
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dispersed nano-MOFs via a stimuli-responsive biohybrid-system with enhanced photocatalytic performance†
Hui-Chun
Lee
 a,
Tobias
Heil
a,
Jian-Ke
Sun
*ab and
Bernhard V. K. J.
Schmidt
*a
a,
Tobias
Heil
a,
Jian-Ke
Sun
*ab and
Bernhard V. K. J.
Schmidt
*a
aDepartment of Colloid Chemistry, Max Planck Institute of Colloids and Interfaces, 14424 Potsdam, Germany. E-mail: bernhard.schmidt@mpikg.mpg.de; jiankesun15@gmail.com
bSchool of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 102488, P. R. China
First published on 15th January 2019
Abstract
MOF-based heterogeneous catalysts with enhanced dispersibility in solution are highly promising for liquid-phase catalysis, yet their preparation remains a great challenge. Herein, via introducing responsive polymer-functionalized hollow pollen as pivots, a general strategy toward preparing MOF@Pollen composites with adjustable dispersibility, environment-responsive activity and significantly enhanced liquid-phase photocatalytic performance is presented.
Conceptual insightsMetal–organic frameworks (MOFs) with diverse pore chemistries have been actively investigated for heterogeneous catalysis. To enhance the catalytic performance, improved dispersibility of MOFs in liquid-phase reactions is highly pursued. Functionalization with polymers directly on the MOF surface is one of the most prevalent strategies. However, this straight-forward method has suffered from a long-term trade-off, that is, the catalytic MOF surface is seriously covered by the freely-joined, entangled polymer chains, resulting in adversely reduced active sites. Herein, a universal approach is presented to prepare MOF/polymer composites with not only stimuli-responsive dispersibility but also enhanced catalytic performance in solution. Via introducing bio-derived hollow pollen as pivots with a 2D-constrained environment-responsive polymer, poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) on the surface, the pollen-PDMAEMA composites (P-pollen) serve intrinsically as “smart” anchors to trap nanoMOFs to enhance dispersibility and to improve liquid-phase photocatalytic performance simultaneously. Moreover, the catalytic activity can be switched “on” and “off” via a stimulable coil-to-globule transition of the PDMAEMA chains exposing or burying the MOF catalytic sites, respectively. |
Introduction
Metal–organic frameworks (MOFs) are highly-crystalline porous materials constructed from organic linkers and metal ions that have recently revolutionized various research fields, including gas separation, sensing technology, drug delivery and energy conversion.1–9 Properties of tunable porous geometry, ultrahigh specific surface area and porosity as well as abundant reactive metallic sites indicate MOFs as unprecedented heterogeneous catalysts with promising catalytic performance.10–19 Although it is well known that liquid-phase catalytic activity can be improved via increasing the accessibility of catalytic sites,20,21 MOF crystals with strong tendency to aggregate in solution inevitably suffer from retarded mass transfer and reduced interfacial area, and this consequently hampers their catalytic capability.22,23 To conquer the problem of limited dispersibility, progress has been continuously made. Due to the complimentary material nature, polymers are prevalently introduced into MOFs,24 forming polymer/MOF composites with refined properties, such as integration of binder-tethered MOF crystals into membranes,25–27 or polymer-covered MOF solids with enhanced dispersibility in solution.28–31 Unfortunately, most of the strategies still suffer from a trade-off, i.e. the MOF surface, which is catalytically active, is covered by polymers, namely, the accessibility to active sites is adversely restricted. Hence, a smart design of MOF stabilizers with strengthened activity is constantly pursued. To break such trade-off, in this study instead of using highly mobile, free-formed polymer chains, surface-anchored polymer brushes on a pivot with constrained two-dimensional (2D) mobility and preserved space in-between are developed in this study. The polymer-functionalized pivot then acts as an anchor to disperse the MOFs for catalytic purposes (Scheme 1). In reactor-design, hollow structures are highly favored due to improved mass transfer, leading to enhanced capacities and catalytic properties.32–34 However, the time-consuming synthesis process and the probable collapse of the structure after template removal are significant issues. In the present work, hierarchically porous pollen grains with naturally-formed, robust hollow chambers are conveniently utilized as an alternative to artificial materials, serving as pivots for grafted polymers. Owing to the hydroxy groups and sporopollenin on the pollen shell, the surface of pollen is not only bio-compatible, but also easily functionalizable with diverse polymers.35–37 Other advantages of high accessibility, light weight, and high mechanical and chemical resistance can further expand the usability.36,38–40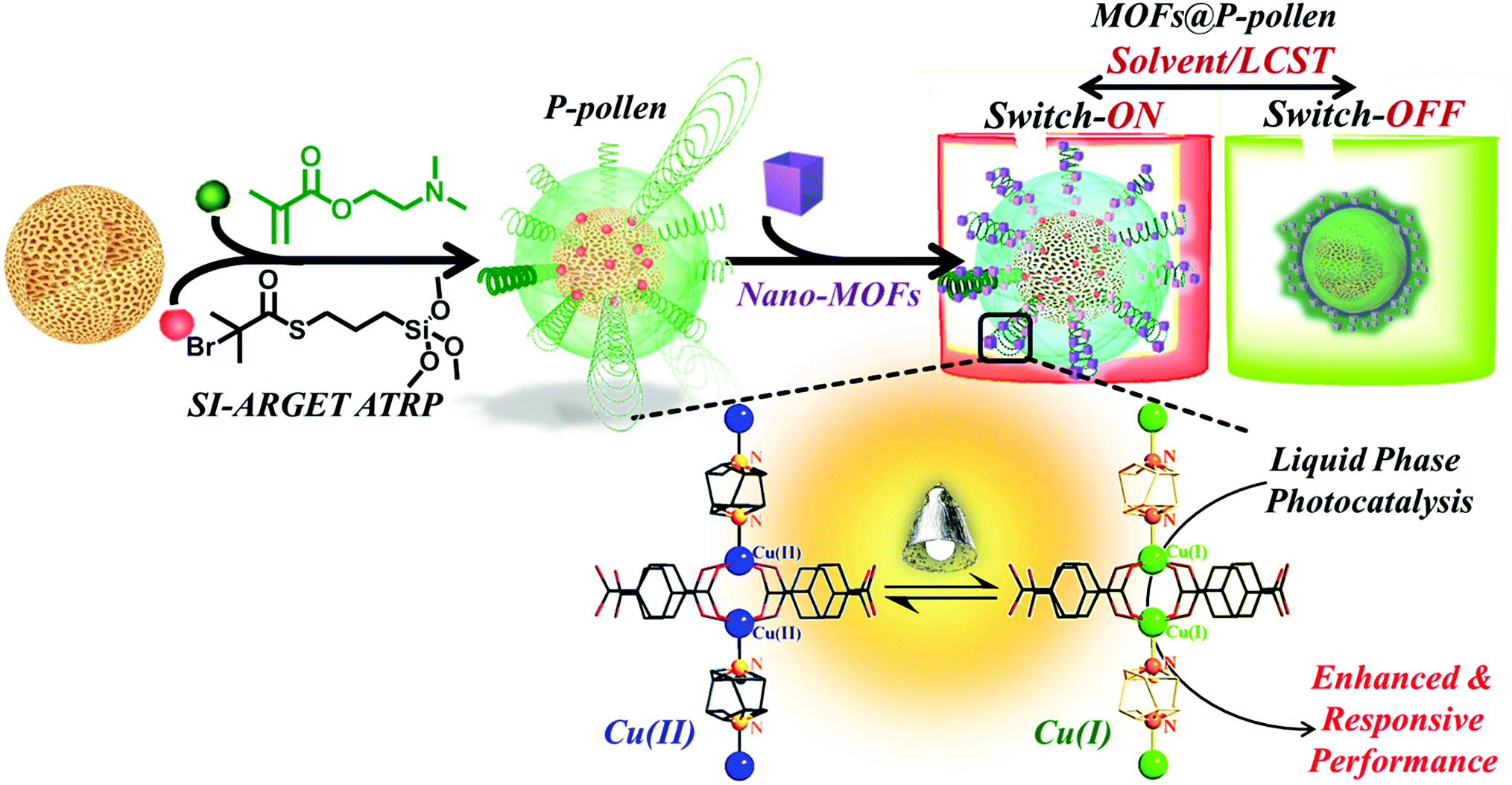 | ||
| Scheme 1 Illustration of the stimuli-responsive MOF@P-pollen catalyst for switchable liquid-phase photocatalytic reactions. | ||
Poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) is well-known for its stimuli-responsive properties.41,42 The chain conformation is strongly dependent on temperature, pH or solvent polarity. Namely, extended coiled chains are observed in good solvent, in an acidic environment (i.e. pH < pKa = 7.4) or at temperature below the lower critical solution temperature (LCST). By contrast, a collapsed globular conformation is presented. In particular, the amine group from each DMAEMA monomer features interfacial activity that enables coordination binding to metallic complexes.43 Taking advantage of this feature, a universal and adjustable MOF-targeting nano-anchor is introduced through combining a biotemplate as a facile, unique supporter and 2D PDMAEMA brushes with responsive properties and localized affinity to MOFs.
The strategy to fabricate environment-responsive dispersed MOF crystals is illustrated in Scheme 1. The surface initiated activators regenerated by electron transfer atom transfer radical polymerization (SI-ARGET ATRP) was applied to graft well-defined PDMAEMA brushes on the surface of washed pollen, giving “smart” PDMAEMA functionalized pollen (P-pollen). Subsequently, by using PDMAEMA as a specific anchor, MOF nanocrystals were trapped on the surface of P-pollen, forming MOF@P-pollen composites with nanoMOFs well dispersed on P-pollen. Referring to the stimulable chain conformations of PDMAEMA (i.e. stretching or recoiling), MOF@P-pollen featuring adjustable dispersibility is demonstrated in various liquid-phase photocatalysis reactions, resulting in a significantly enhanced catalytic performance. Moreover, an artificial switchable on-off catalysis system is achieved by external stimuli, a reminiscent process of biomimetic catalysis that is an ongoing challenge for synthetic chemistry.44–46
Results and discussion
Incorporation of Cu2(bdc)2(dabco) on P-pollen
The detailed functionalization process and incorporation of MOFs on P-pollen are illustrated in the ESI.† Generally, due to the controlled SI-ARGET ATRP polymerization, pollen grains with varied grafting density were prepared via using different concentrations of grafted initiator, for example, 0.5, 1, 2, 3 and 5 wt%. As shown in the ICP-OES results (Fig. S1, ESI†), an increasing incorporation of initiator on the pollen grains is detected, indicating that P-pollen with varied grafting density can be prepared consistently. Due to the controlled property of SI-ARGET ATRP, the grafting density of polymer brushes (σp) can be assessed by eqn (S1) (ESI†),47 giving σp = 0.32 chain nm−2. An adjustable grafting density is of particular advantage for chain length control of grafted brushes. Based on exponential scaling behavior shown in eqn (S2) (ESI†), a higher grafting density (σp ≥ 0.3 nm−2) is a core factor for extended polymers with increased steric hindrance. Thus, polymer chains can act as steric stabilizers to prevent the agglomeration of dispersed particles.48,49Successful trapping of Cu2(bdc)2(dabco) by P-pollen is revealed by energy-dispersive X-ray (EDX) mapping of the obtained composite, where the signals assigned to Cu with homogenous distribution are observed on the P-pollen (Fig. 1a). The morphology of the Cu2(bdc)2(dabco)@P-pollen composite is investigated by field emission scanning electron microscopy (FE-SEM) (Fig. 1b). The Cu2(bdc)2(dabco) crystals with sizes around 100 nm anchored on the hierarchically porous P-pollen are clearly observed. An obvious transition in the powder X-ray diffraction (PXRD) from amorphous pollen to crystalline Cu2(bdc)2(dabco)@P-pollen with consistent peak positions with the parent Cu2(bdc)2(dabco) further evidences the fabrication of Cu2(bdc)2(dabco)@P-pollen (Fig. 1c). The slight variation of relative peak intensity in the forming hybrid systems is attributed to the effect of preferred orientation of MOF crystals in the Cu2(bdc)2(dabco)@P-pollen composite, the solvent effect on metal sites or the interaction between the MOF crystals and P-pollen.50–53 Based on the PXRD profiles and the Debye-Scherrer equation (eqn (S3), ESI†),54,55 the average crystal size of the as-synthesized Cu2(bdc)2(dabco) and Cu2(bdc)2(dabco)@P-pollen is calculated to be ∼85 nm and ∼75 nm, respectively, which is consistent with the FE-SEM images.
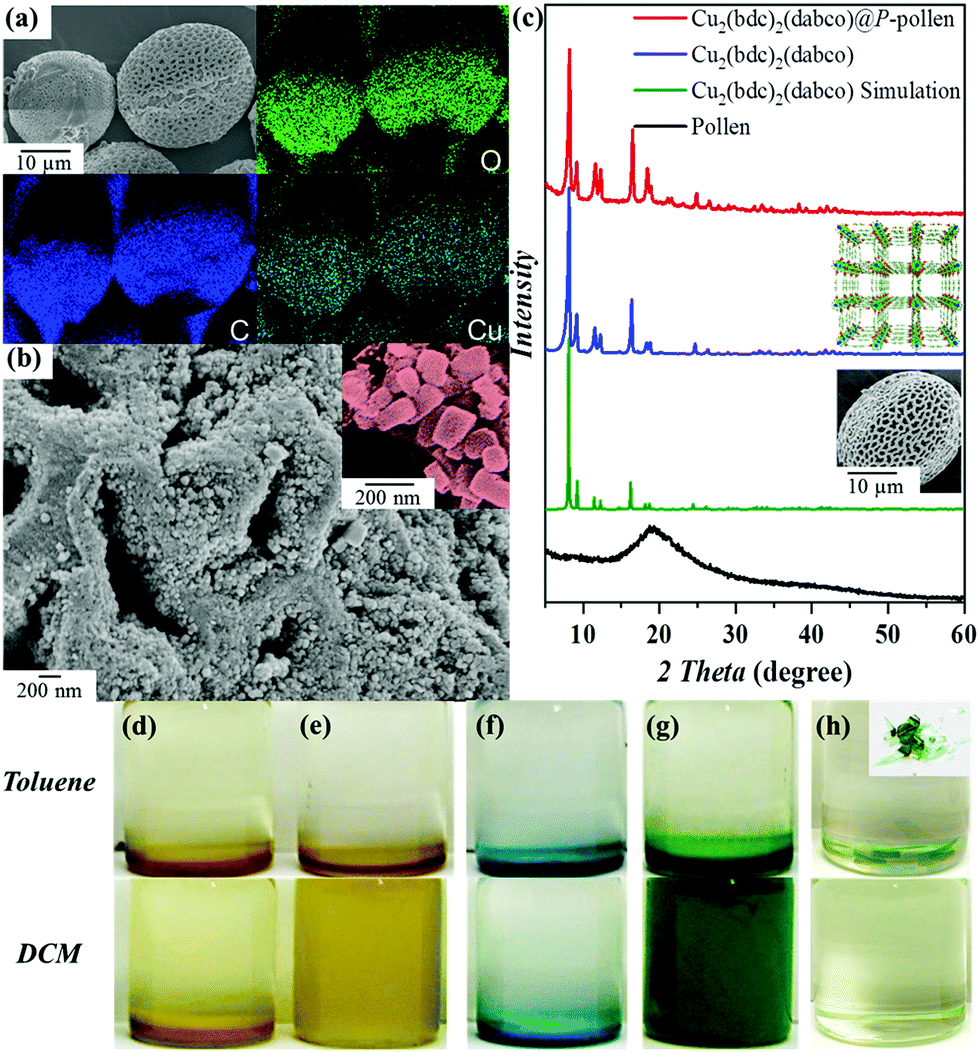 | ||
| Fig. 1 (a) FE-SEM EDX mapping of Cu2(bdc)2(dabco)@P-pollen. (b) Magnification revealing well-distributed MOF nanoparticles on the P-pollen surface. (c) PXRD of the as-synthesized Cu2(bdc)2(dabco)@P-pollen with innate Cu2(bdc)2(dabco) and washed pollen as a reference. Note: the microstructure of Cu2(bdc)2(dabco) is shown on the right side top and washed pollen on the right side bottom. Images of varied dispersibility in toluene (top, non-dispersed) vs. DCM (bottom, well-dispersed): (d) pollen; (e) P-pollen; (f) Cu2(bdc)2(dabco); (g) Cu2(bdc)2(dabco)@P-pollen; (h) Cu2(bdc)2(dabco)@PDMAEMA, the inset is the homogeneous hybrid bulk. Note: in the dispersibility test, the same mass of materials is used for each image. | ||
The remaining hollow structure of Cu2(bdc)2(dabco)@P-pollen which facilitates mass transfer in catalytic reactions is further advocated by FE-SEM cross section images (Fig. S2, ESI†). The association amount of Cu2(bdc)2(dabco) was determined by ICP-OES, giving a mass loading of 28 wt% on P-pollen (Table S1, ESI†).
The overall functionalization process is traced by ATR-FTIR spectroscopy showing the combined signals from pollen, PDMAEMA and Cu2(bdc)2(dabco) in the final composite (Fig. S3, ESI†). The shift of the NH stretching band (2768 and 2819 cm−1) towards higher wavenumbers (2819 and 2823 cm−1) after incorporation of Cu2(bdc)2(dabco) indicates the direct association between PDMAEMA and MOF nanocrystals.56
The interaction between MOF and P-pollen is further revealed by solid-state UV-vis spectroscopy to assess the individual optical band gap, EG, by the Tauc equation (eqn (S4), ESI†).57 As shown in Fig. S4 (ESI†), the Cu2(bdc)2(dabco) has a major absorption in the range of 250–350 nm with EG = ∼3.5 eV. However, upon coordination with P-pollen (EG = ∼1.7 eV), the spectrum of the Cu2(bdc)2(dabco)@P-pollen composite shows a broad absorption in the visible-light region (400–550 nm), with the corresponding band gap around 1.4 eV. Such improved absorption ability of the MOF@P-pollen composite is attributed to the strong coordination between P-pollen and Cu2(bdc)2(dabco). Compared to Cu2(bdc)2(dabco), the shifting absorption assigned for the d–d band of Cu2+ (600–800 nm) in Cu2(bdc)2(dabco)@P-pollen further evidences the slightly varied configuration after association.58,59 Considering the broad absorption of pollen (200–900 nm),60 such high absorption in Cu2(bdc)2(dabco)@P-pollen could result from the overlap spectra of Cu2(bdc)2(dabco) and P-pollen. Therefore, the absorption of different MOF-polymer complexes (i.e. Cu2(bdc)2(dabco)@PDMAEMA and MOF/P-pollen blending in a solid state) was further measured for comparison (Fig. S5, ESI†). Compared to the innate MOF, a slightly shifted absorption and prominently enhanced d–d absorption within 600–700 nm is observed in the Cu2(bdc)2(dabco)@PDMAEMA, advocating the association between Cu(II) ions in Cu2(bdc)2(dabco) and amine groups from DMAEMA monomers. Regardless of the minor shifted absorption, the light absorption ability of Cu2(bdc)2(dabco)@PDMAEMA is still restricted in the low wavelength area (i.e. <400 nm) which is distinct from Cu2(bdc)2(dabco)@P-pollen with major absorption in the visible-light area. Again, despite the same composition ratio in Cu2(bdc)2(dabco)@P-pollen, the solid-state blending complex (i.e. Cu2(bdc)2(dabco)/P-pollen Blending) exhibits absorption distinct from Cu2(bdc)2(dabco)@P-pollen with a weaker d–d absorption band. Actually, a noticeable color change in Cu2(bdc)2(dabco)@P-pollen indicates metal-ligand interaction as often encountered in coordination chemistry (Fig. S4 and S5, ESI†). Consequently, based on the distinct absorption behavior of different complexes, it suggests that a synergic effect between the grafted PDMAEMA brushes, pollen pivots and MOFs contributes this promising absorption behavior.
Universal strategy to diverse MOF@P-pollen composites
Surprisingly, the proposed strategy is highly applicable to a large variety of MOF@P-pollen with environment-responsive dispersibility (Fig. 2). Here, diverse MOFs, such as MOF-5, MOF-74-Zn, Zn2(bdc)2(dabco), Cu(bdc), HKUST-1 and ZIF-67, with mass loading of 18–36 wt% on P-pollen are demonstrated to present the versatility of the current strategy (Table S1, ESI†). The association and preservation of the specific MOF structure are confirmed by FE-SEM and PXRD (Fig. S6, ESI†). Accordingly, the eminent compatibility between P-pollen and various MOFs can be thus stated and further extended to other systems. | ||
| Fig. 2 Adjustable dispersibility of various MOFs@P-pollen: (a) MOF-5; (b) Zn2(bdc)2(dabco); (c) MOF-74-Zn; (d) Cu(bdc); (e) HKUST-1; (f) ZIF-67 in toluene vs. DCM. | ||
Adjustable dispersibility of MOF@P-pollen composites
The incorporated PDMAEMA in MOF@P-pollen allows investigation of environment-responsive dispersibility. The solvent-dependent dispersibility is widely probed by dynamic light scattering (DLS) in various solvents and pH (i.e. DCM, THF, acetone, methanol, toluene and water with pH = 1, 7 or 14). Considering the large particle size of pollen (∼30 μm), PDMAEMA chains cut from P-pollen were utilized in DLS analysis. As shown in Fig. S7 (ESI†), the decreasing hydrodynamic diameter (d, nm) is correlated with increasing solubility of PDMAEMA brushes in specific solvent. That is, the smallest d is detected in DCM or acid (13.5 or 8.7 nm) due to the highly dissolved PDMAEMA chains, and the size increases in toluene and neutral water (712 or 255 nm) owing to the recoiled morphology. Severe aggregation is eventually observed in basic solution (d = 1.7 μm). For the stability of MOFs, DCM and toluene are utilized to test the responsive dispersibility of the as-fabricated MOF@P-pollen composites. As shown in Fig. 1d–h, dispersibility of native MOF crystals and pollen grains is detected neither in DCM nor in toluene. However, by association on P-pollen, the Cu2(bdc)2(dabco)@P-pollen becomes well dispersed in DCM for more than 3 weeks due to highly extended polymer chains in the good solvent. Conversely, in the poor solvent, toluene, precipitation was observed in 3 min. Such solvent-dependent dispersibility is reproducible in the other MOF@P-pollen composites as well (Fig. 2).Because the dissolution of polymer chains in solvent is mostly related to hydrogen-bonding or electrostatic interactions61 with weaker interaction strength (10–40 kJ mol−1) than coordination bonds (∼102 kJ mol−1), the leakage of coordinated MOFs within solvent transition could be very limited.62 Furthermore, during MOF synthesis intensive washing was conducted to select robust MOF crystals with a larger particle size and higher stability for MOF@P-pollen preparation. Thus, as a prerequisite for catalysis, merely robust composites are prepared. The detailed pretreatment is illustrated in the ESI.† The high stability and sustainability of Cu2(bdc)2(dabco)@P-pollen is further reported by negligible Cu2+ leaching in solution in the following catalytic reaction.
Environment-responsive visible light-triggered Cu(I)-catalyzed alkyne-azide cycloaddition (CuAAC) via Cu2(bdc)2(dabco)@P-pollen
The MOF@P-pollen with high dispersibility in DCM is investigated for liquid-phase heterogeneous catalysis. Here, the copper(I) catalyzed azide–alkyne cycloaddition (CuAAC) reaction was chosen as a model reaction. As Cu2(bdc)2(dabco) is sensitive to visible light, by which Cu(II) can be in situ reduced to Cu(I) to catalyze reactions,63 the visible light-mediated CuAAC reaction is especially demonstrated (Fig. 3a). The synthesis of the starting materials and detailed processes are illustrated in the ESI.† The initial catalytic test advocates the photocatalytic property of Cu2(bdc)2(dabco)@P-pollen under visible light irradiation at ambient temperature (Fig. 3b). The time-dependent gas chromatography–mass spectrometry (GC-MS) profiles reveal the gradual consumption of starting alkyne, with the accumulation of triazole product (Fig. 3c and Fig. S8, ESI†). As the pollen and PDMAEMA are photoinert in the reaction (Fig. S9, ESI†), Cu2(bdc)2(dabco) is assumed to be the only catalytic active species. To ascertain the role of light in triggering Cu2(bdc)2(dabco)@P-pollen for photocatalysis, the characteristic temporal-controlled reactivity is then conducted. Namely, the on/off reaction was conducted by using intermittent light irradiation and dark treatment. As shown in Fig. S10 (ESI†), the photo-CuAAC reaction proceeded much faster under light irradiation, and conversely exhibited very limited activity during the “dark” periods.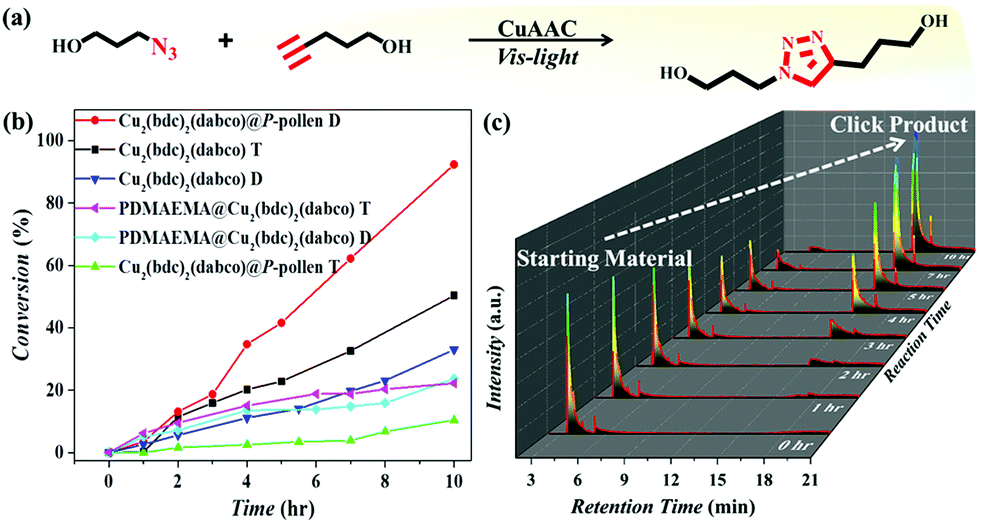 | ||
| Fig. 3 (a) The conducted photo-CuAAC reaction. (b) Time-dependent conversion of various Cu2(bdc)2(dabco) MOF-mediated photo-CuAAC reactions in DCM (D) or toluene (T). (c) Time-dependent GC-MS profiles of a photo-CuAAC reaction catalyzed by Cu2(bdc)2(dabco)@P-pollen. | ||
As shown in Fig. 4a and Table S2 (ESI†), the catalytic activity of the composites is evaluated by turnover number (TON) and turnover frequency (TOF). Under the condition of irradiation by visible light at ambient temperature, the best activity is achieved by Cu2(bdc)2(dabco)@P-pollen in DCM, giving conversion of ∼93% in 10 h with TON and TOF of 2078 and 208 h−1 respectively. Compared to the previously reported TOF of around 5–10 h−1,64–66 the given reactivity is – to the best of our knowledge – amongst the highest values for heterogeneous catalysts in a visible light-triggered CuAAC reaction at ambient temperature without the addition of reducing agents.29,64–66 Such eminent catalytic efficiency suggests that by introducing the P-pollen with well-constrained polymer brushes and hollow pollen pivots, the dispersibility and accessibility of the MOF's catalytic sites can be significantly improved, leading to surprisingly high activity of the MOFs in liquid-phase photocatalytic reactions.
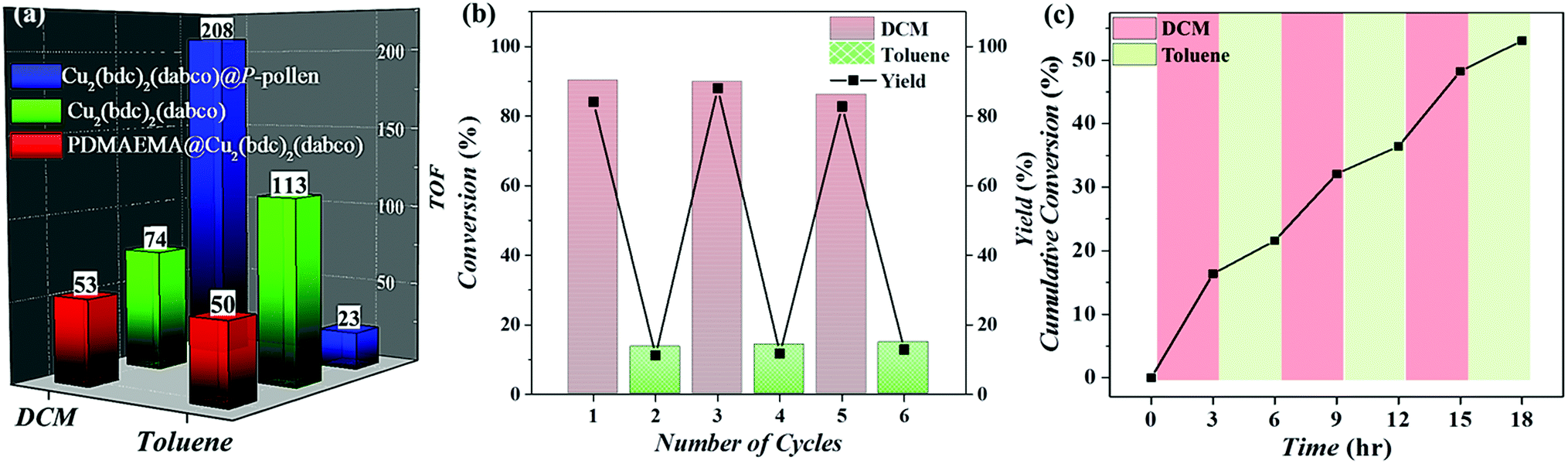 | ||
| Fig. 4 (a) The calculated TOF based on Fig. 3b. (b) The three-cycle on-off catalytic reusability. (c) Dynamic online temporal control of reactivity through alternate addition of DCM and toluene. | ||
A control experiment catalyzed by innate Cu2(bdc)2(dabco) with only ∼33% of conversion and 74.3 h−1 of TOF in DCM after 10 h supports this hypothesis. The restrained activity of the innate MOF is ascribed to the aggregated crystals with less accessible catalytic sites. To ascertain the improved accessibility of MOF catalytic sites in Cu2(bdc)2(dabco)@P-pollen, the catalytic complex comprised by free-formed polymers, but without the pivot (i.e. Cu2(bdc)2(dabco)@PDMAEMA) was also prepared and applied as a comparison. Although the dispersion of Cu2(bdc)2(dabco)@PDMAEMA in DCM is excellent (Fig. 1h), the slowest reaction rate is detected with conversion of merely ∼23% and TOF = 53 h−1 after 10 h. Presumably, the highly-dissolved polymer chains that initially enhance dispersion lead to less accessible catalytic sites (i.e. Cu ions) due to the freely-jointed/folding conformation, resulting in the most restricted accessibility and low catalytic activity. In summary, by comparing to the other catalytic complexes, the outstanding activity observed in Cu2(bdc)2(dabco)@P-pollen could be attributed to the synergistic effect contributed by the MOF catalysts, the hierarchical hollow pollen structure with favorable mass transfer and, most importantly, the enhanced accessibility of catalytic sites efficiently distributed by polymers anchored on the pivot.
Subsequently, two other solvents, methanol and THF, were applied to demonstrate the wide applicability and adjustable reactivity of MOF@P-pollen. As shown in Fig. S11 (ESI†), corresponding to DCM and toluene, different reactivity of Cu2(bdc)2(dabco)@P-pollen is detected in methanol and THF. Compared to DCM, a slower reaction rate is observed from methanol to THF, resulting in a decreased TOF from 207.8 h−1 in DCM to 72.6 h−1 in methanol and 48.8 h−1 in THF. However, the catalytic ability and TOF in these solvents is still higher than that in the poor solvent, toluene (TOF = 23.6 h−1). The reaction rate declines in the order of DCM > methanol > THF > toluene, which can be attributed to the PDMAEMA brushes getting contracted with gradually buried catalytic sites on the MOF surface. The solvent-dependent catalytic behavior is in line with the DLS results (Fig. S7, ESI†), indicating that the catalytic property of Cu2(bdc)2(dabco)@P-pollen is highly tunable based on the environment.
Switching and reusability of MOF@P-pollen composites
As shown in Fig. 4a, by introducing MOF@P-pollen, the catalysis can be switched “on” (i.e. speed up) in DCM (TOF: 208 h−1), and switched “off” (i.e. slow down) in toluene (TOF: 23 h−1). Owing to the heterogeneous and environment-responsive dispersibility of Cu2(bdc)2(dabco)@P-pollen, a switchable catalysis system with high reusability is further attempted through continuously and alternatively immerging in DCM or toluene. This process was repeated three times without noticeable change in the activity (Fig. 4b). ICP-OES revealed a negligible leakage (∼15 ppm) after three cycles (Table S3, ESI†), and together with the preserved MOF microstructure shown in the PXRD profiles (Fig. S12, ESI†) points out the robustness and high sustainability of the as-prepared bio-hybrid composites. The current system can even be extended to at least five cycles with consistent activity and product yield (Fig. S13, ESI†). The switching catalytic performance (i.e. fast/slow reaction) should stem from the alternative exposure and burying of MOF catalytic sites by reversible extension/recoiling of the PDMAEMA brushes.The sensitivity of the catalytic activity can then be demonstrated by using a mixed solvent. As shown in Fig. 4c, when the catalytic reaction started in DCM solution, 19% conversion is observed in the first 3 h. However, after two-third of the DCM mixture was replaced with toluene mixture (i.e. fresh reagents dissolved in toluene), a deactivated behavior is presented in the toluene-dominant solution (i.e. DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :toluene = 1:2 equiv.), resulting in a slowed reaction in the next 3 h. Following the same strategy, the activity of the catalyst can be regained by replacing two-thirds of the given toluene-dominated solvent with fresh DCM mixture, forming again the DCM-enriched solution with enhanced reactivity. In the mixed-solvent test, before each measurement the reactions were centrifuged to keep the catalysts at the bottom of the vial, and then two-thirds of the solution was exchanged with the next test mixture (i.e. DCM or toluene). Thus, the concentration of an individual catalyst was consistent throughout the reaction, and only the solvent ratio was changed (see Experimental section in ESI† for detail). Compared to the conditions using pure solvent in Fig. 4a and b, the switching phenomenon is less significant in the mixed-solvent system (Fig. 4c) due to the mixed solvent with a ratio of 2:1. The varied reactivity reveals the high adjustability and sensitivity of Cu2(bdc)2(dabco)@P-pollen towards the environment.
:toluene = 1:2 equiv.), resulting in a slowed reaction in the next 3 h. Following the same strategy, the activity of the catalyst can be regained by replacing two-thirds of the given toluene-dominated solvent with fresh DCM mixture, forming again the DCM-enriched solution with enhanced reactivity. In the mixed-solvent test, before each measurement the reactions were centrifuged to keep the catalysts at the bottom of the vial, and then two-thirds of the solution was exchanged with the next test mixture (i.e. DCM or toluene). Thus, the concentration of an individual catalyst was consistent throughout the reaction, and only the solvent ratio was changed (see Experimental section in ESI† for detail). Compared to the conditions using pure solvent in Fig. 4a and b, the switching phenomenon is less significant in the mixed-solvent system (Fig. 4c) due to the mixed solvent with a ratio of 2:1. The varied reactivity reveals the high adjustability and sensitivity of Cu2(bdc)2(dabco)@P-pollen towards the environment.
Temperature control of Cu2(bdc)2(dabco)@P-pollen
Due to the typical LCST character, the PDMAEMA brush is highly extended at temperatures below the LCST, and seriously contracts above the LCST. Because of such dramatic morphology change, a distinct thermal-switching catalyst reverse to conventional thermal-active catalysts can be designed. The LCST of PDMAEMA depends on molecular weight, pH, salt concentration, solvent hydrophobicity and so on.41,67 In general, the LCST of PDMAEMA is around 65 °C, and decreases according to the solubility in different solvents. Namely, the LCST in DCM should be higher than that in toluene due to the stronger solvent–polymer interactions by which the polymer chains remain extended in a wider temperature range.68 The cloud point of PDMAEMA was determined in the reaction mixture (azide, alkyne and DCM) to be around 55 °C by UV-vis (Fig. S14, ESI†). Therefore, to investigate the LCST-responsive catalytic property of Cu2(bdc)2(dabco)@P-pollen and to act as a comparison to ambient temperature, a photoCuAAC reaction is conducted at 65 °C, a temperature higher than the cloud point. As summarized in Fig. S15 and Table S4 (ESI†), instead of thermal activation observed under blank conditions and for innate Cu2(bdc)2(dabco) (TOF: 802 h−1), the MOF@P-pollen acts as a thermal switch which deactivates the reaction (TOF: 243 h−1) at high temperature. The restrained reactivity is due to seriously collapsed PDMAEMA blocking the catalytic sites temporarily from reagents at temperatures above the LCST. It is worth mentioning that the activity can be restored at ambient temperature.Environment-responsive visible light-triggered photodegradation via Cu2(bdc)2(dabco)@P-pollen
Cu2(bdc)2(dabco)@P-pollen with switchable catalytic activity can be extended to other photocatalytic reactions, for example the photodegradation of Rhodamine B (RhB). The reaction was easily monitored by UV-vis spectroscopy, recording the varied absorbance at 554 nm assigned to RhB. As shown in Fig. 5 and Fig. S16a (ESI†), in DCM the catalyst achieves quantitative degradation in 165 min, while the catalysis is restricted in toluene, giving only ∼20% degradation within the same time. The pseudo-first-order rate constant (k) estimated from time-dependent UV-vis spectra reveals that the catalytic activity is modulated by more than ten times (kDCM = 1.26 h−1; ktoluene = 0.1 h−1) due to the controlled aggregation/dispersion of MOF nanocrystals. Furthermore, insignificant degradation of RhB is observed in the dark (Fig. S16b, ESI†) in DCM, evidencing the light-dependent catalytic activity of Cu2(bdc)2(dabco)@P-pollen toward RhB degradation instead of physical adsorption. Distinct from the prompt and responsive reactivity in Cu2(bdc)2(dabco)@P-pollen, a lower and non-adjustable reactivity between DCM (kDCM = 0.43 h−1) and toluene (ktoluene = 0.56 h−1) is observed by using Cu2(bdc)2(dabco) as a catalyst (Fig. S17a and b, ESI†). The photodegradation result is in line with the photo-CuAAC reaction (Fig. 3), advocating the prominent catalytic and environment-responsive property of the as-designed Cu2(bdc)2(dabco)@P-pollen. No obvious degradation is detected under blank conditions, indicating that the decomposition of RhB is catalyzed by the applied MOF catalyst (Fig. S17c and d, ESI†). Furthermore, the stability of Cu2(bdc)2(dabco) and the MOF@P-pollen composite can be revealed by the highly consistent PXRD profiles before and after the photodegradation (Fig. S18, ESI†). It should be noted that a shift of UV-vis spectrum in Fig. 5a is observed, which is due to the N-deethylation of RhB.69–72 As illustrated by other researchers, the hyposochromic shift in the absorption peak results from the step-by-step cleavage and deethylation of RhB. The variation in the degradation mechanism is ascribed to different surface properties of the MOF photocatalysts, leading to different intermediates.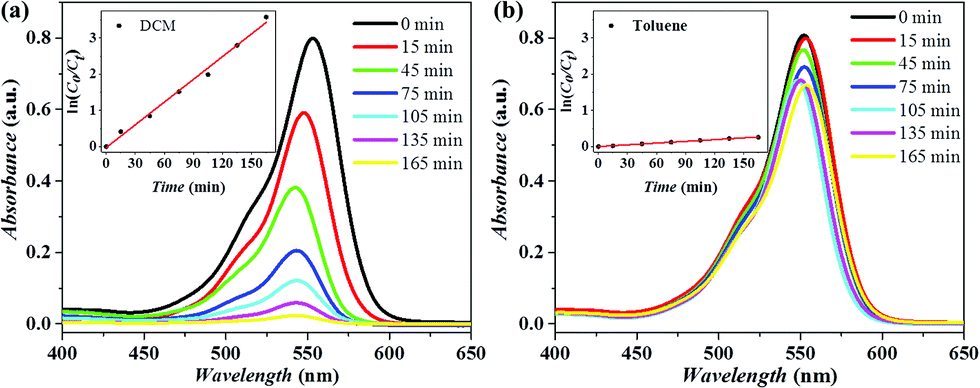 | ||
| Fig. 5 UV-vis spectra monitoring the photodegradation of RhB via a Cu2(bdc)2(dabco)@P-pollen catalyst in (a) DCM and (b) toluene at ambient temperature. Inset: The corresponding slope of the plot ln(c0/ct) vs. time. | ||
Conclusions
In summary, a facile and universal strategy to improve the dispersibility of MOF crystals without debilitating the catalytic potential is presented. Surpassing the previously-established methods which improve MOF dispersibility under a sacrifice of reactivity, via introducing PDMAEMA-functionalized pollen as “smart” anchors, nanoMOFs can be effectively trapped and stabilized with not only enhanced dispersibility, but enhanced catalytic performance in liquid-phase photocatalysis. Owing to the stimuli-responsive polymers, the associated nanoMOFs can be reversibly exposed (catalytic active) or buried (catalytic inactive) to exhibit a switchable catalytic behavior. Considering the eminent compatibility between P-pollen and MOFs, the current work opens up a new perspective in developing stimuli-responsive MOF heterogeneous catalysts with restrengthened activity.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge financial support from the Max Planck Society. The authors further acknowledge the German Academic Exchange Service (DAAD) (HL), and Alexander von Humboldt (AvH) Foundation (JS). The authors thank Marlies Gräwert for SEC experiments, Heike Runge for electron microscopy, and Jeannette Steffen for ICP-OES analysis. Open Access funding was provided by the Max Planck Society.References
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- T. Kitao, Y. Zhang, S. Kitagawa, B. Wang and T. Uemura, Chem. Soc. Rev., 2017, 46, 3108–3133 RSC.
- R. J. Kuppler, D. J. Timmons, Q.-R. Fang, J.-R. Li, T. A. Makal, M. D. Young, D. Yuan, D. Zhao, W. Zhuang and H.-C. Zhou, Coord. Chem. Rev., 2009, 253, 3042–3066 CrossRef CAS.
- Z. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC.
- R.-B. Lin, F. Li, S.-Y. Liu, X.-L. Qi, J.-P. Zhang and X.-M. Chen, Angew. Chem., Int. Ed., 2013, 52, 13429–13433 CrossRef CAS PubMed.
- B. Chen, S. Xiang and G. Qian, Acc. Chem. Res., 2010, 43, 1115–1124 CrossRef CAS PubMed.
- S. Han, Y. Wei, C. Valente, I. Lagzi, J. J. Gassensmith, A. Coskun, J. F. Stoddart and B. A. Grzybowski, J. Am. Chem. Soc., 2010, 132, 16358–16361 CrossRef CAS PubMed.
- E. Gkaniatsou, C. Sicard, R. Ricoux, J.-P. Mahy, N. Steunou and C. Serre, Mater. Horiz., 2017, 4, 55–63 RSC.
- J. Hwang, T. Heil, M. Antonietti and B. V. K. J. Schmidt, J. Am. Chem. Soc., 2018, 140, 2947–2956 CrossRef CAS PubMed.
- A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- J.-D. Xiao, Q. Shang, Y. Xiong, Q. Zhang, Y. Luo, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 9389–9393 CrossRef CAS PubMed.
- D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS PubMed.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- M. A. Nasalevich, R. Becker, E. V. Ramos-Fernandez, S. Castellanos, S. L. Veber, M. V. Fedin, F. Kapteijn, J. N. H. Reek, J. I. van der Vlugt and J. Gascon, Energy Environ. Sci., 2015, 8, 364–375 RSC.
- M. Meilikhov, K. Yusenko, D. Esken, S. Turner, G. Van Tendeloo and R. A. Fischer, Eur. J. Inorg. Chem., 2010, 3701–3714 CrossRef CAS.
- H.-C. Lee, M. Antonietti and B. V. K. J. Schmidt, Polym. Chem., 2016, 7, 7199–7203 RSC.
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC.
- L. Zhu, X.-Q. Liu, H.-L. Jiang and L.-B. Sun, Chem. Rev., 2017, 117, 8129–8176 CrossRef CAS PubMed.
- C. A. Witham, W. Huang, C.-K. Tsung, J. N. Kuhn, G. A. Somorjai and F. D. Toste, Nat. Chem., 2010, 2, 36–41 CrossRef CAS PubMed.
- J.-K. Sun, W.-W. Zhan, T. Akita and Q. Xu, J. Am. Chem. Soc., 2015, 137, 7063–7066 CrossRef CAS PubMed.
- X. Yang, S. Kattel, S. D. Senanayake, J. A. Boscoboinik, X. Nie, J. Graciani, J. A. Rodriguez, P. Liu, D. J. Stacchiola and J. G. Chen, J. Am. Chem. Soc., 2015, 137, 10104–10107 CrossRef CAS PubMed.
- E. D. Metzger, R. J. Comito, C. H. Hendon and M. Dincă, J. Am. Chem. Soc., 2017, 139, 757–762 CrossRef CAS PubMed.
- Q. Sun, H. He, W.-Y. Gao, B. Aguila, L. Wojtas, Z. Dai, J. Li, Y.-S. Chen, F.-S. Xiao and S. Ma, Nat. Commun., 2016, 7, 13300 CrossRef CAS PubMed.
- W.-L. Jiang, L.-G. Ding, B.-J. Yao, J.-C. Wang, G.-J. Chen, Y.-A. Li, J.-P. Ma, J. Ji, Y. Dong and Y.-B. Dong, Chem. Commun., 2016, 52, 13564–13567 RSC.
- Y. Chen, S. Li, X. Pei, J. Zhou, X. Feng, S. Zhang, Y. Cheng, H. Li, R. Han and B. Wang, Angew. Chem., Int. Ed., 2016, 55, 3419–3423 CrossRef CAS PubMed.
- A. U. Czaja, N. Trukhan and U. Muller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC.
- G. Huang, Q. Yang, Q. Xu, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 7379–7383 CrossRef CAS PubMed.
- K. Xie, Q. Fu, Y. He, J. Kim, S. J. Goh, E. Nam, G. G. Qiao and P. A. Webley, Chem. Commun., 2015, 51, 15566–15569 RSC.
- S. Nagata, K. Kokado and K. Sada, Chem. Commun., 2015, 51, 8614–8617 RSC.
- S. Wang, W. Morris, Y. Liu, C. M. McGuirk, Y. Zhou, J. T. Hupp, O. K. Farha and C. A. Mirkin, Angew. Chem., Int. Ed., 2015, 54, 14738–14742 CrossRef CAS PubMed.
- Z. Dandan, C. Xu-Ning and W. Xinchen, Angew. Chem., Int. Ed., 2016, 55, 11512–11516 CrossRef PubMed.
- L.-H. Chen, X.-Y. Li, G. Tian, Y. Li, J. C. Rooke, G.-S. Zhu, S.-L. Qiu, X.-Y. Yang and B.-L. Su, Angew. Chem., Int. Ed., 2011, 50, 11156–11161 CrossRef CAS.
- S. Peng, L. Li, H. Tan, R. Cai, W. Shi, C. Li, S. G. Mhaisalkar, M. Srinivasan, S. Ramakrishna and Q. Yan, Adv. Funct. Mater., 2014, 24, 2155–2162 CrossRef CAS.
- O. O. Fadiran and J. C. Meredith, J. Mater. Chem. A, 2014, 2, 17031–17040 RSC.
- M. G. Potroz, R. C. Mundargi, J. J. Gillissen, T. Ee-Lin, M. Sigalit, J. H. Park, J. Haram, P. Soohyun, C. Daeho, B. Sa-Ik and C. Nam-Joon, Adv. Funct. Mater., 2017, 27, 1700270 CrossRef.
- M. Jenik, A. Seifner, P. Lieberzeit and F. L. Dickert, Anal. Bioanal. Chem., 2009, 394, 523–528 CrossRef CAS.
- S. R. Hall, H. Bolger and S. Mann, Chem. Commun., 2003, 2784–2785 RSC.
- S. R. Hall, V. M. Swinerd, F. N. Newby, A. M. Collins and S. Mann, Chem. Mater., 2006, 18, 598–600 CrossRef CAS.
- B. J. R. Thio, J.-H. Lee and J. C. Meredith, Environ. Sci. Technol., 2009, 43, 4308–4313 CrossRef CAS.
- F. Schacher, M. Ulbricht and A. H. E. Müller, Adv. Funct. Mater., 2009, 19, 1040–1045 CrossRef CAS.
- H.-C. Lee, H.-Y. Hsueh, U. S. Jeng and R.-M. Ho, Macromolecules, 2014, 47, 3041–3051 CrossRef CAS.
- S. C. Warren, L. C. Messina, L. S. Slaughter, M. Kamperman, Q. Zhou, S. M. Gruner, F. J. DiSalvo and U. Wiesner, Science, 2008, 320, 1748–1752 CrossRef CAS PubMed.
- S. Yuan, L. Zou, H. Li, Y.-P. Chen, J. Qin, Q. Zhang, W. Lu, M. B. Hall and H.-C. Zhou, Angew. Chem., Int. Ed., 2016, 55, 10776–10780 CrossRef CAS PubMed.
- Y. Wei, S. Han, J. Kim, S. Soh and B. A. Grzybowski, J. Am. Chem. Soc., 2010, 132, 11018–11020 CrossRef CAS PubMed.
- V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341–5370 RSC.
- T. W. Vasicek, S. V. Jenkins, L. Vaz, J. Chen and J. A. Stenken, J. Colloid Interface Sci., 2017, 506, 338–345 CrossRef CAS PubMed.
- L. C. H. Moh, M. D. Losego and P. V. Braun, Langmuir, 2011, 27, 3698–3702 CrossRef CAS PubMed.
- M. W. Hahn, D. Abadzic and C. R. O'Melia, Environ. Sci. Technol., 2004, 38, 5915–5924 CrossRef CAS PubMed.
- J. Hafizovic, M. Bjørgen, U. Olsbye, P. D. C. Dietzel, S. Bordiga, C. Prestipino, C. Lamberti and K. P. Lillerud, J. Am. Chem. Soc., 2007, 129, 3612–3620 CrossRef CAS PubMed.
- T. Devic, P. Horcajada, C. Serre, F. Salles, G. Maurin, B. Moulin, D. Heurtaux, G. Clet, A. Vimont, J.-M. Grenèche, B. L. Ouay, F. Moreau, E. Magnier, Y. Filinchuk, J. Marrot, J.-C. Lavalley, M. Daturi and G. Férey, J. Am. Chem. Soc., 2010, 132, 1127–1136 CrossRef CAS PubMed.
- T. Uemura, N. Uchida, M. Higuchi and S. Kitagawa, Macromolecules, 2011, 44, 2693–2697 CrossRef CAS.
- J.-K. Sun, H.-J. Lin, W.-Y. Zhang, M.-R. Gao, M. Antonietti and J. Yuan, Mater. Horiz., 2017, 4, 681–687 RSC.
- J. I. Langford and A. J. C. Wilson, J. Appl. Crystallogr., 1978, 11, 102–113 CrossRef CAS.
- H. Borchert, E. V. Shevchenko, A. Robert, I. Mekis, A. Kornowski, G. Grübel and H. Weller, Langmuir, 2005, 21, 1931–1936 CrossRef CAS PubMed.
- J. Coates, in Encyclopedia of Analytical Chemistry, John Wiley & Sons, Ltd, 2006 DOI:10.1002/9780470027318.a5606.
- J. Tauc, R. Grigorovici and A. Vancu, Phys. Status Solidi B, 1966, 15, 627–637 CrossRef CAS.
- C. Wang, D. Wang, S. Yu, T. Cornilleau, J. Ruiz, L. Salmon and D. Astruc, ACS Catal., 2016, 6, 5424–5431 CrossRef CAS.
- A. Das, R. I. Kureshy, N. C. Maity, P. S. Subramanian, N.-U. H. Khan, S. H. R. Abdi, E. Suresh and H. C. Bajaj, Dalton Trans., 2014, 43, 12357–12364 RSC.
- S. L. Atkin, S. Barrier, Z. Cui, P. D. I. Fletcher, G. Mackenzie, V. Panel, V. Sol and X. Zhang, J. Photochem. Photobiol., B, 2011, 102, 209–217 CrossRef CAS PubMed.
- B. A. Miller-Chou and J. L. Koenig, Prog. Polym. Sci., 2003, 28, 1223–1270 CrossRef CAS.
- S. Kitagawa, S.-i. Noro and T. Nakamura, Chem. Commun., 2006, 701–707 RSC.
- H.-C. Lee, M. Fantin, M. Antonietti, K. Matyjaszewski and B. V. K. J. Schmidt, Chem. Mater., 2017, 29, 9445–9455 CrossRef CAS.
- Q. Fu, K. Xie, S. Tan, J. M. Ren, Q. Zhao, P. A. Webley and G. G. Qiao, Chem. Commun., 2016, 52, 12226–12229 RSC.
- C. K. Karan, M. C. Sau and M. Bhattacharjee, Chem. Commun., 2017, 53, 1526–1529 RSC.
- B. Wang, J. Durantini, J. Nie, A. E. Lanterna and J. C. Scaiano, J. Am. Chem. Soc., 2016, 138, 13127–13130 CrossRef CAS PubMed.
- F. A. Plamper, M. Ballauff and A. H. E. Müller, J. Am. Chem. Soc., 2007, 129, 14538–14539 CrossRef CAS PubMed.
- S. B. Lee, S.-C. Song, J. Jung-Il. and Y. S. Sohn, Polym. Bull., 2000, 45, 389–396 CrossRef CAS.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2010, 26, 3894–3901 CrossRef CAS PubMed.
- T. Watanabe, T. Takizawa and K. Honda, J. Phys. Chem., 1977, 81, 1845–1851 CrossRef CAS.
- T. Wu, G. Liu, J. Zhao, H. Hidaka and N. Serpone, J. Phys. Chem. B, 1998, 102, 5845–5851 CrossRef CAS.
- Y. Chen, R. Huang, D. Chen, Y. Wang, W. Liu, X. Li and Z. Li, ACS Appl. Mater. Interfaces, 2012, 4, 2273–2279 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experiment section, ATR-FTIR spectra, UV-vis spectra, characterization of the polymer and incorporated MOFs, DLS results, NMR profiles and SEC chromatograms are illustrated. See DOI: 10.1039/c8mh01342j |
| This journal is © The Royal Society of Chemistry 2019 |