
Algae–mussel-inspired hydrogel composite glue for underwater bonding†
Aleksander
Cholewinski‡
,
Fut (Kuo)
Yang‡
and
Boxin
Zhao
 *
*
University of Waterloo, Department of Chemical Engineering, Waterloo Institute for Nanotechnology, Institute for Polymer Research, Centre for Bioengineering and Biotechnology, 200 University Avenue West, Waterloo, Ontario N2L 3G1, Canada. E-mail: zhaob@uwaterloo.ca
First published on 21st November 2018
Abstract
Inspired by the adhesive strategies of brown algae and marine mussels, we developed a novel hydrogel composite glue formed from initially separate adhesive and polymer precursors. These components then formed a network during application by coordinating with ferric ions. This approach enabled us to turn a non-adhesive polymer, alginate, into an adhesive gel with good performance, which was not possible with conventional methods utilizing chemical conjugation of catechol functionality. Sequential deposition of precursors, mimicking algae, was found to outperform the direct mixing of components before application. The resulting glue does not require chemical conjugation, and yet can strongly bond dissimilar materials completely submerged in water.
Conceptual insightsFormulation of synthetic underwater adhesives conventionally requires the adhesive functionalities to be covalently attached to the polymer backbone. However, nature demonstrates that this need not always be the case. When brown algae stick to surfaces, their adhesive and polymer components are initially separate. By applying this concept to catechol-based adhesives, we fabricated a novel underwater adhesive that could not be created by chemical conjugation of catechol, overcoming the limitations associated with chemical synthesis. We utilized dopamine, a small catecholic molecule, with alginate polymer. These components were initially separate in the composite glue, after which bonding was achieved through in situ coordination. This strategy offers a new avenue for the development of underwater adhesives, potentially lowering the cost and complexity of their fabrication. |
Introduction
To establish bonding between two adherends, the adhesive needs to penetrate surface boundary layers, spread and develop intimate interfacial contacts with the adherends’ surfaces, and cure and set within a reasonable period. These requirements are especially difficult to meet in the presence of water because water as a boundary layer can weaken the interfacial adhesion of the adhesive and as a solvent can undermine the adhesive's integrity. This is the reason why synthetic adhesives developed for dry applications perform poorly on wet surfaces or underwater. However, adhesion in wet and moist environments is an important concern for many construction, biomedical and marine applications.1–3 There has been an interestingly large amount of research activities on the development of adhesive materials that can work effectively in wet and even underwater conditions.In nature, various sessile marine organisms, including benthic algae and marine mussels, have developed their own strategies for adhering to a variety of wet surfaces in these environments.4–6 Brown algae obtain adhesion from polyphenol aggregates, with phenolic residues possessing two or three hydroxyl groups that enhance adhesion to a substrate by forming hydrogen bonds, as well as displacing water molecules at the surface. However, the bulk of the adhesive is a separate network of alginate, which is gelled by calcium ions to provide cohesion; these phenolic and alginate groups are secreted separately, then crosslinked together to form the final adhesive.4,5 Marine mussels use catechol groups (which are also phenolic residues with multiple hydroxyl groups) as part of their adhesive strategy. Unlike with alginate, these adhesive phenolic groups are present in proteins that also serve as the structural fiber of the adhesive plaque; L-3,4-dihydroxyphenylalanine (DOPA) in particular is a major component in adhesive mussel foot proteins.6,7
So far, there have been very few studies mimicking algal adhesion.8–10 Part of the reason for this has been the weak adhesion associated with the polyphenols specifically used by algae, with the resulting biomimetic adhesive utilizing phloroglucinol only outperforming pure alginate by less than twice the strength.8 In contrast, mussel chemistry has been widely studied as an adhesive strategy due to the high underwater adhesive strength of catechol,11 which has inspired a large number of works in developing underwater adhesives. These works achieve adhesion either by incorporating catechol into polymers12–14 or by expressing recombinant mussel adhesive proteins.15,16 The main focus of mussel-based adhesion has been covalent attachment of catechol groups, either to end- or side-groups of existing polymers,13,14,17,18 or through polymerization of monomers with existing or modified catechol functionality.19,20 However, chemical synthesis can be complex and time-consuming, and can have its own set of limitations for mussel-inspired adhesives. For example, the protection and deprotection of catechol during synthesis imposes strict restrictions that can be difficult to meet.21 For catechol–alginate, the limiting factor has been the low degree of modification (typically around 5%).14,22–30 This restricts its applicability as an adhesive, as found by ourselves and others.14 On the other hand, the adhesive strategies of algae demonstrate that adhesive functional groups do not need to initially be present on the polymer.
We believed that by synergistically combining the two strategies of algae and mussels, we could overcome the above limitations of biomimetic glues of algae and mussels. We hypothesized that alginate, dopamine, and ferric ions could be used together to form a novel hydrogel composite glue.31 This is because these components, when combined, could serve to mimic beneficial aspects of each organism's adhesive strategy. Dopamine mimics the adhesive components of both organisms, since it has functionalities (catechol and amine) that can duplicate mussel chemistry. Under alkaline conditions, it auto-oxidizes to form polydopamine, which can coat virtually any surface;32 this polydopamine formation is also reminiscent of the polyphenols present in algae adhesive. Alginate serves as the cohesive backbone of the adhesive secreted by algae, and its ionic crosslinking ability is particularly useful in this work. Fe3+ ions were used to link together the adhesive and cohesive elements of the gel. This exploits the ionic crosslinking of alginate, used by algae (Ca2+ is used by algae, but Fe3+ is also known to crosslink alginate chains33), as well as the formation of catechol–Fe3+ complexes within the mussel cuticle.34,35 These catechol–Fe3+ complexes have been exploited to create injectable polymers with tunable elastic moduli and degradation behaviors,36 as well as to incorporate reversible crosslinks into self-assembling networks to improve mechanical properties and retain self-healing behaviour.37 Based on this, we believed Fe3+ ions could link together the adhesive and cohesive elements of our gel. Fe3+ ions were expected to be able to provide cohesion through crosslinking alginate and coordinating with dopamine, as well as providing sites that could interact with both the alginate and dopamine components of the adhesive gel. A schematic detailing the way the dual inspirations of algae and mussels are combined together is presented in Fig. 1a–c, with 1c illustrating the expected interactions between alginate and catechol, linked by Fe3+. This form of crosslinking is in contrast to typical methods for incorporating catechol adhesion, which frequently involve chemical conjugation of catechol groups to polymer backbones.13,14,17 It should offer a simpler method for introducing catechol adhesion that does not require chemical conjugation or modification.
 | ||
| Fig. 1 (a–c) Illustration of the algae–mussel hydrogel composite adhesive: components of brown algae adhesive system (a) and marine mussel adhesive system (b) were combined to form algae–mussel glue (c). The zoomed view shows the hypothesized molecular interactions between the components, namely, the coordination bonds between the catechol functional group of dopamine with the ferric ion, the ionic bonds between the alginate and the ferric ion, the self-polymerization of dopamine, and the chemical bonding of polydopamine to the adherend's surface through its catechol functionality. (d) Illustration of the sequential application of algae–mussel glue. The two solutions used are a dopamine–iron–Tris solution (D–Fe–Tris) and a 5 wt% alginate solution in deionized water. The adherends’ surfaces are exposed to the D–Fe–Tris solution, and alginate solution is injected in between, then the adherends are pressed together; this takes place while the system is exposed to a bulk solution of 10 mM Tris. | ||
In this work, we also take inspiration from both benthic algae and marine mussels for applying the adhesive. Adhesive dopamine and alginate polymer components are initially separate when added; polydopamine formation and ferric ion coordination give structure to the adhesive and link the components. This resembles the nature of the components in algae adhesive, which are separate, then linked together. This also follows the two stages of algae adhesion, where the adhesive is first in liquid form to spread over the surface, then hardened in a second stage through polymerization and crosslinking.5 Further mimicking the exposed nature of algae adhesion (which takes place in seawater for both stages of adhesion), application of the adhesive takes place between two adherends, but in an open environment in bulk solution (in contrast, mussels secrete their adhesive proteins in a closed and controlled environment at a low pH, only exposing the components to the higher pH of seawater for the final hardening step38). While this makes the glue innovative, stronger, and easier to use, it does make it more difficult to control exact amounts at the surface, and will be more sensitive to environmental factors in solution. To account for this, care was taken to ensure each set of experiments was performed under the same conditions. Additionally, the adhesive dopamine/polydopamine components are injected at the surface of the adherends, providing adhesion where it is needed most – mussels also localize the adhesive proteins at the surface they are trying to adhere to, with the rest of the plaque and thread providing cohesive strength. The overall concept of this adhesive application method is illustrated in Fig. 1d: dopamine is localized at the surface to provide adhesion, alginate is injected in between to provide bulk cohesion, Fe3+ ions link everything together, and the components diffuse and harden to form the final glue.
We performed analytical microscopic examination of the adhesive materials and elucidated the roles of each component, as well as their effects on the final adhesive performance. Environmental factors relating to this open delivery system were also investigated. Through these techniques, we demonstrated the effectiveness of this approach, particularly that of sequential delivery. The resulting composite glue outperforms pure alginate by almost two orders of magnitude, effectively turning alginate into an adhesive, which was not possible through traditional chemical conjugation of alginate with catechol.
Results and discussion
X-ray photoelectron spectroscopy (XPS), Raman spectroscopy, scanning electron microscopy (SEM), and energy-dispersive X-ray spectroscopy (EDX) were used to examine the Fe3+ linking. XPS was used to determine if the alginate–iron–dopamine interactions were indeed present, with elemental atomic percents presented in Table 1, and an example of the resulting high-resolution Fe 2p spectra visible in Fig. 2g. Examining the relative levels of each element, the dopamine–iron–alginate (D–Fe–Alg) gel shares more in common with polydopamine film rather than with the iron–alginate (Fe–Alg) gel. This may indicate that a majority of the final adhesive is polydopamine. In examining the high-resolution Fe spectra (Fig. 2g) for Fe–Alg and D–Fe–Alg gels, the two systems appear to share similar peak locations, which may suggest that Alg–Fe interactions are present.| Atomic% | C1s | N1s | O1s | Fe2p3 |
|---|---|---|---|---|
| Alginate | 60.6 | 1.6 | 37.8 | |
| Polydopamine | 66.9 | 6.7 | 26.4 | |
| Fe–Alg | 54.5 | 3.7 | 40.2 | 1.6 |
| D–Fe–Alg | 66.2 | 6.6 | 26.7 | 0.5 |
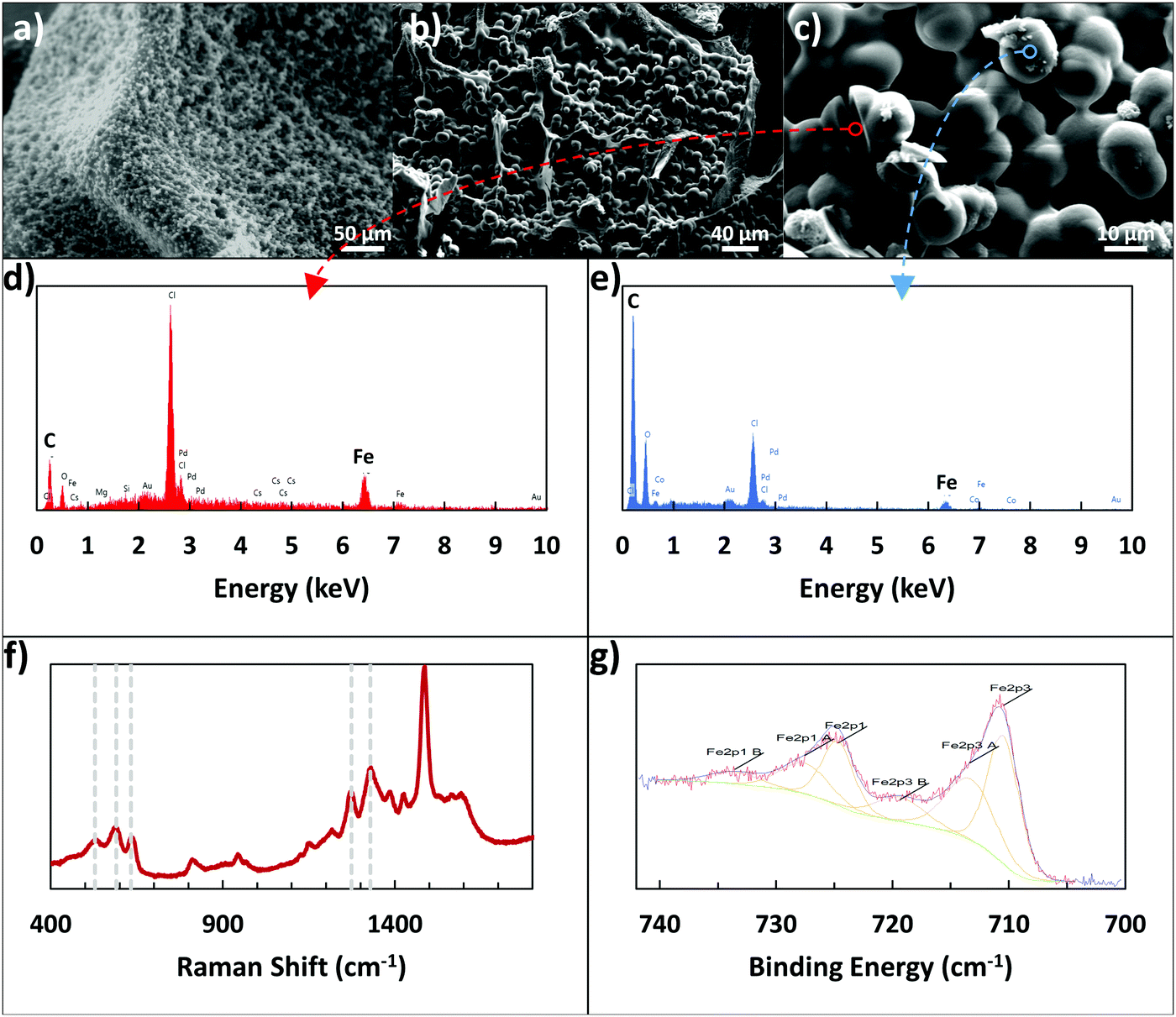 | ||
| Fig. 2 (a) ESEM image and (b and c) SEM images of spherical structures present in dopamine–iron–alginate gel. (d and e) Show EDX analysis of the locations marked in (c), with (d) focusing on the interior of a cracked sphere, and (e) examining the surface of a sphere. (f) Shows a Raman spectrum of dopamine–iron–alginate gel. (g) Shows a high-resolution XPS spectrum of the Fe 2p peaks for iron–alginate gel. | ||
A clearer indication of interactions between dopamine and Fe3+ ions was seen when using Raman spectroscopy. Resonance Raman spectra of dopamine–iron–alginate gel provided evidence of chelation of Fe3+ ions by the catechol group of dopamine, primarily visible in the 470–670 cm−1 Raman band (Fig. 2f). In particular, peaks at ∼590 cm−1 and ∼633 cm−1 mark interactions between Fe3+ ions and individual oxygen atoms in the catechol group, while the peak present at ∼528 cm−1 can be attributed to charge transfer interactions of the catechol–metal bidentate chelate.34,39 The height of the charge transfer peak, compared with the other two peaks, indicates a high level of bidentate complexation, suggesting the dopamine–Fe3+ complexes are predominantly tris-catechol–Fe3+. This is supported by the observation that the dopamine–iron solution was dark red in color, which is typical of tris-catechol–Fe3+.34
One interesting evidence for alginate–iron–dopamine interactions was seen when the microscale structure of the adhesive gel was examined. To do so, the gel was formed by mixing together the components of dopamine, alginate, iron, and tris(hydroxymethyl)aminomethane (Tris). These gels were immersed in 10 mM Tris–HCl buffer overnight, and then examined using optical microscopy and environmental SEM (ESEM). In addition, a portion was freeze-dried and examined using SEM. Three main components are visible with optical microscopy in the resulting hydrogel: pieces of the gel itself (alginate crosslinked by Fe3+ ions), irregular polydopamine particles, and other particles that are highly spherical in shape. These spherical particles were observable through optical microscopy, ESEM, and SEM, as seen in Fig. 2a–c, and were determined to be 12.26 ± 0.75 μm in size. By excluding individual components from the hydrogel-forming pre-mixture (e.g., no addition of dopamine, or no alginate), it was determined that all components – dopamine, iron, alginate, and Tris – were required to form the spherical particles. Without alginate or iron present, only smaller, irregular particles could be seen, which are expected to be polydopamine particles. Without dopamine or Tris present, the solution became a solid gel rapidly, lacking the presence of any of the spherical particles.
Additionally, EDX was used to provide elemental analysis of the spherical particles, both on their surface and in the center, utilizing a large crack on the particles. Examples of the resulting EDX plots can be seen in Fig. 2d and e, with their corresponding locations visible in Fig. 2c. While preliminary images from optical microscopy and ESEM suggested the spheres may be mostly iron, the SEM images and EDX results clarify their nature. The surface of the spheres appears to be polymer, with the majority consisting of carbon and oxygen, and only very small amounts (1–2 atomic%) of iron present. In contrast, the cracked sphere, with analysis closer to the center, showed much higher levels of iron present (8.55 atomic%). This additional iron may be in a solid form, or present as an ion, interacting with the alginate and polydopamine in the rest of the particles. In particular, the polymer content of the spheres has a low amount of oxygen present, bearing more in common with the carbon![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :oxygen ratio of polydopamine than that of alginate. This further reinforces the suggestion from XPS results that the adhesive surface consists mostly of polydopamine.
:oxygen ratio of polydopamine than that of alginate. This further reinforces the suggestion from XPS results that the adhesive surface consists mostly of polydopamine.
While there is evidence of both Alg–Fe3+ and dopamine–Fe3+ interactions, the overall cohesion of the system is a balance of these interactions, as well as Alg–Fe3+–dopamine linkages. In addition, there is another balance, one between cohesion and adhesion resulting from the Fe3+ ions; while their interactions with alginate and dopamine provide the system with cohesive strength, dopamine that is complexed with Fe3+ is unlikely to be able to directly contribute to adhesion. With the current composition of algae–mussel glue, the presence of Fe3+ appears to be more beneficial than harmful (Fig. S1, ESI†), but attention should be paid when modifying the ratio of Fe3+ ions to the other components, since changing concentrations of Fe3+ ions has previously been used to manipulate mechanical properties and degradation behaviour of dopamine–Fe3+-crosslinked elastomer.36
After confirming the interactions between all components, tensile pull-off tests were used to evaluate the bonding performance of the composite glue for underwater joining of aluminum stubs to glass slides. The glue was applied by using two base solutions: (1) a solution of dopamine, ferric nitrate nonahydrate, and Tris in deionized (DI) water; (2) a solution of 5 wt% alginate in DI water. These two solutions were applied separately, here referred to as the ‘sequential’ method, which is illustrated in Fig. 1d (with a detailed schematic in Fig. S2, ESI†). This separate addition means that the dopamine and alginate solutions were never directly mixed before application; however, there is time for the components to diffuse and interact during the curing stage, which mimics the adhesive deposition process of algae.
The tensile adhesive strength obtained from pulling apart these glued components can be seen in Fig. 3. In all cases where dopamine was not present (whether iron was present to cross-link alginate or not), adhesion of the gel was minimal (4–6 kPa). This is unsurprising, as alginate and its crosslinked gels are generally low in adhesion;28 this also shows that the viscous nature of pure alginate does not contribute significantly to adhesion. Alginate with conjugated catechol groups also has poor adhesion, indicating the functionalization had little effect on the final adhesion. The gel also exhibited extremely low adhesion when no alginate was present in the system, which emphasizes how all the components must be present to achieve the adhesive properties of this composite glue. One interesting point of note was the surprising strength of the sequential application with no iron present. While still significantly lower than the full system, the strength of the iron-free case suggests that dopamine and alginate may still be interacting in some way, especially since there were no ions available to crosslink alginate. It is possible that the basic conditions from Tris induced covalent crosslinking between alginate and catechol. With a tensile adhesive strength of 400 kPa, the composite glue achieves an improvement of nearly two orders of magnitude over pure alginate. This algae–mussel glue also provided great improvements over a commercial aquarium glue (Fig. S3, ESI†).
 | ||
| Fig. 3 Performance of algae–mussel adhesive, shows the effects of varying formulations, including: using only 5 wt% alginate (pure alginate); using alginate, iron, and tris, but without dopamine (no dopamine); using iron, dopamine, and tris, but without alginate (no alginate); using catechol-modified alginate with iron and tris, but no additional dopamine (catechol–alginate); using alginate, tris, and dopamine, but without iron (no iron); and the complete system with all components, applied using the pre-mixed (full system pre-mixed) or sequential (full system sequential) application methods. ** refers to a p-value <0.01 between the pair of conditions. | ||
While the sequential method was inspired by algae and mussels, it is also possible to simply mix the components together. We tested a ‘pre-mixed’ system, which more closely resembles traditional two-part adhesives (where solution (1) and (2) were directly mixed before application to the adherends). The pre-mixed method only demonstrated an adhesive strength of 70 kPa, compared to the 400 kPa strength of the sequential method. Based on our observations, we suspect the difference in performance could be attributed to the gelling of the pre-mixed solution occurring before application; the surface localization of the adhesive components in the sequential case could also be playing a role in improving overall adhesion.
In contrast to the controlled environment mussels maintain for adhesion, brown algae attach to surfaces in more exposed and open conditions. For this reason, we wanted to investigate the effects of dilution or concentration of the glue on its adhesive capabilities. In order to do so, solution (1) was prepared with differing initial quantities of DI water. While previous strengths using 70 μL of water are overall higher than values acquired while varying water content, this is due to the variability of batches of the glue being fabricated. All tests over a set of conditions are performed on the same batch, meaning an overall trend can still be observed. As shown in Fig. 4a, a peak in strength was visible using a water quantity of 50 μL. This adhesive strength was clearly higher than that when the water content was decreased further (30 μL). In addition, both the 50 μL and 70 μL cases demonstrated greater tensile strength than conditions where solution (1) was diluted further, particularly those of 120 μL and 180 μL. The effect of low water content can be explained by the high viscosity of solution (1), making mixing difficult and the application less homogeneous. The dilution from high water content would reduce the concentration of both adhesive and cohesive components, namely dopamine and Fe3+, as well as causing solution (1) to spread more rapidly upon application, diluting it further. The combination of these effects would lead to a significant reduction in the overall adhesive strength. This suggests that concentration of the initial adhesive solution, as well as its ability to avoid spreading too quickly, are essential factors in maximizing adhesion.
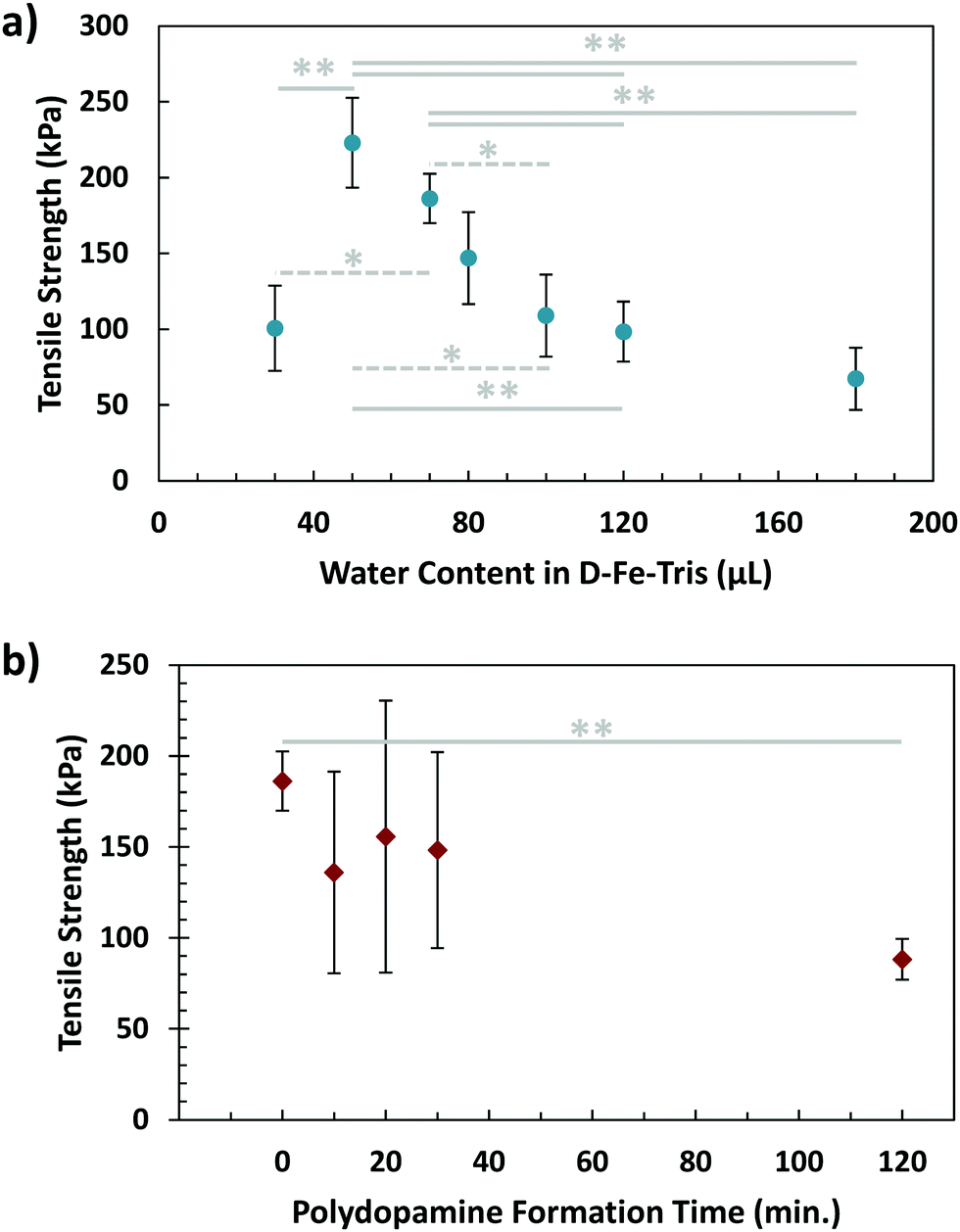 | ||
| Fig. 4 Effects on final underwater adhesive tensile strength of (a) water content of dopamine–Fe3+–Tris (D–Fe–Tris) solution (effectively diluting or concentrating these three components before application) and (b) polydopamine formation time before application. In both plots, * refers to a p-value <0.05, while ** refers to a p-value <0.01, each between the pair of conditions. | ||
Another factor to consider in algal adhesion is the nature of the polyphenol adhesive component. This is important for the composite glue because brown algae use polyphenol molecules in their adhesive strategy, while the adhesive presented in this work uses the small molecule of dopamine. The state of polydopamine depends on time, as dopamine self-polymerizes into polydopamine, growing over time. To investigate this, a waiting step was incorporated into the sequential procedure, where solution (1) was left for varying periods of time before application to the adherends. This was to allow the dopamine molecules to partially form polydopamine, resembling the polyphenols in algae. After this waiting step, the adhesive gel was formed using the sequential procedure, with results of tensile testing visible in Fig. 4b. While variability was lower for waiting times of 0 and 120 minutes, samples tested at 10, 20, and 30 minutes demonstrated large variations. This could be from the dynamic process of polydopamine formation occurring during this timeframe, resulting in a broad dispersity of polydopamine, which could lead to greater inhomogeneity in samples. By 2 hours, polydopamine formation has nearly completed, leading to more stable and consistent results. Additionally, there is a significant difference in adhesion measured between waiting times of 0 and 120 minutes, indicating that polydopamine forming before application could weaken overall adhesion of gel. This makes sense considering the two-stage adhesive of algae, where hardening/crosslinking only occurs after the adhesive has spread.
Further tensile adhesive tests were carried out on the composite glue in different environmental conditions: in 100% humidity, as well as in air on wet and dry glass substrates, with results shown in Fig. 5. Samples in 100% humidity were left overnight, while those in ambient conditions were left to dry over three days. The dry-applied glue demonstrated an adhesive strength almost twice that of the immersed, providing a high maximum strength to the glue. Application to a wet substrate resulted in adhesion similar to that of dry substrates. This indicates that the composite glue can tolerate varying levels of exposure to water. In addition, as we have shown previously, the glue can also tolerate varying pH levels, temperatures, and different surface conditions.31 Overall, the performance of this algae–mussel composite glue indicates that it could potentially find use in a variety of applications, including bonding and patching of soft and rigid materials. These potential applications are illustrated in Fig. 6, where the algae–mussel glue is sequentially injected between varying materials to hold them together. These include rigid inorganic (aluminum to glass) or flexible organic (bonding two sheets of polymer film) materials, or even hydrogels.
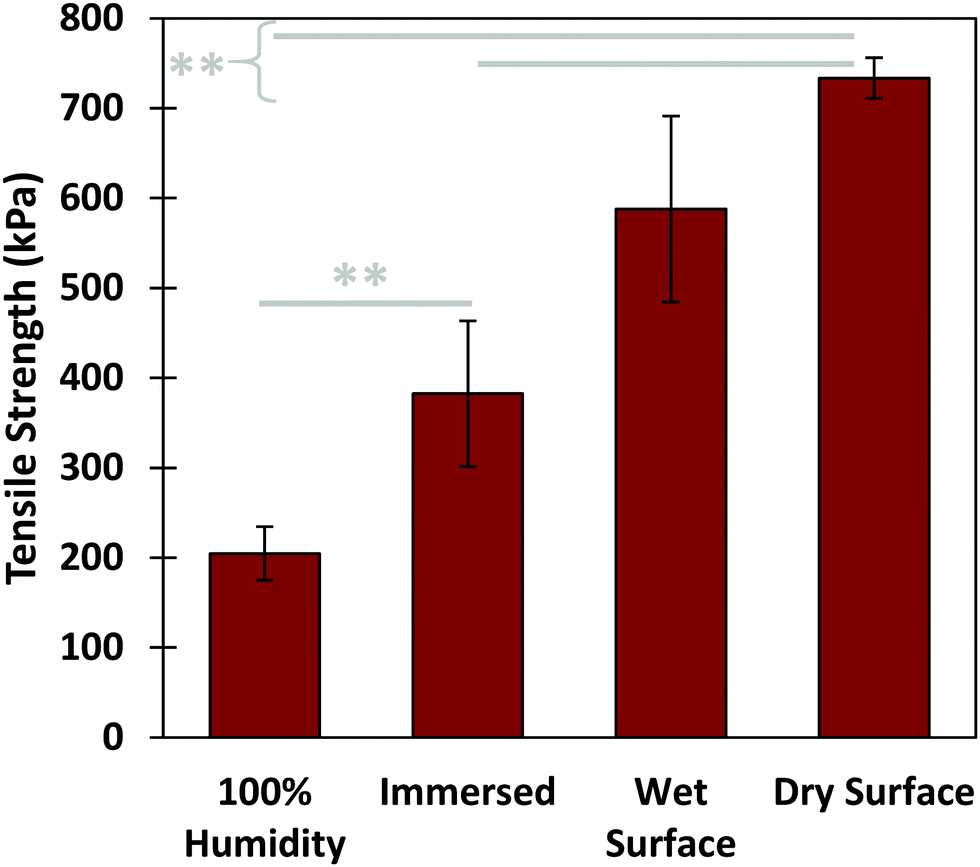 | ||
| Fig. 5 Performance of algae–mussel adhesive in varying environmental conditions, including: applied to a wet substrate and kept in a 100% humidity environment overnight (100% humidity); applied underwater, kept in aqueous conditions for 2 hours (immersed); applied in air to a surface wetted by water, and left to dry at ambient conditions for 3 days (wet surface); and applied to air to a dry surface, and allowed to dry at ambient conditions for 3 days (dry surface). ** refers to a p-value <0.01 between the pair of conditions. | ||
 | ||
| Fig. 6 Photos of (a) solutions used for sequential adhesion, used to join together: (b) two rigid aluminum SEM stubs; (c) two pieces of soft PVA hydrogel; (d) two flexible plastic (PET) films; (e) a plastic film to a plastic Petri dish patch a hole in the Petri dish, preventing the oil from leaking into the water; and (f) an aluminum stub to a rock. All cases were joined by the dopamine–alginate hybrid hydrogel adhesive in 10 mM Tris–HCl buffer at pH 8.5 for 2 hours of curing time. Note that the buffer solution was replaced by water for clarity. | ||
Conclusions
In summary, we have demonstrated a new strategy for constructing underwater adhesive, combining elements of algal and mussel adhesion. This adhesive does not require complicated chemical modification, instead utilizing Fe3+ ions as a bridge between the adhesive dopamine and cohesive alginate components. Good adhesive performance was achieved underwater, with reasonable tolerance to other environmental conditions. Sequential delivery of the components to focus adhesion at the interface, mimicking algal and mussel adhesive strategies, greatly strengthened the adhesive capabilities of the gel. The role of individual components was investigated, demonstrating the importance of incorporating dopamine for adhesion. Overall, our results demonstrate that the adhesive functionality does not have to be initially part of the polymer backbone, and connecting the components without chemical synthesis can still lead to strong bonding performance.Experimental section
Preparation of gel precursor solutions
Adhesive gel was formed in two parts. Solution (1) was prepared by mixing together 17.8 mg of iron(III) nitrate nonahydrate (Sigma-Aldrich), 50 mg of tris(hydroxymethyl) aminomethane (Tris, Sigma-Aldrich), and 100 mg of dopamine hydrochloride (Sigma-Aldrich) in 70 μL of deionized (DI) water. This solution was used immediately after mixing. Solution (2) was prepared by dissolving 5 wt% sodium alginate (HF 120RBS, FMC Biopolymer) in DI water. This solution was either used immediately after mixing or prepared in advance and refrigerated at 4.4 °C to store for up to a month.Fabrication and testing of adhesive gel
For the sequential application method, directly after mixing solution (1), 20 μL of solution (1) was taken up by a syringe and 18 gauge needle, then dropped onto a substrate (typically a glass slide) immersed in 50 mL of 10 mM Tris–HCl solution (prepared by acidifying 100 mM Tris solution with HCl to a pH of 8.56, then diluting 10×). The second adherend (typically an aluminum SEM stub) was then immersed and agitated within the concentrated region of solution (1), which has a higher density and remains at the bottom of the Tris–HCl solution. Finally, the second adhered was brought up out of the Tris–HCl solution, 30 μL of solution (2) was dropped (via a syringe and 18 gauge needle) onto its surface, and it was then pressed onto the surface of the first adherend. This glued-together system was left in the Tris–HCl solution for 2 hours before tensile testing.The pre-mixed application method utilized the same substrates, solutions, and final wait time before testing as the sequential method. The major difference is that instead of applying solutions (1) and (2) in separate steps, these two solutions were added together and mixed by vortex to form a viscous pre-gel solution. Approximately 40 μL of this pre-gel solution was then spread onto the surface of the second adherend, which was then pressed onto the immersed surface of the first adherend.
Tensile pull-off testing was performed using a universal materials tester (UMT, CETR), using a glass slide and an aluminum SEM stub (6.6 mm head, Ted Pella) as the first and second adherends, respectively. The aluminum stub was fitted into a custom holder to be attached to the UMT system, and then glued to the glass slide using either the sequential or pre-mixed application methods. After the 2 hour waiting period in Tris–HCl solution, samples were immediately withdrawn and attached to the UMT. The stub was then pulled away from the glass slide substrate (which was restrained from moving) at a rate of 500 mm min−1, until the two surfaces were fully separated from one another. The force was recorded during this time, and the maximum force achieved at pull-off was used as the adhesive pull-off force, then normalized by the contact area to determine the tensile strength.
For varying conditions, all tests except that labeled “full system pre-mixed” used the sequential application method, with deviations from this technique listed below. For the “pure alginate” case, only solution (2) was used. For the “no dopamine” case, solution (1) did not contain dopamine. For the “catechol–alginate” case, solution (2) used 5 wt% alginate that had been chemically modified with catechol groups, as in our previous work;14 also, solution (1) did not contain dopamine. For the “no iron” case, solution (1) did not contain ferric nitrate nonahydrate. For the “100% humidity” case, the substrate was immersed in Tris–HCl solution and withdrawn prior to application, then the adhesive system was left at 100% humidity overnight. For the wet surface, the glass slide was immersed in DI water, then removed from the water immediately prior to gluing on the aluminum stub, with this system left at room temperature for three days. For the dry surface, the aluminum stub was glued to a clean, dry glass slide, with the system left at room temperature for three days. For water content tests, solution (1) had varying quantities of DI water instead of the standard 70 μL. For polydopamine formation time, solution (1) was left for a varying amount of time after mixing before being dropped onto the glass slide surface for the sequential application.
Microstructure characterization
For examining the microstructure, solutions (1) and (2) were directly added together in a separate mini-centrifuge tube and mixed by vortex. This tube was immersed in 50 mL of 10 mM Tris–HCL solution overnight, then the gel within was removed and either examined directly or freeze-dried. Direct examination used an optical microscope, an environmental scanning microscope (ESEM), and Raman spectroscopy. Freeze-dried samples were examined using scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDX), and X-ray photoelectron spectroscopy (XPS).Statistics
All experiments were carried out with n ≥ 3 data points; the only exceptions are the “pure alginate” and “catechol–alginate” cases in Fig. 3, which only have 2 data points, as most samples were so weak as to detach before testing. For all figures, error bars represent the standard error of the mean.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors want to thank Catherine Roy for her assistance in preparing and analyzing alginate–iron–dopamine samples for examining microstructures in the gel. The authors also want to thank the Natural Sciences and Engineering Research Council of Canada (NSERC) for the financial support on this project (grant no. RGPIN-2014-04663).References
- L. Li, W. Smitthipong and H. Zeng, Polym. Chem., 2015, 6, 353–358 RSC.
- S. A. Burke, M. Ritter-Jones, B. P. Lee and P. B. Messersmith, Biomed. Mater., 2007, 2, 203–210 CrossRef CAS PubMed.
- M. Mehdizadeh, H. Weng, D. Gyawali, L. Tang and J. Yang, Biomaterials, 2012, 33, 7972–7983 CrossRef CAS PubMed.
- V. Vreeland, J. H. Waite and L. Epstein, J. Phycol., 1998, 34, 1–8 CrossRef CAS.
- E. R. Tarakhovskaya, Russ. J. Plant Physiol., 2014, 61, 19–25 CrossRef CAS.
- J. H. Waite, Int. J. Adhes. Adhes., 1987, 7, 9–14 CrossRef CAS.
- B. P. Lee, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Annu. Rev. Mater. Res., 2011, 41, 99–132 CrossRef CAS PubMed.
- R. Bitton and H. Bianco-Peled, Macromol. Biosci., 2008, 8, 393–400 CrossRef CAS PubMed.
- R. Bitton, E. Josef, I. Shimshelashvili, K. Shapira, D. Seliktar and H. Bianco-Peled, Acta Biomater., 2009, 5, 1582–1587 CrossRef CAS PubMed.
- Y. Rozen and H. Bianco-Peled, J. Adhes., 2014, 90, 667–681 CrossRef CAS.
- H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999–13003 CrossRef CAS PubMed.
- H. Shao and R. J. Stewart, Adv. Mater., 2010, 22, 729–733 CrossRef CAS PubMed.
- E. Faure, C. Falentin-Daudré, C. Jérôme, J. Lyskawa, D. Fournier, P. Woisel and C. Detrembleur, Prog. Polym. Sci., 2013, 38, 236–270 CrossRef CAS.
- A. Cholewinski, F. K. Yang and B. Zhao, Langmuir, 2017, 33, 8353–8361 CrossRef CAS PubMed.
- S. Lim, Y. S. Choi, D. G. Kang, Y. H. Song and H. J. Cha, Biomaterials, 2010, 31, 3715–3722 CrossRef CAS PubMed.
- B. J. Kim, D. X. Oh, S. Kim, J. H. Seo, D. S. Hwang, A. Masic, D. K. Han and H. J. Cha, Biomacromolecules, 2014, 15, 1579–1585 CrossRef CAS PubMed.
- J. H. Ryu, Y. Lee, W. H. Kong, T. G. Kim, T. G. Park and H. Lee, Biomacromolecules, 2011, 12, 2653–2659 CrossRef CAS PubMed.
- M. Cencer, Y. Liu, A. Winter, M. Murley, H. Meng and B. P. Lee, Biomacromolecules, 2014, 15, 2861–2869 CrossRef CAS PubMed.
- H. Lee, B. P. Lee and P. B. Messersmith, Nature, 2007, 448, 338–341 CrossRef CAS PubMed.
- B. P. Lee, K. Huang, F. N. Nunalee, K. R. Shull and P. B. Messersmith, J. Biomater. Sci., Polym. Ed., 2004, 15, 449–464 CrossRef CAS PubMed.
- J. Yang, I. Bos, W. Pranger, A. Stuiver, A. H. Velders, M. A. Cohen Stuart and M. Kamperman, J. Mater. Chem. A, 2016, 4, 6868–6877 RSC.
- S. Kim, J.-M. Moon, J. S. Choi, W. K. Cho and S. M. Kang, Adv. Funct. Mater., 2016, 26, 4099–4105 CrossRef CAS.
- S. H. Hong, M. Shin, J. Lee, J. H. Ryu, S. Lee, J. W. Yang, W. D. Kim and H. Lee, Adv. Healthcare Mater., 2015, 75–79 Search PubMed.
- C. J. Kastrup, M. Nahrendorf, J. L. Figueiredo, H. Lee, S. Kambhampati, T. Lee, S.-W. Cho, R. Gorbatov, Y. Iwamoto, T. T. Dang, P. Dutta, J. H. Yeon, H. Cheng, C. D. Pritchard, A. J. Vegas, C. D. Siegel, S. MacDougall, M. Okonkwo, A. Thai, J. R. Stone, A. J. Coury, R. Weissleder, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 21444–21449 CrossRef CAS PubMed.
- C. Lee, J. Shin, J. S. Lee, E. Byun, J. H. Ryu, S. H. Um, D.-I. Kim, H. Lee and S.-W. Cho, Biomacromolecules, 2013, 14, 2004–2013 CrossRef CAS PubMed.
- F. Ponzio, V. Le Houerou, S. Zafeiratos, C. Gauthier, T. Garnier, L. Jierry and V. Ball, Langmuir, 2017, 33, 2420–2426 CrossRef CAS PubMed.
- X. Wang, Z. Jiang, J. Shi, C. Zhang, W. Zhang and H. Wu, Ind. Eng. Chem. Res., 2013, 52, 14828–14836 CrossRef CAS.
- J. Hou, C. Li, Y. Guan, Y. Zhang and X. X. Zhu, Polym. Chem., 2015, 6, 2204–2213 RSC.
- C. Tian, C. Zhang, H. Wu, Y. Song, J. Shi, X. Wang, X. Song, C. Yang and Z. Jiang, J. Mater. Chem. B, 2014, 2, 4346 RSC.
- X. Wang, Z. Jiang, J. Shi, Y. Liang, C. Zhang and H. Wu, ACS Appl. Mater. Interfaces, 2012, 4, 3476–3483 CrossRef CAS PubMed.
- B. Zhao and F. Yang, US Pat., US9982169B2, 2018.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- K. J. Sreeram, H. Y. Shrivastava and B. U. Nair, Biochim. Biophys. Acta, Gen. Subj., 2004, 1670, 121–125 CrossRef CAS.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed.
- J. Yang, M. A. Cohen Stuart and M. Kamperman, Chem. Soc. Rev., 2014, 43, 8271–8298 RSC.
- B. Mizrahi, S. A. Shankarappa, J. M. Hickey, J. C. Dohlman, B. P. Timko, K. A. Whitehead, J. J. Lee, R. Langer, D. G. Anderson and D. S. Kohane, Adv. Funct. Mater., 2013, 23, 1527–1533 CrossRef CAS PubMed.
- H. Ceylan, M. Urel, T. S. Erkal, A. B. Tekinay, A. Dana and M. O. Guler, Adv. Funct. Mater., 2013, 23, 2081–2090 CrossRef CAS.
- J. H. Waite, J. Exp. Biol., 2017, 220, 517–530 CrossRef PubMed.
- I. Michaud-Soret, K. K. Andersson, L. Que and J. Haavik, Biochemistry, 1995, 34, 5504–5510 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8mh01421c |
| ‡ Aleksander Cholewinski and Fut (Kuo) Yang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |