
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioinspired interface design modulates pathogen and immunocyte responses in biomaterial-centered infection combination therapy†
Jinhua
Li‡
 ab,
Wei
Liu‡
c,
David
Kilian
a,
Xianlong
Zhang
*c,
Michael
Gelinsky
a and
Paul K.
Chu
*b
ab,
Wei
Liu‡
c,
David
Kilian
a,
Xianlong
Zhang
*c,
Michael
Gelinsky
a and
Paul K.
Chu
*b
aCentre for Translational Bone, Joint and Soft Tissue Research, University Hospital and Faculty of Medicine Carl Gustav Carus, Technische Universität Dresden, Fetscherstraβe 74, Dresden 01307, Germany
bDepartment of Physics and Department of Materials Science and Engineering, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong, China. E-mail: paul.chu@cityu.edu.hk
cDepartment of Orthopaedics, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai Jiao Tong University, Shanghai 200233, China. E-mail: dr_zhangxianlong@163.com
First published on 29th March 2019
Abstract
The rise of multidrug-resistant pathogens and the dearth of novel antibiotic development means that breakthrough strategies that go beyond the classical antibiotic mechanism are urgently required to fight this approaching human health cataclysm. Herein, inspired by the metabolism cascade of pathogens and the clearance of infection by the host immune system, a new three-step strategy is reported to achieve combination therapy of biomaterial-centered infections. Band-structure-tunable cobalt doped TiO2 semiconductor can effectively restrain bacterial biofilm formation and enhance phagocytosing and killing of bacteria by immune cells. Released Co2+ ions can produce a proinflammatory microenvironment and potentiate the antibacterial capability of immune cells. This design concept can be applied to develop other antibacterial biomaterials, and holds great promise for advanced biomaterial-centered infection therapy.
New conceptsAn alarming and persistent rise in the antibiotic resistance among many important pathogenic bacterial species poses one of the greatest contemporary challenges to public health. Whereas most of the previous work developing antibacterial biomaterials is solely focused on inhibiting bacterial biofilm formation, herein, we demonstrate a new design concept of “infection combination therapy” for biomaterial-centered bacterial infections. This concept is achieved by three-step tactics: (1) band-structure-tailorable TiO2 semiconductor through cobalt doping can act as the extracellular electron acceptors to interrupt the bacterial electron transfer (discharge process) and proton transfer (hydrogenation process), jeopardize the energy metabolism causing bacteria starvation, and thereby suppress bacterial biofilm formation (bacteria starvation therapy). (2) Innate immune cells (including macrophages and neutrophils) can effectively phagocytose and kill the biofilm-formation-blocked bacteria using the divide-and-conquer strategy. (3) Released cobalt ions can create a proinflammatory local environment and potentiate the phagocytosing and killing of bacteria by the innate immune cells (infection immunotherapy). This concept of infection combination therapy can contribute to the advanced design of biomaterials for antibacterial treatment. We anticipate that this novel design concept can be extended to developing other antibacterial biomaterials and find a wide range of applications in the advanced therapy of biomaterial-centered bacterial infections. |
Nowadays it is increasingly necessary to understand the interaction mechanism between biomaterials and host tissues in a more sophisticated manner. Biomaterial-centered infections involve the interactions among biomaterials, bacteria/biofilms, and host innate immunity in the local environment.1,2 In this context, it is highly required to investigate the impact of biomaterials on innate immunity more than the mere relationship between bacteria/biofilms and biomaterials. It is believed that the inflammatory response is highly beneficial for the host innate immune defense against bacterial infection; macrophages and neutrophils are important innate immune cells, and play a central role as effector cells in the immune response by initiating and maintaining inflammation.3 Therefore, strategies that can incorporate, modulate, and even encourage the host immune response could provide a new paradigm for biomaterial-centered infection therapy. This may be manipulated via the modification and functionalization of biomaterials.4,5 For example, in response to varied tissue microenvironment cues, highly plastic macrophages can switch phenotypes and functions to the classically activated proinflammatory M1 and alternatively activated antiinflammatory M2 forms,6 and the macrophage polarization can be modulated by the physical and chemical cues of biomaterials.7,8 It has been reported that biofilms can impair host innate immunity through skewing macrophages to the M2 phenotype and inhibiting the inflammatory reaction around the infective site, thereby impairing the clearance of bacteria by immune cells.9 Therefore, modulating the macrophage polarization response to biomaterials may be a promising strategy for eliciting desired outcomes of biomaterial-centered infection therapy. In fact, bacteria are exposed to a rapid and profound host innate immune response characterized by the recruitment of neutrophils and macrophages to sites of infection; these cells are essential for the initiation and execution of the inflammatory response and subsequent resolution of infection.10 Furthermore, an in-depth understanding of the mechanism underlying the immune response mediated by biomaterials would aid in the development of novel biomaterials that can create a beneficial host immune microenvironment for combating bacterial infection.
Extracellular electron transfer (EET) is the description of the bacterial bioelectrochemical process that electrons are transferred from the cytosol to the cellular exterior.11,12 Redox-active minerals are abundant in soils and in aquatic or subsurface sediments; they can electrically support bacterial growth by serving as electron sinks for heterotrophy-based respiration.13,14 Such minerals include the solid oxides/hydroxides of Fe(III) and Mn(IV)15,16 with relatively high redox potentials as extracellular electron acceptors, including Fe(OH)3/Fe2+ (E° = +1.06 V), FeOOH/Fe2+ (E° = +0.67 V), Fe2O3/Fe2+ (E° = +0.66 V) and MnO2/Mn2+ (E° = +1.29 V).17 Inspired by these biological processes, we hypothesize that (i) tuning the band structure of the TiO2 semiconductor by cobalt doping can inhibit bacterial biofilm formation through interrupting extracellular electron transfer and proton transfer by acting as extracellular electron acceptors, (ii) innate immune cells e.g. macrophages and neutrophils can destroy the biofilm-formation-blocked bacteria using the divide-and-conquer strategy, and (iii) released cobalt ions can reinforce the antibacterial function of innate immune cells when maintaining biocompatibility. We demonstrate herein the capability to modulate the behaviors of bacteria and innate immune cells on cobalt-doped TiO2 coating by facile plasma modification, thereby leading to combination therapy for biomaterial-centered infections.
The samples were prepared using a plasma electrolytic oxidation (PEO) apparatus. The preparation parameters are shown in Table S1 (ESI†). The pristine sample was undoped TiO2 coating (TiO2:Co-0), while a series of Co-doped TiO2 coatings (TiO2:Co-0.5, TiO2:Co-1, TiO2:Co-2, TiO2:Co-4) were acquired by tuning the Co dopant concentration. Surface morphologies of the samples were determined by scanning electron microscopy (SEM). TiO2:Co-0 had a homogeneous porous coating on its surface with smooth morphology at higher magnification (inset) and Co doping did not sharply change the surface topographies of the coatings (Fig. 1A). Crystal structures of the samples were characterized by X-ray diffraction (Fig. 1B). In TiO2:Co-0, the coating mainly consisted of anatase TiO2 phase (JPCDS No. 21-1272). When performing PEO in Co-containing electrolyte, Co2+ might occupy the substitutional sites to replace Ti4+ in the host lattice or enter into the interstitial lattice sites due to the fact that, for the hexacoordinated central metal with octahedron geometry, Co2+ and Ti4+ have very similar ionic radii of 79 pm and 74.5 pm, respectively. Hence, X-ray photoelectron spectroscopy (XPS), that is a surface-sensitive characterization technique, was utilized to determine the surface chemistry. XPS full spectra and elemental content data (Fig. S1 and Table S2, ESI†) of the coatings reveal that increasing the Co2+ concentration of the electrolyte can increase the Co doping content in the TiO2 coating. Fig. 1C gives the high-resolution Co2p XPS spectra and the fitted spectra are shown in Fig. S2 (ESI†). The absence of a metallic Co2p3/2 peak at 778 eV with Δ = 14.86 eV indicated the inexistence of Co metal phase. There are clear characteristics in the 2p core level photoemission spectra that allow unambiguous distinguishing between Co2+ and Co3+. First, the Co2+-2p core level spectrum possesses strong shakeup satellite peaks toward higher binding energies, different from the very weak ones of the Co3+-2p core level spectrum.18,19 Second, the splitting of the Co2p1/2–Co2p3/2 orbital component Δ is usually 16 eV for the high-spin Co2+, while it is 15 eV for the low-spin Co3+.18 The existence of strong shakeup satellite peaks at 786.4 eV and 802.6 eV indicated the oxidation state of 2+ of the Co atom in the TiO2 coating. Meanwhile, the measured Δ (spin–orbit splitting) value from the main peaks at 781.0 eV and 796.9 eV was 15.9 eV, which was accordant with the valence state of 2+. Following the increase of Co2+ doping level in TiO2 coating, new diffraction peaks gradually appeared for the CoTiO3 phase (JCPDS No. 29-0516) and Co2TiO4 phase (JCPDS No. 39-1410). Both anatase TiO2 (l41/amd) and rutile TiO2 (P42/mnm) have a tetragonal crystal structure, but the rutile phase has a smaller lattice parameter (c-axis) and unit cell volume.20 During the formation of new CoTiO3 and Co2TiO4 phases, crystal lattice rearrangement occurred and might impose extrusion stress on the surrounding TiO2 unit cell and cause the shrink of the c-axis. As a result, a phase transformation from anatase to rutile might occur upon CoIITiIVO3 and CoII2TiIVO4 phase formation. Meanwhile, increasing amounts of oxygen vacancies were created in the TiO2 lattice upon subvalent Co2+ replacing Ti4+ sites of the lattice for charge compensation, which also promoted the rutile formation.21 In fact, new rutile phase (JPCDS No. 21-1276) emerged in the TiO2 coating when increasing the Co2+ dopant content, especially in TiO2:Co-2 and TiO2:Co-4. In addition, there were no peaks of metallic Co or binary cobalt oxides in the coatings even at a high cobalt content of 6.03 at%. Therefore, Co2+ largely substituted for the Ti4+ sites of the TiO2 lattice and the phase transformation of anatase to rutile implied that the solubility limit of Co2+ in TiO2 was ∼2 at%, which was quite consistent with the determined value for Co2+-doped TiO2 nanopowders.22 Besides, the fitted Ca2p XPS spectra (Fig. S3, ESI†) included the predominant peaks at 347.1 eV and 350.7 eV belonging to Ca3(PO4)2 and the third speak at 347.5 eV assigned to CaHPO4; accordingly, the fitted P2p XPS peaks at 133.6 eV and 132.8 eV (Fig. S4, ESI†) pertain to PO43− and HPO42−, respectively.23 Hence, Co2+ was substitutionally doped and homogeneously distributed in the TiO2 matrix.
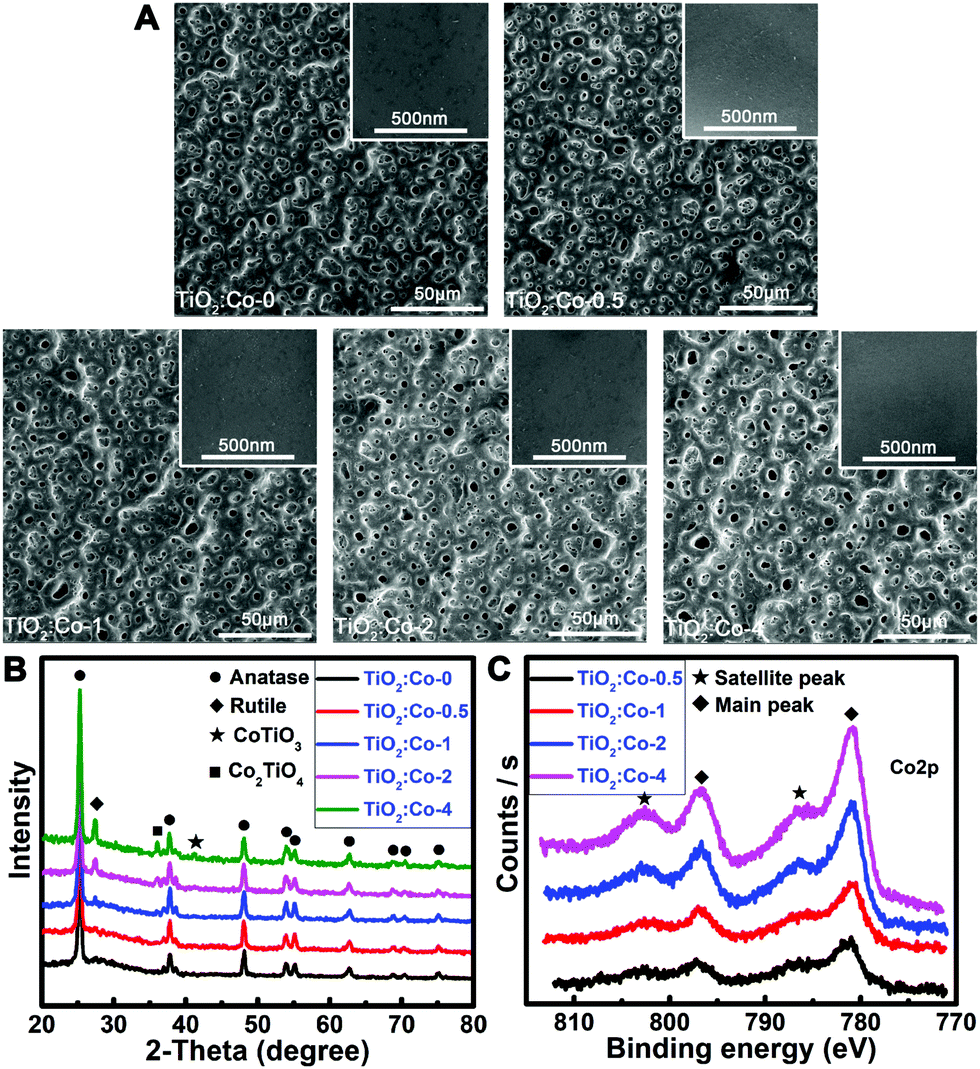 | ||
| Fig. 1 Surface topographies and compositions of the coating samples. (A) SEM images of the coatings with inserted high resolution images on the top-right. (B) XRD patterns of the coatings. (C) High resolution Co2p XPS spectra acquired from the surfaces of Co-doped coatings. | ||
UV-Vis absorption spectra of the samples were determined (Fig. 2A). A strong absorption below 400 nm for TiO2:Co-0 was attributed to the absorption edge of anatase TiO2. Increasing the Co doping content shifted the absorption edge to a longer wavelength range. Meanwhile, there existed a clear broad peak between 500 nm and 700 nm, which resulted from the CoTiO3 and Co2TiO4 phases,24 and the intensity increased with the Co2+ dopant content. The homogeneous absorption in the visible light range reflects the homogeneous distribution of the additional energy levels, and the shoulder at ∼530 nm and band at ∼605 nm come from respective free excitons and bound excitons.25Fig. 2B depicts the (αhν)1/2 plot versus hν, and the vertical segments of the spectra were extended to intersect with the hν axis to acquire the band gap (Eg) of various samples. The Eg of TiO2:Co-0 was 3.10 eV, but the Eg of the Co-doped samples gradually decreased from 2.69 eV of TiO2:Co-0.5 to 1.55 eV of TiO2:Co-4 with increasing Co dopant content. Hexacoordinated Co2+ and Ti4+ have very similar ionic radii and Co2+ doping will produce defects in the TiO2 lattice and create additional energy levels (impurity levels or trap levels) between the conduction band (CB) and valence band (VB),25,26 thereby leading to the narrowed band gap and enhanced visible light absorption with incremental Co doping content. Ultraviolet photoelectron spectroscopy (UPS) was employed to determine the VB maximum and work function (Fig. S5–S9, ESI†). Based on the data of VB maximum and band gap, the CB minimum was acquired (Table S3, ESI†). Fig. 2C summarizes the energy band information, including CB minimum (red), VB maximum (blue) and Fermi level (EF, green) with incremental content of Co doping. The CB minimum for TiO2:Co-0, TiO2:Co-0.5, TiO2:Co-1, TiO2:Co-2, and TiO2:Co-4 was −4.36, −4.81, −5.14, −5.63, and −6.03 eV, respectively. Table S4 (ESI†) lists the biological redox potentials (BRP) for the redox couples in the electron transfer system of the respiration chain located on the bacterial membrane (ESI†) and they are depicted in Fig. 2D according to the conversion equation of standard hydrogen electrode (E°, V) and absolute potential level (E, eV) as: E° = −4.44 − E,27 in the range of −4.12 to −4.84 eV. The BRP range is also indicated in Fig. 2C (light blue region). Interestingly, the CB minimum decreased dramatically from overlap of TiO2:Co-0 (−4.36 eV) with the BRP range (−4.12 to −4.84 eV) to levels of TiO2:Co-2 (−5.63 eV) and TiO2:Co-4 (−6.03 eV) far below the BRP minimum (−4.84 eV), which are able to allow the doped samples to accept extracellular electrons from bacterial redox couples, just as served by Fe(OH)3/Fe2+ (−5.5 eV), FeOOH/Fe2+ (−5.11 eV), Fe2O3/Fe2+ (−5.1 eV), and MnO2/Mn2+ (−5.73 eV).
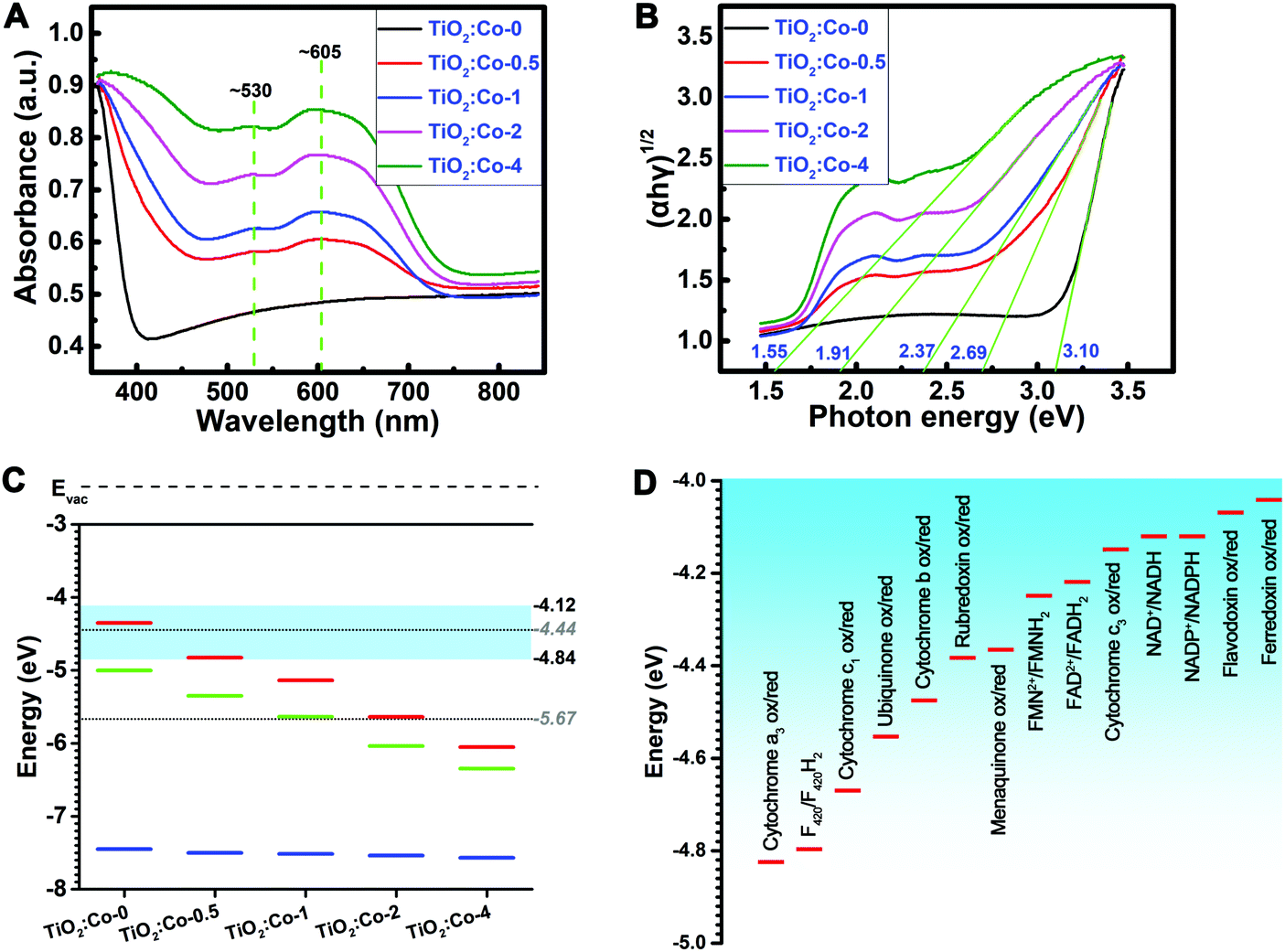 | ||
| Fig. 2 The analysis of the band structure of the coatings and the redox couples. (A) UV-Vis diffuse reflectance spectra of the coatings. (B) The Kubelka–Munk transformed reflectance spectra from (A). (C) Distribution of the CB minimum (EC, red line), Fermi level (EF, green line), and VB maximum (EV, blue line) of the coatings relative to the standard levels (dotted lines) of hydrogen production (4H+ + 4e− → 2H2(g), E = −4.44 eV) and water oxidation (2H2O + 4h+ → O2(g) + 4H+, E = −5.67 eV). Please notice that the light blue area shows the range of biological redox potentials (BRP, −4.12 to −4.84 eV) resulting from (D). (D) Demonstration of the sequential redox couples that contribute to the extracellular electron transfer (EET) of bacteria in the BRP range of −4.12 to −4.84 eV. | ||
To test our hypothesis, we first explored the in vitro antibacterial performance of doped samples. As shown in Fig. 3A, the viability of adhering MRSA (methicillin-resistant Staphylococcus aureus) was visualized by live/dead fluorescence staining after being cultured on samples for 24 h. The green fluorescence intensity for live MRSA sharply decreased with incremental Co doping content, demonstrating the significant inhibition effect on bacterial biofilm formation; meanwhile, the red fluorescence intensity for dead MRSA increased clearly, thereby indicating the apparent loss of MRSA viability when in contact with doped samples (especially on TiO2:Co-2 and TiO2:Co-4). The morphology of adhering MRSA and their biofilm production on samples were further examined by SEM. A large number of MRSA were adhering on TiO2:Co-0 with a continuous dense biofilm totally covering the surface micropores. By contrast, the number of adhering MRSA was significantly reduced on the doped samples upon increasing the Co content, thereby showing the sharply inhibited biofilm formation (especially on TiO2:Co-2 and TiO2:Co-4). Hence, the SEM observations agreed well with the live/dead staining results. To quantify the number of adhering MRSA on the samples, the spread plate method was utilized and the average Log10CFU per ml values (Fig. 3B) were in the order of TiO2:Co-0 (7.13 ± 0.07) > TiO2:Co-0.5 (6.80 ± 0.09) > TiO2:Co-1 (6.55 ± 0.12) > TiO2:Co-2 (6.19 ± 0.17) > TiO2:Co-4 (5.92 ± 0.12). Even at a low Co doping content, TiO2:Co-0.5 has shown good anti-biofilm ability compared to TiO2:Co-0 (P < 0.05). Further increasing the Co content can endow the doped samples with better anti-biofilm outcomes (P < 0.01). Noticeably, TiO2:Co-2 and TiO2:Co-4 exhibited a more powerful anti-biofilm effect when compared with TiO2:Co-0 and TiO2:Co-0.5 (P < 0.001) and TiO2:Co-1 (P < 0.05). In addition, broad-spectrum biofilm inhibition capability was ascertained by using Staphylococcus epidermidis (S. epidermidis, Fig. S10–S12, ESI†), Pseudomonas aeruginosa (P. aeruginosa, Fig. S13–S15, ESI†) and Escherichia coli (E. coli, Fig. S16–S18, ESI†). We sought to establish whether doped samples could modulate the expression levels of biofilm-related genes to block biofilm formation and development. By virtue of quantitative polymerase chain reaction (qPCR) analysis, the expression levels of fnbA and fnbB genes participating in MRSA biofilm formation are shown in Fig. 3C and D. TiO2:Co-0.5 can effectively inhibit the fnbA gene expression (P < 0.05), which was further remarkably downregulated on doped samples (P < 0.01) with incremental Co content. Regarding TiO2:Co-2 and TiO2:Co-4, the statistical difference even ascended to a value of P < 0.001 compared with other groups, especially for the fnbB gene expression. Similar results were also obtained by analyzing the expression levels of biofilm-related genes icaA and icaR for S. epidermidis (Fig. S19, ESI†). Therefore, it is concluded that the expression inhibition of biofilm-related genes resulted in the blocked biofilm formation on the doped samples. Prior to in vivo study, the biocompatibility was evaluated in vitro on rat bone mesenchymal stem cells (rBMSCs). Fig. 3E displays the fluorescence staining images of the actin cytoskeleton (green) and cell nuclei (blue) of rBMSCs after 24 h culture on the samples. The rBMSCs can adhere and spread very well with polygonal morphology and abundant actin on TiO2:Co-0, TiO2:Co-0.5, TiO2:Co-1 and TiO2:Co-2; however, apparently fewer rBMSCs can adhere on TiO2:Co-4. Fig. 3F and G show the cell viability results using a Cell Counting Kit-8 (CCK-8) assay. At day 1 and 3 of culture, the rBMSCs maintained high viability on TiO2:Co-0.5, TiO2:Co-1 and TiO2:Co-2 with negligible cytotoxicity. Nevertheless, TiO2:Co-4 caused significant toxicity to rBMSCs in comparison with other groups (P < 0.001). SEM observations also showed that, apart from TiO2:Co-4, the majority of sample surfaces (even micropores) were covered by the spreading rBMSCs (Fig. S20, ESI†). These concordant results indicated the excellent biocompatibility of the doped samples except TiO2:Co-4. In accordance with the endosymbiotic theory (symbiogenesis), eukaryotic mitochondrion is an analogue of prokaryote bacteria. However, the electron transport chain of mammalian cells for respiration is located in the intracellular mitochondria. As a result, the electron transport chain is not interrupted and the mammalian cells can survive on the doped samples. In fact, previous work showed that a low content of Co2+ (<5 at%) incorporation into a mesoporous bioactive glass (MBG) scaffold had no significant cytotoxicity and dramatically promoted angiogenesis and osteogenesis,28 which supported our work very well since the Co doping contents of TiO2:Co-0.5, TiO2:Co-1, TiO2:Co-2 and TiO2:Co-4 were 1.91, 2.45, 4.07 and 6.03 at%, respectively (Table S2, ESI†).
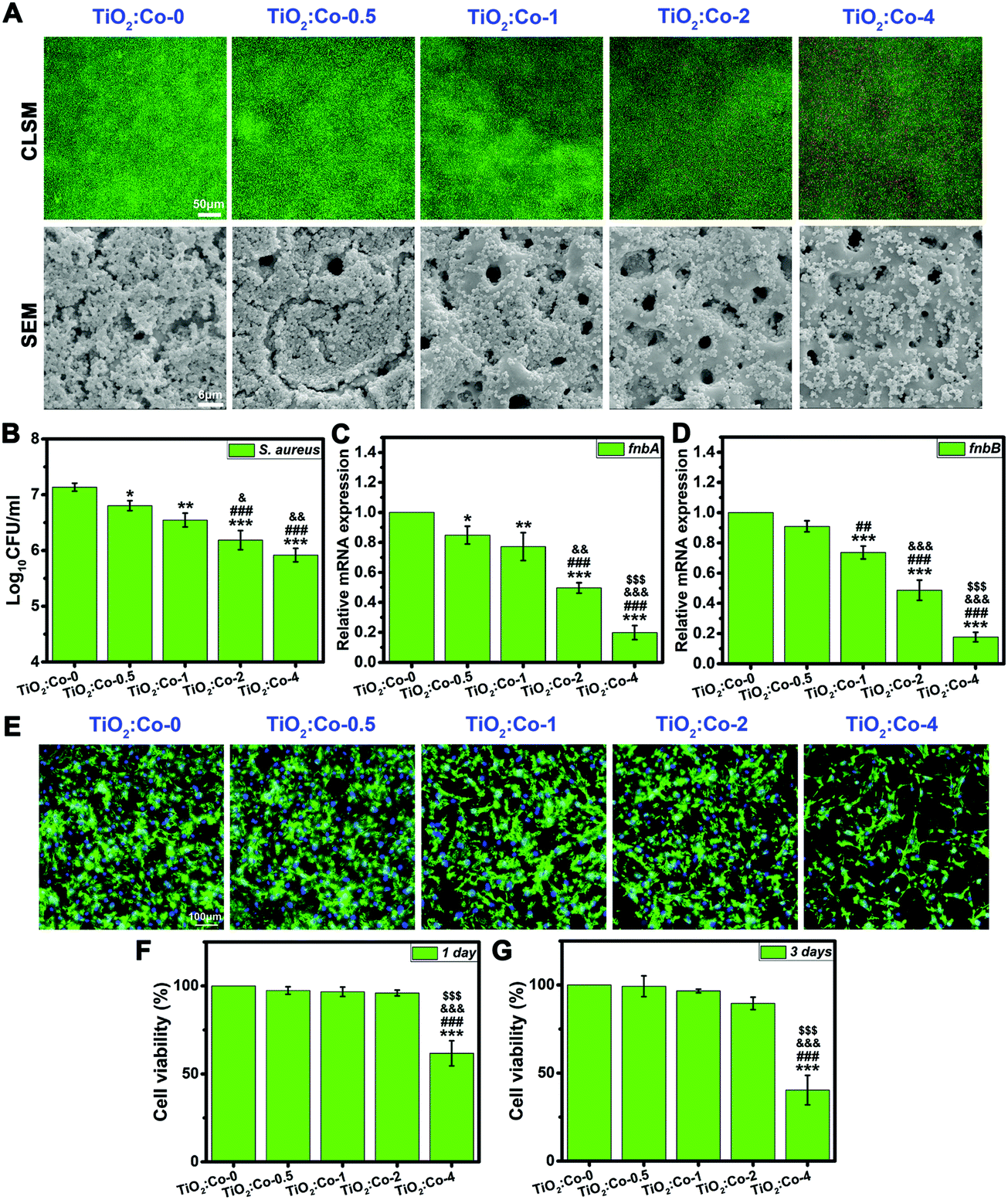 | ||
| Fig. 3 In vitro antibacterial effect and biocompatibility. (A) Bacterial viability and biofilm formation on the coatings examined by live/dead fluorescence staining and SEM methods after overnight culture. (B) Bacteria colony-forming units (CFUs) recovered from the coatings. (C and D) The effect of the coatings on the biofilm genes fnbA (C) and fnbB (D) transcription by qRT-PCR, normalized to the 16S rRNA gene. (E) The fluorescence staining images of the actin cytoskeleton (green) and cell nuclei (blue) of the rBMSCs on the coatings at 24 h post-culture. (F and G) The cellular viability results of the rBMSCs on the coatings at 1 day (F) and 3 days (G) post-culture using CCK-8 assay. Significant differences for all pairwise comparisons are indicated by the symbols over the bars: *P < 0.05, **P < 0.01, and ***P < 0.001 vs. TiO2:Co-0; ##P < 0.01 and ###P < 0.001 vs. TiO2:Co-0.5; &P < 0.05, &&P < 0.01, and &&&P < 0.001 vs. TiO2:Co-1; $$$P < 0.001 vs. TiO2:Co-2. | ||
After confirming the favorable biocompatibility of TiO2:Co-0, TiO2:Co-0.5, TiO2:Co-1 and TiO2:Co-2, we next explored their in vivo anti-infectious performance. A femoral osteomyelitis model was established on rats using MRSA. Fig. 4A shows the X-ray images of femurs. At week 0 (the 2nd day post surgery), there were no radiographic signs of osteolysis or periosteal reaction detected in each group. Nevertheless, at week 2, we detected the apparent radiographic signs of osseous destruction (blue arrow) and periosteal reaction (red arrow) around TiO2:Co-0 and the slight situations for TiO2:Co-0.5, while little osteolytic lesions were detected around TiO2:Co-1 and TiO2:Co-2. At week 4 post surgery, the osteolysis exacerbation and periosteal reaction progress indicated the development of bacterial infection around TiO2:Co-0, while other groups especially TiO2:Co-2 showed relatively mild osseous destruction or periosteal reaction. Micro-CT analyses were further performed at week 4 post surgery to validate the radiographic results. As seen in Fig. 4B, TiO2:Co-0 showed evident periprosthetic osteolysis and porous change throughout the femur. Although TiO2:Co-1 displayed slight loosening and femur porous change, TiO2:Co-1 and TiO2:Co-2 demonstrated beneficial osseointegration and cortical integrity. Further supported by the quantitative results of bone volume/total volume (Fig. 4C) and cortical bone mineral density (Fig. 4D), they showed the ascending trend in order of TiO2:Co-0, TiO2:Co-0.5, TiO2:Co-1 and TiO2:Co-2. Compared with TiO2:Co-0 and TiO2:Co-0.5, TiO2:Co-1 and TiO2:Co-2 can produce the statistical differences of P < 0.05 and P < 0.01, respectively. To visualize femur histopathological change and bacterial residue, hematoxylin and eosin (H&E) staining and Giemsa staining were conducted as shown in Fig. 4E. We observed from H&E staining for TiO2:Co-0 conspicuous cancellous bone destruction, intramedullary abscess and inflammatory cell infiltration at the bone–implant interface, as the typical indicators of femur infection; meanwhile, Giemsa staining indicated many bacterial residues in the intramedullary tissue. By contrast, TiO2:Co-0.5 displayed slight bone destruction, moderate abscess lesion and less inflammatory cell infiltration as well as decreased bacterial residues. Regarding TiO2:Co-1 and especially TiO2:Co-2, they can contribute to the mildest bone infection and fewest bacterial residues. Fig. 4F shows the number of bacterial colonies dislodged from implants in the order of TiO2:Co-2 < TiO2:Co-1 < TiO2:Co-0.5 < TiO2:Co-0 with a statistical difference among groups of P < 0.01. For TiO2:Co-2, the difference can increase to P < 0.001 versus TiO2:Co-0.5 or TiO2:Co-0. Therefore, with Co content increase, doped samples can significantly decrease the adhering bacterial burden. We also assayed the bacterial burden from powder bone of each group and obtained a similar trend of CFU number in the femur (Fig. 4G). TiO2:Co-2 can even exhibit a statistical difference of P < 0.01 versus TiO2:Co-1. Hence, Co doped samples can also exert a powerful inhibition effect on planktonic bacteria in peri-implant femur. Besides, although the mechanical properties of the coating samples decreased slightly after the in vivo study, they can maintain good mechanical properties in the in vivo environment (Fig. S33 and S34, ESI†).
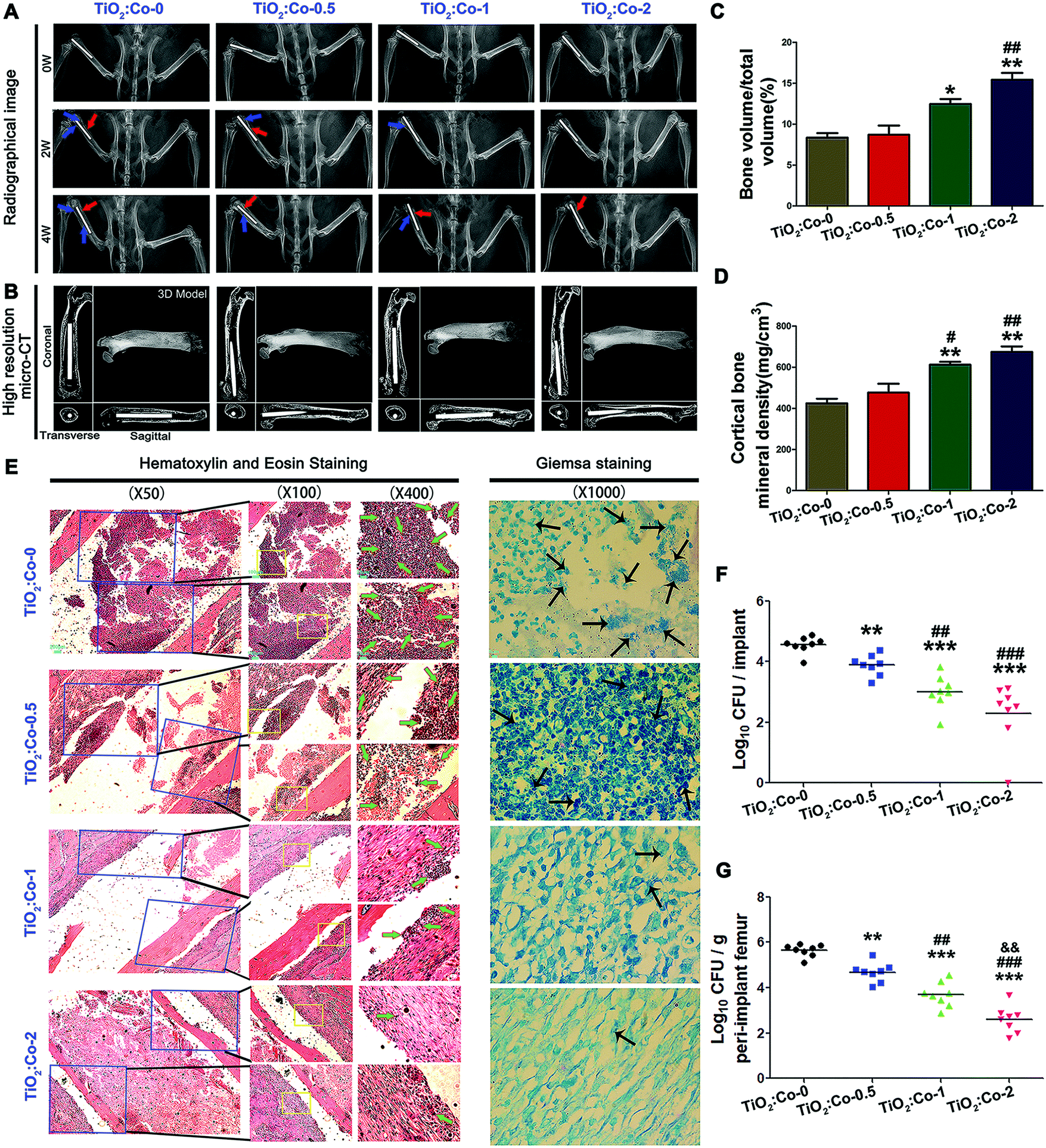 | ||
| Fig. 4 In vivo antibacterial effect using a rat osteomyelitis model. (A) Radiological images of the rat femurs at 0, 2, and 4 weeks post surgery. The red arrow and blue arrow show the periosteal reaction and osteolytic lesions, respectively. (B) The micro-CT reconstruction images of the rat femurs at the time of sacrifice. (C and D) The bone volume/total volume (C) and the cortical bone mineral density (D) of the rat femurs. (E) Hematoxylin and eosin staining and Giemsa staining images of the infected femur slices. The green arrow shows the neutrophil infiltration, and the black arrow shows the bacterial residues. (F and G) Bacteria colony-forming units (CFUs) recovered from the explanted implant (F) and from the infected femur (G). Significant differences for all pairwise comparisons are indicated by the symbols over the bars: *P < 0.05, **P < 0.01, and ***P < 0.001 vs. TiO2:Co-0; #P < 0.05, ##P < 0.01, and ###P < 0.001 vs. TiO2:Co-0.5; &&P < 0.01 vs. TiO2:Co-1. | ||
Encouraged by the excellent in vivo anti-infectious outcome, we further tested whether Co doped samples can combat planktonic bacteria in vitro. As verified in the ESI,† doped samples have much more inferior inhibition capability on planktonic MRSA (Fig. S21 and S22, ESI†), S. epidermidis (Fig. S23 and S24, ESI†), P. aeruginosa (Fig. S25 and S26, ESI†) and E. coli (Fig. S27 and S28, ESI†). Although a very low concentration of Co ions can be released from the doped samples (<3 ppm within 8 weeks, Fig. S29, ESI†), the Co element itself has no strong antibacterial property29–31 and Co ions released from the MBG scaffold (∼17.5 ppm within 1 week) did not show any antibacterial effect and had to load ampicillin to kill bacteria.28 Therefore, some other mechanism should account for the excellent suppressive effect on planktonic bacteria in vivo. We thus conclude that the anti-biofilm capability of doped samples is irrelevant to the release of Co ions, but strongly dependent on the tailorable band structure of TiO2 by Co doping. Scheme 1A illustrates the extracellular electron transfer process along the bacterial membrane respiration chain through a sequence of highly redox-active transmembrane protein complexes (Fig. 2D and Table S4, ESI†). It is reasonable that electron leakage from the electron transfer chain should endanger bacterial energy metabolism and life activity. From the biomimetic viewpoint, Co doped TiO2 can serve as an extracellular electron acceptor like natural redox-active Fe(III) and Mn(IV) minerals. Upon physical contact with a bacterium, doped TiO2 can extract electrons from the respiration chain by acting as an electron sink or trap to “short-circuit” the EET chain, as illustrated in Scheme 1B; such extraction is destructive and energetically favorable driven by redox potentials.32,33 To further clarify this process, we draw the energy band diagram of the interaction of the bacterial EET chain with undoped TiO2 (Scheme 1C) and Co doped TiO2 (Scheme 1D). As a result of the overlap of the CB minimum with the BRP range, undoped TiO2 can hardly extract electrons from the EET chain and perturb BRP. In contrast, Co doped TiO2 can spontaneously extract electrons from the EET chain and disrupt BRP, thereby interrupting the EET chain. And as verified herein, the larger the gap between the CB minimum and BRP minimum is, the more powerful the interface electron trap and the EET chain interruption will be. Parallel to the EET process, extracellular proton transfer can generate proton motive force across the bacterial membrane; these protons will re-enter the bacterium interior through adenosine triphosphate (ATP) synthase and the transmembrane proton movement will release energy to synthesize ATP (Scheme 1A).34 In this context, the interruption of bacterial proton transfer can hinder the ATP synthesis. It is well known that the TiO2 semiconductor has been widely used for water splitting and hydrogen (H2) production.35,36 Hydrogen can strongly influence the structural and electronic properties of a wide range of as-doped host materials including semiconductors, insulators and even aqueous solutions.37 Considering the fact of the electrostatic attraction between electrons and protons, the extracted electrons by TiO2:Co from a bacterium will accumulate in the titania layer (electron transfer); such excess electrons will thereafter electrostatically attract the protons surrounding the bacterial membrane especially ATP synthase to penetrate into the titania layer (proton diffusion), thereby generating a hydrogenated surface titania layer (hydrogenation). A recent study showed experimentally and theoretically that a self-hydrogenated shell was produced first on the TiO2 surface before forming H2 bubbles; this shell was generated through the subsurface diffusion of photo-reduced water protons at the aqueous TiO2 interface and can improve H2 production by lowering the activation barrier of H–H bond formation.38 Excess electrons at the aqueous anatase TiO2(101) interface are able to trigger water splitting and trap protons into a stable hydrogenated surface.39 Interestingly, as shown in Fig. 1B, the designed coating mainly contained anatase TiO2 phase with a large percentage of (101) facet. For a spontaneous photoelectrochemical water splitting process, the oxygen and hydrogen reactions must lie between the CBM and VBM of the TiO2 semiconductor, i.e. having a CBM higher than the H2/H2O level (−4.44 eV) and a VBM lower than the H2O/O2 level (−5.67 eV).35,36 We have indicated the standard levels of H2 production and H2O oxidation in Fig. 2C (dotted lines). Except TiO2:Co-0, the CBM of doped TiO2 is increasingly lower than the H2/H2O level with incremental Co doping content. Therefore, H2 production is theoretically impossible at the bacterium-TiO2:Co interface, and we have verified this by gas chromatography-mass spectrometry analysis. Since hydrogenation can enhance the electrical conductivity of TiO2:Co coating,40 this will further facilitate the electron transfer to the adjacent coating and subsequent proton diffusion. The proposed hydrogenation process can be illustrated in Scheme 1E. As a consequence, Co doping can tailor the band structure of TiO2 and interrupt the extracellular electron transfer and proton transfer; this will impede bacterial ATP synthesis and energy metabolism, thus contributing to the inhibited biofilm formation on the TiO2:Co coating.
 | ||
| Scheme 1 Schematic diagram for elucidating the mechanism of band-structure-tailorable TiO2 semiconductor coating to thwart bacterial biofilm formation through acting as an extracellular electron acceptor. (A) The interaction of TiO2 coating with a bacterium, showing that the bacterium can normally implement extracellular electron transfer (EET) and proton transfer for energy metabolism through respiration. (B) The interaction of TiO2:Co coating with a bacterium, showing that TiO2:Co can serve as an extracellular electron acceptor to interrupt the EET and proton transfer, thereby leading to energy starvation and biofilm formation inhibition. (C and D) The energy band diagrams showing the interactions of TiO2-bacterium (C) and TiO2:Co-bacterium (D) upon contact. In (C), TiO2 can rarely disrupt the EET and biological redox potential (BRP). On the contrary, in (D), TiO2:Co can spontaneously disrupt the EET and BRP, thereby leading to electron leakage. (E) The proposed hydrogenation process at the TiO2:Co-bacterium interface. First, the electrons are transferred from a bacterium and accumulate in the titania layer when the bacterium contacts TiO2:Co (electron transfer). Then the protons surrounding the bacterium membrane penetrate into the titania layer to meet the excess electrons (proton diffusion). Therefore, this can finally lead to the generation of a hydrogenated surface titania layer (hydrogenation). | ||
Due to the excellent repression effect on planktonic bacteria in vivo and the inconsistency in vitro, we then sought to assess whether innate immune cells (macrophages and neutrophils) could be involved in eradicating bacteria. Fig. 5A and B shows the cellular viability results of RAW 264.7 macrophages using CCK-8 assay. At day 1 and 3 of culture, RAW 264.7 sustained higher viability on TiO2:Co-0.5 and TiO2:Co-1 than TiO2:Co-0, and TiO2:Co-2 had no significant cytotoxicity. However, TiO2:Co-4 exerted apparent cytotoxicity to RAW 264.7 at day 1 (P < 0.001) and day 3 (P < 0.01). Fig. 5C indicates the labelling of the activated macrophages using immunofluorescence staining (M1, green for iNOS positive; M2, red for Arg1 positive). Incremental activated M1 macrophages were observed in TiO2:Co coating at day 1 and 3, and the proportion of M1 macrophages increased with Co doping content (Fig. 5C and Fig. S30, S31, ESI†), revealing that released Co2+ ions can boost the activation of M1 macrophages in a dose-dependent manner.41Fig. 5D–G give the concentrations of TNF-α, IL-6, IL-10, and IL-4 cytokines in the supernatants using ELISA at day 1, 2, and 3. Overall, the secretion of inflammatory cytokines (TNF-α and IL-6) augmented with Co2+ release of TiO2:Co coating, consistent with the immunofluorescence staining results. Interestingly, the secretion of antiinflammatory cytokines (IL-10) also augmented on TiO2:Co coating, which can antagonize proinflammatory cytokines and prevent uncontrolled inflammatory response.42 Therefore, overall TiO2:Co coating can trigger a local inflammatory response. Highly plastic macrophages can respond to external stimuli and display the classically activated proinflammatory M1 and alternatively activated antiinflammatory M2 phenotypes.43 M1 macrophages can secrete inflammatory cytokines to combat bacterial infection. However, the biofilms formed at the infection site can induce macrophages polarizing to M2 phenotype, thereby leading to an immunosuppressive environment, impairing phagocyte influx, and restraining bacterial clearance.9,44 In this context, developing an immunomodulatory biomaterial surface that can activate M1 macrophages could be promising for biomaterial-centered infection therapy.
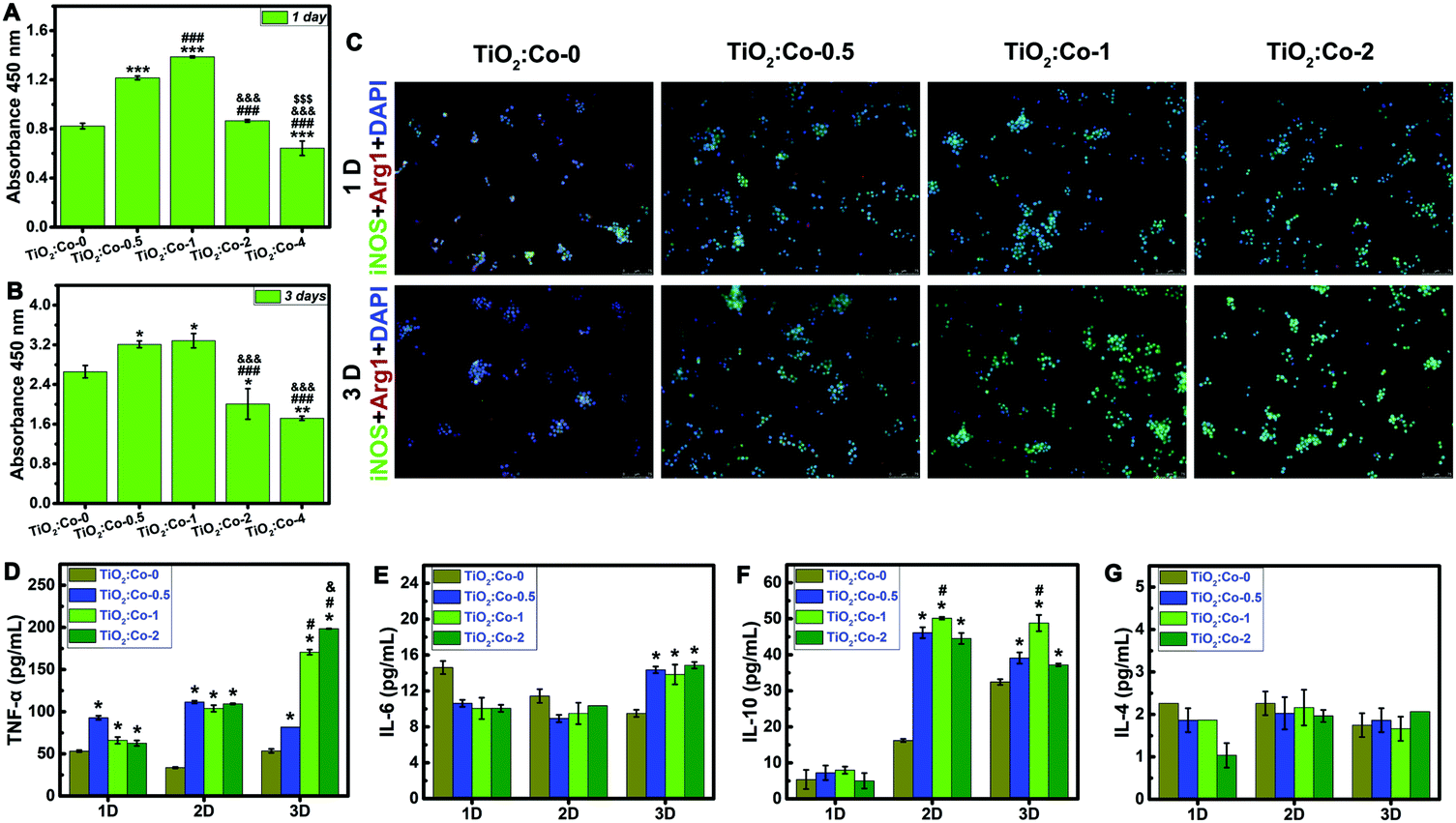 | ||
| Fig. 5 Cell viability, activation, and cytokine secretion of the RAW264.7 macrophages cultured on the coatings. (A and B) The cellular viability results of the RAW264.7 macrophages on the coatings at 1 day (A) and 3 days (B) post-culture using CCK-8 assay. (C) The immunofluorescent staining images of the RAW 264.7 macrophages scratched from the coatings at 1 and 3 days post-culture. The green fluorescence shows iNOS positive for M1 phenotype macrophages, the red fluorescence shows Arg1 positive for M2 phenotype macrophages, and the blue fluorescence shows cell nuclei. (D–G) The concentrations of the cytokines TNF-α (D), IL-6 (E), IL-10 (F), and IL-4 (G) secreted by the RAW 264.7 macrophages on the coatings using ELISA at 1, 2, and 3 days post-culture. Significant differences for all pairwise comparisons are indicated by the symbols over the bars: *P < 0.05, **P < 0.01, and ***P < 0.001 vs. TiO2:Co-0; #P < 0.05 and ###P < 0.001 vs. TiO2:Co-0.5; &P < 0.05 and &&&P < 0.001 vs. TiO2:Co-1; $$$P < 0.001 vs. TiO2:Co-2. | ||
To test if creating such an inflammatory microenvironment could be beneficial for bacterial clearance, we first evaluated the phagocytosis of MRSA by sample-conditioned macrophages. Fig. 6A and Fig. S32 (ESI†) show the fluorescence staining images of phagocytosed MRSA (green) in macrophages (red for cytoskeleton), indicating that released Co2+ ions from TiO2:Co coating can facilitate more phagocytosed MRSA in macrophages in a dose-dependent manner. Fig. 6B gives the representative SPM images of CFU of phagocytosed MRSA in macrophages, revealing the following order: TiO2:Co-0 < TiO2:Co-0.5 < TiO2:Co-1 < TiO2:Co-2. Fig. 6C depicts the MRSA phagocytosis by sample-conditioned macrophages using flow cytometry, and the phagocytosis rates of MRSA by macrophages were in the following order: TiO2:Co-0 (16.12 ± 2.03%) < TiO2:Co-0.5 (23.98 ± 2.24%) < TiO2:Co-1 (29.33 ± 1.42%) < TiO2:Co-2 (35.63 ± 1.90%). Therefore, we have demonstrated that TiO2:Co coating can skew the majority of macrophages polarizing to M1 phenotype, and enhance the phagocytic ability of macrophages on MRSA. We further assessed the phagocytosis of MRSA in human whole blood by polymorphonuclear leukocytes (i.e. neutrophils), the major white blood cells playing an important role in innate immunity.3Fig. 6E shows that the number of phagocytosed MRSA in neutrophils increased gradually with the incubation period; furthermore, at each time point, TiO2:Co coating contributed in a dose-dependent manner to the stronger phagocytic activity of neutrophils with a larger number of engulfed MRSA. This was in accordance with the MRSA phagocytosis by macrophages. Therefore, it is clear that TiO2:Co coating can reinforce the phagocytosis of MRSA by neutrophils in human blood. When phagocytes sense a local infection, they emigrate from blood vessels and migrate actively through tissue to the infection site in a well-controlled fashion. Once arrived, phagocytes avidly phagocytose bacteria and kill them intracellularly by releasing reactive oxygen species (ROS) and bactericidal products contained within granules.45 In the meantime, bacteria produce toxins e.g. leukocidin to combat phagocytes.3 Therefore, an increase in the capability to phagocytose or kill bacteria will contribute to a reduced risk of infection. Typically, phagocytes show the normal respiratory burst and produce the bactericidal agents of superoxide (˙O2−) and hydrogen peroxide (H2O2).46 Cobalt is a redox-active element and the standard redox potential (E°) of Co3+/Co2+ is 1.808 V. Considering the standard redox potential of H2O2/˙OH (0.38 V), according to the relationship between the thermodynamic function and redox potential: ΔG = −nFE = −nF [E°(Co3+/Co2+) − E°(H2O2/˙OH)], where ΔG, F and E are the free energy change, Faraday constant and redox potential, respectively; obviously, ΔG is <0, which confirms the spontaneity that Co2+ can elicit the generation of highly toxic hydroxyl radicals (˙OH) via the Fenton-like reaction: Co2+ + H2O2 → Co3+ + OH− + ˙OH. This is similar to the Fenton reaction of ferrous ions (Fe2+) with E°(Fe3+/Fe2+) = 0.771 V to induce ˙OH generation.47 As a result, the generated ˙O2− and ˙OH radicals can enhance the ability of phagocytes to kill bacteria.
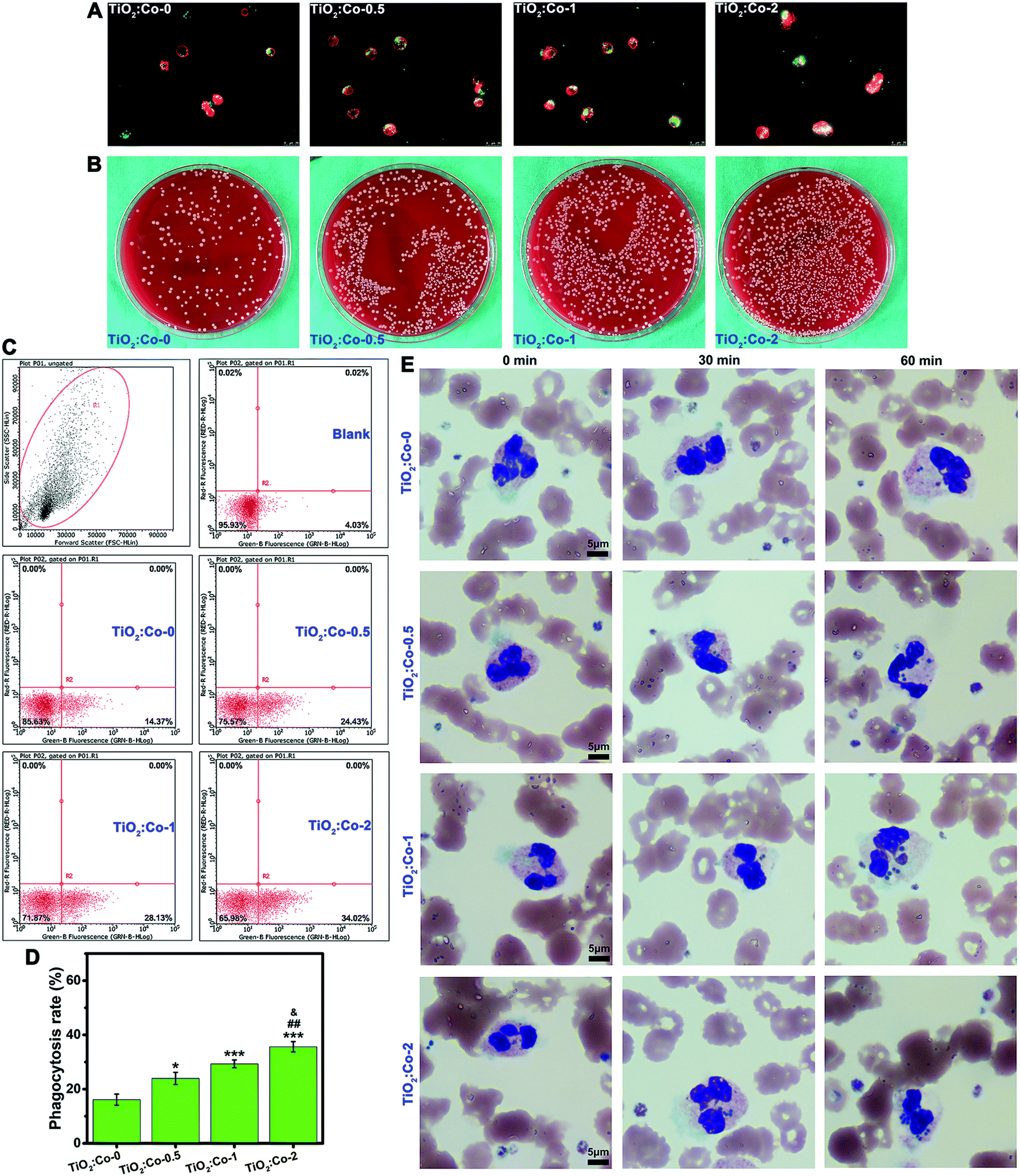 | ||
| Fig. 6 Phagocytosis of bacteria by the sample-conditioned RAW264.7 macrophages and polymorphonuclear leukocytes (neutrophils). (A) The fluorescence staining images of phagocytosis of bacteria by the sample-conditioned RAW264.7 macrophages. The green fluorescence shows phagocytosed bacteria, and the red fluorescence shows macrophages (cytoskeleton). (B) Representative images of phagocytosed bacteria colony-forming units (CFUs) recovered from the RAW264.7 macrophages. (C) The flow cytometry analysis of phagocytosis of labelled bacteria by the sample-conditioned RAW264.7 macrophages. (D) Quantitative results of phagocytosis rate of the RAW264.7 macrophages. (E) Wright–Giemsa staining images of phagocytosis of bacteria in human whole blood using a bacteria-blood-biomaterial co-culture model. The round blue granules show bacteria. Significant differences for all pairwise comparisons are indicated by the symbols over the bars: *P < 0.05 and ***P < 0.001 vs. TiO2:Co-0; ##P < 0.01 vs. TiO2:Co-0.5; &P < 0.05 vs. TiO2:Co-1. | ||
Bacteria form biofilms to resist leukocytes due to a lack of penetration of leukocytes into bacterial biofilms and a reduced capability of phagocytes to kill biofilm-encased bacteria. A mature biofilm has a dense polymeric matrix that is difficult to engulf by phagocytes, which leads to the ‘frustrated phagocytosis’.9,48 The biofilms can skew the host innate immune response from a proinflammatory bactericidal response to an antiinflammatory profibrotic response,9 and shift macrophage polarization from the M1 to M2 phenotype.49 Therefore, using the biofilm formation strategy, bacteria can evade host innate immunity. We have validated that TiO2:Co coating can block the formation of bacterial biofilms. It is thus reasonable to speculate that phagocytes (e.g. macrophages, neutrophils) can phagocytose and kill the biofilm-formation-blocked bacteria using a divide-and-conquer strategy because in this situation biofilms cannot well protect bacteria from phagocytes. When bacterial infection occurs, innate immune cells, particularly macrophages, will be first recruited to the site of infection and polarize to the proinflammatory M1 phenotype, secrete cytokines to boost inflammation reaction, and then phagocytose and kill bacteria.50 We have verified that the majority of macrophages in the TiO2:Co coating can be activated and polarize to the M1 phenotype, and then secrete a high level of inflammatory cytokines to create a proinflammatory microenvironment; the TiO2:Co coating can facilitate the MRSA phagocytosis by sample-conditioned macrophages and neutrophils. Therefore, such an immunomodulatory ability of the TiO2:Co coating can enhance the capability of phagocytes to phagocytose and kill bacteria, and contribute to the superior antiinfective function. This immunoregulatory antibacterial strategy can be called infection immunotherapy. In fact, cancer immunotherapy has been widely used to inhibit tumor growth.47,51 The promoted local inflammatory response can recruit more immune cells to combat bacteria; the enhanced phagocytosis of bacteria and release of proinflammatory cytokines by phagocytes can jointly orchestrate local innate immunity and following adaptive immunity to fight infection.52,53
On the other hand, it was observed in vivo that TiO2:Co-1 and TiO2:Co-2 showed beneficial osseointegration. As an essential element in human physiology, cobalt has been verified to promote osteogenesis and angiogenesis/vascularization.28,54 Hypoxia-inducible factor (HIF) plays a central role in the cellular adaptation to low oxygen availability. In normoxia, HIF-α is targeted for rapid degradation upon prolyl hydroxylation by HIF prolyl-hydroxylases (PHDs).55 PHDs need oxygen and act as primary oxygen sensors in the cell repertoire. Reduced oxygen levels will impair PHDs and stabilize HIF-α.56,57 Under hypoxic conditions, the hydroxylation of HIF-α can be decreased and lead to the transactivation and induction of HIF-α target genes,58,59 thereby in favour of neovascularization and bone regeneration in wound healing responses. As a result, inhibition of PHDs can stabilize HIF-α in bone and wound tissues, enhance neovascularization, and further promote bone repair and wound healing. It has been suggested that the activation of HIF forms the base for ischemic tolerance.60–62 Cobalt chloride (CoCl2) is a hypoxia mimetic agent possessing the capability to upregulate the HIF level by inhibiting PHDs (i.e. inhibiting the prolyl hydroxylation of HIF),57,59 thus having the ability to enhance vascularization and bone formation. Cobalt can induce ischemic tolerance in a variety of tissues and cells, and thereby endow tissues with resistance to subsequent ischemia/hypoxia through HIF activation.60,63 The hypoxia will increase the expression of pro-angiogenic factors (e.g. VEGF) by BMSCs that are able to improve angiogenesis/vascularization in ischemic tissues64,65 and osteogenesis.66 Therefore, the observed favorable osseointegration in vivo may be attributed to the effect of released Co on the inhibition of PHDs.
Conclusions
In summary, we devised a novel three-step strategy for biomaterial-centered infection combination therapy based on Co doped TiO2 semiconductor. The first step is that Co doping can tailor the band structure of TiO2 to serve as extracellular electron acceptors; in this way TiO2:Co can interrupt extracellular electron transfer and proton transfer, and bacterial energy metabolism, thereby able to block bacterial biofilm formation. Secondly, innate immune cells e.g. macrophages and neutrophils can adopt divide-and-conquer tactics to annihilate the biofilm-formation-blocked bacteria. In the meantime, TiO2:Co can release Co2+ ions to create a proinflammatory microenvironment, and potentiate the ability of phagocytes to phagocytose and kill bacteria, thus leading to the enhanced antibacterial capability of innate immune cells. We anticipate that this design concept of infection combination therapy can be applied to develop other advanced antibacterial biomaterials and find wide applications in antibacterial research.Experimental section
Details on the experimental procedures are provided in the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Dr Jinhua Li gratefully acknowledges the funding support from the Alexander von Humboldt Stiftung/Foundation. This work was financially supported by Hong Kong Research Grant Council General Research Funds (CityU 11205617), City University of Hong Kong Donation Research Grant (9220061), and the National Natural Science Foundation of China (81472109).References
- C. R. Arciola, D. Campoccia and L. Montanaro, Nat. Rev. Microbiol., 2018, 16, 397–409 CrossRef CAS PubMed.
- J. Liao, W. Chen, M. Yang, J. Zhou, Z. Wang, Y. Zhou, C. Ning and H. Yuan, Mater. Horiz., 2018, 5, 545–552 RSC.
- S. D. Kobayashi, K. R. Braughton, A. R. Whitney, J. M. Voyich, T. G. Schwan, J. M. Musser and F. R. DeLeo, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10948–10953 CrossRef CAS PubMed.
- H. J. Busscher, H. C. van der Mei, G. Subbiahdoss, P. C. Jutte, J. J. A. M. van den Dungen, S. A. J. Zaat, M. J. Schultz and D. W. Grainger, Sci. Transl. Med., 2012, 4, 153rv110 Search PubMed.
- R. Sridharan, A. R. Cameron, D. J. Kelly, C. J. Kearney and F. J. O’Brien, Mater. Today, 2015, 18, 313–325 CrossRef CAS.
- A. Mantovani, A. Sica, S. Sozzani, P. Allavena, A. Vecchi and M. Locati, Trends Immunol., 2004, 25, 677–686 CrossRef CAS PubMed.
- D. M. Mosser and J. P. Edwards, Nat. Rev. Immunol., 2008, 8, 958–969 CrossRef CAS PubMed.
- S. Chen, J. A. Jones, Y. Xu, H.-Y. Low, J. M. Anderson and K. W. Leong, Biomaterials, 2010, 31, 3479–3491 CrossRef CAS PubMed.
- L. R. Thurlow, M. L. Hanke, T. Fritz, A. Angle, A. Aldrich, S. H. Williams, I. L. Engebretsen, K. W. Bayles, A. R. Horswill and T. Kielian, J. Immunol., 2011, 186, 6585–6596 CrossRef CAS PubMed.
- C. Nathan, Nature, 2002, 420, 846–852 CrossRef CAS PubMed.
- S. H. Light, L. Su, R. Rivera-Lugo, J. A. Cornejo, A. Louie, A. T. Iavarone, C. M. Ajo-Franklin and D. A. Portnoy, Nature, 2018, 562, 140–144 CrossRef CAS PubMed.
- R. S. Hartshorne, C. L. Reardon, D. Ross, J. Nuester, T. A. Clarke, A. J. Gates, P. C. Mills, J. K. Fredrickson, J. M. Zachara, L. Shi, A. S. Beliaev, M. J. Marshall, M. Tien, S. Brantley, J. N. Butt and D. J. Richardson, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22169–22174 CrossRef CAS PubMed.
- L. Shi, H. Dong, G. Reguera, H. Beyenal, A. Lu, J. Liu, H.-Q. Yu and J. K. Fredrickson, Nat. Rev. Microbiol., 2016, 14, 651 CrossRef CAS PubMed.
- R. K. Thauer, K. Jungermann and K. Decker, Bacteriol. Rev., 1977, 41, 100–180 CAS.
- C. R. Myers and K. H. Nealson, Science, 1988, 240, 1319 CrossRef CAS PubMed.
- K. A. Weber, L. A. Achenbach and J. D. Coates, Nat. Rev. Microbiol., 2006, 4, 752 CrossRef CAS PubMed.
- J. M. Zachara, J. K. Fredrickson, S.-M. Li, D. W. Kennedy, S. C. Smith and P. L. Gassman, Am. Mineral., 1998, 83, 1426–1443 CrossRef CAS.
- M. Oku and Y. Sato, Appl. Surf. Sci., 1992, 55, 37–41 CrossRef CAS.
- A. C. Tuan, J. D. Bryan, A. B. Pakhomov, V. Shutthanandan, S. Thevuthasan, D. E. McCready, D. Gaspar, M. H. Engelhard, J. W. Rogers, K. Krishnan, D. R. Gamelin and S. A. Chambers, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 054424 CrossRef.
- D. H. Hanaor and C. Sorrell, J. Mater. Sci., 2011, 46, 855–874 CrossRef CAS.
- X. H. Wang, J. G. Li, H. Kamiyama, M. Katada, N. Ohashi, Y. Moriyoshi and T. Ishigaki, J. Am. Chem. Soc., 2005, 127, 10982–10990 CrossRef CAS PubMed.
- J.-G. Li, R. Büchel, M. Isobe, T. Mori and T. Ishigaki, J. Phys. Chem. C, 2009, 113, 8009–8015 CrossRef CAS.
- X. Zhu, J. Chen, L. Scheideler, R. Reichl and J. Geis-Gerstorfer, Biomaterials, 2004, 25, 4087–4103 CrossRef CAS PubMed.
- M. Iwasaki, M. Hara, H. Kawada, H. Tada and S. Ito, J. Colloid Interface Sci., 2000, 224, 202–204 CrossRef CAS PubMed.
- L. Zhou, S.-y. Zhang, J.-c. Cheng, L.-d. Zhang and Z. Zeng, Mater. Sci. Eng., B, 1997, 49, 117–122 CrossRef.
- S. George, S. Pokhrel, Z. Ji, B. L. Henderson, T. Xia, L. Li, J. I. Zink, A. E. Nel and L. Mädler, J. Am. Chem. Soc., 2011, 133, 11270–11278 CrossRef CAS.
- Z. Xuming, C. Yu Lim, L. Ru-Shi and T. Din Ping, Rep. Prog. Phys., 2013, 76, 046401 CrossRef PubMed.
- C. Wu, Y. Zhou, W. Fan, P. Han, J. Chang, J. Yuen, M. Zhang and Y. Xiao, Biomaterials, 2012, 33, 2076–2085 CrossRef CAS PubMed.
- K. Boldbaatar, K. Dashnyam, J. C. Knowles, H.-H. Lee, J.-H. Lee and H.-W. Kim, Acta Biomater., 2019, 83, 322–333 CrossRef CAS PubMed.
- J. Li, J. Wang, D. Wang, G. Guo, K. W. K. Yeung, X. Zhang and X. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 27475–27490 CrossRef CAS PubMed.
- Q. Shi, X. Luo, Z. Huang, A. C. Midgley, B. Wang, R. Liu, D. Zhi, T. Wei, X. Zhou, M. Qiao, J. Zhang, D. Kong and K. Wang, Acta Biomater., 2019, 86, 465–479 CrossRef CAS PubMed.
- Y. Xu and M. A. A. Schoonen, Am. Mineral., 2000, 85, 543–556 CrossRef CAS.
- M. T. Greiner, M. G. Helander, W.-M. Tang, Z.-B. Wang, J. Qiu and Z.-H. Lu, Nat. Mater., 2012, 11, 76–81 CrossRef CAS PubMed.
- P. Mitchell, Biol. Rev., 1966, 41, 445–501 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed.
- S. U. M. Khan, M. Al-Shahry and W. B. Ingler, Science, 2002, 297, 2243 CrossRef CAS PubMed.
- C. G. Van de Walle and J. Neugebauer, Nature, 2003, 423, 626 CrossRef CAS PubMed.
- Y. Lu, W.-J. Yin, K.-L. Peng, K. Wang, Q. Hu, A. Selloni, F.-R. Chen, L.-M. Liu and M.-L. Sui, Nat. Commun., 2018, 9, 2752 CrossRef PubMed.
- S. Selcuk and A. Selloni, Nat. Mater., 2016, 15, 1107 CrossRef CAS PubMed.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 1200448 Search PubMed.
- Z. Chen, J. Yuen, R. Crawford, J. Chang, C. Wu and Y. Xiao, Biomaterials, 2015, 61, 126–138 CrossRef CAS PubMed.
- A. Eckhardt, T. Harorli, J. Limtanyakul, K.-A. Hiller, C. Bosl, C. Bolay, F.-X. Reichl, G. Schmalz and H. Schweikl, Biomaterials, 2009, 30, 1665–1674 CrossRef CAS PubMed.
- Z. Chen, T. Klein, R. Z. Murray, R. Crawford, J. Chang, C. Wu and Y. Xiao, Mater. Today, 2016, 19, 304–321 CrossRef CAS.
- C. E. Heim, D. Vidlak, T. D. Scherr, C. W. Hartman, K. L. Garvin and T. Kielian, J. Immunol., 2015, 194, 3861–3872 CrossRef CAS PubMed.
- P. Friedl and B. Weigelin, Nat. Immunol., 2008, 9, 960–969 CrossRef CAS PubMed.
- B. M. Babior, J. Clin. Invest., 1984, 73, 599–601 CrossRef CAS PubMed.
- S. Zanganeh, G. Hutter, R. Spitler, O. Lenkov, M. Mahmoudi, A. Shaw, J. S. Pajarinen, H. Nejadnik, S. Goodman, M. Moseley, L. M. Coussens and H. E. Daldrup-Link, Nat. Nanotechnol., 2016, 11, 986–994 CrossRef CAS PubMed.
- T. J. Foster, Nat. Rev. Microbiol., 2005, 3, 948 CrossRef CAS PubMed.
- C. M. Gries and T. Kielian, J. Am. Acad. Orthop. Surg., 2017, 25(suppl. 1), S20–S24 CrossRef PubMed.
- S. L. Brandt, N. E. Putnam, J. E. Cassat and C. H. Serezani, J. Immunol., 2018, 200, 3871 CrossRef CAS PubMed.
- X. Zhang, C. Wang, J. Wang, Q. Hu, B. Langworthy, Y. Ye, W. Sun, J. Lin, T. Wang, J. Fine, H. Cheng, G. Dotti, P. Huang and Z. Gu, Adv. Mater., 2018, 30, 1707112 CrossRef PubMed.
- A. Aderem, J. Infect. Dis., 2003, 187, S340–345 CrossRef CAS PubMed.
- D. M. Underhill and H. S. Goodridge, Nat. Rev. Immunol., 2012, 12, 492 CrossRef CAS PubMed.
- W. Fan, R. Crawford and Y. Xiao, Biomaterials, 2010, 31, 3580–3589 CrossRef CAS PubMed.
- E. Berra, E. Benizri, A. Ginouvès, V. Volmat, D. Roux and J. Pouysségur, EMBO J., 2003, 22, 4082 CrossRef CAS PubMed.
- R. H. Wenger, FASEB J., 2002, 16, 1151–1162 CrossRef CAS PubMed.
- K. S. Hewitson, L. A. McNeill, M. V. Riordan, Y.-M. Tian, A. N. Bullock, R. W. Welford, J. M. Elkins, N. J. Oldham, S. Bhattacharya, J. M. Gleadle, P. J. Ratcliffe, C. W. Pugh and C. J. Schofield, J. Biol. Chem., 2002, 277, 26351–26355 CrossRef CAS PubMed.
- S. M. Jones, A. E. Novak and J. P. Elliott, Neuroscience, 2013, 252, 420–430 CrossRef CAS PubMed.
- E. Pacary, H. Legros, S. Valable, P. Duchatelle, M. Lecocq, E. Petit, O. Nicole and M. Bernaudin, J. Cell Sci., 2006, 119, 2667 CrossRef CAS PubMed.
- M. Bergeron, J. M. Gidday, A. Y. Yu, G. L. Semenza, D. M. Ferriero and F. R. Sharp, Ann. Neurol., 2000, 48, 285–296 CrossRef CAS.
- N. M. Jones and M. Bergeron, J. Cereb. Blood Flow Metab., 2001, 21, 1105–1114 CrossRef CAS PubMed.
- M. Bernaudin, Y. Tang, M. Reilly, E. Petit and F. R. Sharp, J. Biol. Chem., 2002, 277, 39728–39738 CrossRef CAS PubMed.
- N. M. Jones, L. Kardashyan, J. K. Callaway, E. M. Lee and P. M. Beart, Pediatr. Res., 2008, 63, 620–624 CrossRef CAS PubMed.
- X. Hu, S. P. Yu, J. L. Fraser, Z. Lu, M. E. Ogle, J.-A. Wang and L. Wei, J. Thorac. Cardiovasc. Surg., 2008, 135, 799–808 CrossRef CAS PubMed.
- T.-S. Li, K. Hamano, K. Suzuki, H. Ito, N. Zempo and M. Matsuzaki, Am. J. Physiol.: Heart Circ. Physiol., 2002, 283, H468–H473 CrossRef CAS PubMed.
- D. Kaigler, Z. Wang, K. Horger, D. J. Mooney and P. H. Krebsbach, J. Bone Miner. Res., 2006, 21, 735–744 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details on the experimental procedures, and supplementary figures (Fig. S1–S34), tables (Tables S1–S5), and references. See DOI: 10.1039/c8mh01606b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |