
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevisiting the charge compensation mechanisms in LiNi0.8Co0.2−yAlyO2 systems†
Zachary W.
Lebens-Higgins
 a,
Nicholas V.
Faenza
b,
Maxwell D.
Radin
c,
Hao
Liu
d,
Shawn
Sallis
ef,
Jatinkumar
Rana
a,
Julija
Vinckeviciute
c,
Philip J.
Reeves
g,
Mateusz J.
Zuba
e,
Fadwa
Badway
b,
Nathalie
Pereira
b,
Karena W.
Chapman
h,
Tien-Lin
Lee
i,
Tianpin
Wu
j,
Clare P.
Grey
g,
Brent C.
Melot
k,
Anton
Van Der Ven
c,
Glenn G.
Amatucci
b,
Wanli
Yang
f and
Louis F. J.
Piper
*ae
a,
Nicholas V.
Faenza
b,
Maxwell D.
Radin
c,
Hao
Liu
d,
Shawn
Sallis
ef,
Jatinkumar
Rana
a,
Julija
Vinckeviciute
c,
Philip J.
Reeves
g,
Mateusz J.
Zuba
e,
Fadwa
Badway
b,
Nathalie
Pereira
b,
Karena W.
Chapman
h,
Tien-Lin
Lee
i,
Tianpin
Wu
j,
Clare P.
Grey
g,
Brent C.
Melot
k,
Anton
Van Der Ven
c,
Glenn G.
Amatucci
b,
Wanli
Yang
f and
Louis F. J.
Piper
*ae
aDepartment of Physics, Applied Physics, and Astronomy, Binghamton University, New York 13902, USA. E-mail: lpiper@binghamton.edu
bEnergy Storage Research Group, Department of Materials Science and Engineering, Rutgers University, North Brunswick, New Jersey 08902, USA
cMaterials Department, University of California Santa Barbara, Santa Barbara, CA 93106, USA
dDepartment of Chemistry, Binghamton University, New York 13902, USA
eMaterials Science & Engineering, Binghamton University, Binghamton, New York 13902, USA
fAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
gDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK
hDepartment of Chemistry, Stony Brook University, 100 Nicolls Rd, Stony Brook, NY 11790, USA
iDiamond Light Source Ltd., Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
jX-ray Science Division, Advanced Photon Source, Argonne National Laboratory, Argonne, Illinois 60439, USA
kDepartment of Chemistry, University of Southern California, Los Angeles, CA 90089, USA
First published on 23rd July 2019
Abstract
Oxygen participation, arising from increased transition metal–oxygen covalency during delithiation, is considered essential for the description of charge compensation in conventional layered oxides. The advent of high-resolution mapping of the O K-edge resonant inelastic X-ray scattering (RIXS) provides an opportunity to revisit the onset and extent of oxygen participation. Combining RIXS with an array of structural and electronic probes for the family of Ni-rich LiNi0.8Co0.2−yAlyO2 cathodes, we identify common charge compensation regimes that are assigned to formal transition metal redox (<4.25 V) and oxygen participation through covalency (>4.25 V). From O K-edge RIXS maps, we find the emergence of a sharp RIXS feature in these systems when approaching full delithiation, which has previously been associated with lattice oxidized oxygen in alkali-rich systems. The lack of transition metal redox signatures and strong covalency at these high degrees of delithiation suggest this RIXS feature is similarly attributed to lattice oxygen charge compensation as in the alkali-rich systems. The RIXS feature's evolution with state of charge in conventional layered oxides is evidence that this feature reflects the depopulation of occupied O 2p states associated with oxygen participation.
New conceptsConventional Ni-rich layered oxides remain at the forefront of research on practical high-energy density Li-ion battery cathodes with a continued focus on increasing the operating voltage window. When accessing higher degrees of delithiation, the abrupt c-lattice contraction gives rise to a large anisotropic volume change. This contraction has long been considered a consequence of oxygen participation in charge compensation arising from strong transition metal (TM)–oxygen covalency. The advent of alkali-rich cathodes has renewed discussion of oxygen charge compensation, where excess capacity beyond traditional TM redox couples has been debated in terms of various localized oxidized oxygen configurations. Novel resonant inelastic X-ray scattering (RIXS) mapping of the O K-edge studies have identified unique signatures attributed to the oxidized lattice oxygen in alkali-rich cathodes that correlate with excess capacity. Extending the RIXS technique to the conventional layered oxides, we find the surprising emergence of the same RIXS signatures at the onset of c-lattice contraction despite no excess capacity. Any universal mechanism of oxygen redox must reconcile the emergence of oxidized oxygen RIXS signatures in both alkali-rich and conventional layered oxides. |
1 Introduction
Conventional Ni-rich layered oxide cathodes remain in focus due to their high reversible capacities, low polarization, and rate performance during long-term cycling for commercial Li-ion batteries.1–3 In pursuit of further closing the gap between practical and theoretical capacities, there has been extensive work investigating performance issues beyond 4.3 V4–7 arising primarily from surface phase transformations4 and particle cracking.5 Engineering solutions have demonstrated success in mitigating these issues through the use of cathode and electrolyte modifications to improve high voltage performance.8–11 From a fundamental perspective, the structure5,12,13 and stability14 of the layered oxides at high voltages can be directly related to oxygen participation in charge compensation. Although the 3d transition metal (TM) redox couples can fully account for the extractable lithium in the textbook electrochemical definition of conventional layered oxides (LiMO2 systems), this assumes a purely ionic description. In reality, strong TM–O covalency will facilitate oxygen participation.12,15–17Extensive theoretical and experimental work on LiCoO2 has revealed the importance of Co–O covalency in facilitating oxygen charge compensation.16,18–20 Over the past 20 years, this mechanism has been extended across various Co- and Ni-rich LiMO2 systems.5,15,21 The advent of alkali-rich oxides, which have demonstrated reversible capacities beyond traditional TM-redox couples, has renewed discussions on oxygen charge compensation.22–26 In alkali-rich systems, lowering the metal coordination around oxygen atoms lifts non-bonding O 2p states towards the top of the valence band and is considered to give rise to accessible oxygen orbitals along Li–O–Li chains.22 This oxygen redox mechanism, deriving from weakly hybridized oxygen orbitals along these Li–O–Li chains, is often highlighted in direct contrast to oxygen participation through covalency in conventional layered oxides.22,25,26
Evaluation of these two distinct mechanisms has been challenging as there are few direct oxygen environment probes and, in comparison to TM probes, their interpretation is not as straightforward. While O K-edge soft X-ray absorption spectroscopy (sXAS) has been extensively used to consider oxygen participation,18,27–30 the oxygen absorption lineshape can be directly affected by TM redox through hybridization, even when the TM is the main redox center, e.g. Fe2+/3+ in LiFePO4.31 When evaluating the origins of spectral changes, this technique alone has clear limitations in its ability to conclusively confirm oxygen charge compensation. Resonant inelastic X-ray scattering (RIXS) at the O K-edge has emerged as a prime technique to directly probe oxidized oxygen states32–37 and has been powerful in driving discussions on the oxygen redox mechanism in alkali-rich systems.31,37–39 A sharp RIXS feature has been found to emerge when there is limited further TM redox activity32–34 and has recently been reported in both Li-rich and heavily Mg doped Na-ion systems.37 Extending the RIXS technique to LiMO2 systems is important for understanding oxygen participation through covalency and its similarities and differences from the oxygen redox mechanism considered in alkali-rich systems.
Here, we primarily focus on the first charge of the state-of-the-art LiNi0.8Co0.15Al0.05O2 (NCA)27,40–43 cathode, along with other Ni-rich LiNi0.8Co0.2−yAlyO2 compounds, to examine the evolution of the TM and oxygen environments throughout delithiation.27,40–43 In contrast to the ubiquitous first charge activation plateau in alkali-rich oxides,26 the first cycle of NCA is representative of subsequent cycles, following a similar electrochemical and structural evolution when surface Li2CO3 is suppressed.42 Despite the widespread practical interest in the NCA system,1–3 there has been limited discussion or direct investigation of oxygen participation in this system.27,44
From our joint hard and soft X-ray spectroscopy studies, we split the electrochemical processes for the Ni-rich systems into two regimes: an ionic (formal TM redox) regime below 4.25 V and covalency regime at higher voltages. The ionic regime is marked by nickel and cobalt redox signatures with concurrent changes in O K-edge spectra linked to the changing TM 3d states and increasing TM–O covalency. Upon further delithiation (>4.25 V), we find the emergence of a RIXS feature in several LiMO2 systems that matches the oxidized oxygen feature reported in alkali-rich oxides. This feature increases in strength with delithiation and becomes pronounced at the highest degrees of delithiation. This sharp RIXS feature is considered to be a direct indication of the lattice oxygen participation in charge compensation in LiMO2 systems given the strong TM–O covalency and lack of further TM redox in this regime.
2 Experimental section
2.1 Material preparation
Commercial LiNi0.8Co0.15Al0.05O2 (NAT1050, TODA America) was used as received in all characterization and electrochemistry experiments. All other materials were synthesized in-house according to previously published procedures.7 Of the range of LiNi1−y−zCoyAlzO2 compositions studied in this previous work, we selected four compositions synthesized in-house to focus on: LiNi0.8Al0.2O2 (LNA), LiNi0.8Co0.15Al0.05O2 (NCA), LiNi0.8Co0.20O2 (LNC), and LiCoO2 (LCO). The LNA sample presented throughout this study has a high phase segregation and cation mixing and was referred to as disordered (DO) in the previous work.7 Inductively Coupled plasma-optical emission spectroscopy (ICP-OES) and inductively coupled plasma atomic emission spectroscopy (ICP-AES) were used to confirm the metal stoichiometry for in-house synthesized and commercial compounds, respectively.7,40 Results on ordered LNA material for an electrode charged to 4.75 V with the same composition (LiNi0.8Al0.2O2)7 are given in ESI.† The commercial and in-house synthesized NCA are referred to as NCA 1 and NCA 2, respectively. The in-house synthesized compounds have smaller primary particles of around 200 nm compared to the commercial NCA compound with particles between 300 nm to 1 μm.40 Scanning electron microscopy images of the NCA 1 (commercial NCA) and NCA 2 can be found in ref. 7. Limited Li2CO3 was present at the surface of all of the compounds used in this study.2.2 Electrochemical testing
Dried LiMO2 powder was mixed in an argon-filled glovebox with 2.5 wt% carbon black (SuperP (SP), MMM) using a mortar and pestle. Powder cells (2032, Hohsen Corp.) were assembled using a lithium metal (FMC Lithium) negative electrode with a combination of glass fiber (Whatman GF/D), polyolefin (Celgard) and poly(vinylidene fluoride-co-hexafluoropropylene) (PVDF-HFP, Kynar 2801, Elf Atochem) depending on the test temperature with 1 M LiPF6 ethylene carbonate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dimethyl carbonate (EC:DMC) (1:1 volume ratio) electrolyte (BASF, <20 ppm H2O). All samples were galvanostatically charged and discharged at 10 mA g−1 (of active material), except for the electrodes tested at 60 °C and used for operando XRD. The electrodes tested at 60 °C were charged to 4.75 V at 25 mA g−1 and then potentiostatically held at 4.75 V for 10 hours. Electrochemical characterization was performed with either a VMP3 (Bio-Logic Science Instruments), or Series 4000 (Maccor) battery cycling system. Electrochemical curves were highly reproducible for the powder cells with the highest variation in delithiation of ∼5% Li.
:dimethyl carbonate (EC:DMC) (1:1 volume ratio) electrolyte (BASF, <20 ppm H2O). All samples were galvanostatically charged and discharged at 10 mA g−1 (of active material), except for the electrodes tested at 60 °C and used for operando XRD. The electrodes tested at 60 °C were charged to 4.75 V at 25 mA g−1 and then potentiostatically held at 4.75 V for 10 hours. Electrochemical characterization was performed with either a VMP3 (Bio-Logic Science Instruments), or Series 4000 (Maccor) battery cycling system. Electrochemical curves were highly reproducible for the powder cells with the highest variation in delithiation of ∼5% Li.
2.3 Characterization techniques
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m crystal structure. Additional information regarding the XRD measurements performed in situ including the determination of LixMO2 phase transformations can be found in ref. 7.
m crystal structure. Additional information regarding the XRD measurements performed in situ including the determination of LixMO2 phase transformations can be found in ref. 7.
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) structure was generated by the ISODISTORT software package47 as a symmetry-mode distortion from the conventional Rm structure. The ISODISTORT software package was also used to express the lattice parameters and the oxygen atomic coordinates as linear functions of the symmetry-mode amplitudes and to export those symbolic relationships in TOPAS.str format. The strain and displacive symmetry-mode amplitudes, instead of the lattice constants and the oxygen atomic coordinates, were refined directly. The symmetry-mode refinement constrains the lattice of the triclinic cell to conform to the geometry of the hexagonal lattice of the Rm structure.
) structure was generated by the ISODISTORT software package47 as a symmetry-mode distortion from the conventional Rm structure. The ISODISTORT software package was also used to express the lattice parameters and the oxygen atomic coordinates as linear functions of the symmetry-mode amplitudes and to export those symbolic relationships in TOPAS.str format. The strain and displacive symmetry-mode amplitudes, instead of the lattice constants and the oxygen atomic coordinates, were refined directly. The symmetry-mode refinement constrains the lattice of the triclinic cell to conform to the geometry of the hexagonal lattice of the Rm structure.
Details of sample preparation and synchrotron X-ray and neutron powder diffraction measurement for the samples charged to x = 0.16 (4.3 V) and x = 0.06 (4.7 V) against a graphite electrode used for the joint X-ray and neutron powder diffraction measurement have been published elsewhere.46
m).50 A random distribution of transition-metal ions Ni, Co and Al on the octahedral site of NCA was modelled by developing a theoretical model consisting of three FEFF calculations, each corresponding to one type of backscattering TM ion.51 These FEFF calculations were weighted according to the nominal stoichiometry LiNi0.8Co0.2−yAlyO2. EXAFS fits at the Ni and Co K-edges involved all single-scattering and relevant multiple-scattering paths up to 5 Å. Details about parameterization of these paths are discussed in ref. 52. Good agreement between the data and theory can be seen in Fig. S1 (ESI†).
For all sXAS, RIXS and XPS measurements, samples were mounted in a glovebox and transported to the measurement chamber using a vacuum suitcase so that there was no air exposure.
2.4 DFT
Density functional theory (DFT) calculations were performed using the Vienna ab initio Simulation Package (VASP)54–57 with projector augmented-wave (PAW) pseudopotentials.58 The PBE59 calculations were spin polarized and were initialized with a ferromagnetic spin ordering. Calculations employed the on-site Coulomb correction for Ni with Ueff = 5 eV.60 The energy cutoff for these calculations was 530 eV and the k-point mesh density was at least 38 Å.3 Results and discussion
The first charge of the primary Ni-rich systems used in this study along with the corresponding evolution of the c- and a-lattice parameters from in situ XRD measurements are given in Fig. 1. Three Ni-rich compositions were selected to compare the effect of cobalt and aluminum incorporation: LiNi0.8Co0.2O2 (LNC), LiNi0.8Co0.15Al0.05O2 (NCA), and LiNi0.8Al0.2O2 (LNA). Commercial and solid-state synthesized NCA compounds are referred to as NCA 1 and NCA 2, respectively. Except for the LNA compound, a solid-solution-like behavior is found throughout the first charge. The LNA material shown here has significant phase segregation and a high voltage plateau that is not found for a more ordered LNA compound.7 The in situ XRD measurements were previously reported along with a detailed examination of the high-voltage cycling performance to 4.5 V and 4.75 V for these compounds.7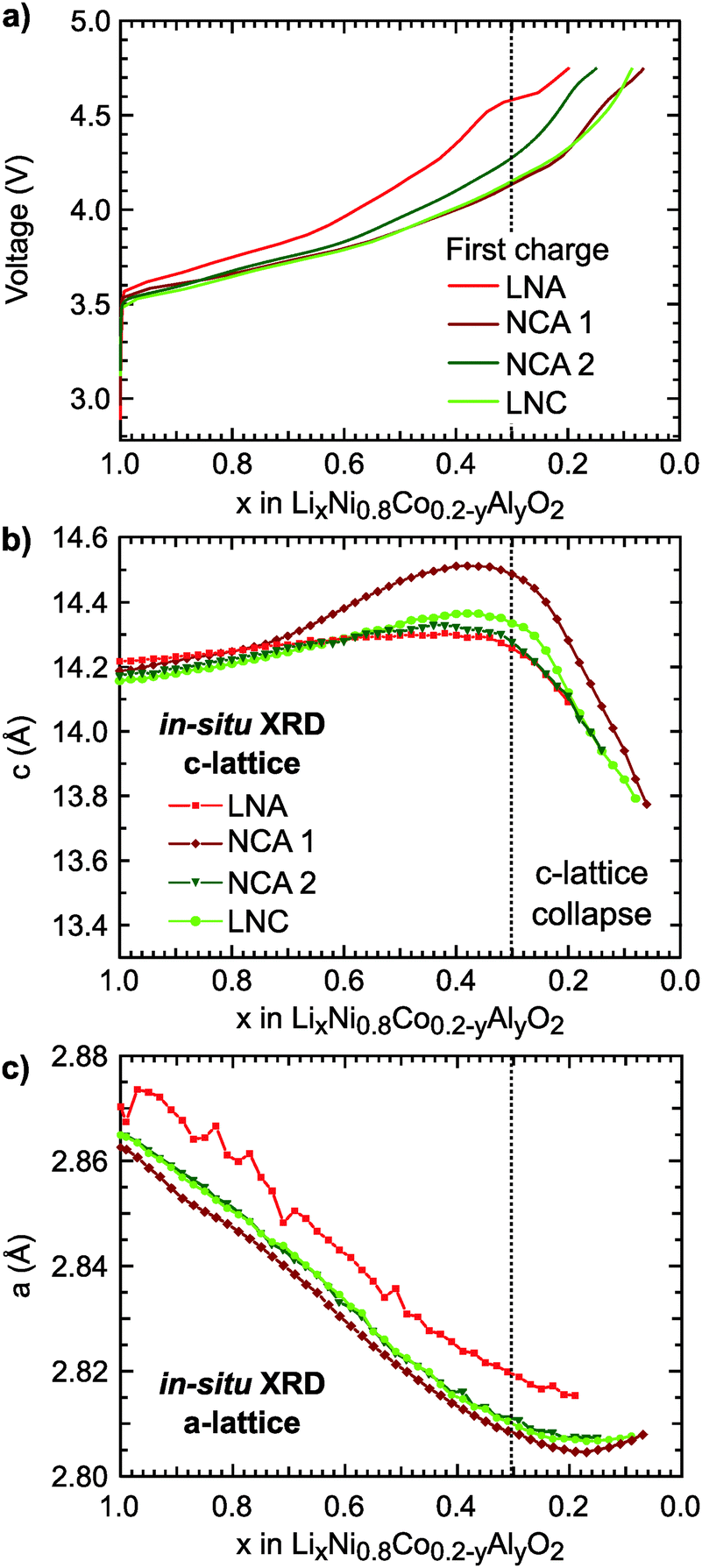 | ||
| Fig. 1 (a) First charge profile for layered oxide cathodes with chemical formula LiNi0.8Co0.2−yAlyO2 (0 ≤ y ≤ 0.2) to 4.75 V at 10 mA g−1 (vs. Li metal) and at RT. In situ (b) c- and (c) a-lattice parameters for the first charge at C/50 to 4.75 V followed by a potentiostatic hold at 4.75 V for the Ni-rich systems. The c- and a-lattice parameters were calculated from the (003) peak and the (101) and (003) peaks, respectively. | ||
Throughout delithiation of these Ni-rich systems, the structural behavior of the c- and a-lattice parameters can be separated into two distinct regimes. An initial c-lattice expansion and a-lattice contraction in the first regime are followed by the collapse of the c-lattice beyond x < ∼0.3 in the second regime (Fig. 1b and c). The a-lattice contraction is attributed to TM redox given the smaller ionic radii and shorter TM–O bonds associated with higher TM oxidation states.62 From monitoring the c-lattice evolution for a similar Ni-rich layered system (LiNi0.8Mn0.1Co0.1O2), A. Kondrakov et al.5 found the drop in the c-lattice could be partially attributed to the collapse in the lithium layer height (hLi–O) near the end of charge. Tarascon et al. in 199912 proposed the shrinkage in the c-lattice parameter results from the depopulation of the oxygen sp band, which has antibonding character, that gives rise to a shortening of the anion–anion separation across the depopulated 3a Li layer for LiCoO2. This model has since been extended to a number of isostructural Ni-rich cathodes.5,13 Although we only evaluate the overall c-lattice collapse, our XRD measurements suggest that oxygen participation in charge compensation may be expected to increase for these Ni-rich systems when x < ∼0.3. This gives a starting point for determining the relationship between the lithium content and the extent of TM and oxygen participation. We primarily focus on the widely reported NCA 1 compound,6,40–43,63 with similar characterization extended to the other Ni-rich systems given in ESI.†
3.1 Formal TM redox regime
For NCA 1, we examined the nickel and cobalt oxidation states for select points along the first charge (Fig. 2a) from a combination of X-ray absorption near edge structure (XANES), extended X-ray absorption fine structure (EXAFS), and soft X-ray absorption (sXAS) measurements. The Ni and Co K-edges (Fig. 2c) for pristine NCA 1 are consistent with the TMs in the trivalent state. Upon delithiation, the oxidation of nickel is evident from the shift of the Ni K-edge between x = 1.00 and x = 0.24. EXAFS fits show shortening of the Ni–O bond in the same delithiation range (Fig. 2b).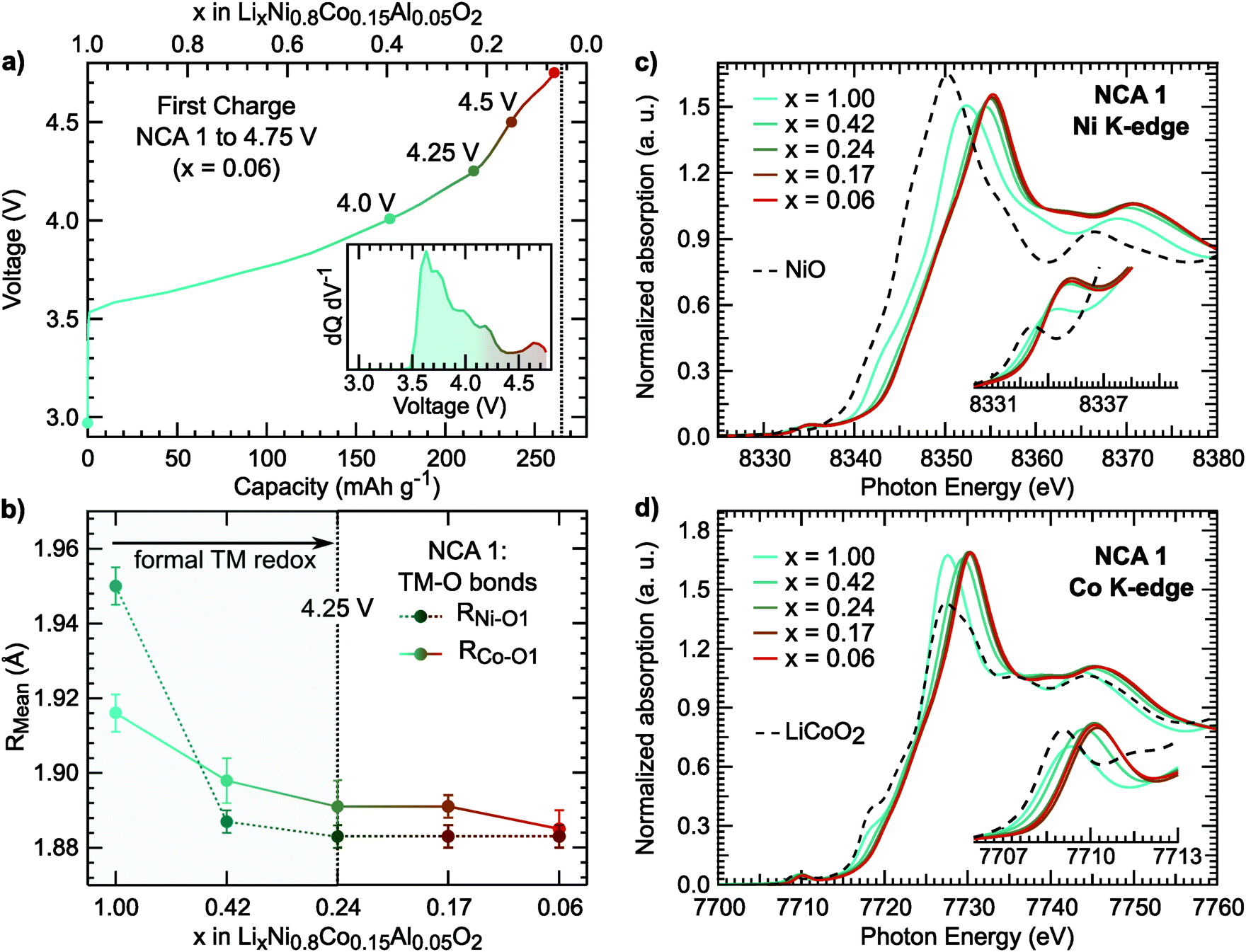 | ||
| Fig. 2 (a) Electrochemical profile of first charge and dQ dV−1 (inset) including the cutoff voltages of the selected NCA 1 electrodes used for X-ray spectroscopy measurements. (b) Ni–O1 and Co–O1 bond lengths derived from EXAFS fits. Example fits for the Ni and Co K-edge EXAFS are given in Fig. S1 (ESI†). The average Ni–O bond length of the pristine sample is similar to previously reported LiNiO261 (c and d) XANES measurements of the Ni and Co K-edges including NiO (Ni2+) and LiCoO2 (Co3+) references. | ||
In LiNi1−yCoyO2 systems, cobalt is considered to activate towards the end of charge at high degrees of delithiation64,65 as discussed in recent layered oxide reviews.1,2 It is important to note limited changes of the Co K-edge can lead to difficulties in assigning cobalt oxidation due to the limited edge shift during delithiation.66 While our measurements similarly show limited edge shift, we find most changes in the pre-edge and main peak up to x = 0.24 and limited changes at higher states of charge.
Rather than relying on just the Co K-edge, we also evaluated the Co–O bond length and Co L3-edge for assignment of cobalt oxidation. Similar to the Ni–O bond evolution, the Co–O bond shows the most pronounced shortening between x = 1.00 and x = 0.24. From surface sensitive total electron yield (TEY) measurements of the Co L3-edge (Fig. 3a), we find the primary shift in the main peak below x = 0.24 and the extent of this shift matches with cobalt oxidation reported for conventional and Li-rich NMC.33,67 By comparing the Co L3-edge shift to the % Co–O bond shortening, given in Fig. 3b, we find a similar evolution from electronic and structural methods indicating formal cobalt oxidation primarily occurs between x = 1.00 and x = 0.24 (<4.25 V). Limited formal TM redox beyond x = 0.24 is further supported by bulk-sensitive Ni and Co L3-edge total fluorescence yield measurements (Fig. S2, ESI†). Evolution of the TM K-edges and TM–O bond lengths for LNA, NCA 2, and LNC show changes related to formal TM redox primarily below 4.25 V (Fig. S3, ESI†). For each of these layered oxide systems, the end of formal TM redox occurs before all available lithium is extracted and shortly after the start of the c-lattice collapse (Fig. 1b).
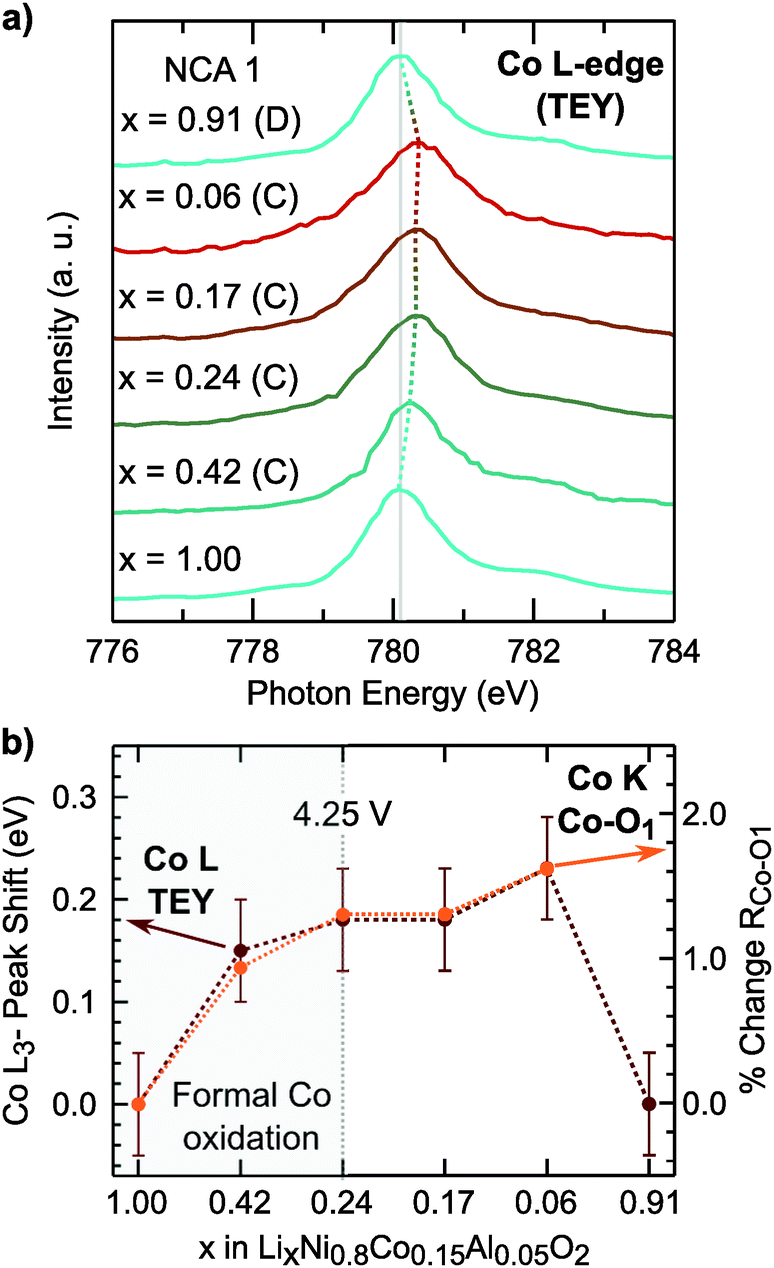 | ||
| Fig. 3 (a) Co L3-edge for NCA 1 electrodes along the first charge and after full discharge to 2.7 V. The dotted line indicates the reversible shift of the main Co L3-edge peak during charge and after discharge. (b) The extent of the Co L3-edge shift is compared to the % Co–O1 bond shortening extracted from EXAFS fits. | ||
For the NCA 1 system, we examined the effect of the formal TM redox on the occupied and unoccupied electronic structure. Hard X-ray photoelectron spectroscopy (HAXPES) and normal X-ray emission spectroscopy (XES) measurements of the occupied total density of states (TDOS) and occupied O 2p partial density of states (PDOS), respectively, are compared to ground state density functional theory (DFT) calculations of the O 2p PDOS of LixNi6/7Co1/7O2 in Fig. 4a. The O K-edge sXAS spectra, given in TEY and bulk-sensitive partial fluorescence yield (PFY) modes in Fig. 4b, provide insight into the unoccupied O 2p PDOS and sensitivity to the hybridized TM 3d–O 2p states.
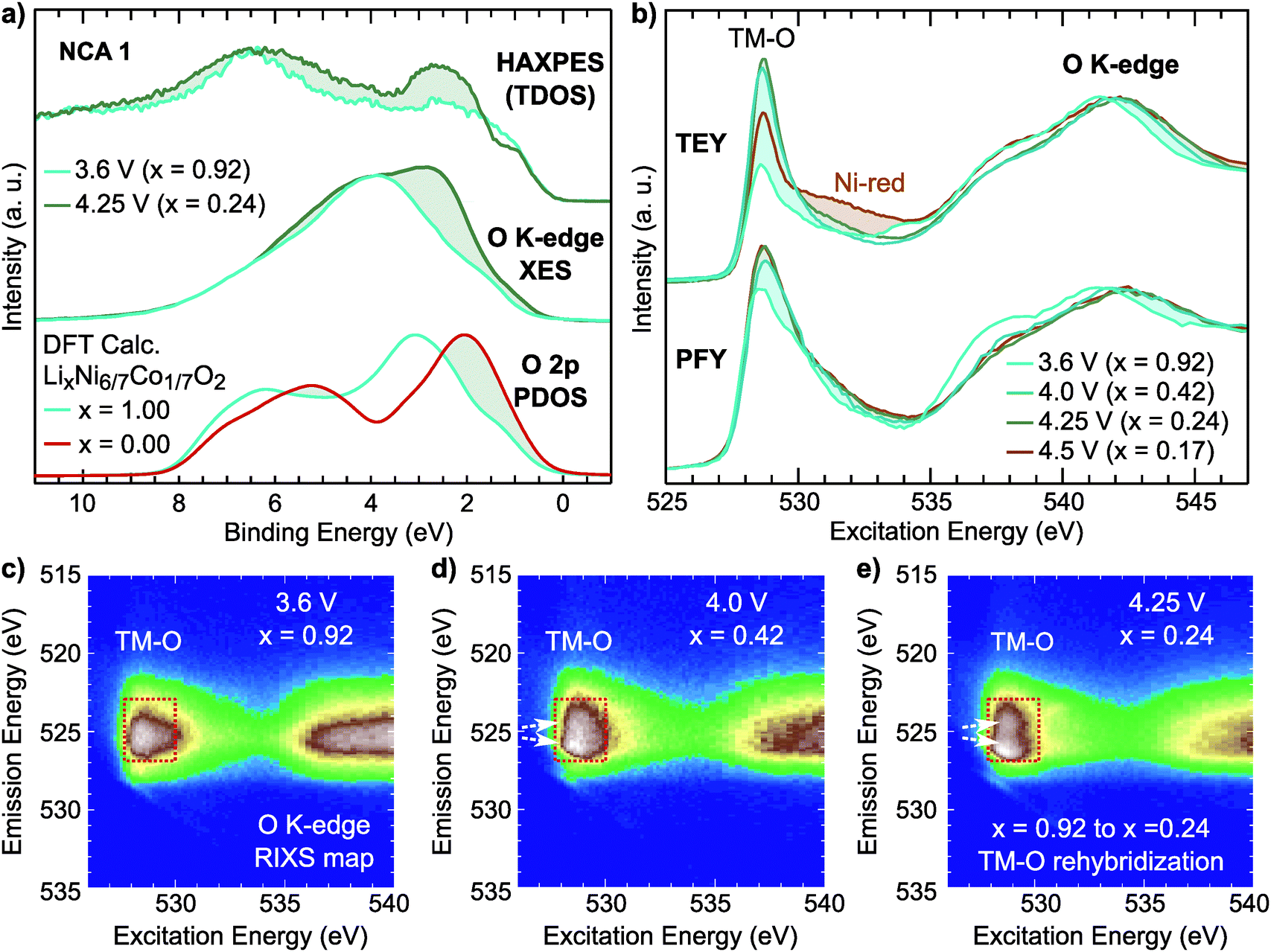 | ||
| Fig. 4 (a) HAXPES and normal O K-edge XES measurements for lithiated (x = 0.92) and delithiated (x = 0.24) states are compared to ground state calculations of the occupied O 2p PDOS for fully lithiated and delithiated LiNi6/7Co1/7O2. (b) O K-edge sXAS measurements in TEY and PFY mode for NCA 1 electrodes. The evolution of the pre-edge region with delithiation is shaded. (c–e) RIXS maps showing the evolution of broad TM–O hybridization features with delithiation for electrodes charged to 3.6 V, 4.0 V and 4.25 V. | ||
Focusing first on O K-edge sXAS spectra (Fig. 4b), we find a pronounced increase in the main pre-edge region peak (527 eV to 534 eV) upon initial delithiation. For NCA, the main pre-edge peak at 528.8 eV is related to the Ni 3d–O 2p hybridized states and its increase between x = 1.00 and x = 0.24 is a direct result of TM oxidation that gives rise to stronger TM–O hybridization. This effect is roughly captured in ground state DFT calculations of the O 2p unoccupied PDOS (Fig. S4, ESI†). Upon further delithiation to x = 0.17, there is no increase in pre-edge intensity and only a change in TEY related to reduced Ni2+ at the surface.43
There are clear changes in the occupied states as well from examination of the HAXPES and O K-edge XES in the formal TM redox regime. By comparing lithiated (x = 0.92) and delithiated (x = 0.24) electrodes, we find a clear redistribution of the states near the top of the valence band, shaded in Fig. 2a. From the O K-edge XES, this results in increased O 2p character near the valence band maximum agreeing with the changes observed in the DFT calculations of the O 2p PDOS.
From examination of the emission decay channels that follow the initial absorption process, resonant inelastic X-ray scattering (RIXS) maps can provide further sensitivity to the occupied and unoccupied oxygen states.31,39,68 The emission processes include an elastic peak that scales linearly with excitation energy, X-ray emission from occupied O 2p PDOS, and energy loss features due to excitations.31 For the RIXS map of the lithiated state (x = 0.92), given in Fig. 4c, there is a broad emission feature in the pre-edge region (527 eV to 530 eV) which is directly associated with emission from Ni3+ 3d–O 2p and Co3+ 3d–O 2p hybridized states (red rectangle). Upon delithiation to x = 0.42 and x = 0.24, this pre-edge feature splits concurrently with the formal TM redox (white arrows). This broadening elegantly shows the increasing O 2p bandwidth associated with shortening of TM–O bond lengths that signifies higher TM–O covalency.
3.2 Covalency regime
For NCA 1 electrodes charged beyond x = 0.24, we find limited evidence of further formal TM redox from XANES, EXAFS and sXAS (Fig. 2, 3, and Fig. S2, ESI†). Combined with the c-lattice evolution (Fig. 1b), this suggests bulk delithiation beyond x = 0.24 is associated with oxygen charge compensation driven by stronger TM–O covalency in the delithiated system. In previous studies,18,29,30 oxygen charge compensation had been assigned from changes in the sXAS lineshape of the O K-edge, such as in LiCoO2, where large (small) oxygen (TM) absorption changes were used to identify oxygen as the main redox center.20,69 For NCA, changes in the O K-edge absorption lineshape occur alongside formal TM-redox and can be primarily attributed to stronger TM–O hybridization rather than oxygen participation. The strong link between the O K-pre-edge intensity and the TM oxidization state indicates that this technique alone cannot provide direct assignment of oxygen participation.As RIXS provides additional sensitivity to the oxygen environment, we turn to this technique to identify any changes that can be linked to increased oxygen participation through covalency at higher degrees of delithiation. RIXS maps of the O K-edge for electrodes charged to 4.5 V and 4.75 V at RT shown in Fig. 5a and b. At x = 0.06, a sharp emission feature at 523.5 eV is clearly distinguished at an excitation energy of 531 eV in the RIXS map (arrow in Fig. 5b) along with a corresponding enhancement of the elastic peak. This sharp feature is distinct from the wide emission ranges associated with the hybridized TM–O states. When revisiting the RIXS measurements of the lower voltage points, we find the RIXS feature for NCA is weakly present for both the 4.25 V and 4.5 V electrodes (Fig. S5, ESI†).
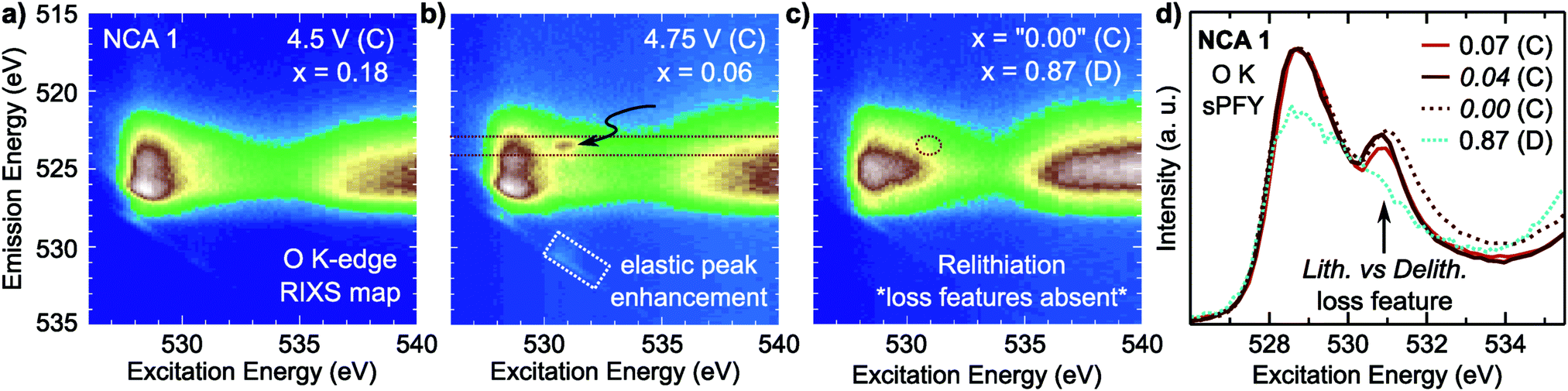 | ||
| Fig. 5 O K-edge RIXS maps for electrodes charged to (a) 4.5 V at RT, (b) 4.75 V at RT and (c) 4.75 V and held at CV for 10 h before discharge to 2.7 V, all at 60 °C. Arrows indicate the emergence of a sharp emission feature and corresponding elastic peak enhancement present at high degrees of delithiation and absent on discharge. (d) O K-edge sPFY measurements for the select emission window between the red dashed lines in part (b), for electrodes charged to 4.75 V at RT (x = 0.07), at 60 °C (x = 0.04), and 60 °C with a 10 h CV hold at 4.75 V (x = 0.00) compared to electrode discharged at 60 °C after the 10 h CV hold. | ||
Focusing on a select emission window from 522.9 to 524.1 eV, given as the super partial fluorescence yield (sPFY) mode, provides additional sensitivity to the presence of this RIXS feature. In Fig. 5d, we compare the sPFY lineshape for electrodes charged to 4.75 V at RT, 60 °C, and 60 °C with a 10 h constant voltage (CV) hold to an electrode discharged after the 10 h CV hold to 2.7 V. The 531 eV peak, associated with the sharp RIXS feature, shows a slight increase when charging at 60 °C compared to RT, but no increase with CV holding. The lack of change during the CV holding when there is an increase in TM reduction and electrolyte decomposition at the surface43,70 is evidence that the feature is associated with bulk oxygen environments. From further studies of highly delithiated NCA 1 electrodes, we found the onset of this sharp RIXS feature was highly reproducible at different dose levels of X-ray radiation and independent of electrolyte salt (Fig. S6, ESI†). For the discharged electrode, the RIXS feature is absent which suggests the feature is reversible upon relithiation.
Compared with the RIXS feature assigned to lattice oxidized oxygen in alkali-rich systems, this feature for NCA 1 is at a similar excitation and emission energy.29,32–35 Recent joint experiment and simulation of the Li2O2 peroxide have shed additional light on the origin of this feature.39 In Li2O2, a loss feature is observed at the same excitation/emission energy which is ascribed to an intra-band transition from the πu to unoccupied 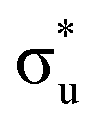 states.39 Although the exact origin of the RIXS feature remains uncertain for the complex TM oxide systems, this work clearly shows that such a RIXS feature stems from excitations to unoccupied O 2p states, i.e. oxidized oxygen states, as O 2p orbitals of O2− are fully occupied. It is important to note that short O–O distances (∼1.5 Å) have not been found in ex situ Raman measurements of delithiated alkali-rich cathodes.29,35,71 For alkali-rich systems, these combined insights into the RIXS feature imply its association with lattice oxygen redox. Considering the distinction often drawn between oxygen participation in conventional and alkali-rich oxides,22,25,26 it is surprising to observe a similar RIXS feature in NCA.
states.39 Although the exact origin of the RIXS feature remains uncertain for the complex TM oxide systems, this work clearly shows that such a RIXS feature stems from excitations to unoccupied O 2p states, i.e. oxidized oxygen states, as O 2p orbitals of O2− are fully occupied. It is important to note that short O–O distances (∼1.5 Å) have not been found in ex situ Raman measurements of delithiated alkali-rich cathodes.29,35,71 For alkali-rich systems, these combined insights into the RIXS feature imply its association with lattice oxygen redox. Considering the distinction often drawn between oxygen participation in conventional and alkali-rich oxides,22,25,26 it is surprising to observe a similar RIXS feature in NCA.
3.3 Compositional dependence
To evaluate the RIXS feature's presence across Ni-rich systems, we conducted O K-edge RIXS maps on the Ni-rich LNA, NCA 2, and LNC electrodes at 3.6 V, 4.25 V, and 4.75 V (Fig. S7 and S8, ESI†). In all three systems, the primary changes in the sXAS O K-edge absorption lineshape and O K-edge RIXS TM–O hybridized feature are alongside formal nickel and cobalt redox. The crystal structure and approximate a- and c-lattice parameters for these systems at 4.75 V, based on the in situ XRD shown in Fig. 1, are given in Table S2 (ESI†). In the example RIXS map of LNA charged to 4.75 V (x = 0.2) shown in Fig. 3c, the RIXS feature is observed along with a corresponding enhancement of the elastic peak. As this LNA material is disordered and has some phase segregation, we also conducted RIXS measurements of ordered LNA when charged to 4.75 V which reached a similar level of delithiation based on the capacity (x = 0.24). We found the RIXS feature was also present in the ordered sample and a similar intensity as the disordered LNA electrode (Fig. S9, ESI†). Even though the RIXS feature emerges with ∼20% lithium remaining in these LNA systems, this is near the maximum extractable lithium expected from just the Ni3+/4+ redox couple (80%) when considering aluminum as inactive.As the elastic peak is well separated from any overlapping emission at an excitation energy of 531 eV, the elastic peak evolution is more easily distinguishable than the RIXS feature at 523.5 eV that can be buried in TM–O hybridization features. Therefore, we can use this dedicated feature away from any broad emission signals for a quasi-quantitative analysis of the RIXS feature. This is particularly viable for comparing the Ni3+/4+ layered oxides as there is minimal contribution from Ni 3d–O 2p hybridized states at an excitation energy of 531 eV (Fig. S4, ESI†).
In the covalency regime, the elastic peak shows a clear increase in intensity when there is limited change in either the O K-edge sXAS spectra or the broad TM–O hybridized RIXS features (Fig. 5 and Fig. S7, S8, ESI†). For the region highlighted in blue in Fig. 6b, a clear RIXS feature is observed for the NCA 1 and LNA systems along with the pronounced increase in the elastic peak. Although LiCoO2 is not the main focus of this study, we found the RIXS feature present at 4.75 V (x = 0.13) (Fig. S8, ESI†), indicating this feature is not restricted to Ni-rich/Al-doped systems. Additionally, the RIXS feature is absent in partially discharged LNA and LCO electrodes (Fig. S8, ESI†), similar to the NCA 1 case. Based on the observation of a RIXS feature in the LCO system, there is no clear link between the RIXS feature and the crystal structure as LCO has transitioned to an H1–3 staged structure by 4.75 V while NCA maintains the Rm phase throughout delithiation (Table S2, ESI†).
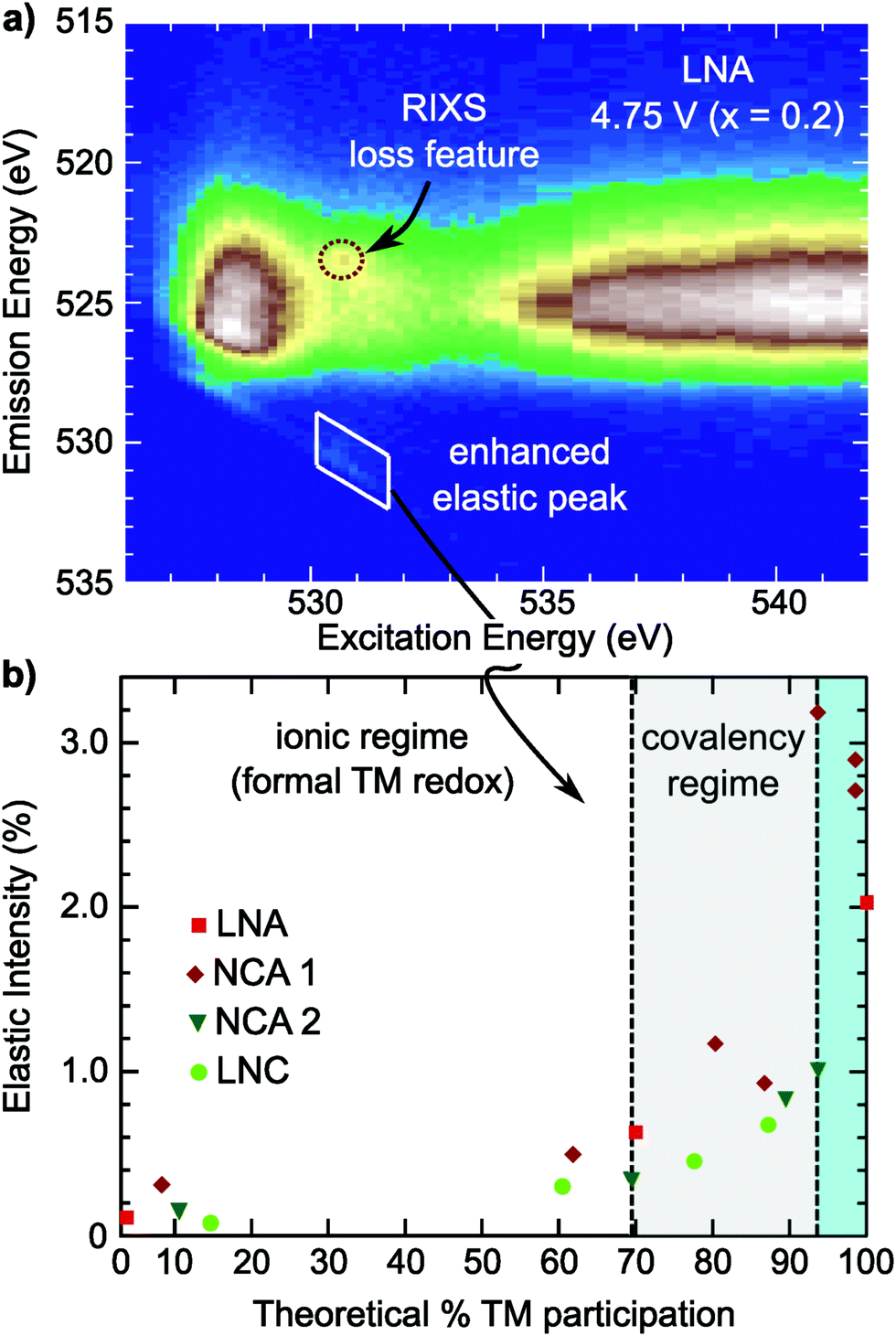 | ||
| Fig. 6 (a) RIXS map of LNA electrode charged to 4.75 V (x = 0.2) showing the weak RIXS feature and corresponding enhancement of the elastic peak at 531 eV. (b) Evolution of the 531 eV elastic peak intensity compared to the expected % TM redox in the ionic viewpoint (Ni3+/4+ & Co3+/4+ redox couples) based on electrode delithiation and taking into account the inactive aluminum in LNA and NCA. | ||
Although both NCA 2 and LNC show an enhancement of the elastic peak in the covalency regime, it is important to note that a sharp RIXS feature was not clearly observed for these systems when charged to 4.75 V. In these systems, there is a benefit to using the elastic peak as weak RIXS features can be obscured by overlying TM–O features. Weaker RIXS features in NCA 2 and LNC that are not clearly distinguished may be related to the smaller particle size of these systems compared to NCA 1 and any differences in actual bulk delithiation not accounted for including excess capacity generated through cathode-electrolyte side reactions beyond 4.3 V.40
3.4 Local oxygen environment
Combining our observations for the LiMO2 systems to this point, we find the RIXS feature emerges with delithiation in the strong covalency regime after the end of formal TM redox and is absent in the discharged state. These observations suggest the origin of the RIXS feature in conventional layered oxides is associated with oxygen participation in charge compensation. To further evaluate this hypothesis, we conducted measurements focusing on the oxygen coordination and chemical environment. Lattice peroxo-like oxygen dimerization has been proposed as the oxygen redox mechanism for the alkali-rich 3d72,73 and 4d systems,74–77 so it is relevant to consider whether this mechanism can be applied to LiMO2 systems. In the alkali-rich systems, the dimerization is considered to lead to shorter O–O separations, though the extent of dimerization is still much larger than the ∼1.5 Å separation in peroxides, e.g. 2.51 Å in Na0.6[Li0.2Mn0.8]O2.73In LiMO2 systems, the onset of the RIXS feature occurs during the c-lattice collapse at high degrees of delithiation so it is informative to examine any concurrent changes in lattice O–O distances in this regime. Based on previously reported ND/XRD Rietveld refinement for NCA 1,40 we evaluated the O–O distances for lithium contents in the onset range of the RIXS feature. Between x = 0.16 and x = 0.06 in Fig. 7a, the main structural change is the drop in the lithium layer height (hLi–O) to 2.71 Å. Within the TM layer, there is a short and a long O–O separation throughout the first charge which remain relatively unchanged between x = 0.16 and x = 0.06. Even when lowering the symmetry to allow for variation in the oxygen coordinate, there is no pronounced shortening of the O–O separation (2.47 Å) from ND/XRD refinement (Fig. S10 and Tables S3, S4, ESI†). From these measurements, which provide an average view of the O–O distances, we do not find evidence for lattice dimerization associated with the RIXS feature.
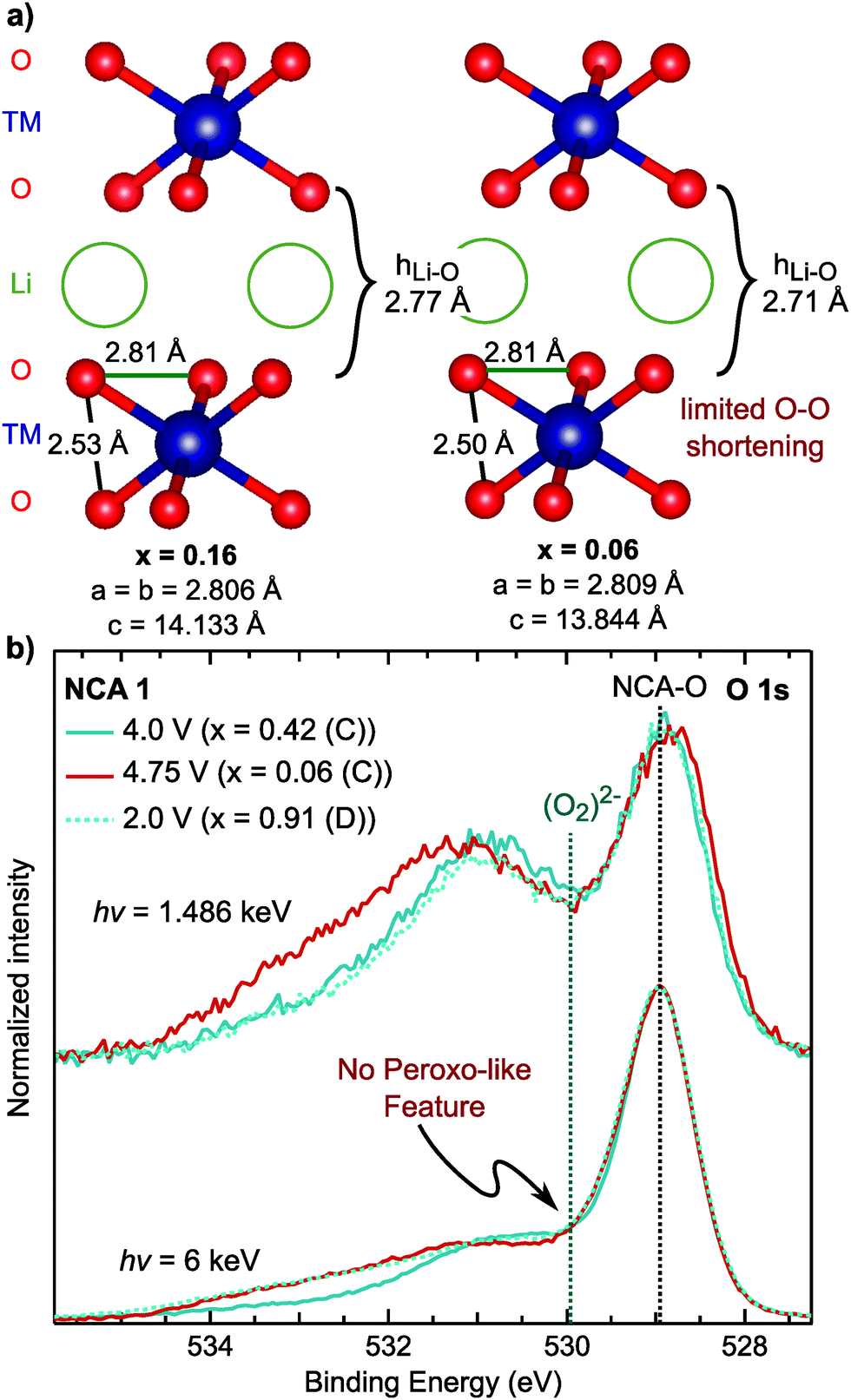 | ||
| Fig. 7 (a) Nearest O–O distances based on previously reported joint XRD/ND Rietveld refinement in the Rm SG for NCA 1 electrodes cycled versus graphite and measured ex situ.40 The lithium contents given here are based on the charge capacity. (b) Depth dependent XPS measurements of the O 1s core region, at 1.486 keV and 6 keV, in the formal TM redox regime (x = 0.42), after the RIXS feature onset (x = 0.06), and discharged to 2.7 V (x = 0.91). | ||
As the XRD/ND refinement does not rule out local O–O dimerization, we used depth dependent X-ray photoelectron spectroscopy (XPS), a primary technique for reporting peroxo-like species,74,76,78 for sensitivity to the oxygen chemical environment. The main NCA O 1s peak is at ∼529 eV so that a peroxo-like peak would be expected around 1 eV higher at ∼530 eV.74,76,78 To identify which peaks >530 eV are related to the cathode electrolyte interface (CEI), we utilized HAXPES to increase bulk sensitivity by a factor of three to ∼15 nm. In comparison to XPS, all peaks >530 eV decrease in intensity indicating these are primarily associated with CEI components. Moreover, there is no change between the lithiated [x = 0.42 (C) and x = 0.91 (D)] and delithiated samples [x = 0.06] related to peroxo-like species formation. In contrast to previous XPS studies,74,78 our HAXPES spectra is not dominated by CEI contributions, providing greater certainty regarding the presence/absence of local peroxo-like species in the bulk. While the RIXS feature is weaker for NCA 1 compared to Li-rich systems, previous work has suggested pronounced peroxo-like formation at the start of the high voltage plateau.78 Additional XPS/HAXPES for LNA shows variation of a CEI peak at 530 eV but no evidence of a bulk peroxo-like species (Fig. S11, ESI†). As the RIXS feature for NCA 1 and LNA is related to the bulk, we find no link for either system between an O 1s asymmetry and the onset of the RIXS feature. While there may be peroxo-like species at the surface, the lack of correlation with the RIXS suggests these O 1s peaks do not relate to the bulk oxidized oxygen states.
3.5 RIXS feature in LiMO2
Extension of O K-edge RIXS maps to conventional layered oxides to directly measure the oxygen environment revealed the onset of a similar RIXS feature at high degrees of delithiation that was previously only found in alkali-rich systems. When considering the c-lattice collapse at the highest degrees of delithiation found across layered oxide cathodes,13 the onset of this RIXS feature matches well with what is expected from the current structural viewpoint of oxygen participation in charge transfer. In contrast to the alkali-rich systems, this RIXS feature emerges within the delithiation range that is accounted for by just the TMs in the ionic viewpoint (Ni3+/4+ & Co3+/4+ redox couples) so that there is no excess capacity. While there are various reports that consider lattice oxygen dimerization as vital for oxygen redox in alkali-rich systems,74 the RIXS feature for LiMO2 systems was observed with no concurrent shortening of average O–O separations in the lattice. In addition, we do not find a clear new O 1s peak in HAXPES associated with a bulk peroxo-like states (Fig. 7), though more work may be needed to conclude that the oxygen states associated with the RIXS feature are distinct from peroxide-like oxygen environments, e.g. Li2O2. These environments have mainly been theorized to form in alkali-rich systems, e.g. Li2MnO3,72,79 but may be important to consider for the conventional layered oxides, given their thermodynamic instability at high degrees of delithiation. Nonetheless, our combined observations point towards lattice oxygen participation through covalency as the origin of the RIXS feature at high degrees of delithiation. We hope this directs further research into oxygen participation in conventional layered oxides including its impact on cathode stability and the formation of new surface phases driven by oxygen loss.3,14,414 Conclusion
For the past 20 years, conventional layered oxides have been widely studied and well characterized with a continued focus on utilizing their full theoretical capacity.1,2,80 From our comprehensive approach combining structural and electronic probes of the transition metal and oxygen environments, we have distinguished two charge compensation regimes: (1) low voltage regime (<4.25 V) where the ionic viewpoint of TM–O bonding is appropriate and (2) high voltage regime (>4.75 V) where oxygen participation has to be considered. In the formal TM-redox regime, shortening of the TM–O bond lengths and evolution of the electronic structure signify increasing TM–O covalency with delithiation. In the covalency regime, where there is strong hybridization between fully oxidized TM ions (Ni4+/Co4+) and oxygen, we observed the onset of RIXS features associated with oxidized oxygen alongside the c-lattice collapse. Taken together, this suggests that RIXS provides a more sensitive probe of the oxygen involvement in charge compensation for the conventional layered oxides. The observation of similar RIXS features associated with oxidized oxygen states in fully delithiated conventional and alkali-rich layered oxides needs to be addressed for any complete description of oxygen participation in charge compensation.Author contributions
The contributions of authors are as follows. Project was conceived by SS, ZWL-H and LFJP and written by ZWL-H and LFJP. The sample preparation and electrochemistry were performed by NVF, FB, NP, GGA, with additional characterization performed by PR, and CPG. Structural measurements and analysis (XRD, EXAFS) was performed by JR, MJZ, NVF, HL, TW, KC, GGA, BCM. X-ray spectroscopy measurements and analysis were performed by ZLH, WY and SS. Computational modeling was performed by MDR, JV, and AVDV. Synchrotron beamline support was provided by TP and T-LL. All authors contributed to the discussion and drafting of this manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported as part of NECCES, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award No. DE-SC0012583. X-ray spectroscopy experiments (HAXPES, XAS, RIXS, XANES) were performed at beamline 8.0.1 at the Advanced Photon Source, beamline I09 at Diamond Light Source, and beamline 9-BM at the Advanced Light Source. The work at ALS was supported by the Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Z. W. L.-H. gratefully acknowledges support from a Doctoral Fellowship in Residence Award from the ALS. S. S. gratefully acknowledges support from an ALS Collaborative Postdoctoral Fellowship. We thank Diamond Light Source for access to beamline I09 (SI19162) that contributed to the results presented here. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.References
- J. Xu, F. Lin, M. Doeff and W. Tong, J. Mater. Chem. A, 2016, 5, 874–901 RSC.
- M. D. Radin, S. Hy, M. Sina, C. Fang, H. Liu, J. Vinckeviciute, M. Zhang, M. S. Whittingham, Y. S. Meng and A. Van der Ven, Adv. Energy Mater., 2017, 7, 1602888 CrossRef.
- S.-T. Myung, F. Maglia, K.-J. Park, C. S. Yoon, P. Lamp, S.-J. Kim and Y.-K. Sun, ACS Energy Lett., 2017, 2, 196–223 CrossRef CAS.
- F. Lin, I. M. Markus, D. Nordlund, T.-C. Weng, M. D. Asta, H. L. Xin and M. M. Doeff, Nat. Commun., 2014, 5, 3529 CrossRef PubMed.
- A. O. Kondrakov, H. Geßwein, K. Galdina, L. De Biasi, V. Meded, E. O. Filatova, G. Schumacher, W. Wenzel, P. Hartmann, T. Brezesinski and J. Janek, J. Phys. Chem. C, 2017, 121, 24381–24388 CrossRef CAS.
- H. Liu, M. Wolf, K. Karki, Y.-S. Yu, E. A. Stach, J. Cabana, K. W. Chapman and P. J. Chupas, Nano Lett., 2017, 17, 3452–3457 CrossRef CAS PubMed.
- N. V. Faenza, N. Pereira, D. M. Halat, J. Vinckeviciute, L. Bruce, M. D. Radin, P. Mukherjee, F. Badway, A. Halajko, F. Cosandey, C. P. Grey, A. Van der Ven and G. G. Amatucci, Chem. Mater., 2018, 30, 7545–7574 CrossRef CAS.
- Y.-K. Sun, S.-T. Myung, B.-C. Park, J. Prakash, I. Belharouak and K. Amine, Nat. Mater., 2009, 8, 320 CrossRef CAS PubMed.
- I. D. Scott, Y. S. Jung, A. S. Cavanagh, Y. Yan, A. C. Dillon, S. M. George and S. H. Lee, Nano Lett., 2011, 11, 414–418 CrossRef CAS PubMed.
- S.-K. Jung, H. Gwon, J. Hong, K.-Y. Park, D.-H. Seo, H. Kim, J. Hyun, W. Yang and K. Kang, Adv. Energy Mater., 2014, 4, 1300787 CrossRef.
- F. Lin, D. Nordlund, Y. Li, M. K. Quan, L. Cheng, T.-C. Weng, Y. Liu, H. L. Xin and M. M. Doeff, Nat. Energy, 2016, 1, 15004 CrossRef CAS.
- J. Tarascon, G. Vaughan, Y. Chabre, L. Seguin, M. Anne, P. Strobel and G. Amatucci, J. Solid State Chem., 1999, 147, 410–420 CrossRef CAS.
- W. Li, H. Y. Asl, Q. Xie and A. Manthiram, J. Am. Chem. Soc., 2019, 141, 5097–5101 CrossRef CAS PubMed.
- M. Gauthier, T. J. Carney, A. Grimaud, L. Giordano, N. Pour, H.-H. Chang, D. P. Fenning, S. F. Lux, O. Paschos, C. Bauer, F. Maglia, S. Lupart, P. Lamp and Y. Shao-Horn, J. Phys. Chem. Lett., 2015, 6, 4653–4672 CrossRef CAS PubMed.
- M. K. Aydinol, A. F. Kohan, G. Ceder, K. Cho and J. Joannopoulos, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 1354–1365 CrossRef CAS.
- A. Van der Ven, M. K. Aydinol, G. Ceder, G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 2975–2987 CrossRef CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- W. S. Yoon, K. B. Kim, M. G. Kim, M. K. Lee, H. J. Shin, J. M. Lee, J. S. Lee and C. H. Yo, J. Phys. Chem. B, 2002, 106, 2526–2532 CrossRef CAS.
- C.-H. Chen, B.-J. Hwang, C.-Y. Chen, S.-K. Hu, J.-M. Chen, H.-S. Sheu and J.-F. Lee, J. Power Sources, 2007, 174, 938–943 CrossRef CAS.
- T. Mizokawa, Y. Wakisaka, T. Sudayama, C. Iwai, K. Miyoshi, J. Takeuchi, H. Wadati, D. G. Hawthorn, T. Z. Regier and G. A. Sawatzky, Phys. Rev. Lett., 2013, 111, 056404 CrossRef CAS PubMed.
- G. Klinser, S. Topolovec, H. Kren, S. Koller, W. Goessler, H. Krenn and R. Würschum, Appl. Phys. Lett., 2016, 109, 213901 CrossRef.
- D.-H. Seo, J. Lee, A. Urban, R. Malik and S. Kang, Nat. Chem., 2016, 8, 692–697 CrossRef CAS PubMed.
- B. Qiu, M. Zhang, Y. Xia, Z. Liu and Y. S. Meng, Chem. Mater., 2017, 29, 908–915 CrossRef CAS.
- Y. Xie, M. Saubanère and M.-L. Doublet, Energy Environ. Sci., 2017, 10, 266–274 RSC.
- B. Li and D. Xia, Adv. Mater., 2017, 29, 1701054 CrossRef PubMed.
- G. Assat and J.-M. Tarascon, Nat. Energy, 2018, 3, 373–386 CrossRef CAS.
- W. S. Yoon, K. Y. Chung, J. McBreen, D. A. Fischer and X. Q. Yang, J. Power Sources, 2007, 174, 1015–1020 CrossRef CAS.
- N. Yabuuchi, M. Takeuchi, M. Nakayama, H. Shiiba, M. Ogawa, K. Nakayama, T. Ohta, D. Endo, T. Ozaki, T. Inamasu, K. Sato and S. Komaba, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7650–7655 CrossRef CAS PubMed.
- K. Luo, M. R. Roberts, R. Hao, N. Guerrini, D. M. Pickup, Y.-S. Liu, K. Edström, J. Guo, A. V. Chadwick, L. C. Duda and P. G. Bruce, Nat. Chem., 2016, 8, 684–691 CrossRef CAS PubMed.
- T. Risthaus, D. Zhou, X. Cao, X. He, B. Qiu, J. Wang, L. Zhang, Z. Liu, E. Paillard, G. Schumacher, M. Winter and J. Li, J. Power Sources, 2018, 395, 16–24 CrossRef CAS.
- W. Yang and T. P. Devereaux, J. Power Sources, 2018, 389, 188–197 CrossRef CAS.
- K. Luo, M. R. Roberts, N. Guerrini, N. Tapia-Ruiz, R. Hao, F. Massel, D. M. Pickup, S. Ramos, Y. S. Liu, J. Guo, A. V. Chadwick, L. C. Duda and P. G. Bruce, J. Am. Chem. Soc., 2016, 138, 11211–11218 CrossRef CAS PubMed.
- W. E. Gent, K. Lim, Y. Liang, Q. Li, T. Barnes, S.-J. Ahn, K. H. Stone, M. Mcintire, J. Hong, J. H. Song, Y. Li, A. Mehta, S. Ermon, T. Tyliszczak, D. Kilcoyne, D. Vine, J.-H. Park, S.-K. Doo, M. F. Toney, W. Yang, D. Prendergast and W. C. Chueh, Nat. Commun., 2017, 8, 2091 CrossRef PubMed.
- J. Xu, M. Sun, R. Qiao, S. E. Renfrew, L. Ma, T. Wu, S. Hwang, D. Nordlund, D. Su, K. Amine, J. Lu, B. D. McCloskey, W. Yang and W. Tong, Nat. Commun., 2018, 9, 947 CrossRef PubMed.
- U. Maitra, R. A. House, J. W. Somerville, N. Tapia-Ruiz, J. G. Lozano, N. Guerrini, R. Hao, K. Luo, L. Jin, M. A. Pérez-Osorio, F. Massel, D. M. Pickup, S. Ramos, X. Lu, D. E. McNally, A. V. Chadwick, F. Giustino, T. Schmitt, L. C. Duda, M. R. Roberts and P. G. Bruce, Nat. Chem., 2018, 10, 288–295 CrossRef CAS PubMed.
- R. A. House, L. Jin, U. Maitra, K. Tsuruta, J. W. Somerville, D. P. Förstermann, F. Massel, L. Duda, M. R. Roberts and P. G. Bruce, Energy Environ. Sci., 2018, 11, 926–932 RSC.
- K. Dai, J. Wu, Z. Zhuo, Q. Li, S. Sallis, J. Mao, G. Ai, C. Sun, Z. Li, W. E. Gent, W. C. Chueh, Y. de Chuang, R. Zeng, Z. xun Shen, F. Pan, S. Yan, L. F. Piper, Z. Hussain, G. Liu and W. Yang, Joule, 2018, 3, 518–541 CrossRef.
- M. Okubo and A. Yamada, ACS Appl. Mater. Interfaces, 2017, 9, 36463–36472 CrossRef CAS PubMed.
- Z. Zhuo, C. D. Pemmaraju, J. Vinson, C. Jia, B. Moritz, I. Lee, S. Sallies, Q. Li, J. Wu, K. Dai, Y.-d. Chuang, Z. Hussain, F. Pan, T. P. Devereaux and W. Yang, J. Phys. Chem. Lett., 2018, 9, 6378–6384 CrossRef CAS PubMed.
- H. Liu, H. Liu, I. D. Seymour, N. Chernova, K. M. Wiaderek, N. M. Trease, S. Hy, Y. Chen, K. An, M. Zhang, O. J. Borkiewicz, S. H. Lapidus, B. Qiu, Y. Xia, Z. Liu, P. J. Chupas, K. W. Chapman, M. S. Whittingham, C. P. Grey and Y. S. Meng, J. Mater. Chem. A, 2018, 6, 4189–4198 RSC.
- S. Hwang, W. Chang, S. M. Kim, D. Su, D. H. Kim, J. Y. Lee, K. Y. Chung and E. A. Stach, Chem. Mater., 2014, 26, 1084–1092 CrossRef CAS.
- A. Grenier, H. Liu, K. M. Wiaderek, Z. W. Lebens-Higgins, O. J. Borkiewicz, L. F. J. Piper, P. J. Chupas and K. W. Chapman, Chem. Mater., 2017, 29, 7345–7352 CrossRef CAS.
- Z. W. Lebens-Higgins, S. Sallis, N. V. Faenza, F. Badway, N. Pereira, D. M. Halat, M. Wahila, C. Schlueter, T.-L. Lee, W. Yang, C. P. Grey, G. G. Amatucci and L. F. J. Piper, Chem. Mater., 2018, 30, 958–969 CrossRef CAS.
- C. H. Chen, J. Liu, M. E. Stoll, G. Henriksen, D. R. Vissers and K. Amine, J. Power Sources, 2004, 128, 278–285 CrossRef CAS.
- J. R. Kim and G. G. Amatucci, Chem. Mater., 2015, 27, 2546–2556 CrossRef CAS.
- H. Liu, H. Liu, S. H. Lapidus, Y. S. Meng, P. J. Chupas and K. W. Chapman, J. Electrochem. Soc., 2017, 164, A1802–A1811 CrossRef CAS.
- H. T. Stokes, D. M. Hatch, B. J. Campbell and D. E. Tanner, J. Appl. Crystallogr., 2006, 39, 607–614 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- A. Ankudinov and B. Ravel, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565–7576 CrossRef CAS.
- J. Akimoto, Y. Gotoh and Y. Oosawa, J. Solid State Chem., 1998, 141, 298–302 CrossRef CAS.
- J. Rana, R. Kloepsch, J. Li, M. Stan, G. Schumacher, M. Winter and J. Banhart, J. Electrochem. Soc., 2016, 163, A811–A820 CrossRef CAS.
- S. Calvin, Relationship Between Electron Delocalization and Asymmetry of the Pair Distribution Function as Determined by X-ray Absorption Spectroscopy, The City University of New York, 2001 Search PubMed.
- R. Qiao, Q. Li, Z. Zhuo, S. Sallis, O. Fuchs, M. Blum, L. Weinhardt, C. Heske, J. Pepper, M. Jones, A. Brown, A. Spucces, K. Chow, B. Smith, P. A. Glans, Y. Chen, S. Yan, F. Pan, L. F. Piper, J. Denlinger, J. Guo, Z. Hussain, Y. D. Chuang and W. Yang, Rev. Sci. Instrum., 2017, 88, 033106 CrossRef PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- J. Rana, S. Glatthaar, H. Gesswein, N. Sharma, J. R. Binder, R. Chernikov, G. Schumacher and J. Banhart, J. Power Sources, 2014, 255, 439–449 CrossRef CAS.
- M. N. Ates, S. Mukerjee and K. M. Abraham, RSC Adv., 2015, 5, 27375–27386 RSC.
- N. M. Trease, I. D. Seymour, M. Radin, H. Liu, H. Liu, S. Hy, N. Chernova, P. Parikh, A. Devaraj, K. M. Wiaderek, P. J. Chupas, K. W. Chapman, M. S. Whittingham, Y. S. Meng, A. Van der Ven and C. P. Grey, Chem. Mater., 2016, 28, 8170–8180 CrossRef CAS.
- M. Balasubramanian, X. Sun, X. Q. Yang and J. Mcbreen, J. Electrochem. Soc., 2000, 147, 2903–2909 CrossRef CAS.
- D. Carlier, M. Ménétrier and C. Delmas, J. Mater. Chem., 2001, 11, 594–603 RSC.
- Y. Makimura, T. Sasaki, H. Oka, C. Okuda, T. Nonaka, Y. F. Nishimura, S. Kawauchi and Y. Takeuchi, J. Electrochem. Soc., 2016, 163, A1450–A1456 CrossRef CAS.
- R. Qiao, J. Liu, K. Kourtakis, M. G. Roelofs, D. L. Peterson, J. P. Duff, D. T. Deibler, L. A. Wray and W. Yang, J. Power Sources, 2017, 360, 294–300 CrossRef CAS.
- T. Tokushima, Y. Horikawa, Y. Harada, O. Takahashi, A. Hiraya and S. Shin, Phys. Chem. Chem. Phys., 2009, 11, 1679 RSC.
- W.-S. Yoon, K.-B. Kim, M.-G. Kim, M.-K. Lee, H.-J. Shin, J.-M. Lee, J.-S. Lee and C.-H. Yo, J. Phys. Chem. B, 2002, 106, 2526–2532 CrossRef CAS.
- N. V. Faenza, Z. W. Lebens-higgins, P. Mukherjee, S. Sallis, N. Pereira, F. Badway, A. Halajko, G. Ceder, F. Cosandey, L. F. J. Piper and G. G. Amatucci, Langmuir, 2017, 33, 9333–9353 CrossRef CAS PubMed.
- R. E. Ruther, A. F. Callender, H. Zhou, S. K. Martha and J. Nanda, J. Electrochem. Soc., 2014, 162, A98–A102 CrossRef.
- H. Chen and M. S. Islam, Chem. Mater., 2016, 28, 6656–6663 CrossRef CAS.
- X. Rong, J. Liu, E. Hu, Y. Liu, Y. Wang, J. Wu, X. Yu, K. Page, Y.-S. Hu, W. Yang, H. Li, X.-Q. Yang, L. Chen and X. Huang, Joule, 2017, 2, 125–140 CrossRef CAS.
- M. Sathiya, G. Rousse, K. Ramesha, C. P. Laisa, H. Vezin, M. T. Sougrati, M. L. Doublet, D. Foix, D. Gonbeau, W. Walker, A. S. Prakash, M. Ben Hassine, L. Dupont and J. M. Tarascon, Nat. Mater., 2013, 12, 827–835 CrossRef CAS PubMed.
- M. Saubanère, E. McCalla, J.-M. Tarascon and M.-L. Doublet, Energy Environ. Sci., 2016, 9, 984–991 RSC.
- G. Assat, A. Iadecola, C. Delacourt, R. Dedryvère and J.-M. Tarascon, Chem. Mater., 2017, 29, 9714–9724 CrossRef CAS.
- N. Yabuuchi, M. Nakayama, M. Takeuchi, S. Komaba, Y. Hashimoto, T. Mukai, H. Shiiba, K. Sato, Y. Kobayashi, A. Nakao, M. Yonemura, K. Yamanaka, K. Mitsuhara and T. Ohta, Nat. Commun., 2016, 7, 13814 CrossRef CAS PubMed.
- D. Foix, M. Sathiya, E. McCalla, J. M. Tarascon and D. Gonbeau, J. Phys. Chem. C, 2016, 120, 862–874 CrossRef CAS.
- A. Marusczyk, J.-M. Albina, T. Hammerschmidt, R. Drautz, T. Eckl and G. Henkelman, J. Mater. Chem. A, 2017, 15183–15190 RSC.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Soft and hard X-ray characterization of the TM and O environments for the LiNi0.8Co0.2−yAlyO2 systems and additional structural characterization. See DOI: 10.1039/c9mh00765b |
| This journal is © The Royal Society of Chemistry 2019 |