
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Suppression of electron trapping by quantum dot emitters using a grafted polystyrene shell†
Elham
Khodabakhshi‡
a,
Benjamin
Klöckner‡
b,
Rudolf
Zentel
 b,
Jasper J.
Michels
a and
Paul W. M.
Blom
*a
b,
Jasper J.
Michels
a and
Paul W. M.
Blom
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: blom@mpip-mainz.mpg.de
bInstitute for Organic Chemistry, Johannes Gutenberg University, Duesbergweg 10–14, 55128 Mainz, Germany
First published on 25th June 2019
Abstract
A fundamental problem of adding chromophores to an organic host is that their smaller band gap leads to severe trapping of either electrons or holes, resulting in strongly unbalanced transport. We demonstrate that electron trapping by an inorganic quantum dot (QD) in a conjugated polymer host can be suppressed by functionalizing its shell with a thin insulating polystyrene layer. The polystyrene shell not only reduces trapping, but also suppresses detrapping of captured electrons, resulting in increased charging of the QDs with subsequent voltage scans, after initial charging, a red-emitting hybrid polymer:QD light-emitting diode is obtained with voltage independent electroluminescence spectrum and equal efficiency as the blue polymer host.
New conceptsIn displays it is important to have small linewidths to make saturated colors. This is why nowadays quantum dots (QDs) are being used as phosphor, meaning that they convert light generated by a blue/white LED backlight. An even more attractive device would be to incorporate the QDs directly in an electroluminescent device such that no backlight is needed, the QD LED. However, until now this approach only works when a very thin emissive layer containing only a few monolayers of QDs is used, surrounded by a number of organic transport and confinement layers, of which the thickness is very critical. A major problem is that in the QD emissive layer the charge transport needs to be exactly balanced, otherwise the layer gets charged with one type of carrier, which blocks the current. The way out to a robust and simple structure would be to use a thicker layer where the QDs are simply blended with an organic host. All attempts in this direction failed due to the problem that due to their high electron affinity QDs are deep electron traps, leading to strongly unbalanced transport. Here we demonstrate that electron trapping by CdSe/CdxZn1−xS core/shell red QDs in a blue-emitting poly(di-octylfluorene) (PFO) host can be strongly suppressed by functionalizing them with a thin insulating shell of polystyrene. The strong reduction of trapping is confirmed by charge transport measurements and a voltage independent electroluminescence spectrum of hybrid polymer:QD blend LEDs. Our results open a new route towards emissive devices with narrow linewidth, where due to the preservation of charge transport there are no limits to the active layer thickness. |
Introduction
Organic light-emitting diodes1 are being applied in displays and are a promising option for large-area lighting panels,2 eventually produced by roll-to-roll processes. A disadvantage of using organic semiconductors for displays and lighting is their relatively broad emission spectrum due to inhomogeneous broadening. The emission spectrum can be considerably narrowed by blending a blue-emitting organic host with a green or red-emitting (phosphorescent) dye with narrow linewidth.3 In polymer based LEDs it has been demonstrated that due to efficient energy transfer from the host to the dye already for 1% dye concentration 95% of the blue excitons are transferred to the dye.4 For display fabrication using ink-jet printing this is convenient since such a small dye concentration does not affect the rheology of the solution, allowing all colors to be printed using the same host and identical conditions.5 A reduction of the dye concentration towards 0.1% or less, giving rise to a partial energy transfer, can be used for the generation of white light.6–8 This allows for the fabrication of lighting devices consisting of only one solution-processed layer. However, for efficient energy transfer the absorption spectrum of the dye has to overlap with the emission spectrum of the host, such that by definition dyes have a smaller bandgap as compared to the host. This will inevitably lead to severe trapping of charge carriers, as indicated in Fig. 1a. For a white-emitting polymer with only 0.02% red dye incorporated in the blue-emitting main chain it has been demonstrated that the electron transport was reduced by several orders of magnitude.9 The resulting strongly imbalanced transport leads to a strong reduction of the efficiency of the corresponding PLED containing dyes. Another unwanted side-effect of the strong electron trapping by the dyes is that the electroluminescence spectrum becomes voltage dependent. The blue emission of the host is governed by bimolecular Langevin recombination, with a rate given by| RL = BLnp | (1) |
| RSRH = BSRHntp | (2) |
| RSRH = BSRHNtp | (3) |
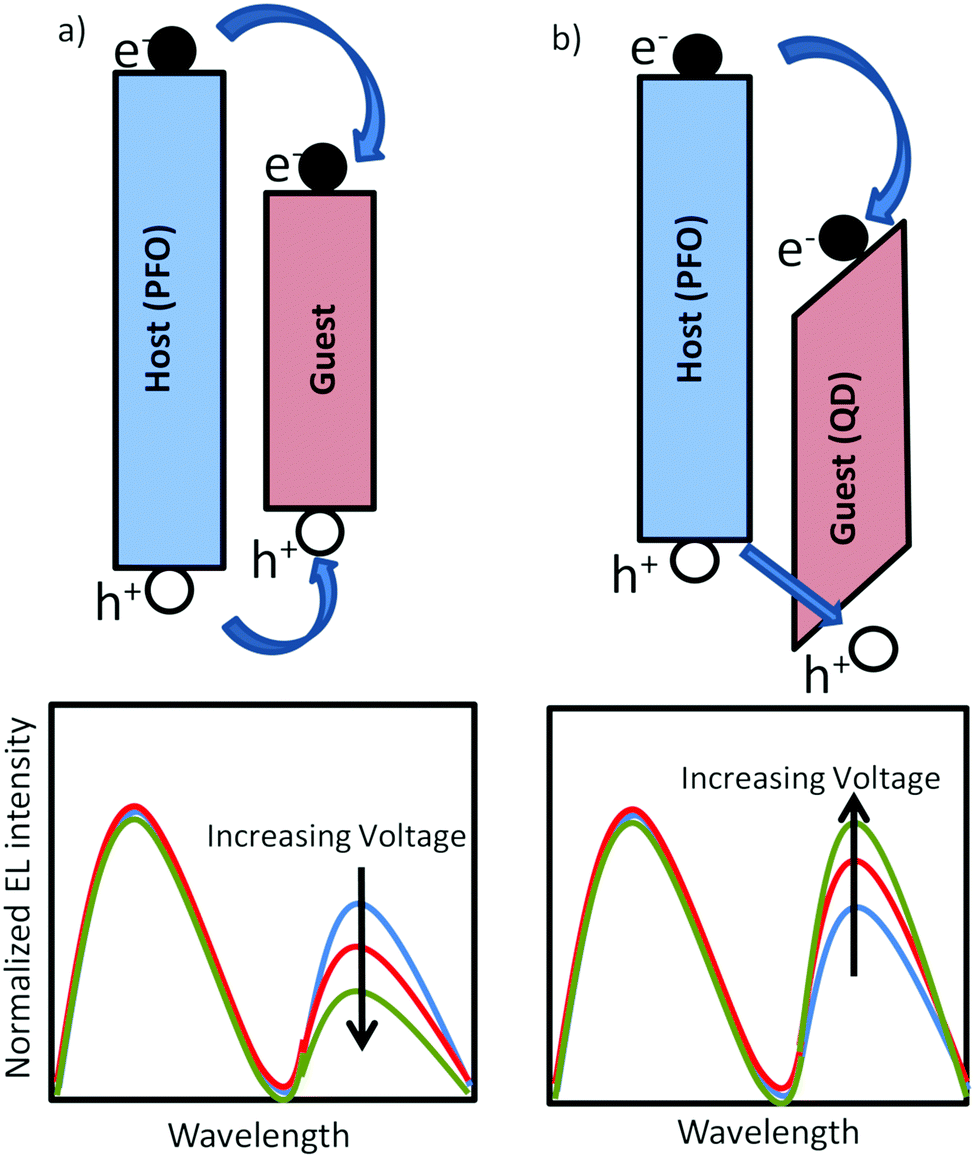 | ||
| Fig. 1 Schematic energy diagram and electroluminescence spectrum of (a) blend of large band gap organic host and an organic dye resulting in severe electron trapping. (b) Blend of large band gap organic host and an inorganic QD, leading to electron trapping and charging effects that enhance hole capture. | ||
An interesting alternative for organic dyes are inorganic quantum dots (QDs). High quality colloidal core/shell semiconductor quantum dots (QDs) offer stable, tunable narrow and intense photoemission as function of size in the visible range.10–15 These properties can be exploited in solution-processed hybrid QD/organic polymer light-emitting diodes, which combine the emitting properties of QDs with the flexibility in device construction of the polymeric materials.16–19 However, the conduction and valence band of inorganic semiconductors as CdSe and ZnS are deeper in energy as compared to the corresponding molecular orbitals of most organic hosts, as schematically indicated in Fig. 1b, resulting in strong electron trapping. Furthermore, the holes have to surmount an energy barrier before they can recombine with the trapped electron on the QD. As a result the QD emission can be a combination of Förster energy transfer and trap-assisted recombination.17 In multilayer organic LEDs based on evaporated small molecules, one or a few monolayers of QDs are sandwiched between electron and hole transporting organic layers.11,12 The trapped electrons are then confined in a narrow QD region, enabling the trapped electrons to build up a large electric field that enhances the hole injection into the deep QD valence band. With increasing voltage this charging effect becomes more dominant such that the QD emission grows relative to the host emission (Fig. 1b), which is the opposite spectral dependence as compared to the typical trap-assisted recombination occurring with organic dyes (Fig. 1a). However, at high voltages the high carrier concentration and electric field in the confined QD layers leads to efficiency losses due to Auger recombination and field-induced spatial separation of the electron and hole wave function.20 For solution processed devices such a multilayer approach combined with a few monolayers of QDs is not feasible. Realization of balanced charge transport remains a fundamental problem due to the severe electron trapping of the QDs.21,22 Next to the mismatch in energy levels between organic semiconductors and inorganic QDs for hybrid polymer:QD blends also the blend morphology plays a crucial role.23 For the incorporation of a larger amount of QDs within a polymer matrix the physical blending of the two components is usually insufficient to obtain a homogeneous distribution of QDs and phase separation typically occurs. By hybridizing QDs with conducting polymer brushes a more homogeneous distribution of QDs in the polymer matrix was obtained,24 leading to a better distribution of charges and excitons across the active layer with a reduced efficiency roll-off.23,25
Ideally, a solution would be preferred where the negative contribution of severe electron trapping by the QDs is reduced. Furthermore, a homogeneous distribution of the QDs in the polymer host matrix is required, which can be achieved by coating them with a suitable polymer.24 Charge transport and electron trapping in conjugated polymers has been extensively studied.26 The hopping distance for charge carriers is determined by the wave function overlap of the localized sites and typically amounts to 1.5–2 nm.26 This distance also governs the charge transfer process from a host polymer into a trap. In contrast, fluorescence resonance energy transfer (FRET) is driven by dipole–dipole interaction between an excited donor molecule and an acceptor molecule, with a Förster radius of 5–8 nm.27 Grafting of a thin insulting shell with thickness 3–5 nm on the QD is therefore expected to have a stronger effect on trapping as compared to energy transfer. In the present study we demonstrate that electron trapping by CdSe/CdxZn1−xS core/shell red QDs in a blue-emitting poly(dioctylfluorene) (PFO) host can be suppressed by functionalizing them with a thin insulating shell of polystyrene. The strong reduction of trapping is confirmed by charge transport measurements. Upon charging of the QDs a voltage independent electroluminescence spectrum, dominated by QD emission, is obtained for the hybrid polymer:QD blend LEDs.
Results and discussion
We designed a novel QD/polymer hybrid material in which the surface of QDs is covered with a thin shell of insulating polystyrene. In addition, the polymer coverage of the QD allows us to tune the miscibility with the host polymer matrix to obtain a homogenous distribution of the QDs. The ligand exchange procedure enables the synthesis of various QD/PS hybrids by replacing the initial stabilizing oleic acid ligands coordinated onto the QDs surface with new polymeric ligands containing anchor units.28 Reversible addition–fragmentation chain transfer (RAFT) radical polymerization and post-polymerization modification techniques were employed for the synthesis of the diblock copolymer poly(styrene-block-cysteaminemethyldisulfide) (P(S-b-SSMe)) containing a chemical and electrochemical robust polystyrene block and cysteaminemethyldisulfide anchoring block, as shown in Fig. 2a. First, the polystyrene block was synthesized and used as a macro-initiator in a second RAFT polymerization for the polymerization of the reactive ester monomer pentafluorophenyl acrylate (PFPA) leading to diblock copolymer. After polymerization, the reactive CTA group was replaced with an inert 2-cyanoisoproyl group by reaction with excess of AIBN to obtain defined end-groups on both sides of the polymer. For the anchor block, cysteaminemethyldisulfide was chosen since due to its high affinity to unsaturated Zn-centres it enables the replacement of pristine oleic acid ligands that are initially grafted on the QDs for stabilization.29,30 The ligand exchange (Fig. 2b) was further monitored with the solubility change of the QDs, IR spectroscopy and thermogravimetric analysis (Fig. S1–S3, ESI†) as well as NMR spectroscopy and size-exclusion chromatography (Fig. S4–S6, ESI†). The obtained molecular weight of the diblock copolymer P(S-b-SSMe) amounts to 2600 g mol−1.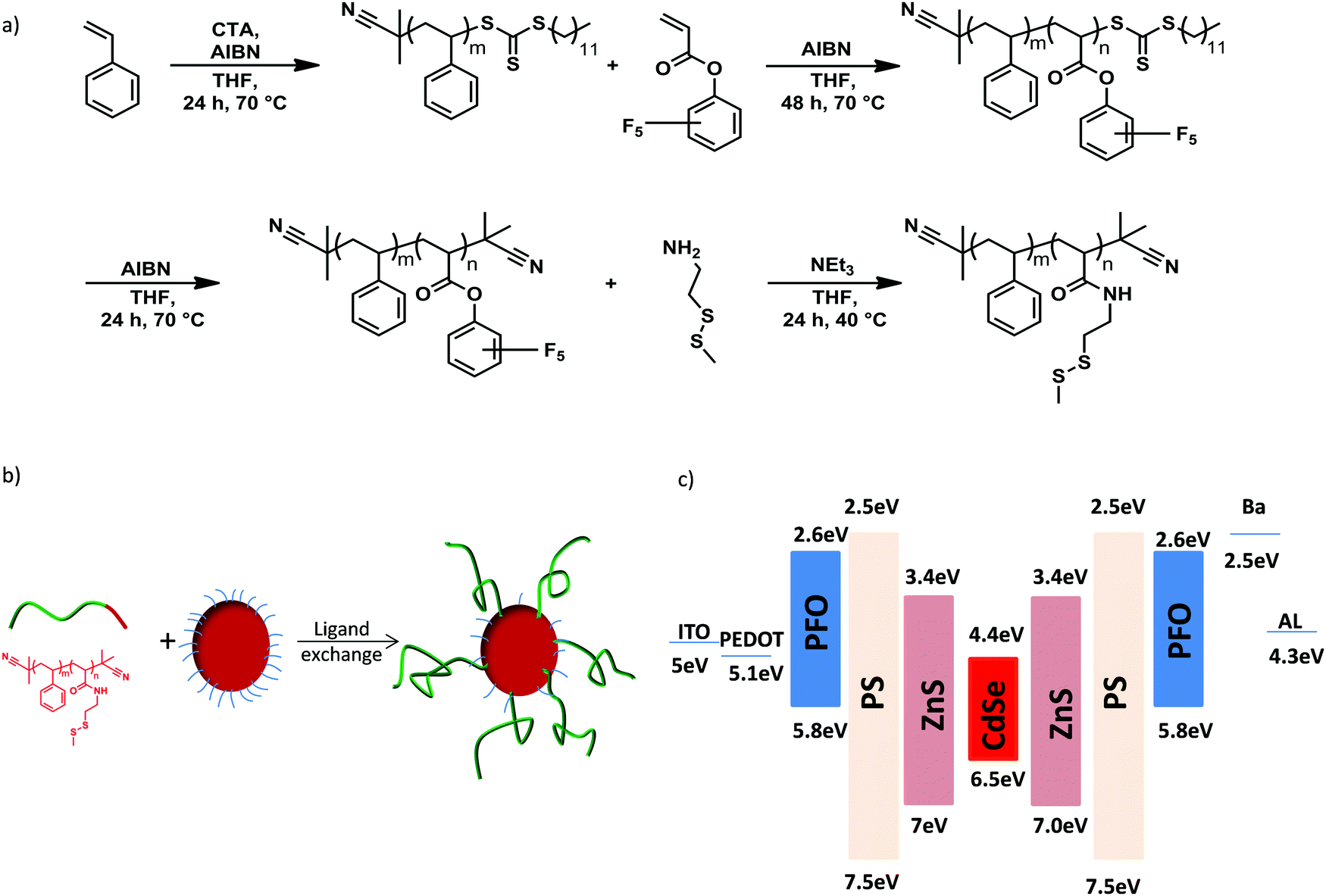 | ||
| Fig. 2 (a) Synthesis of diblock copolymer poly(styrene-block-cysteaminemethyldisulfide) (P(S-b-SSMe)), (b) the ligand exchange procedure and (c) energy-band diagram of PFO:QD system. | ||
The thickness of the grafted polystyrene shell can be estimated from the dimensions of the polymer chain. The persistence length of polystyrene amounts to lp ∼ 1 nm. Our polystyrene blocks consist of about 20 repeat units. We hence estimate the contour length of the polymer to be Rmax ≈ 5 nm, based on an inter-monomer spacing of ∼2.5 Å. For the determination of the shell thickness the root-mean-square end-to-end distance of the chain is needed, which according to the worm-like chain model31 can be written as a function of lp and Rmax:
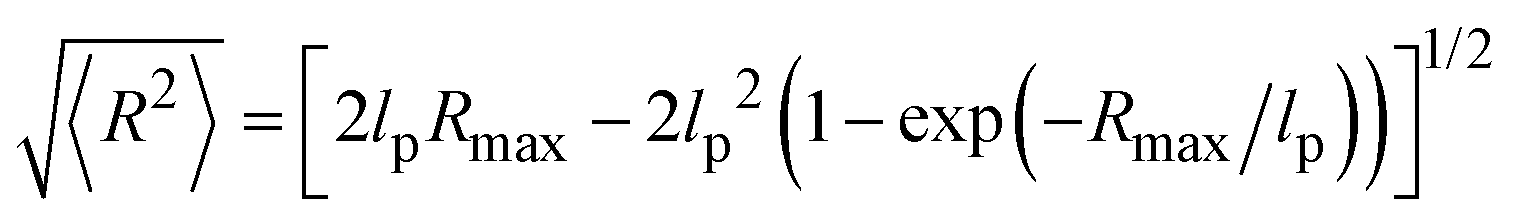 | (4) |
Plugging in the above given estimates for lp and Rmax gives 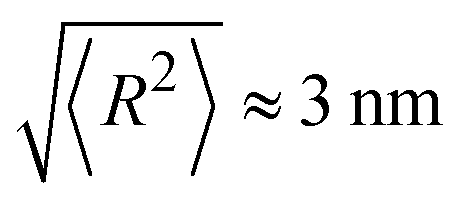 , which should be sufficient to suppress charge transfer to the QDs.
, which should be sufficient to suppress charge transfer to the QDs.
To investigate the effect of the PS shell on performance of PFO:QDs hybrid LEDs we first compare the electroluminescence spectra of blends of PFO with the unmodified QDs, containing oleic acid ligands (oa-QDs), and with the QD/PS-hybrids, respectively. As is evident from the energy diagram, (Fig. 2c) QDs with a ZnS shell are expected to act as an electron trapping center in PFO. To investigate the mechanism of the QD emission we chose a QD concentration of 3 wt%, in which part of the blue emission from the host can still be observed.
The voltage-dependence of the normalized (to the blue emission) EL spectra of the PLEDs with 3% QD are shown in Fig. 3a and b (unmodified oa-QD) and Fig. 3c–f (QD/PS-hybrid), respectively. As expected, the FWHM linewidth of the red QD emission is significantly smaller as compared to the blue PFO emission (0.1 eV vs. 0.5 eV, Fig. S7, ESI†). For PLEDs based on the PFO:oa-QD blend the contribution of the red QD emission reduces at higher voltages, both in the up-scan (Fig. 3a) and down-scan (Fig. 3b), which is a fingerprint for trap-assisted recombination. For every subsequent voltage scan the magnitude and voltage dependence of the EL spectrum remains identical (not shown). The spectra are not dependent on the bias history of the device, indicating that the QDs charge and discharge with every up- and down scan, respectively. Furthermore, we observe that for the unmodified oa-QD a significant blue emission remains. In contrast, for the QD/PS-hybrid blends in the first up-scan the contribution from the QD is very small and voltage independent, whereas during the down-scan the relative contribution from the red QD emission strongly grows. This behaviour, a voltage independent EL spectrum in the up-scan and larger but voltage dependent QD emission in the down-scan repeats in subsequent scans (Fig. S8, ESI†). Also, with every new scan the overall QD contribution to the EL spectrum is becoming slightly larger. After five scans (Fig. 3e and f) the contribution from the QD emission is comparable to the spectra from the PFO:oa-QD blend (Fig. 3a and b), but now the spectrum is nearly voltage independent. These EL spectra and corresponding voltage independence are maintained in subsequent scans, even when taken after ten days. We note that the lower noise level in the EL spectra of the PFO:oa-QD blends (Fig. 3a and b) is the result of an increased integration time of the detector.
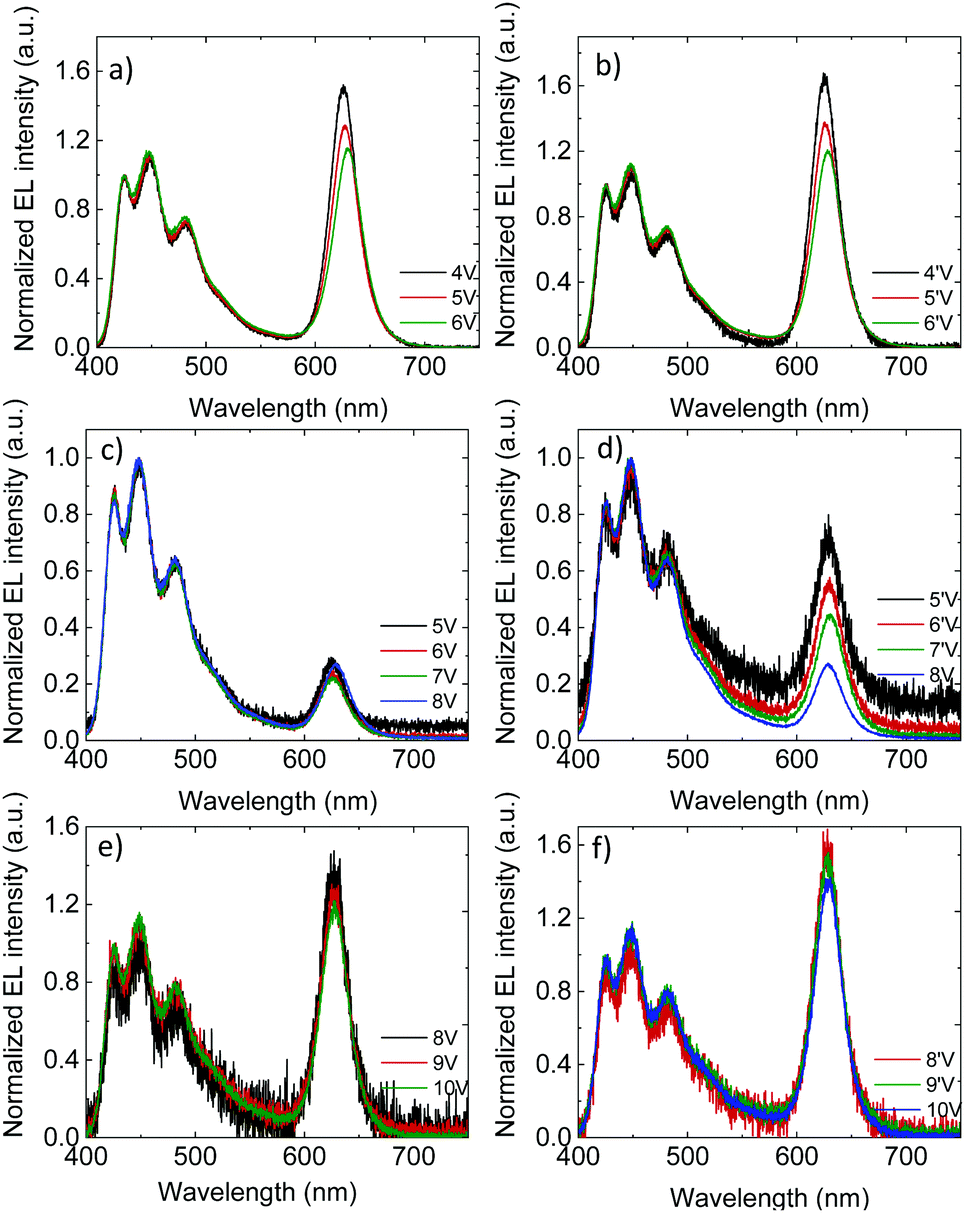 | ||
| Fig. 3 Voltage dependence of normalized EL spectra for PLED with 100 nm PFO:3% oa-QD/PS blend active layer (a) first up-scan and (b) consecutive down-scan. Voltage dependence of normalized EL spectra for PLED with 100 nm PFO:3% QD/PS-hybrid blend active layer (c) first up-scan and (d) consecutive down-scan, (e) fifth up-scan and (f) corresponding down-scan. | ||
The voltage dependence of the EL spectra of the PFO:oa-QD blend already indicates that the red QD emission is dominated by trap-assisted recombination. Clearly, coverage of the QD with an oleic-acid ligand, which is representative for an insulating shell of only ∼1.5 nm, does not suppress electron trapping. The near absence of energy transfer is further confirmed by photoluminescence measurements, where the contribution of the QD is hardly visible (Fig. S9, ESI†). The very small contribution of red QD emission during the first voltage up-scan of the QD/PS-hybrid blend indicates that now the trap-assisted recombination of the QDs is strongly suppressed due to reduced electron trapping. By varying the molecular weight of the PS from 2600 g mol−1 to 9800 g mol−1, corresponding to a variation in PS shell thickness from 3 nm to 6 nm, we demonstrate that the contribution of red QD emission to the EL spectra strongly decreases with increasing PS layer thickness (Fig. S10, ESI†). This strong dependence is the result of the exponential dependence of charge transfer on distance.
One could argue that the relative large blue contribution to the electroluminescence of the PFO:oa-QD and QD/PS-hybrid blends could also have a morphological origin. Strong phase separation leading to pure PFO regions would also enhance the blue emission. However, as shown in the Scanning Electron Microscopy (SEM) images of Fig. 4a and b for both types of QD with 5 wt% there is no significant macroscopic phase separation observed and the QDs are well dispersed (Fig. S11, ESI†). By further increasing the QD content we found that for the QD/PS-hybrid phase separation starts to occur for loadings of about 7% as shown in Fig. S12 (ESI†).
 | ||
| Fig. 4 (a) SEM image of PLED active layer with PFO:5% oa-QDs. (b) SEM image of PLED active layer with PFO:5% QD/PS-hybrids. | ||
To verify the reduced electron trapping in the QD/PS-hybrid blends further we have carried out charge transport measurements on pristine PFO and PFO blended with oa-QDs and QD/PS-hybrids, respectively. In Fig. 5a the electron current is shown using Al/blend/Ba/Al electron-only devices. It is observed that the incorporation of 5% of QD/PS-hybrids leads to a reduction of the electron current of only one order of magnitude. In contrast, the incorporation of 5% of unmodified oa-QDs reduces the electron current nearly by 3 orders of magnitude and clearly shows strong trapping behaviour. The trap-limited electron current of pristine PFO can be described using a Gaussian trap distribution26 and a universal trap density of 2 × 1017 cm−3. Estimating the effect of electron trapping by the QDs is difficult since it is not known how many electrons are trapped by a single QD. However, we can make a relative estimation by calculating the amount of traps that would be required to explain the observed J–V characteristics under the assumption that each trap captures one electron. From numerical device modelling we then obtain that a reduction by one order of magnitude of the electron current of pristine PFO corresponds to 4 × 1016 cm−3 additional single level traps with trap depth of 0.8 eV (Fig. 2c). For the oa-QDs 2 × 1017 cm−3 additional traps are required to describe the electron current. From this relative comparison we obtain that the PS shell suppresses the trapping efficiency by approximately a factor of 5.
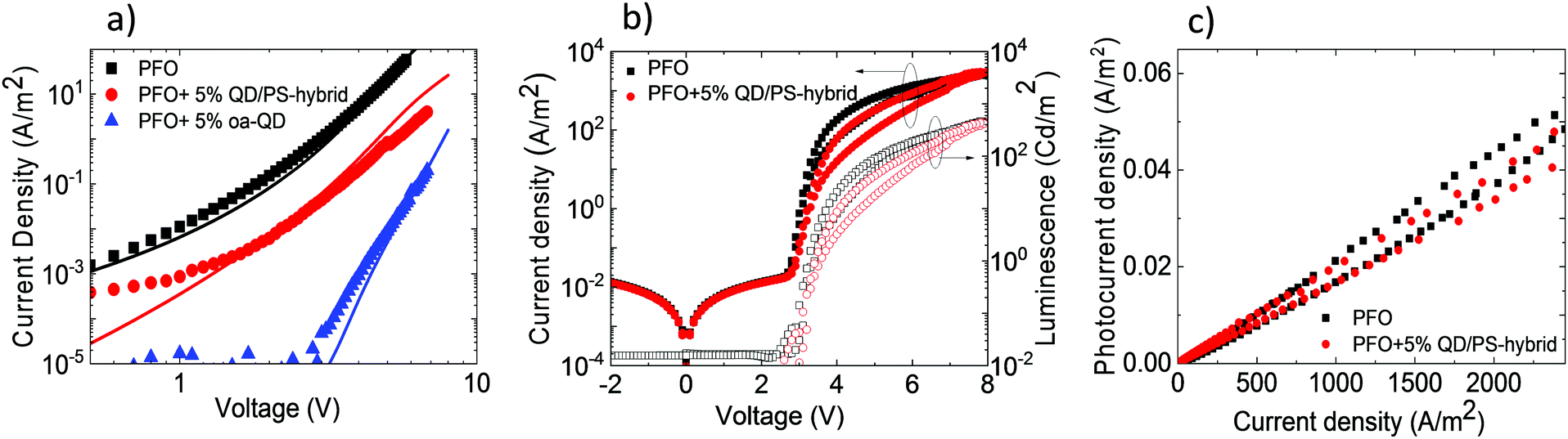 | ||
| Fig. 5 (a) Experimental electron-only currents (symbols) and modelled currents using additional single-level traps (lines) as function of voltage for pristine PFO and blends of PFO with 5% oa-QD and QD/PS-hybrid with 110 nm active layer, (b) current density and photocurrent density vs. voltage after initial charging, (c) photocurrent efficiency vs. current density of PLED. | ||
We furthermore verified that the incorporation of the QD/PS-hybrids does not significantly affect the hole transport (Fig. S13, ESI†).
As mentioned above, the voltage dependence of the EL spectra for PFO:oa-QD blend PLEDs is characteristic for trap-assisted recombination. Due to the severe trapping the trap-assisted recombination mainly takes place close to the cathode where most of the traps are filled, such that eqn (3) applies, giving rise to a linear dependence on carrier density. However, due to the strongly reduced electron trapping in the PFO:QD/PS-hybrid blends the traps are only partially filled, such that eqn (2) applies. Since QDs have a well-defined conduction band level, the QDs can be considered as single level traps. For a single level trap nt is proportional to n, such that eqn (1) and (2) exhibit an identical voltage dependence. This is the reason why during the up-scan the EL spectrum is voltage independent. However, next to the reduced trapping the PS shell also inhibits escape from electrons that are trapped in a QD. As a result, during the back scan the electrons that were being trapped at 8 V remain in the QDs, such that the trap-assisted recombination can be approximated by RSRH = BSRH × nt(@8 V) × p, leading again to a linear dependence on carrier density. This not only leads to a voltage dependence in the EL spectrum, but also to a stronger contribution of the QD emission at lower voltages as compared to the up-scan, due to the larger amount of trapped electrons. This process repeats in subsequent scans. Since in the subsequent up-scans the amount of trapped electrons is further increased the red emission of the QDs gets more pronounced after each scan.
We note that due to the ‘permanently’ trapped electrons the system is out of thermal equilibrium. The increased amount of trapped electrons also enhances the injection of holes into the QDs due to the build-up of an electric field in the QDs, This charging effect typically leads to an enhanced contribution of QD emission. After about five scans a steady-state is reached, where the effect of trap-assisted recombination and charging on the EL spectra cancel each other, leading to nearly voltage independent EL spectra for both up and down scan.
Finally, PLEDs with 5% QD/PS-hybrids were fabricated and after initial charging (five J–V scans) compared with pristine PFO PLEDs. As can be seen in Fig. 5b the current and light-output measured by the photocurrent density of a Si photodiode remain almost unchanged. We note that the measured photocurrent density is corrected for the difference in sensitivity for blue (PFO) and red (QD) emission of the Si-photodiode. As a result (Fig. 5c) we were able to obtain enhanced voltage independent red QD emission without losing efficiency, as is typical for standard polymer:QD blends due to imbalanced transport as result of severe electron trapping. We verified that the concept of electron trapping suppression also applies to other blue-emitting polymers as polyspirobifluorene (PSF), copolymerized with N,N,N′,N′-tetraaryldiamino biphenyl (TAD), as shown in Fig. S14 (ESI†) However, we note that in this PSF-TAD polymer the hole transporting TAD units lift the HOMO level up to 5.2 eV, thereby increasing the energy barrier for hole transfer to the QD. As a result, the contribution of the red QD decreases when blended with PSF-TAD as compared to PFO, which has a deeper HOMO of 5.8 eV (Fig. 2c).
Furthermore, initial lifetime experiments show that the decrease of light-output under current stress is reduced for the QD/PS-hybrid blends as compared to pristine PFO PLEDs (Fig. S15, ESI†), which is a subject of further study.
Conclusions
In conclusion, we have presented a new concept how to incorporate highly fluorescent quantum dots into a conjugated polymer matrix without inducing severe electron trapping. Trapping of electrons is prevented by shielding of the quantum dot by a thin insulating shell. The reduced trapping is demonstrated by the realization of a red-emitting hybrid polymer:QD light-emitting diode with voltage independent electroluminescence spectrum and no efficiency loss as compared to the matrix.Experimental section
Materials
Styrene (Acros) was distilled before use, 2-(Dodecylthiocarbonothioylthio)-2-methylpropionic acid (CTA) was used as purchased (Sigma-Aldrich). 2,2-Azobis(2-methyl-propionitrile) (AIBN) was purchased from Acros and recrystallized from diethyl ether prior to use. Cysteaminemethyldisulfide, pentafluorophenyl acrylate (PFPA) and quantum dots with CdSe core, CdxZn1−xS shell (core diameter 4 nm, total diameter 16 nm with oleic acid surface ligands) were synthesized according to literature.30,32,33 PFO was synthesized via the Yamamato method according to literature (Mw = 230 kDa, PDI = 3.02).34 THF was dried over sodium and distilled prior to use and all other solvents were used without further purification.Materials characterization
For all polymers, size exclusion chromatography (SEC) was performed in THF with polystyrene as external and toluene as an internal standard to calculate the molecular weights. Both a refractive index detector (G 1362A RID, Jasco) and a UV/vis detector (UV-2075 Plus, Jasco) were used to detect the polymer. TGA measurements were performed at a PerkinElmer Pyris 6 TGA under nitrogen flow. Heating rate was 10 °C min−1 from 50 to 700 °C. Infrared spectroscopy was performed on a Jasco FT/IR-4100 with an ATR sampling accessory (MIRacle, Pike Technologies) using 16 scans per measurement. IR spectra were analyzed using Spectra Manager 2.0 (Jasco).Synthesis of poly(styrene-block-cysteamine methyl disulfide) (P(S-b-SSMe))
At first, the macro chain transfer agent (macro-CTA) was synthesized: styrene (60 eq.), 2-cyano-2-propyl dodecyl trithiocarbonate (CTA) (1 eq.) and 2,2′-azobis(2-methylpropionitrile) (AIBN) (0.1 eq.) were dissolved in dry THF. After three freeze–pump–thaw cycles the flask was filled with nitrogen and the solution was heated to 70 °C for 24 hours. The macro-CTA was purified by dissolution in THF and precipitation from methanol three times and dried under vacuum for 24 h. Mn,SEC: 2000 g mol−1, Mw/Mn = 1.17. Second, the reactive diblock copolymer was synthesized: the macro-CTA (1 eq.), pentafluorophenyl acrylate (PFPA) (30 eq.) and AIBN (0.1 eq.) were dissolved in dry THF. After three freeze–pump–thaw cycles the flask was filled with nitrogen and the solution was heated to 70 °C for 48 hours. The diblock copolymer was purified by dissolution in THF and precipitation from hexane three times. Then, the CTA end group was removed directly. The diblock copolymer (1 eq.) was dissolved in dry THF. AIBN (70 eq.) was added and the mixture was stirred at 70 °C for 24 hours. The reaction solution was cooled down and precipitated into hexane three times. The polymer appeared as white powder. Mn,SEC: 3300 g mol−1, Mw/Mn = 1.19. Third, the diblock copolymer (1 eq.), cysteaminemethyldisulfide (30 eq.) and triethylamine (60 eq.) were dissolved in dry THF and heated to 40 °C for 24 hours under an argon atmosphere. The solution was purified by precipitation from hexane three times. The desired diblock copolymer P(S-b-SSMe) was dried under vacuum for 24 h. Mn,SEC: 2600 g mol−1, Mw/Mn = 1.18.Quantum dot/polymer hybridization
The QD/PS-hybrids were prepared using CdSe/CdxZn1−xS core/shell Type-I hetero-structured QDs with a core diameter of 4 nm and a total diameter of 16 nm and the corresponding diblock copolymer. This specific type of QDs has extensively been used in our previous works in the field of QLED and QD-semiconducting polymer hybrid studies.24,28 The QD/PS-hybrids were surface functionalized with polymer chains by the ligand exchange procedure:35,36 diblock copolymer P(S-b-SSMe) and quantum dots (QD, red, CdSe core, core diameter 4 nm, CdxZn1−xS shell, total diameter 16 nm, oleic acid ligands) were separately dispersed in chlorobenzene (each 100 μL) and subsequently combined (weight ratio polymer to QD 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The reaction mixture was sonicated for one hour and ethanol (1.5 mL) was added to precipitate the functionalized QDs. The precipitate was dispersed in chlorobenzene (250 μL), sonicated for one additional hour and left at room temperature overnight. Ethanol (1.5 mL) was added to precipitate the functionalized QDs. The precipitate was dispersed in chlorobenzene (250 μL) and sonicated for one additional hour at room temperature. Hexane (1.5 mL) was added to remove the remaining QDs which were not coated with the diblock copolymer and to precipitate the functionalized QDs. Finally, the precipitate was dispersed in chlorobenzene to obtain the desired QD-/polymer hybrid solution.
:1). The reaction mixture was sonicated for one hour and ethanol (1.5 mL) was added to precipitate the functionalized QDs. The precipitate was dispersed in chlorobenzene (250 μL), sonicated for one additional hour and left at room temperature overnight. Ethanol (1.5 mL) was added to precipitate the functionalized QDs. The precipitate was dispersed in chlorobenzene (250 μL) and sonicated for one additional hour at room temperature. Hexane (1.5 mL) was added to remove the remaining QDs which were not coated with the diblock copolymer and to precipitate the functionalized QDs. Finally, the precipitate was dispersed in chlorobenzene to obtain the desired QD-/polymer hybrid solution.
Device fabrication
PFO and QD/PS-hybrid solutions were prepared by dissolving PFO and QD/PS-hybrids in chlorobenzene and consequently blending the solutions using different weight ratios. Films of the pristine conducting polymer and also the hybrid solution were applied using standard spin-coating methods.The electron current through the blends was measured using electron-only devices having a glass/Al (30 nm)/polymer-QD/Ba (5 nm)/Al (100 nm) architecture. For hole-only and light emitting (dual carrier) devices, a 60 nm hole-injection layer of poly(3,4 ethylenedioxythiophene):poly(styrene sulfonic acid) (PEDOT:PSS) (Heraeus Clevios 4083) was spin-coated on ITO-patterned substrates and annealed at 140 °C. Subsequently, the hybrid solution was deposited by spin coating. As top contacts, for hole-only devices MoO3 (10 nm)/Al (100 nm) and for PLEDs Ba (5 nm)/Al (100 nm) were thermally evaporated. After device preparation, steady-state current–voltage measurements were performed in inert (N2) atmosphere using a Keithley 2400 source meter.
Light output was recorded with a calibrated Si photodiode, and electroluminescence (EL) spectra were recorded with a USB4000 UV-Vis-ES Ocean Optics spectrometer. In order to calibrate the photometric brightness we used a luminance meter Konica Minolta LS-110. The measured intensities in candelas were used to convert the photocurrent of the Si diode to luminance (cd m−2).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Michelle Beuchel, Christian Bauer, Frank Keller, and Hanspeter Raich for technical support. Open Access funding provided by the Max Planck Society.References
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS.
- S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lüssem and K. Leo, Nature, 2009, 459, 234 CrossRef CAS PubMed.
- S. R. Forrest, Nature, 2004, 428, 911 CrossRef CAS PubMed.
- T. Virgili, D. G. Lidzey and D. D. Bradley, Adv. Mater., 2000, 12, 58 CrossRef CAS.
- S.-C. Chang, J. Bharathan, Y. Yang, R. Helgeson, F. Wudl, M. B. Ramey and J. R. Reynolds, Appl. Phys. Lett., 1998, 73, 2561 CrossRef CAS.
- M. Berggren, O. Inganäs, G. Gustafsson, J. Rasmusson, M. R. Andersson, T. Hjertberg and O. Wennerström, Nature, 1994, 372, 444 CrossRef CAS.
- J. Kido, H. Shionoya and K. Nagai, Appl. Phys. Lett., 1995, 67, 2281 CrossRef CAS.
- C. Ego, D. Marsitzky, S. Becker, J. Zhang, A. C. Grimsdale, K. Müllen, J. D. MacKenzie, C. Silva and R. H. Friend, J. Am. Chem. Soc., 2003, 125, 437 CrossRef CAS PubMed.
- H. T. Nicolai, A. Hof and P. W. M. Blom, Adv. Funct. Mater., 2012, 22, 2040 CrossRef CAS.
- X. Dai, Z. Zhang, Y. Jin, Y. Niu, H. Cao, X. Liang, L. Chen, J. Wang and X. Peng, Nature, 2014, 515, 96 CrossRef CAS PubMed.
- Y. Yang, Y. Zheng, W. Cao, A. Titov, J. Hyvonen, J. R. Manders, J. Xue, P. H. Holloway and L. Qian, Nat. Photonics, 2015, 9, 259–266 CrossRef CAS.
- B. S. Mashford, M. Stevenson, Z. Popovic, C. Hamilton, Z. Zhou, C. Breen, J. Steckel, V. Bulovic, M. Bawendi and S. Coe-Sullivan, Nat. Photonics, 2013, 7, 407 CrossRef CAS.
- Y. Shirasaki, G. J. Supran, W. A. Tisdale and V. Bulović, Phys. Rev. Lett., 2013, 110, 217403 CrossRef PubMed.
- Y. Shirasaki, G. J. Supran, M. G. Bawendi and V. Bulović, Nat. Photonics, 2013, 7, 13 CrossRef CAS.
- K.-H. Lee, J.-H. Lee, H.-D. Kang, B. Park, Y. Kwon, H. Ko, C. Lee, J. Lee and H. Yang, ACS Nano, 2014, 8, 4893 CrossRef CAS PubMed.
- Q. Sun, Y. A. Wang, L. S. Li, D. Wang, T. Zhu, J. Xu, C. Yang and Y. Li, Nat. Photonics, 2007, 1, 717 CrossRef CAS.
- P. T. Chin, R. A. Hikmet and R. A. Janssen, J. Appl. Phys., 2008, 104, 013108 CrossRef.
- M.-L. Tu, Y.-K. Su and R.-T. Chen, Nanoscale Res. Lett., 2014, 9, 611 CrossRef PubMed.
- Y. Liu, F. Li, Z. Xu, C. Zheng, T. Guo, X. Xie, L. Qian, D. Fu and X. Yan, ACS Appl. Mater. Interfaces, 2017, 9, 25506 CrossRef CAS PubMed.
- D. Bozyigit and V. Wood, MRS Bull., 2013, 38, 731 CrossRef CAS.
- Y. R. Park, H. Y. Jeong, Y. S. Seo, W. K. Choi and Y. J. Hong, Sci. Rep., 2017, 7, 46422 CrossRef CAS PubMed.
- Y. R. Park, J. H. Doh, K. Shin, Y. S. Seo, Y. S. Kim, S. Y. Kim, W. K. Choi and Y. J. Hong, Org. Electron., 2015, 19, 131 CrossRef CAS.
- W. K. Bae, J. Lim, M. Zorn, J. Kwak, Y.-S. Park, D. Lee, S. Lee, K. Char, R. Zentel and C. Lee, J. Mater. Chem. C, 2014, 2, 4974 RSC.
- J. Kwak, W. K. Bae, M. Zorn, H. Woo, H. Yoon, J. Lim, S. W. Kang, S. Weber, H. J. Butt and R. Zentel, Adv. Mater., 2009, 21, 5022 CrossRef CAS PubMed.
- A. Fokina, Y. Lee, J. H. Chang, M. Park, Y. Sung, W. K. Bae, K. Char, C. Lee and R. Zentel, Adv. Mater. Interfaces, 2016, 3, 1600279 CrossRef.
- M. Kuik, G. J. A. Wetzelaer, H. T. Nicolai, N. I. Craciun, D. M. De Leeuw and P. W. Blom, Adv. Mater., 2014, 26, 512 CrossRef CAS PubMed.
- M. Anni, L. Manna, R. Cingolani, D. Valerini, A. Creti and M. Lomascolo, Appl. Phys. Lett., 2004, 85, 4169 CrossRef CAS.
- F. Mathias, A. Fokina, K. Landfester, W. Tremel, F. Schmid, K. Char and R. Zentel, Macromol. Rapid Commun., 2015, 36, 959 CrossRef CAS PubMed.
- B. Klöckner, K. Niederer, A. Fokina, H. Frey and R. Zentel, Macromolecules, 2017, 50, 3779 CrossRef.
- L. zur Borg, D. Lee, J. Lim, W. K. Bae, M. Park, S. Lee, C. Lee, K. Char and R. Zentel, J. Mater. Chem. C, 2013, 1, 1722 RSC.
- M. Rubinstein and R. H. Colby, Polym. Phys., vol. 23, Oxford University Press, New York, 2003 Search PubMed.
- J. Choi, P. Schattling, F. D. Jochum, J. Pyun, K. Char and P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4010 CrossRef CAS.
- J. Lim, B. G. Jeong, M. Park, J. K. Kim, J. M. Pietryga, Y. S. Park, V. I. Klimov, C. Lee, D. C. Lee and W. K. Bae, Adv. Mater., 2014, 26, 8034 CrossRef CAS PubMed.
- W. J. Li, B. Liu, Y. Qian, L. H. Xie, J. Wang, S. B. Li and W. J. P. C. Huang, Polym. Chem., 2013, 4, 1796 RSC.
- B. Klöckner, P. Daniel, M. Brehmer, W. Tremel and R. Zentel, J. Mater. Chem. C, 2017, 5, 6688 RSC.
- A. Fokina, Y. Lee, J. H. Chang, L. Braun, W. K. Bae, K. Char, C. Lee and R. Zentel, Polym. Chem., 2016, 7, 101 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9mh00551j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |