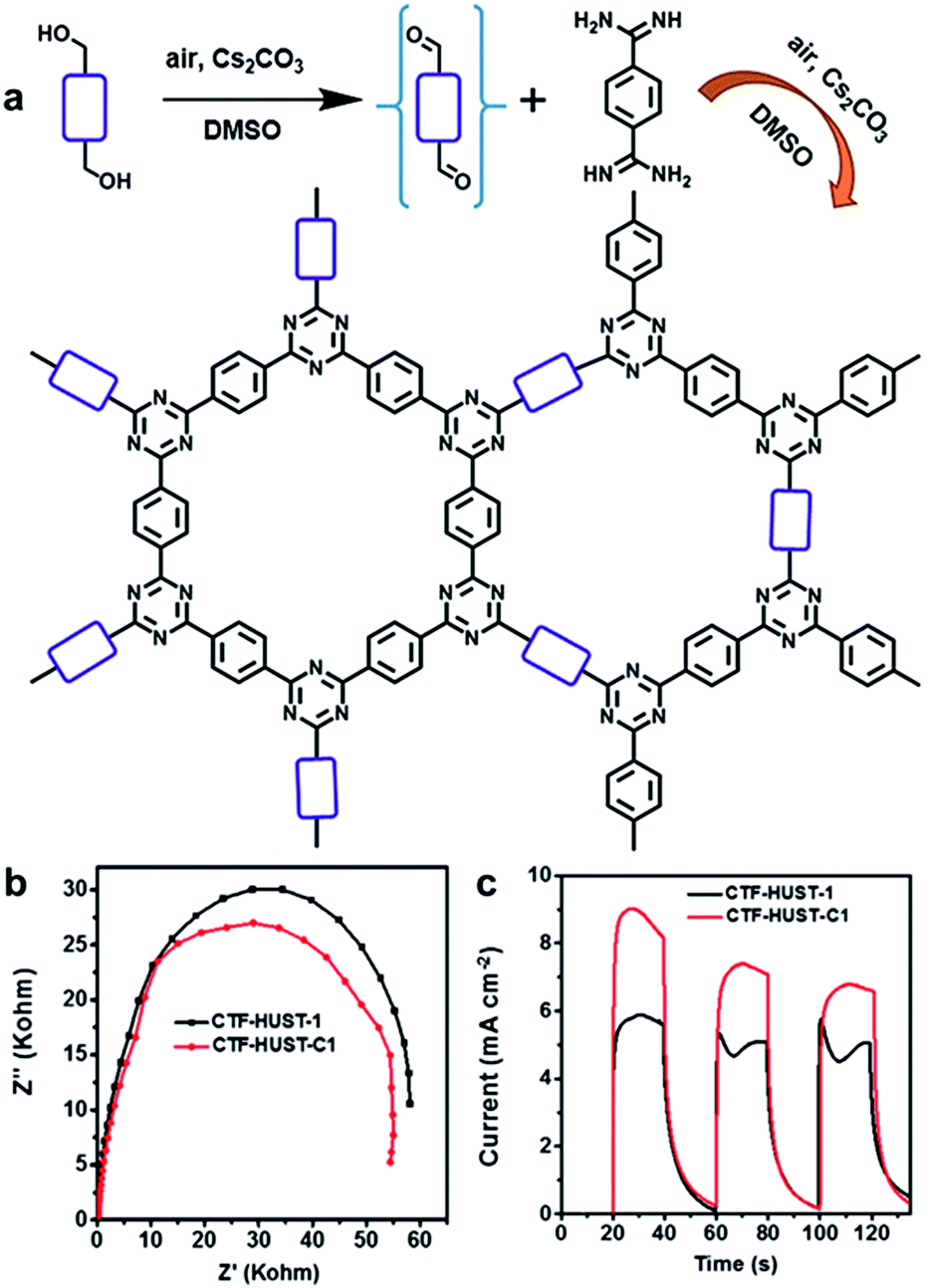 | ||
| Fig. 9 Crystallinity of CPPs for charge transport. (a) Synthetic scheme of crystalline CTF-HUST-1 through in situ formation of aldehyde monomer from alcohol monomer. (b) Electrochemical impedance spectroscopy Nyquist plots and (c) photocurrent responses of the crystalline CTF-HUST-C1 and the layered CTF-HUST-1. Reproduced with permission.95 Copyright 2018 Wiley-VCH. | ||
In a typical system of D–A based CPPs, Zhang et al. introduced benzobisthiadiazole as a strong electron acceptor, and they found that varying the amount of the acceptor was effective to tune the band structure of CPPs for the oxidative cyclization of N,N-dimethylaniline with N-phenylmaleimide under visible-light irradiation.98 As the amount of benzobisthiadiazole increased, the resulting HOMO–LUMO levels of the CPPs could be strategically aligned with narrower optical band gaps (Fig. 10). The reaction mechanism involved the oxidation of N,N-dimethylaniline (EOx = 0.384 V vs. SCE)99 and the generation of perhydroxyl radicals from superoxide (ERed = −0.86 V vs. SCE) to oxidize the cyclized intermediate. The CPP containing 10 mol% of benzobisthiadiazole (P-BBT-10) with the second highest redox potential to bracket the redox potentials of substrates therefore showed the best photocatalytic performance among the four structures. Despite the higher redox potential than P-BBT-10, the structure without benzobisthiadiazole (P-BBT-0) exhibited a large energy band gap up to 3.12 eV, yielding less conversion efficiency due to the limited visible light absorption.
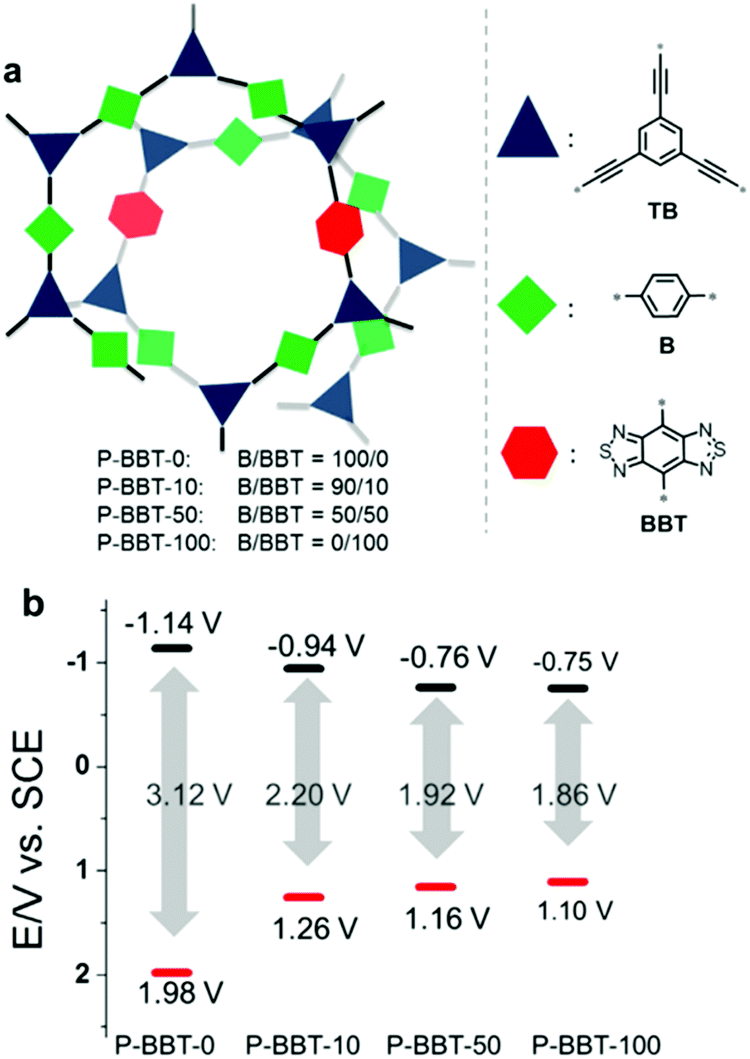 | ||
| Fig. 10 HOMO/LUMO controls in CPPs by varying the amount of electron withdrawing units. (a) Synthetic compositions of D–A based conjugated nanoporous polymers, P-BBTs and (b) their redox potentials measured by cyclic voltammetry vs. SCE (Saturated Calomel Electrode). Reproduced with permission.98 Copyright 2016 Wiley-VCH. | ||
Even a single change of heteroatom in the acceptor unit can alter the band position of CPPs, as displayed in Fig. 11a. Poly(benzochalcogenadiazole)s were constructed with different types of chalcogen atoms (i.e. O, S, and Se) on acceptor-type monomers, giving B-BO, B-BT, and B-BS, respectively.100 It has been earlier reported that the stabilizing effect of a heteroatom on the LUMO can correlate with the ionization potential (IP) of the heteroatom, in other words, a smaller IP of the heteroatom leads to larger stabilization of the LUMO level in a macromolecule.101 The resulting CPPs exhibited a stabilizing LUMO level from B-BO to B-BS, since the degree of IP is decreasing from oxygen to selenium, resulting in the lower LUMO level of CPPs. Unexpectedly, however, the HOMO level of CPPs was significantly affected by the change in heteroatoms on the acceptor unit, making B-BS with sulfur of benzothiadiazole have the strongest oxidation potential. This implies that the band positions of CPPs cannot be simply predicted by the original properties of the D–A units;102 nonetheless, the higher surface area and the stronger response in the EPR spectrum of B-BS reflected the structural differences among the three CPPs for the discrepancy in their HOMO levels. The sulfur-containing B-BT showed the strongest oxidative potential, largest band gap, and most long-lived electron–hole pair production (Fig. 11b and c), resulting in the highest conversion for oxidative coupling of benzyl amines among the three CPPs.
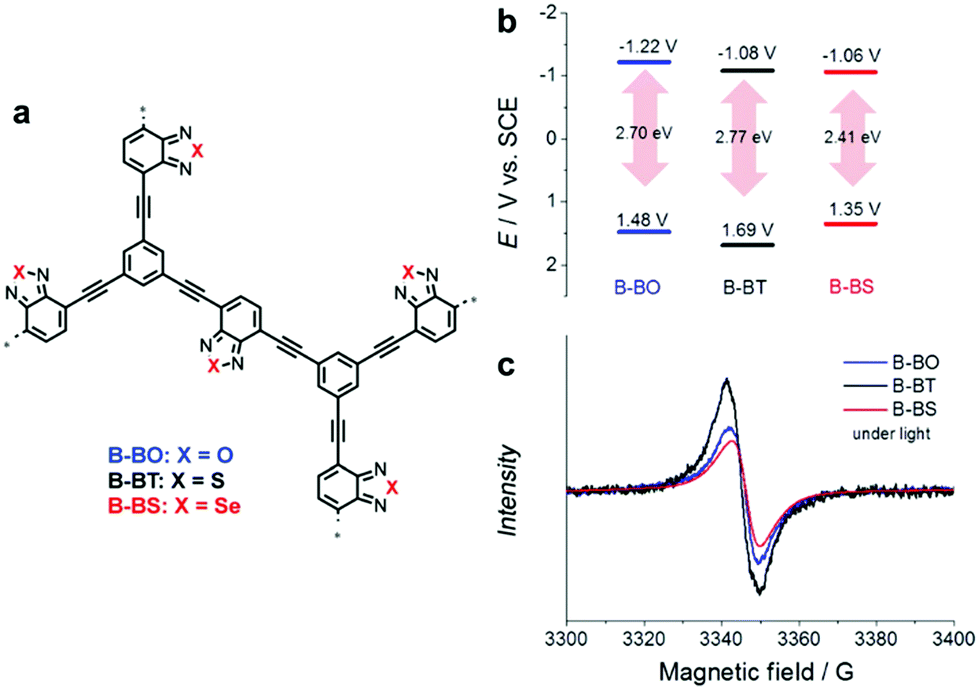 | ||
| Fig. 11 Role of heteroatoms in electron acceptor units. (a) Chemical structures of poly(benzochalcogenadiazole)s, (b) their redox potentials, and (c) EPR spectra under light irradiation. Reproduced with permission.100 Copyright 2015 Wiley-VCH. | ||
In parallel to the acceptor control, donor-type monomers have been introduced to optimize the band structure of CPPs for target applications. Typically, carbazole-based porous organic framework (Cz-POF-1) has been successfully synthesized by Zhang et al.45 The Cz-POF-1 exhibited strong electron-donating properties and extremely high surface area (∼2065 m2 g−1), accelerating photocatalytic reactions, i.e. dehalogenation of phenacyl bromides, hydroxylation of arylboronic acids, and α-alkylation of aldehydes. Similar carbazole-based conjugated microporous polymer (C-CMP) has been created by Loh et al., where the C-CMP photocatalyst was utilized for oxidative coupling of amine, aerobic dehydrogenation of heterocycles, and oxidation of sulfides.105 Recent trial of varying D–A ratio in the carbazolic copolymers (CzCP) showed a clear tendency of downshift in the HOMO/LUMO levels with respect to the increasing amount of acceptor units by the stabilizing effect, where the oxidation potential gradually increased from 1.23 V to 1.83 V vs. SCE for CzCP0 (D![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :A = 100:0) and CzCP100 (D:A = 0:100), respectively (Fig. 12a).103 With the strongest oxidation capability, the CzCP100 consisting of 100% acceptor unit exhibited the highest efficiency for the oxidation of benzylic β-O-4 alcohols. For the reductive cleavage of β-O-4 ketones, on the other hand, the CzCP33 (D:A = 66:33) gave the highest yield owing to the largest reductive capability for the reduction of lignin oxide as a target substrate. Likewise, Pan and Yu et al. demonstrated that the oxidation potentials of carbazole-based conjugated microporous polymers could be altered by incorporating different D–A units as a core (Fig. 12b).104 When consisting of all donor units with benzene as a core, the polymer exhibited the strongest oxidation potential of 1.42 V vs. SCE (CMP-SCU6), while the polymer with triazine as a core resulted in the oxidation potential of 1.19 V vs. SCE (CMP-SCU7). The stronger oxidation potential of CMP-SCU6 allowed the effective C-3 formylation and thiocyanation of indoles owing to the greater overpotential toward the target substrates.
:A = 100:0) and CzCP100 (D:A = 0:100), respectively (Fig. 12a).103 With the strongest oxidation capability, the CzCP100 consisting of 100% acceptor unit exhibited the highest efficiency for the oxidation of benzylic β-O-4 alcohols. For the reductive cleavage of β-O-4 ketones, on the other hand, the CzCP33 (D:A = 66:33) gave the highest yield owing to the largest reductive capability for the reduction of lignin oxide as a target substrate. Likewise, Pan and Yu et al. demonstrated that the oxidation potentials of carbazole-based conjugated microporous polymers could be altered by incorporating different D–A units as a core (Fig. 12b).104 When consisting of all donor units with benzene as a core, the polymer exhibited the strongest oxidation potential of 1.42 V vs. SCE (CMP-SCU6), while the polymer with triazine as a core resulted in the oxidation potential of 1.19 V vs. SCE (CMP-SCU7). The stronger oxidation potential of CMP-SCU6 allowed the effective C-3 formylation and thiocyanation of indoles owing to the greater overpotential toward the target substrates.
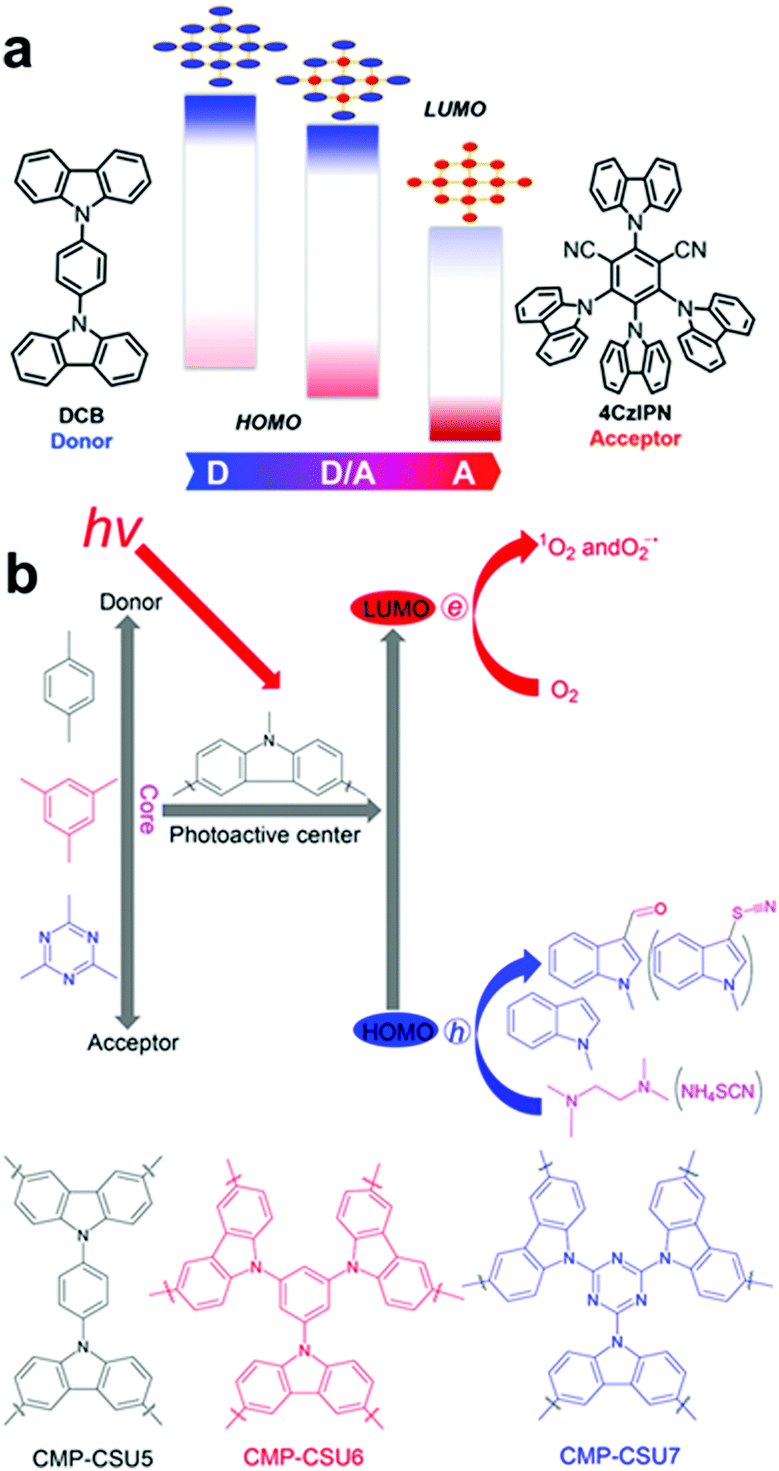 | ||
| Fig. 12 HOMO/LUMO controls in CPPs by varying the amount of electron donating units. Schematic illustration of (a) D–A based carbazolic porous organic frameworks CzCP. Adapted with permission.103 Copyright 2017 American Chemical Society. (b) Carbazole-based conjugated microporous polymers CMP-CSU. Adapted with permission.104 Copyright 2018 American Chemical Society. | ||
Elemental doping on CPPs can change the chemical composition of D–A units, thus resulting in the shift of the band positions. The doping method on CPPs has been much conducted by using elemental sulfur. Su et al. reported sulfur-doped covalent triazine-based frameworks (CTFSx, where x indicates the amount of injected sulfur, x = 0, 5, 10, 20 and 30 wt%), where the electronic structure and energy band positions of CTF were effectively modified by the loading of sulfur up to 0.52 atom% (Fig. 13).106 The doped sulfur atom on CTF replaced the nitrogen atom on the triazine ring to form a C–S bond, and led to a narrower band gap of CTF by a drastic upshift of the HOMO level (being shifted from 1.61 V to 0.55 V vs. Ag/AgCl) through the electronic coupling between the sulfur-doped triazine ring as an acceptor and the neighboring phenyl rings as a donor. The smaller band gap of sulfur-doped CTF could harvest more visible light with a clear red shift in light absorbance, and the improved photocurrent was further observed on CTFSx, giving 5 times higher H2 evolution rate than the bare CTF structure. Similarly, Wang et al. produced a sulfur-doped polyimide (SPI) through thermal copolymerization with sublimed sulfur, giving 0.38 atom% sulfur content within the structure.107 The nitrogen atom in polyimide was replaced with the doped sulfur to form a S–C bond, resulting in improved light absorbance. In contrast to the sulfur-doped CTF, however, the HOMO of SPI was lowered by 0.7 eV compared to the bare PI. The authors explained that PI consisted of pyromellitic dianhydride as an acceptor and triazine as a donor, and thus the sulfur doping on triazine promoted the stronger electron pull–push effect to lower the HOMO level of the SPI. The SPI thereby showed a faster degradation kinetic of organic dyes due to the stronger oxidation potential.
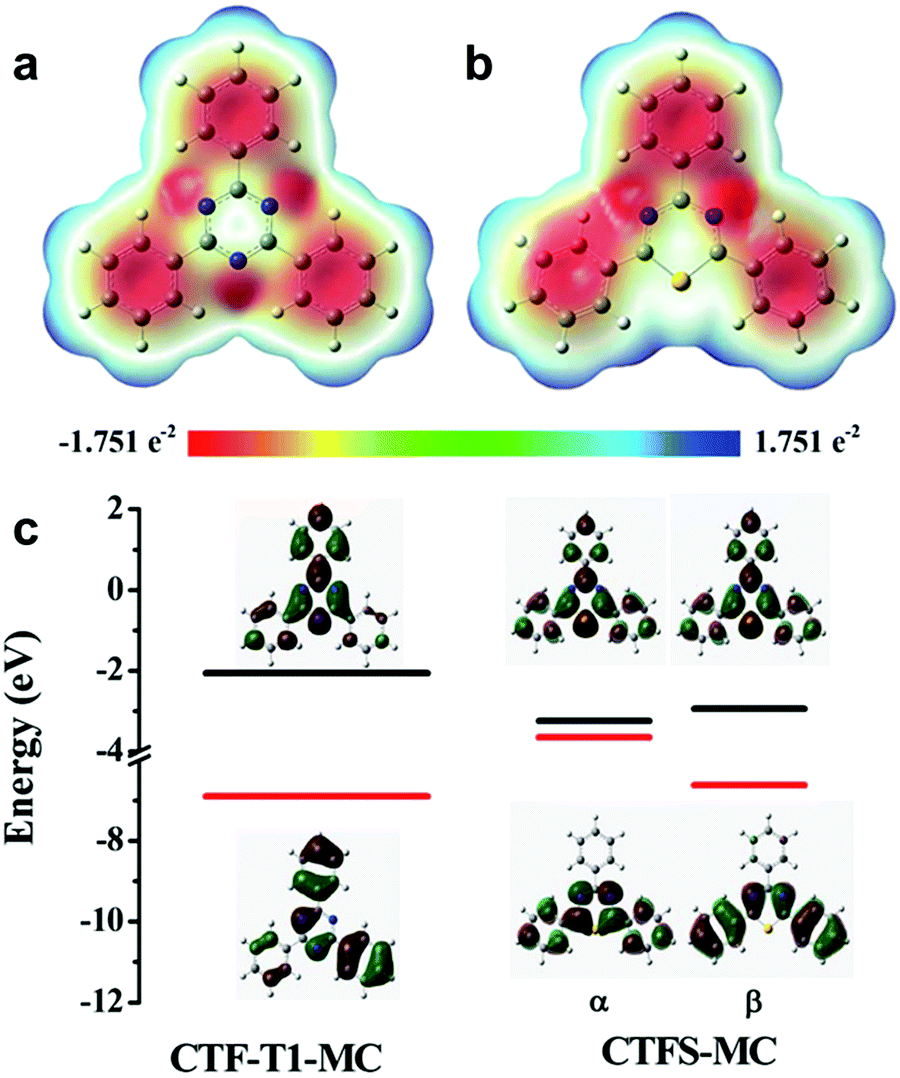 | ||
| Fig. 13 Elemental doping on CPPs for controlling the energy band structure. Charge distributions of cluster models from structures of (a) covalent triazine framework (CTF-T1-MC) and (b) sulfur-doped CTF (CTFS-MC). (c) The calculated HOMO/LUMO orbitals of the cluster models. Adapted with permission.106 Copyright 2016 The Royal Society of Chemistry. | ||
Utilization of two (or more) semiconductors as a composite is a way to adjust the band positions of photocatalysts by the charge transfer between the photocatalysts. The formation of a heterojunction in the hybrid structure facilitates the efficient dissociation of photo-generated charge carriers at the interface of the photocatalyst, leading to an increase in the number of holes and electrons in the photocatalytic system and the decrease in charge recombination rate, as described in Section 2.1.2. As a result, one photocatalyst could have much more photogenerated electrons collected on the LUMO level for enhanced reduction reaction, while the other photocatalyst with more holes on the HOMO level can play a role in an oxidation reaction. Huang et al. prepared an organic/organic hybrid photocatalyst with carbon nitride (C3N4) and linear-type poly(3-hexylthiophene) (P3HT) for H2 evolution.108 The photoluminescence (PL) quenching experiment of the composites revealed the electron transfer between two photocatalysts, showing a drastic decrease in PL intensity of g-C3N4 peak with the increasing amount of P3HT in the composite. The evolution of new PL peaks further confirmed the recombination of the photogenerated electron in P3HT with the hole in g-C3N4 and vice versa, indicating the charge transfer between two organic photocatalysts. By incorporating 3 wt% of P3HT on C3N4, the H2 production rate increased up to 300 times compared to bare g-C3N4, giving 2.9% of quantum yield at 420 nm. In the identical regime of P3HT/C3N4, Peng et al. demonstrated that the PL lifetime of the P3HT was significantly prolonged by adding C3N4 to the P3HT solution, demonstrating the electron transfer from P3HT to C3N4.109 With ascorbic acid as a sacrificial agent, the composite exhibited a quantum yield of H2 evolution up to 77.4% at 420 nm. Three-dimensional poly(benzothiadiazole) was also introduced in the hybrid system with C3N4 (BBT–C3N4).110 The BBT–C3N4 composite exhibited enhanced oxidation and reduction ability for photocatalytic removal of sulfathiazole and Cr(VI) owing to the efficient charge carrier transfer for the band alignment to the target substrates. Recently, the van der Waals heterostructure of aza-conjugated microporous polymer (aza-CMP) and C2N nanosheets was shown by Xiong and Xu et al., accomplishing the Z-scheme overall water splitting to produce a 2:1 stoichiometric ratio of H2 and O2 (Fig. 14).111 The proper alignment of the band structure was obtained by two polymer nanosheets, where the aza-CMP took part for water oxidation and C2N for proton reduction, leading to the Z-scheme photocatalysis. The rapid charge carrier separation and transfer were also observed by PL quenching and photocurrent measurement, obviously due to the overlapping interfaces between the ultrathin layers of two polymers.
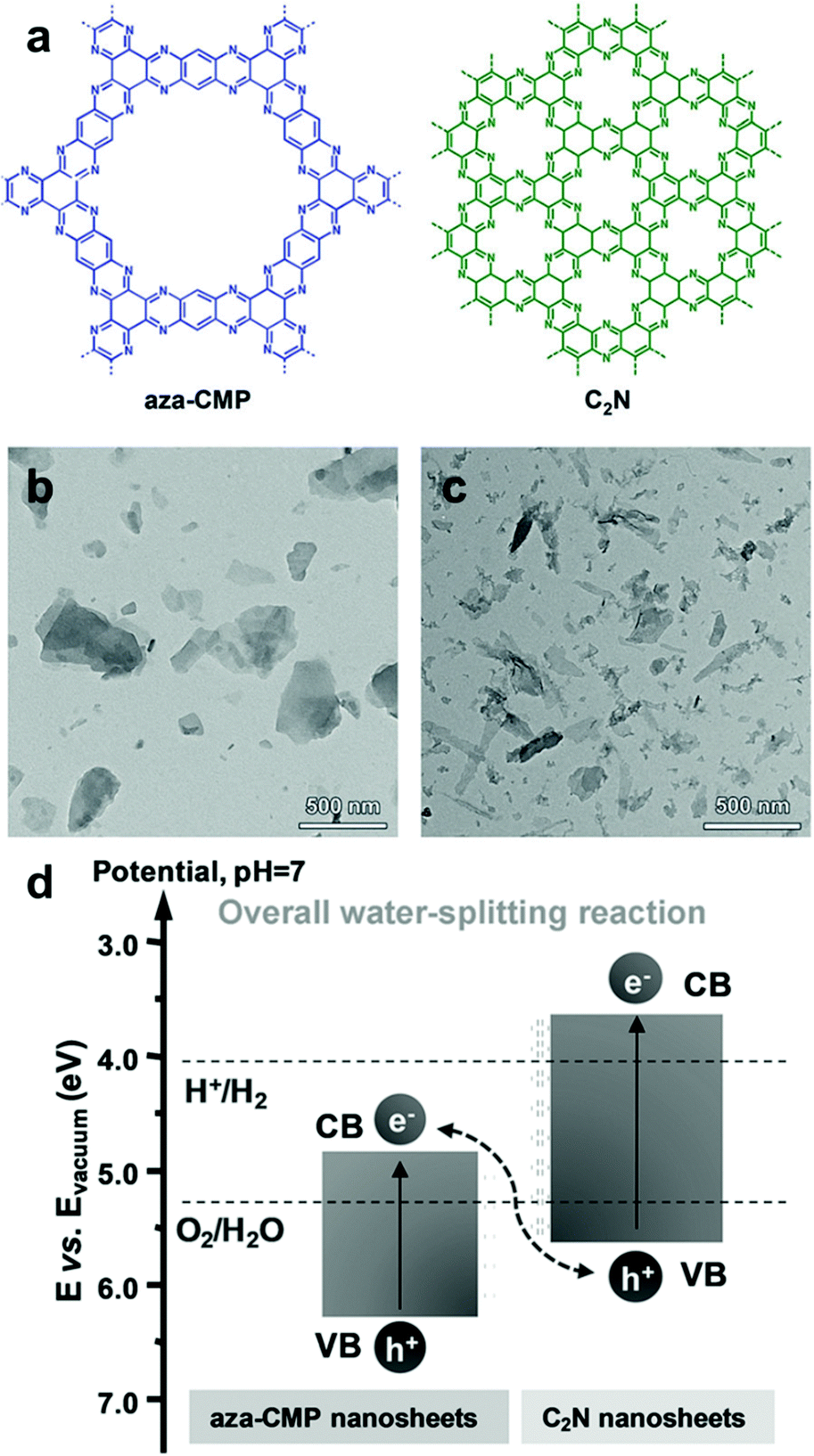 | ||
| Fig. 14 Organic hybrid structure of CPPs for energy band alignment. (a) Chemical structure of aza-conjugated microporous polymer (aza-CMP) and C2N nanosheets. TEM images of the exfoliated (b) aza-CMP and (c) C2N nanosheet. (d) Electronic band structures of aza-CMP and C2N nanosheet for a Z-scheme heterostructure. Reproduced with permission.111 Copyright 2018 Wiley-VCH. | ||
Inorganic/organic hybrid systems have been developed in a row. Zhang et al. immobilized Pd nanoparticles (5–10 nm in size, 3 wt%) on a conjugated microporous poly(benzoxadiazole) (B-BO3) for photocatalytic Suzuki coupling between aryl halide and arylboronic acid (Fig. 15).112 The formation of a hybrid heterojunction on the Pd metal and B-BO3 interface resulted in a positively-charged region in the polymer and negatively-charged region in the metal nanoparticles due to the Schottky effect.113 When irradiated, the charge carrier generated within the B-BO3 migrated into the Pd nanoparticles, and thus the excited electron could further amplify the Schottky effect to activate hard substrates such as iodobenzene with a C–I bond even at room temperature. The pure B-BO3 without Pd content exhibited no conversion, proving the indispensable role of Pd nanoparticles. Yu et al. reported that the residual Pd content in CPPs after the polymer synthesis could affect the catalytic activity of CPPs, where the H2 evolution rate could be increased by having max. ∼2 wt% Pd remaining in the structure.92 Linear conjugated polymers shown by Kosco et al. demonstrated that the remaining Pd concentration in the polymers (∼0.1 wt%) had a critical effect on the photocatalytic H2 production rate, and the pure polymer after the complete removal of Pd species was inactive for H2 evolution.114 This implies that the residual Pd species should be considered to evaluate the photocatalytic behavior of CPPs.
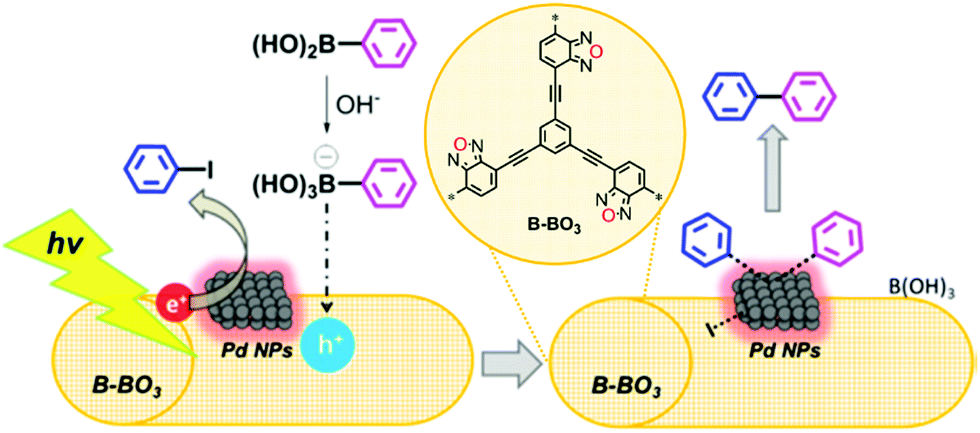 | ||
| Fig. 15 Organic/inorganic hybrid structure of CPPs for electron transfer. Heterojunction formation between poly(benzoxadiazole) and palladium nanoparticles. Reproduced with permission.112 Copyright 2015 American Chemical Society. | ||
Incorporation of UV-active TiO2 nanoparticles was recently carried out upon CPP photocatalysts. Banerjee et al. reported a hybrid system of porous porphyrin organic polymer (TpTph) and TiO2 nanoparticles.115 The composite structure of TpTph–TiO2 in a 1:2 ratio won over both bare TpTph and TiO2 nanoparticles for photocatalytic H2 production under visible-light illumination. The photo-excited electron generated by a porphyrin unit was transferred into the conduction band of TiO2, which reduced a water molecule into H2 in the presence of a Pt co-catalyst. The polymer/TiO2 hybrid was also applied for photocatalytic organic transformations.116 Zhang et al. showed that poly(benzothiadiazole) containing 80 wt% of TiO2 nanoparticles displayed an excellent photocatalytic behavior for oxidative coupling of amine and oxidation of sulfides, exceeding the efficiency of pristine poly(benzothiadiazole). More examples of inorganic/organic hybrid photocatalysts have been recently shown, for instance, MoO3/polyimide,117 MoS2/polyimide,118 WO3/polyimide,119 and phosphotungstic acid/polyimide.120
2.2. Macroscopic design of CPPs
The design of the CPP structure on a molecular level has induced intrinsic optical and electronic structure modulation of CPPs, affecting their photocatalytic behavior. Besides, there exist macroscopic design criteria of CPPs, i.e. morphology, porosity, and chemical functionalities, which can help to promote the photocatalytic reaction efficiency. Those criteria ought to be a secondary consideration after the structural control of CPPs, however, there have been a number of studies showing that the macroscopic factors boost up the photocatalytic efficiency of CPPs.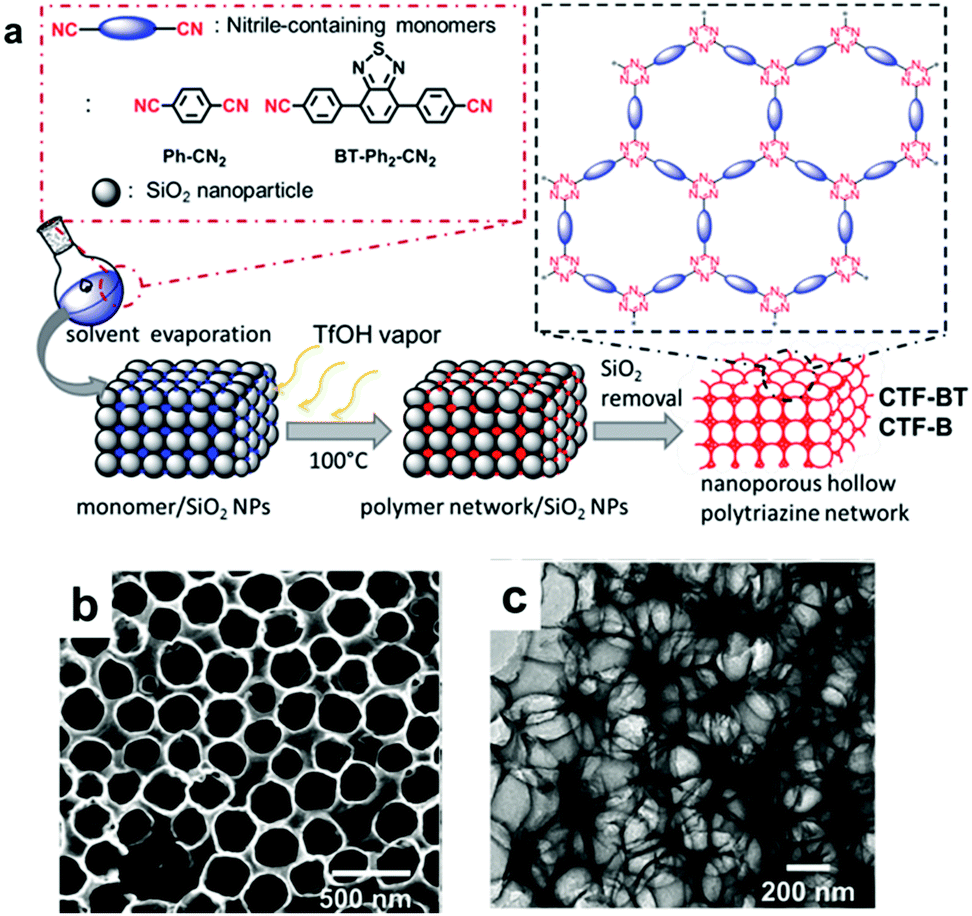 | ||
| Fig. 16 Morphology control of CPPs by silica template. (a) Solid vapor synthesis of a hollow covalent triazine framework, CTF. (b) SEM and (c) TEM image of photoactive CTF-BT with a hollow structure. Reproduced with permission.34 Copyright 2016 The Royal Society of Chemistry. | ||
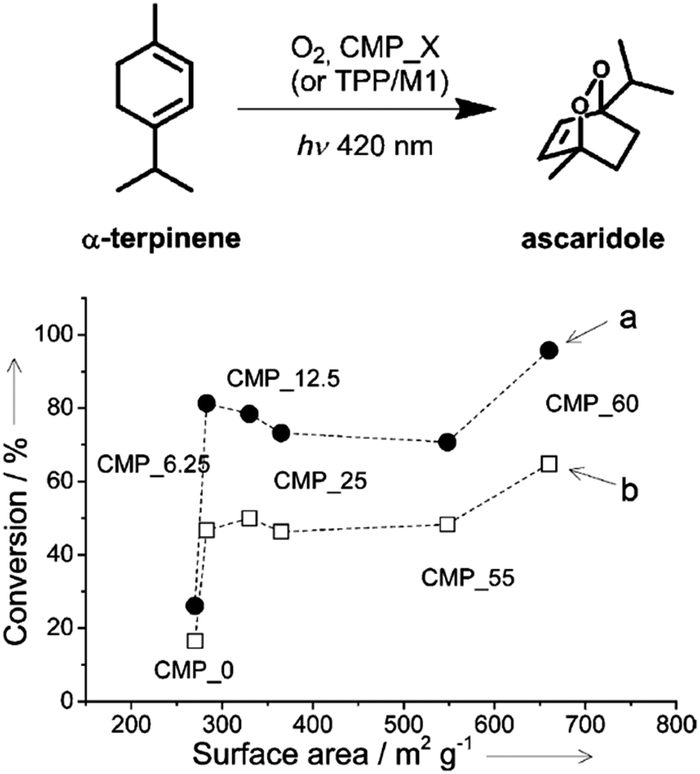 | ||
| Fig. 17 Porosity control of CPPs by the silica template. Photocatalytic conversion of α-terpinene into ascaridole with poly(benzothiadiazole)s having difference surface areas. Photo-reactor set-up with (a) lower O2 flow rate (5 mL min−1) and (b) higher O2 flow rate (10 mL min−1). Adapted with permission.22 Copyright 2013 Wiley-VCH. | ||
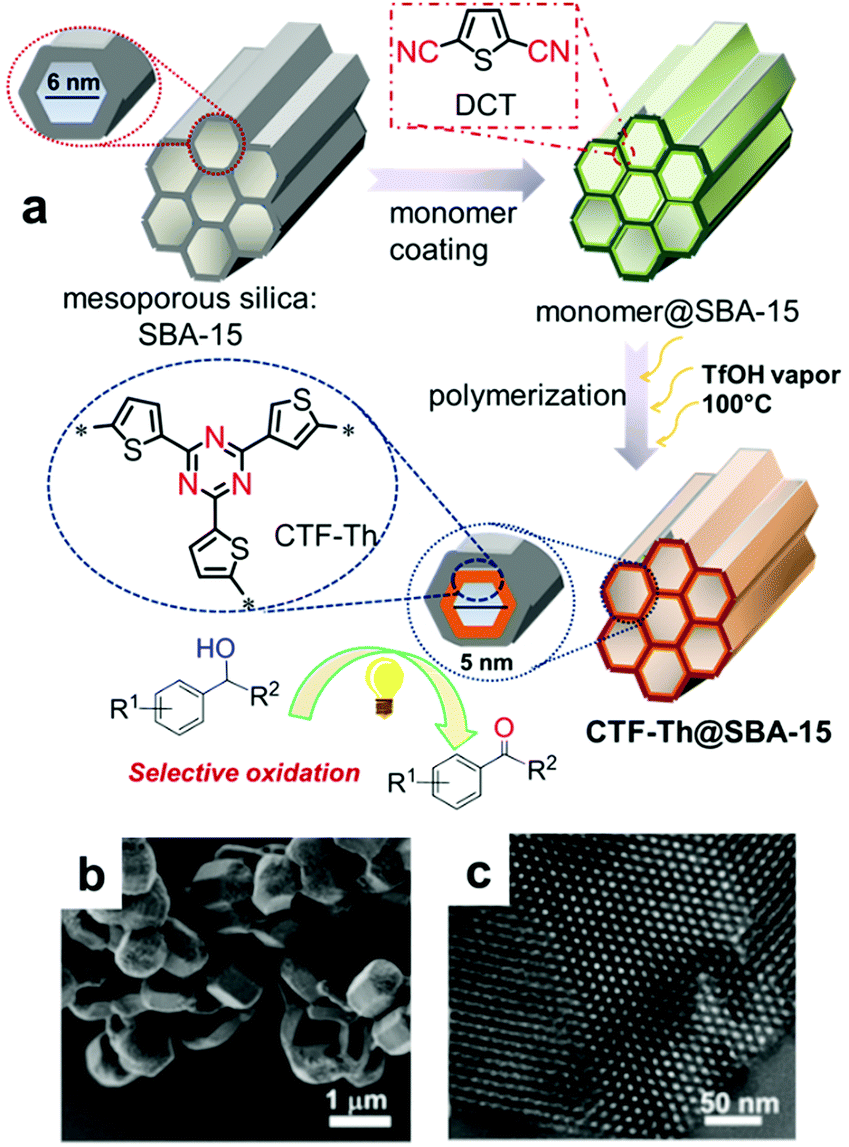 | ||
| Fig. 18 Support-induced high porosity of CPPs. (a) Thiophene-based covalent triazine framework on mesoporous silica SBA-15 (CTF-Th@SBA-15). (b) SEM and (c) TEM image of CTF-Th@SBA-15. Reproduced with permission.124 Copyright 2017 American Chemical Society. | ||
A large-scale porosity of CPPs was further achieved via a polymerized high internal phase emulsion (polyHIPE) synthetic protocol. The π-conjugated polyHIPEs were firstly developed by Vilela and coworkers,125 in which benzothiadiazole and fluorene moieties were copolymerized with 1,3,5-tribromobenzene in different ratios. The resulting polyHIPEs exhibited the BET surface areas of 35–50 m2 g−1, and the monolithic structures could be utilized for a continuous flow set-up for photocatalytic oxidation of α-terpinene. Various CPP photocatalysts were produced in polyHIPE form, showing excellent performance for free radical polymerization24 and reductive dehalogenation of haloketones.25 The low surface area of polyHIPE-based CPP was reformed by thermal deprotection of a tert-butyl carboxylate (BOC) functional group in the polymer network (Fig. 19).23 In a typical set-up, the π-conjugated polyHIPE (B-CB3) showed 8 times increase in BET surface area from 27 m2 g−1 to 230 m2 g−1 by the removal of the BOC group. The improved porosity led to increase in conversion efficiency and selectivity for photocatalytic oxidation of sulfides.
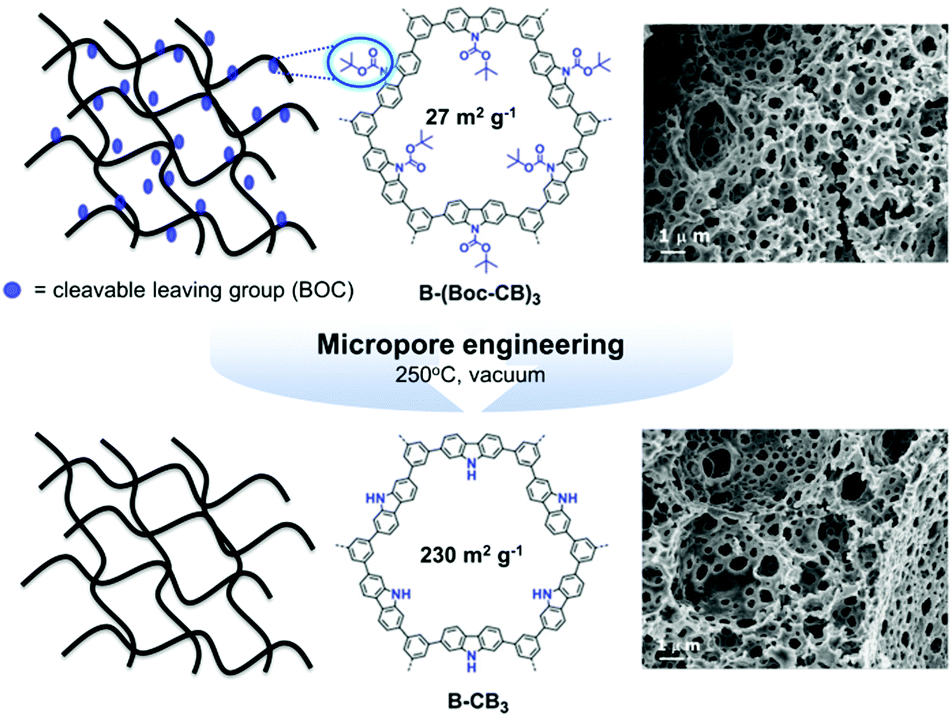 | ||
| Fig. 19 Porosity control of CPPs by thermal deprotection of the functional group. Generation of additional porosity via a thermal deprotection of the BOC group in π-conjugated polyHIPEs. Reproduced with permission.23 Copyright 2014 The Royal Society of Chemistry. | ||
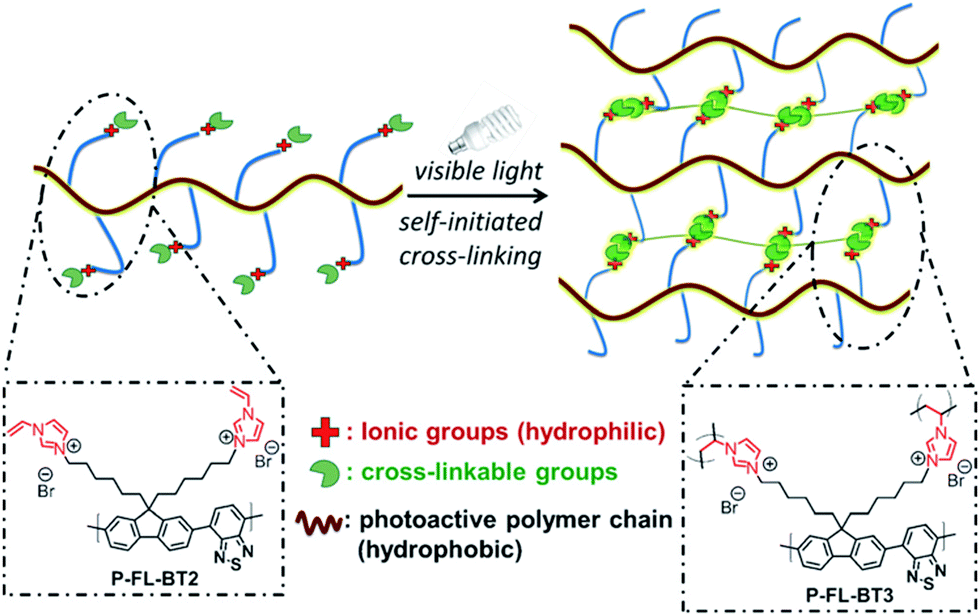 | ||
| Fig. 20 Functionality control of CPPs for water compatibility. Conjugated polymer networks with vinylimidazolium group for self-initiated cross-linking. Adapted with permission.128 Copyright 2015 Wiley-VCH. | ||
2.3. Process design of CPPs
Future research direction of CPPs will rely on their integration into a physical device for field applications. At the current stage, most of the research is heavily focused on material development over process design. Early series of photo-reactors using CPPs were carried out with the π-conjugated polyHIPEs (shown in Section 2.2.2).23–25,125 In a typical condition, the porous polyHIPE monoliths were packed in a glass column, and the mixture of substrates was pumped through the column, which was irradiated with visible-light. The monolithic photocatalyst, however, has limitations in absorbing light through the thick body, so that a large number of photoactive sites hidden inside of the polymer are often wasted. A commercial photochemical flow reactor consisting of a coil reactor and LED module has also been used.76 The photocatalyst dispersion and liquid substrate were pumped through the coil reactor repeatedly, thus improving the contact between the photocatalyst and substrate. But it may take more time and energy to treat a large amount of substrate in the coil reactor system. One way of fully utilizing the CPPs in the continuous flow system would be employing a packing support to create inner flow paths and expose the photoactive polymer on the surface. In this regard, a fixed-bed flow reactor was designed by Zhang et al., incorporated with photoactive CPP-coated glass fiber (Fig. 21).37 The CPP supported with glass fiber showed good light harvesting ability, which was attributed to the thin layer of the CPP (80 nm) on glass fiber, allowing facile light penetration through the fibers. The CPP-coated glass fiber showed an excellent photocatalytic activity for enantioselective α-alkylation of aldehyde and α-bromoketone in the photo-reactor. In consideration of the amount of polymer on the glass fiber (∼3.2 wt%), the photocatalytic efficiency exceeded that from a flow system with a monolithic photocatalyst, using 15 times less amount of the photoactive polymer. Therefore, processing of CPPs would be of importance to maximize their photocatalytic performance for industrial applications. The coating of CPPs upon glass beads131 and films132,133 may also be good options to bring CPPs into well-designed photo-reactor systems.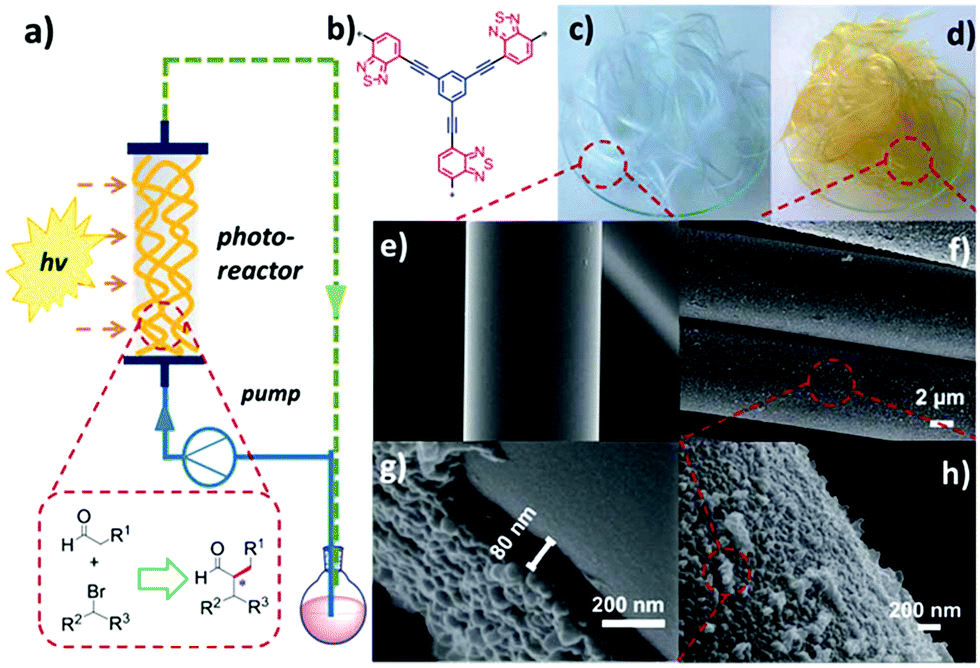 | ||
| Fig. 21 Processing of CPPs for a flow column photo-reactor. (a) Schematic set-up of a fixed-bed flow photo-reactor with CPP-coated glass fibers. (b) The chemical structure of photoactive CPP. Photographs of (c) pure glass fiber and (d) CPP-coated glass fiber. SEM images of (e) pure glass fiber and (f–h) CPP-coated glass fiber. Adapted with permission.37 Copyright 2017 The Royal Society of Chemistry. | ||
3. Conclusions and outlook
Given the designability, various efforts have been made to optimize the structure and properties of CPPs for target-specific photocatalytic reactions under visible light irradiation. Fine control over the molecular structure of CPPs has been implemented by considering the type, amount, and geometry of monomers for the optimal band structure and charge transport/transfer within CPPs. Morphology and porosity of CPPs could be intensively controlled when a template and porogen are utilized. Chemical functionality of CPPs offers hydrophilicity manipulation. The processing of CPPs to fit in industrial standard still has a long way to go, however, the embedment of CPPs with light and transparent supports could be an option for integrating CPPs into photo-reactors.Future challenges in designing CPPs lie ahead. One of the open questions is how far we can tune the energy levels of CPPs to tackle highly demanding reactions such as CO2 activation and methane oxidation. The next urgent task would be the process engineering of CPPs for large-scale photocatalysis, i.e. commercialization of CPPs. Keeping a balance between photocatalytic activity and cost of CPPs is thus becoming a key issue. New synthetic protocols of CPPs, cost effective and good in performance, have to be considered. Nevertheless, in terms of the photocatalytic activity along with tunable structure and properties, CPPs have demonstrated explosive growth in recent years and great promise for future applications. Through a limitless structural combination of CPPs, we envision that remaining challenges upon CPPs will be dissolved in the near future by best-tailoring CPPs with the design strategies that we have summarized in this review.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors sincerely thank Prof. Katharina Landfester for continuous and strong support. J. B. acknowledges the Alexander von Humboldt Foundation for the postdoctoral research fellowship and KIST for the institutional program grant (Project No. 2E29660). K. A. I. Z. thanks the Deutsche Forschungsgesellschaft (DFG). The authors thank the Max Planck Society for financial support. Open Access funding provided by the Max Planck Society.Notes and references
- R. C. Evans, P. Douglas and H. D. Burrow, Applied Photochemistry, Springer, Netherlands, 2014 Search PubMed.
- K. R. Reddy, M. S. Jyothi, A. V. Raghu, V. Sadhu, S. Naveen and T. M. Aminabhavi, in Nanophotocatalysis and Environmental Applications: Detoxification and Disinfection, ed. I. Inamuddin, A. M. Asiri and E. Lichtfouse, Springer International Publishing, Cham, 2020, pp. 139–169, DOI:10.1007/978-3-030-12619-3_6.
- S. B. Patil, P. S. Basavarajappa, N. Ganganagappa, M. S. Jyothi, A. V. Raghu and K. R. Reddy, Int. J. Hydrogen Energy, 2019, 44, 13022–13039 CrossRef CAS.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed.
- M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed.
- F. Guo, P. Yang, Z. Pan, X.-N. Cao, Z. Xie and X. Wang, Angew. Chem., Int. Ed., 2017, 56, 8231–8235 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2008, 8, 76 CrossRef PubMed.
- Y. Zhang, T. Mori, J. Ye and M. Antonietti, J. Am. Chem. Soc., 2010, 132, 6294–6295 CrossRef CAS PubMed.
- Z. Pan, G. Zhang and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 7102–7106 CrossRef CAS PubMed.
- Y. Fang, Y. Xu, X. Li, Y. Ma and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 9749–9753 CrossRef CAS PubMed.
- H. Ou, X. Chen, L. Lin, Y. Fang and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 8729–8733 CrossRef CAS PubMed.
- J. Lin, Z. Pan and X. Wang, ACS Sustainable Chem. Eng., 2014, 2, 353–358 CrossRef CAS.
- K. Wang, Q. Li, B. Liu, B. Cheng, W. Ho and J. Yu, Appl. Catal., B, 2015, 176-177, 44–52 CrossRef CAS.
- G. Zhang, G. Li, T. Heil, S. Zafeiratos, F. Lai, A. Savateev, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 3433–3437 CrossRef CAS.
- J. Ji, J. Wen, Y. Shen, Y. Lv, Y. Chen, S. Liu, H. Ma and Y. Zhang, J. Am. Chem. Soc., 2017, 139, 11698–11701 CrossRef CAS.
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18–21 CrossRef CAS.
- H. Arazoe, D. Miyajima, K. Akaike, F. Araoka, E. Sato, T. Hikima, M. Kawamoto and T. Aida, Nat. Mater., 2016, 15, 1084 CrossRef CAS.
- C. Huang, J. Wen, Y. Shen, F. He, L. Mi, Z. Gan, J. Ma, S. Liu, H. Ma and Y. Zhang, Chem. Sci., 2018, 9, 7912–7915 RSC.
- F. Vilela, K. Zhang and M. Antonietti, Energy Environ. Sci., 2012, 5, 7819–7832 RSC.
- K. Zhang, D. Kopetzki, P. H. Seeberger, M. Antonietti and F. Vilela, Angew. Chem., 2013, 125, 1472–1476 CrossRef.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Chem. Commun., 2014, 50, 8177–8180 RSC.
- Z. J. Wang, K. Landfester and K. A. I. Zhang, Polym. Chem., 2014, 5, 3559–3562 RSC.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, J. Mater. Chem. A, 2014, 2, 18720–18724 RSC.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. Zhang, Adv. Mater., 2015, 27, 6265–6270 CrossRef CAS.
- S. Bi, Z.-A. Lan, S. Paasch, W. Zhang, Y. He, C. Zhang, F. Liu, D. Wu, X. Zhuang, E. Brunner, X. Wang and F. Zhang, Adv. Funct. Mater., 2017, 27, 1703146 CrossRef.
- R. S. Sprick, J.-X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS.
- V. S. Vyas, V. W.-h. Lau and B. V. Lotsch, Chem. Mater., 2016, 28, 5191–5204 CrossRef CAS.
- K. Wang, L.-M. Yang, X. Wang, L. Guo, G. Cheng, C. Zhang, S. Jin, B. Tan and A. Cooper, Angew. Chem., Int. Ed., 2017, 56, 14149–14153 CrossRef CAS.
- C. Yang, B. C. Ma, L. Zhang, S. Lin, S. Ghasimi, K. Landfester, K. A. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 9202–9206 CrossRef CAS.
- L. Wang, Y. Wan, Y. Ding, Y. Niu, Y. Xiong, X. Wu and H. Xu, Nanoscale, 2017, 9, 4090–4096 RSC.
- L. Wang, Y. Wan, Y. Ding, S. Wu, Y. Zhang, X. Zhang, G. Zhang, Y. Xiong, X. Wu, J. Yang and H. Xu, Adv. Mater., 2017, 29, 1702428 CrossRef.
- W. Huang, Z. J. Wang, B. C. Ma, S. Ghasimi, D. Gehrig, F. Laquai, K. Landfester and K. A. I. Zhang, J. Mater. Chem. A, 2016, 4, 7555–7559 RSC.
- S. Ghasimi, S. A. Bretschneider, W. Huang, K. Landfester and K. A. I. Zhang, Adv. Sci., 2017, 4, 1700101 CrossRef.
- R. Li, B. C. Ma, W. Huang, L. Wang, D. Wang, H. Lu, K. Landfester and K. A. I. Zhang, ACS Catal., 2017, 7, 3097–3101 CrossRef CAS.
- W. Huang, B. C. Ma, D. Wang, Z. J. Wang, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, J. Mater. Chem. A, 2017, 5, 3792–3797 RSC.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS.
- L. Sun, Z. Liang, J. Yu and R. Xu, Polym. Chem., 2013, 4, 1932–1938 RSC.
- J. Schmidt, M. Werner and A. Thomas, Macromolecules, 2009, 42, 4426–4429 CrossRef CAS.
- A. Li, H.-X. Sun, D.-Z. Tan, W.-J. Fan, S.-H. Wen, X.-J. Qing, G.-X. Li, S.-Y. Li and W.-Q. Deng, Energy Environ. Sci., 2011, 4, 2062–2065 RSC.
- G. Lu, H. Yang, Y. Zhu, T. Huggins, Z. J. Ren, Z. Liu and W. Zhang, J. Mater. Chem. A, 2015, 3, 4954–4959 RSC.
- V. Senkovskyy, I. Senkovska and A. Kiriy, ACS Macro Lett., 2012, 1, 494–498 CrossRef CAS.
- J. Luo, X. Zhang and J. Zhang, ACS Catal., 2015, 5, 2250–2254 CrossRef CAS.
- J. Schmidt, J. Weber, J. D. Epping, M. Antonietti and A. Thomas, Adv. Mater., 2009, 21, 702–705 CrossRef CAS.
- M. G. Schwab, M. Hamburger, X. Feng, J. Shu, H. W. Spiess, X. Wang, M. Antonietti and K. Müllen, Chem. Commun., 2010, 46, 8932–8934 RSC.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS.
- Z. J. Wang, R. Li, K. Landfester and K. A. I. Zhang, Polymer, 2017, 126, 291–295 CrossRef CAS.
- S. Chu, C. Wang, Y. Yang, Y. Wang and Z. Zou, RSC Adv., 2014, 4, 57153–57158 RSC.
- Y. Kou, Y. Xu, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2011, 50, 8753–8757 CrossRef CAS.
- Y. L. Wong, J. M. Tobin, Z. Xu and F. Vilela, J. Mater. Chem. A, 2016, 4, 18677–18686 RSC.
- G. Zhang, Z.-A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef.
- K. S. Lakhi, D.-H. Park, K. Al-Bahily, W. Cha, B. Viswanathan, J.-H. Choy and A. Vinu, Chem. Soc. Rev., 2017, 46, 72–101 RSC.
- J. Liu, H. Wang and M. Antonietti, Chem. Soc. Rev., 2016, 45, 2308–2326 RSC.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS.
- Z. Zhou, Y. Zhang, Y. Shen, S. Liu and Y. Zhang, Chem. Soc. Rev., 2018, 47, 2298–2321 RSC.
- B. Muktha, G. Madras, T. N. Guru Row, U. Scherf and S. Patil, J. Phys. Chem. B, 2007, 111, 7994–7998 CrossRef CAS PubMed.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, J. Chem. Soc., Chem. Commun., 1985, 474–475 RSC.
- T. Amaya, D. Hata, T. Moriuchi and T. Hirao, Chem. – Eur. J., 2015, 21, 16427–16433 CrossRef CAS PubMed.
- S. Ghosh, N. A. Kouamé, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P.-H. Aubert and H. Remita, Nat. Mater., 2015, 14, 505–511 CrossRef CAS PubMed.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef CAS PubMed.
- R. S. Sprick, B. Bonillo, M. Sachs, R. Clowes, J. R. Durrant, D. J. Adams and A. I. Cooper, Chem. Commun., 2016, 52, 10008–10011 RSC.
- M. Zhang, W. D. Rouch and R. D. McCulla, Eur. J. Org. Chem., 2012, 6187–6196 CrossRef CAS.
- A. Pron and P. Rannou, Prog. Polym. Sci., 2002, 27, 135–190 CrossRef CAS.
- C. K. Song, B. J. Eckstein, T. L. D. Tam, L. Trahey and T. J. Marks, ACS Appl. Mater. Interfaces, 2014, 6, 19347–19354 CrossRef CAS PubMed.
- T. M. McCormick, C. R. Bridges, E. I. Carrera, P. M. DiCarmine, G. L. Gibson, J. Hollinger, L. M. Kozycz and D. S. Seferos, Macromolecules, 2013, 46, 3879–3886 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- C. B. Meier, R. S. Sprick, A. Monti, P. Guiglion, J.-S. M. Lee, M. A. Zwijnenburg and A. I. Cooper, Polymer, 2017, 126, 283–290 CrossRef CAS.
- Z.-A. Lan, Y. Fang, Y. Zhang and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 470–474 CrossRef CAS PubMed.
- J.-X. Jiang, Y. Li, X. Wu, J. Xiao, D. J. Adams and A. I. Cooper, Macromolecules, 2013, 46, 8779–8783 CrossRef CAS.
- S. Bandyopadhyay, A. G. Anil, A. James and A. Patra, ACS Appl. Mater. Interfaces, 2016, 8, 27669–27678 CrossRef CAS.
- M. Liras, M. Iglesias and F. Sánchez, Macromolecules, 2016, 49, 1666–1673 CrossRef CAS.
- M. Liras, M. Pintado-Sierra, M. Iglesias and F. Sanchez, J. Mater. Chem. A, 2016, 4, 17274–17278 RSC.
- J. M. Tobin, J. Liu, H. Hayes, M. Demleitner, D. Ellis, V. Arrighi, Z. Xu and F. Vilela, Polym. Chem., 2016, 7, 6662–6670 RSC.
- C.-A. Wang, Y.-W. Li, X.-L. Cheng, J.-P. Zhang and Y.-F. Han, RSC Adv., 2017, 7, 408–414 RSC.
- L. Pan, M.-Y. Xu, L.-J. Feng, Q. Chen, Y.-J. He and B.-H. Han, Polym. Chem., 2016, 7, 2299–2307 RSC.
- J.-L. Wang, C. Wang, K. E. deKrafft and W. Lin, ACS Catal., 2012, 2, 417–424 CrossRef CAS.
- A. Nagai, X. Chen, X. Feng, X. Ding, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2013, 52, 3770–3774 CrossRef CAS.
- Z. Xiao, Y. Zhou, X. Xin, Q. Zhang, L. Zhang, R. Wang and D. Sun, Macromol. Chem. Phys., 2016, 217, 599–604 CrossRef CAS.
- X. Ding and B.-H. Han, Angew. Chem., Int. Ed., 2015, 54, 6536–6539 CrossRef CAS PubMed.
- Z. Xie, C. Wang, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 2056–2059 CrossRef CAS PubMed.
- X. Fan, L. Zhang, R. Cheng, M. Wang, M. Li, Y. Zhou and J. Shi, ACS Catal., 2015, 5, 5008–5015 CrossRef CAS.
- K. Kailasam, M. B. Mesch, L. Möhlmann, M. Baar, S. Blechert, M. Schwarze, M. Schröder, R. Schomäcker, J. Senker and A. Thomas, Energy Technol., 2016, 4, 744–750 CrossRef CAS.
- W. Huang, Q. He, Y. Hu and Y. Li, Angew. Chem., Int. Ed., 2019, 58, 8676–8680 CrossRef CAS.
- C. Ayed, L. Caire da Silva, D. Wang and K. A. I. Zhang, J. Mater. Chem. A, 2018, 6, 22145–22151 RSC.
- C. Yang, W. Huang, L. C. da Silva, K. A. I. Zhang and X. Wang, Chem. – Eur. J., 2018, 24, 17454–17458 CrossRef CAS PubMed.
- Y. S. Kochergin, D. Schwarz, A. Acharjya, A. Ichangi, R. Kulkarni, P. Eliášová, J. Vacek, J. Schmidt, A. Thomas and M. J. Bojdys, Angew. Chem., Int. Ed., 2018, 57, 14188–14192 CrossRef CAS.
- W. Huang, J. Byun, I. Rörich, C. Ramanan, P. W. M. Blom, H. Lu, D. Wang, L. Caire da Silva, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, Angew. Chem., Int. Ed., 2018, 57, 8316–8320 CrossRef CAS.
- L. Li and Z. Cai, Polym. Chem., 2016, 7, 4937–4943 RSC.
- L. Li, Z. Cai, Q. Wu, W.-Y. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681–7686 CrossRef CAS PubMed.
- S. Kuecken, A. Acharjya, L. Zhi, M. Schwarze, R. Schomäcker and A. Thomas, Chem. Commun., 2017, 53, 5854–5857 RSC.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS.
- M. Liu, Q. Huang, S. Wang, Z. Li, B. Li, S. Jin and B. Tan, Angew. Chem., Int. Ed., 2018, 57, 11968–11972 CrossRef CAS.
- L. Wang, J. Byun, R. Li, W. Huang and K. A. I. Zhang, Adv. Synth. Catal., 2018, 360, 4312–4318 CrossRef CAS.
- A. Casey, S. D. Dimitrov, P. Shakya-Tuladhar, Z. Fei, M. Nguyen, Y. Han, T. D. Anthopoulos, J. R. Durrant and M. Heeney, Chem. Mater., 2016, 28, 5110–5120 CrossRef CAS.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Adv. Synth. Catal., 2016, 358, 2576–2582 CrossRef CAS.
- R. N. Renaud, C. J. Stephens and G. Brochu, Can. J. Chem., 1984, 62, 565–569 CrossRef CAS.
- Z. J. Wang, K. Garth, S. Ghasimi, K. Landfester and K. A. I. Zhang, ChemSusChem, 2015, 8, 3459–3464 CrossRef CAS.
- M. Heeney, W. Zhang, D. J. Crouch, M. L. Chabinyc, S. Gordeyev, R. Hamilton, S. J. Higgins, I. McCulloch, P. J. Skabara, D. Sparrowe and S. Tierney, Chem. Commun., 2007, 5061–5063 RSC.
- B.-G. Kim, X. Ma, C. Chen, Y. Ie, E. W. Coir, H. Hashemi, Y. Aso, P. F. Green, J. Kieffer and J. Kim, Adv. Funct. Mater., 2013, 23, 439–445 CrossRef CAS.
- J. Luo, X. Zhang, J. Lu and J. Zhang, ACS Catal., 2017, 7, 5062–5070 CrossRef CAS.
- W. Zhang, J. Tang, W. Yu, Q. Huang, Y. Fu, G. Kuang, C. Pan and G. Yu, ACS Catal., 2018, 8, 8084–8091 CrossRef CAS.
- C. Su, R. Tandiana, B. Tian, A. Sengupta, W. Tang, J. Su and K. P. Loh, ACS Catal., 2016, 6, 3594–3599 CrossRef CAS.
- L. Li, W. Fang, P. Zhang, J. Bi, Y. He, J. Wang and W. Su, J. Mater. Chem. A, 2016, 4, 12402–12406 RSC.
- C. Wang, Y. Guo, Y. Yang, S. Chu, C. Zhou, Y. Wang and Z. Zou, ACS Appl. Mater. Interfaces, 2014, 6, 4321–4328 CrossRef CAS.
- H. Yan and Y. Huang, Chem. Commun., 2011, 47, 4168–4170 RSC.
- X. Zhang, B. Peng, S. Zhang and T. Peng, ACS Sustainable Chem. Eng., 2015, 3, 1501–1509 CrossRef CAS.
- S. Wang, X. Yang, H. Hou, X. Ding, S. Li, F. Deng, Y. Xiang and H. Chen, Catal. Sci. Technol., 2017, 7, 418–426 RSC.
- L. Wang, X. Zheng, L. Chen, Y. Xiong and H. Xu, Angew. Chem., Int. Ed., 2018, 57, 3454–3458 CrossRef CAS.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Chem. Mater., 2015, 27, 1921–1924 CrossRef CAS.
- X.-H. Li and M. Antonietti, Chem. Soc. Rev., 2013, 42, 6593–6604 RSC.
- J. Kosco, M. Sachs, R. Godin, M. Kirkus, L. Francas, M. Bidwell, M. Qureshi, D. Anjum, J. R. Durrant and I. McCulloch, Adv. Energy Mater., 2018, 8, 1802181 CrossRef.
- G. Mukherjee, J. Thote, H. B. Aiyappa, S. Kandambeth, S. Banerjee, K. Vanka and R. Banerjee, Chem. Commun., 2017, 53, 4461–4464 RSC.
- C. Ayed, W. Huang, R. Li, L. C. da Silva, D. Wang, O. Suraeva, W. Najjar and K. A. I. Zhang, Part. Part. Syst. Charact., 2017, 35, 1700234 CrossRef.
- C. Ma, J. Zhou, H. Zhu, W. Yang, J. Liu, Y. Wang and Z. Zou, ACS Appl. Mater. Interfaces, 2015, 7, 14628–14637 CrossRef CAS.
- C. Ma, H. Zhu, J. Zhou, Z. Cui, T. Liu, Y. Wang, Y. Wang and Z. Zou, Dalton Trans., 2017, 46, 3877–3886 RSC.
- J. Dasgupta, J. Sikder, S. Chakraborty, U. Adhikari, V. P. Reddy B, A. Mondal and S. Curcio, ACS Sustainable Chem. Eng., 2017, 5, 6817–6826 CrossRef CAS.
- P. Meng, H. Heng, Y. Sun, J. Huang, J. Yang and X. Liu, Appl. Catal., B, 2018, 226, 487–498 CrossRef CAS.
- N. Park, D. Kang, M. C. Ahn, S. Kang, S. M. Lee, T. K. Ahn, J. Y. Jaung, H.-W. Shin and S. U. Son, RSC Adv., 2015, 5, 47270–47274 RSC.
- B. C. Ma, S. Ghasimi, K. Landfester, F. Vilela and K. A. I. Zhang, J. Mater. Chem. A, 2015, 3, 16064–16071 RSC.
- L. Wang, R. Fernández-Terán, L. Zhang, D. L. A. Fernandes, L. Tian, H. Chen and H. Tian, Angew. Chem., Int. Ed., 2016, 55, 12306–12310 CrossRef CAS.
- W. Huang, B. C. Ma, H. Lu, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, ACS Catal., 2017, 7, 5438–5442 CrossRef CAS.
- K. Zhang, Z. Vobecka, K. Tauer, M. Antonietti and F. Vilela, Chem. Commun., 2013, 49, 11158–11160 RSC.
- H. Urakami, K. Zhang and F. Vilela, Chem. Commun., 2013, 49, 2353–2355 RSC.
- K. Schwinghammer, S. Hug, M. B. Mesch, J. Senker and B. V. Lotsch, Energy Environ. Sci., 2015, 8, 3345–3353 RSC.
- S. Ghasimi, S. Prescher, Z. J. Wang, K. Landfester, J. Yuan and K. A. I. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14549–14553 CrossRef CAS.
- Q. Cui, X. Wang, Y. Yang, S. Li, L. Li and S. Wang, Chem. Mater., 2016, 28, 4661–4669 CrossRef CAS.
- S. Ghasimi, K. Landfester and K. A. I. Zhang, ChemCatChem, 2016, 8, 694–698 CrossRef CAS.
- K. T. Oppelt, J. Gasiorowski, D. A. M. Egbe, J. P. Kollender, M. Himmelsbach, A. W. Hassel, N. S. Sariciftci and G. Knör, J. Am. Chem. Soc., 2014, 136, 12721–12729 CrossRef CAS PubMed.
- R. Irigoyen-Campuzano, M. González-Béjar, E. Pino, J. B. Proal-Nájera and J. Pérez-Prieto, Chem. – Eur. J., 2017, 23, 2867–2876 CrossRef CAS.
- D. J. Woods, R. S. Sprick, C. L. Smith, A. J. Cowan and A. I. Cooper, Adv. Energy Mater., 2017, 7, 1700479 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |