
Etching silver nanoparticles using DNA†
Shengqiang
Hu
ab,
Tiantian
Yi
a,
Zhicheng
Huang
b,
Biwu
Liu
 b,
Jianxiu
Wang
*a,
Xinyao
Yi
*a and
Juewen
Liu
*b
b,
Jianxiu
Wang
*a,
Xinyao
Yi
*a and
Juewen
Liu
*b
aCollege of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China. E-mail: jxiuwang@csu.edu.cn; yixinyao@csu.edu.cn
bDepartment of Chemistry, Waterloo Institute for Nanotechnology, University of Waterloo, Waterloo, Ontario, N2L 3G1, Canada. E-mail: liujw@uwaterloo.ca
First published on 9th October 2018
Abstract
While DNA has been widely used for directing the growth and assembly of nanomaterials, the reverse reaction, etching nanoparticles using DNA, has yet to be demonstrated. We herein communicate that poly-cytosine (poly-C) DNA can efficiently etch silver nanoparticles (AgNPs) followed by Ostwald ripening at higher DNA concentrations. The etching process was precisely controlled by varying the length, sequence, and concentration of DNA, and the number of consecutive cytosines is particularly important for the efficacy of etching. In addition to spherical AgNPs, etching also occurred for silver nanoplates displaying interesting color changes. Compared to other chemical etching agents such as H2O2 and ferricyanide, DNA is highly biocompatible, allowing biological applications. Poly-C etching enhanced the cytotoxicity of AgNPs against cancer cells, and Gram-positive and Gram-negative bacterial cells. This study will stimulate many related studies in DNA nanotechnology, bioanalytical sensors and nanomedicine.
Conceptual insightsUsing DNA to control the growth of nanomaterials is a well-developed concept, but the reverse reaction of etching is rarely explored. Since etching agents are often highly toxic, using them for bio-related applications is difficult. In this work, biocompatible DNA-directed etching is demonstrated on silver nanoparticles. This observation is complementary to the notion that biopolymers can protect nanoparticles. Taking advantage of the excellent programmability of DNA, precisely tunable etching was realized on both spherical silver nanoparticles and plates. The excellent biocompatibility of DNA allows the application of this process in controlled killing of cancer and bacterial cells. This concept can be expanded to other nanomaterials and biopolymers to impact both fundamental nanoscience and applications. |
With excellent programmability and versatile surface interactions,1–5 DNA has been widely used for directing the growth of nanomaterials.6–10 For example, DNA can template the growth of gold, silver, and palladium nanoparticles and nanoclusters.11–16 DNA nanostructures can also serve as templates for deposition of gas phase materials.17 In addition, DNA can direct the assembly of DNA-functionalized nanomaterials.18–23 These reactions allow exquisite control of material synthesis with a very high spatial resolution.
An interesting question is whether DNA can be used in the reverse reaction, such as the dissolution of nanoparticles. Etching or dissolution of inorganic nanomaterials can be a method for controlled release for biomedical and analytical applications.24 Controlled etching might be attractive for fabrication and synthesis as well. Coupled with the programmability of DNA, a new field may be created by such studies. However, DNA-based interactions are often non-covalent and relatively weak.25–28 Therefore, using DNA to dissolve nanoparticles seems a big challenge. In fact, DNA-capped zinc oxide nanoparticles are even more resistant to dissolution.29
Silver is a very useful material due to its plasmonic, Raman enhancement, and anti-microbial properties. Etching of silver nanomaterials by many chemicals has been reported, including metal cations,30,31 anions,32,33 small biomolecules,34 and oxidants.35–37 These studies have enabled various silver nanostructures and colorimetric biosensors. Cytosine is known to strongly bind to Ag+,38 and C-rich DNA was extensively used to template fluorescent silver nanoclusters.14 Lu and coworkers reported that oligo-C DNA could mediate the growth of Ag tetrahedrons.12 Poly-C DNA was also used as an anchor to functionalize AgNPs.39 All these studies assumed the stability of related silver nanomaterials. Herein, we communicate the first example of DNA-directed etching. Poly-C DNA can etch AgNPs in a highly controllable manner, and this process was used for material transformation and enhanced killing of bacterial and cancer cells.
To test whether DNA can etch AgNPs, the optical properties of the AgNPs were studied in the presence of various DNA sequences. The starting 20 nm AgNPs had a yellow color (Fig. 1A, top inset) with a strong surface plasmon peak at 402 nm. Adding A18 (e.g. 18 mer poly-adenine) or T18 DNA had almost no effect on the color of the AgNPs. G18 DNA caused a slight decrease in this peak. With C18 DNA, the color changed to light pink (bottom panel), which was reflected in the decreased and red shifted 402 nm peak (Fig. 1A). According to a previous report, AgNPs had different affinities for the nucleobases in the order of T < A < G < C.40 The decrease of the 402 nm peak of our AgNPs followed a similar order. It is quite possible that the strong interaction between cytosine and AgNPs played an important role here. Previous work also indicated the effect of DNA binding affinity in control of the morphological evolution of AgNPs.12
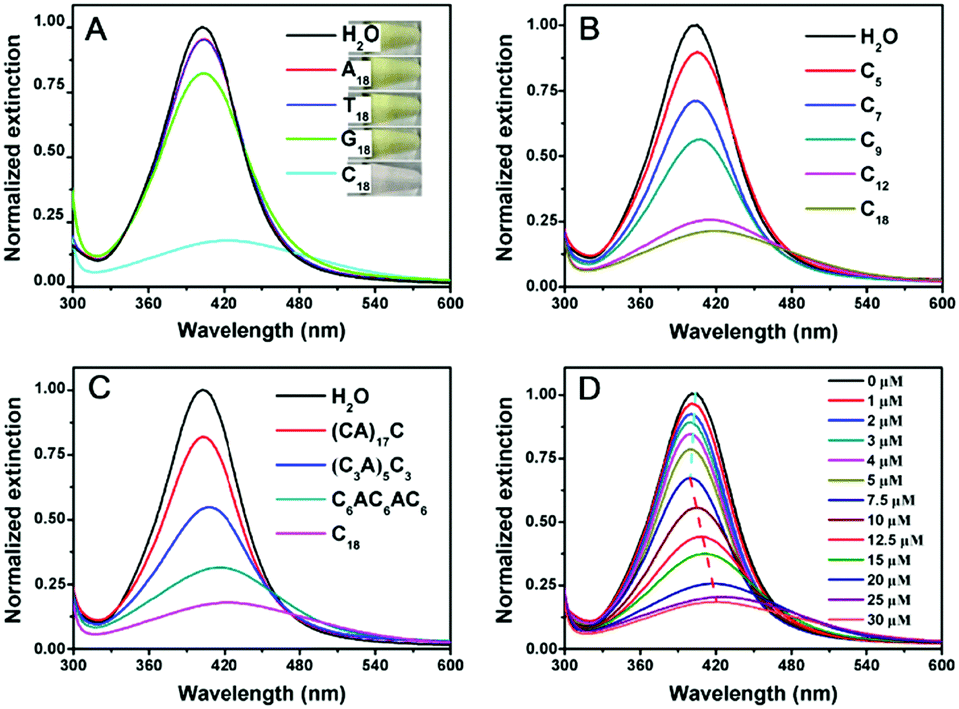 | ||
| Fig. 1 (A) UV-vis spectra of AgNPs before and after adding 25 μM of 18 mer homo-DNAs for 1.5 h. Inset: The photographs of these samples. DNA-mediated etching of AgNPs as a function of (B) poly-C DNA length, and (C) the number of base insertions. (D) C18-concentration-dependent etching of AgNPs for 1.5 h. The initial blue shift and then the red shift of the surface plasmon peak at high DNA concentrations are highlighted by the dashed lines. | ||
We then focused on poly-C DNA and performed further studies to gain mechanistic insights. The length of poly-C DNA was varied from 5 to 18 (Fig. 1B). In each case, the total number of cytosine base was maintained to be the same (e.g. the molar concentration of C9 was twice of that of C18). Interestingly, a longer DNA caused a more obvious decrease in the peak intensity, and saturation occurred at around C12 and C18. Therefore, most of our subsequent experiments were performed with the C18 DNA. A lot of work has been carried out on DNA adsorption by AuNPs, and the results from those studies might be useful for understanding DNA adsorption by AgNPs here. For example, shorter DNA strands were adsorbed faster than longer ones.41 The fact that longer poly-C, even with a lower molar concentration and slower adsorption kinetics, still showed a more obvious change suggested that the stronger adsorption affinity of longer DNA derived from polyvalent binding was vital.
To further understand the effect of DNA sequence, we maintained the total number of cytosines to be 18 but inserted other nucleotides (Fig. 1C). This way, the number of consecutive cytosines was reduced from 18 to 6, 3, and 1, respectively. The peak decreased inversely scaled with the number of consecutive cytosines, suggesting a neighboring effect of cytosine. Binding of Ag+ by cytosine is well-established to be chelation by two cytosine bases.38 The fact that consecutive cytosines are required suggested that the adsorption of DNA on the AgNPs was crucial.
Finally, we varied the concentration of C18 DNA (Fig. 1D). The peak intensity decreased and blue-shifted when the DNA concentration was lower than 7.5 μM, while adding more DNA induced a red-shift with further decrease of the peak intensity. This suggested that the AgNPs might be etched to become smaller particles with a low concentration of C18 DNA, but some grew larger with higher DNA concentrations. This is reminiscent of the Ostwald ripening process. With 12.5 μM C18 DNA, a typical ripening process occurred, where larger AgNPs grew at the expense of smaller ones (Fig. 2A). Most previous studies on DNA/AgNPs used a much lower concentration of DNA, which may explain why the etching effect of DNA was not noticed until this work.
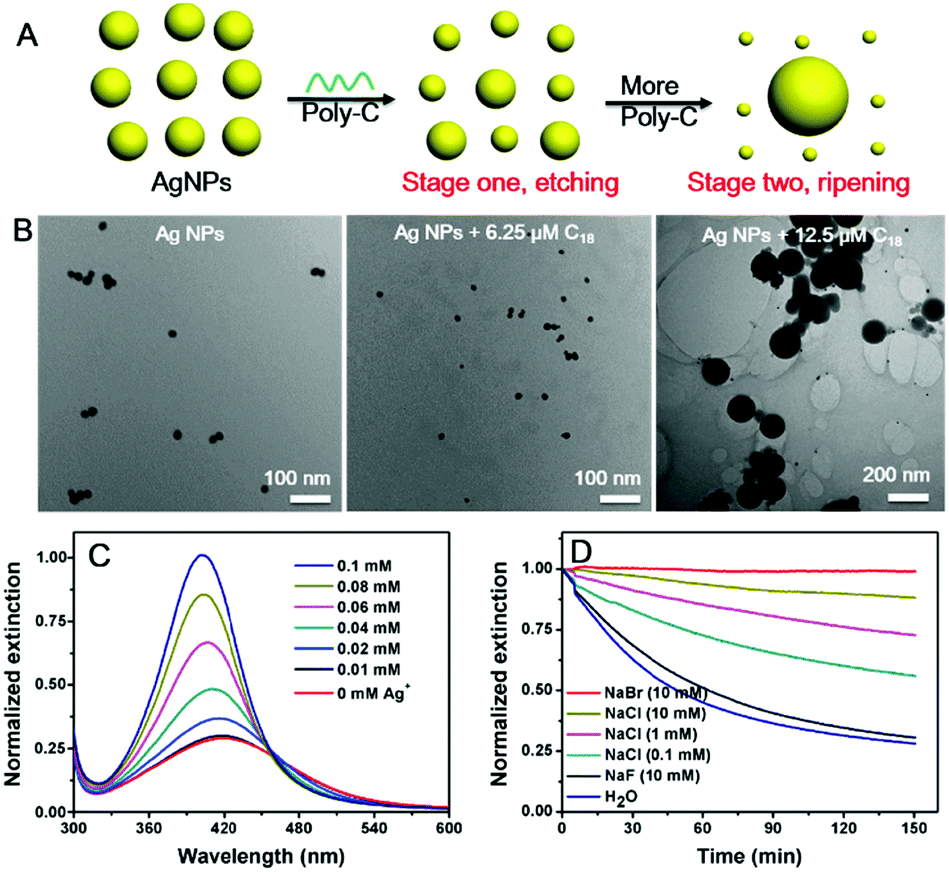 | ||
| Fig. 2 (A) Etching and ripening of AgNPs by low and high concentrations of poly-C DNA. (B) TEM micrographs of 20 nm AgNPs mixed with different concentrations of C18 DNA. (C) Free Ag+ ions inhibited DNA-mediated etching of AgNPs. (D) Kinetics of the C18-mediated etching of halide-capped AgNPs. | ||
To confirm the mechanism, we then monitored the reaction by transmission electron microscopy (TEM, Fig. 2B and Fig. S1, ESI†). With 6.25 μM C18 DNA, the AgNPs became smaller but maintained their spherical shape, suggesting that the DNA etched the AgNPs. With 12.5 μM C18 DNA, many larger size AgNPs of over 100 nm were observed. These two stages of the DNA concentration dependent reaction were also confirmed by dynamic light scattering (DLS, Fig. S2, ESI†).
It needs to be noted that AgNPs are susceptible to aggregation, and aggregation can also decrease their plasmonic peak. For example, adsorption of individual DNA bases can induce aggregation of AgNPs.40 However, the following evidence argued against aggregation in our system. First, when salt was added to the AgNPs to induce aggregation, the plasmonic peak only red shifted (Fig. S3, ESI†), while at low DNA concentrations, our sample blue-shifted (Fig. 1D). Second, our DLS and TEM data showing smaller AgNPs also disproved aggregation, and only etching of AgNPs can explain it. Our sample did not aggregate possibly due to protection from DNA oligonucleotides, while free DNA bases can induce aggregation of AgNPs.
We suspected that the need of a high DNA concentration was due to binding to the etching products, Ag+, and sufficient DNA was needed to increase the solubility limit of the AgNPs to observe etching. To confirm this, we mixed various concentrations of free Ag+ ions with the AgNPs before adding the C18 DNA (Fig. 2C). Indeed, the more free Ag+ ions, the less etching of AgNPs, and thus the etched silver species were likely associated with the DNA.
With 10 μg mL−1 of 20 nm AgNPs, the total concentration of silver atoms was 92.7 μM. The critical C18 DNA concentration was 7.5 μM with a total of 135 μM cytosine base. At this DNA concentration, the sample had the maximal blue shift, and the red shift started with 10 μM DNA. Assuming that two cytosine bases bind to one Ag+, 7.5 μM DNA can bind a total of 67.5 μM Ag+. The actual binding capacity could be lower due to folding of the DNA and adsorption of some DNA molecules on the remaining AgNPs. To test whether this critical DNA concentration depends on the concentration of AgNPs, we further reduced the initial AgNPs by half (5 μg mL−1). In this case, 7.5 μM C18 DNA showed a red shift (Fig. S4, ESI†). Therefore, the concentration of DNA required for ripening was a function of the AgNP concentration or total Ag+ species in the system. As long as the DNA can take a sufficient fraction of the silver species, ripening can take place. We further performed ICP on the supernatant after centrifugation of the AgNPs (10 μg mL−1 AgNPs incubated with 0, 7.5 and 25 μM C18 DNA, respectively), and the Ag+ concentration was 0.26 ± 0.01, 3.54 ± 0.01 and 9.13 ± 0.09 μg mL−1, respectively. Indeed, a large fraction of silver was solubilized (or became very small particles or clusters that could not be centrifuged).
Faster ripening would require increased solubility of AgNPs. To further confirm this mechanism, we added various halides to compete with DNA and to decrease the solubility of silver. The kinetics of the C18-mediated etching was monitored at various NaCl concentrations. Without extra salt (Fig. 2D, blue curve), it took around 2 h for the etching to reach equilibrium. Indeed, a higher NaCl concentration resulted in slower etching of the AgNPs by C18 DNA. At the same 10 mM halide concentration, NaBr fully protected the AgNPs from etching, while NaF showed a negligible effect. This trend might be due to the decreased solubility of Ag+ by halide capping and restricted adsorption of C18 on the AgNPs.42
The above work was performed with uniform 20 nm commercial AgNPs and they might contain some stabilizing agents. To confirm the generality of etching, we also prepared citrate-capped AgNPs in the laboratory with sizes in the range of 4.5 nm to 11.6 nm, and an obvious etching by the C18 DNA was also observed for these AgNPs (Fig. S5, ESI†). Then, AgNPs with different capping ligands were further tested (Fig. S6, ESI†). The C18 DNA could effectively etch the polyvinyl pyrrolidone (PVP)-capped AgNPs, while it induced aggregation of cetyltrimethyl ammonium bromide (CTAB)-capped AgNPs attributable to the electrostatic attraction between the negatively charged DNA and the positively charged CTAB. Finally, we tested gold nanoparticles with a high concentration of 18 mer DNAs, but no obvious change was observed (Fig. S7, ESI†). In earlier studies, iodide-mediated etching of AuNPs has been reported, and it appears DNA binding was too weak for AuNPs.43
An interesting question is whether etching can also take place with silver nanomaterials of other morphologies, which are readily available by chemical synthesis.44–48 Here, we chose Ag nanoplates as a model. The as-synthesized Ag nanoplates had a blue color (Fig. 3B) with an absorption peak at 647 nm (Fig. 3A, black trace).49 After incubation with 2.5 to 10 μM of the C18 DNA, the peak gradually blue shifted with damped intensity, and the color changed to purple and light red. Meanwhile, the triangular nanoplates transformed to be spherical AgNPs with smaller size as observed from TEM (Fig. 3C and Fig. S8, ESI†). Therefore, DNA can not only direct the growth of nanomaterials, but also produce AgNPs by etching. With a high surface energy, the atoms on the edges of the Ag nanoplates were likely more prone to be diffused away from the surface carried by the adsorbed poly-C DNA, leading to the formation of less faceted and more spherical particles.
 | ||
| Fig. 3 (A) UV-vis spectra, (B) photographs and (C) TEM micrographs of Ag nanoplates before and after mixing with different concentrations of C18 DNA. | ||
Although AgNPs can be etched by many chemicals such as hydrogen peroxide,37 and ferricyanide,30 most of them are highly toxic, confining their applications to materials synthesis. DNA, on the other hand, is a highly biocompatible molecule, and this may allow us to explore biological applications of this process.3,50–53 AgNPs are known for their anti-microbial activities, and etching by poly-C DNA may result in silver containing species with a higher biological activity and toxicity (Fig. 4A).
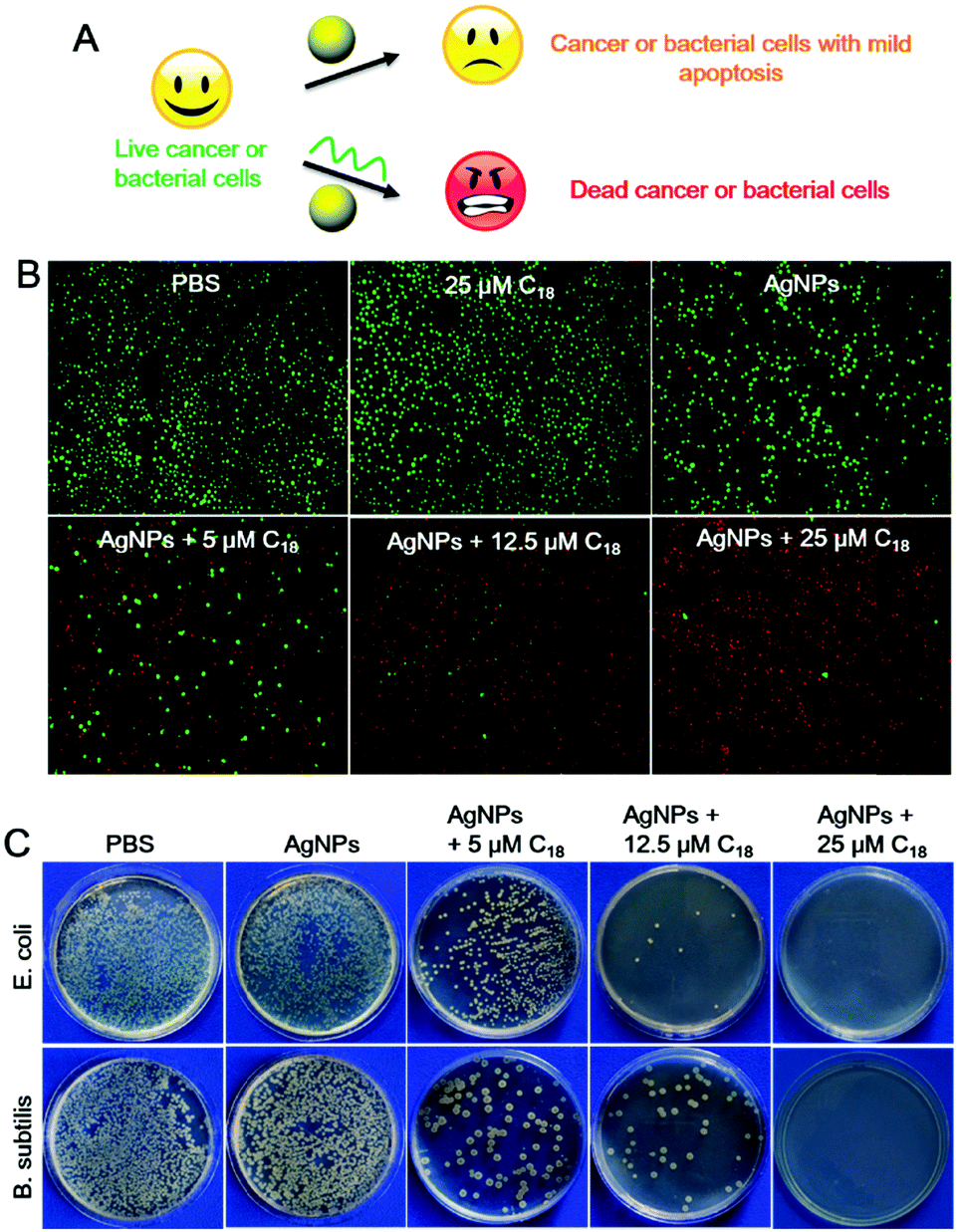 | ||
| Fig. 4 (A) DNA-mediated etching of AgNPs for enhanced toxicity to cancer and bacterial cells. (B) Fluorescence live/dead stain micrographs of Ramos cells after treating with AgNPs and C18 DNA of different concentrations, where red is indicative of dead cells. (C) Photographs of plated E. coli and B. subtilis cells after different treatments. | ||
To investigate the therapeutic effect of the C18 DNA treated AgNPs towards cancer, Ramos cells (a human lymphoma cell line) were tested as a model cancer cell line. To assess cytotoxicity, fluorescent dyes including calcein-AM and propidium iodide (PI) were employed. Live cells were stained green after permeation of calcein-AM into the cytoplasm,54,55 while dead cells were stained with the red DNA binding PI dye due to their compromised cell membrane.56 The cells retained full viability when treated with PBS or the C18 DNA alone (no AgNPs, Fig. 4B, the first two panels). After exposing the cells to free AgNPs (no C18 DNA), the viability of Ramos cells decreased to 77.2% (Fig. S9, ESI†), and the viability was also reflected from the stain assay showing scattered red spots (Fig. 4B). The amount of red stained dead cells increased drastically with increase of the C18 DNA concentration for the AgNP containing samples. With 25 μM C18 DNA, only 4.5% of the Ramos cells were viable (the last panel in Fig. 4B and Fig. S9, ESI†). The C18 DNA dose-dependent viability suggested that etching of AgNPs by the C18 DNA was the major contributor for the enhanced toxicity. These results confirmed that cell apoptosis could be controlled by the concentration of the C18 DNA.
We then studied the toxicity of AgNPs for bacterial cells. Two kinds of bacterial strains including Gram-negative Escherichia coli (E. coli) and Gram-positive Bacillus subtilis (B. subtilis) were tested. Free AgNPs exhibited a low anti-bacterial activity against both cells, shown by the colony-forming units (CFUs) of the bacteria on agar plates (Fig. 4C), which might be due to the relatively large particle size. After treatment with 5 μM C18 DNA, the number of CFUs decreased greatly. Further increasing the C18 DNA to 12.5 μM and 25 μM, a dose-dependent inhibition was observed. With 25 μM of the C18 DNA, no notable bacterial growth was observed for either strain. The C18 DNA alone did not show any antibacterial activity (data not shown), demonstrating that the enhanced toxicity was from DNA interacting with the AgNPs.
To gain a more quantitative understanding of the enhanced antibacterial activity, the growth kinetics of the cells were monitored for the optical density at 600 nm (OD600) in the LB media (Fig. S10, ESI†). Consistent with the colony counting assay, the increase of C18 concentration resulted in a delayed growth of both strains, although a better antimicrobial efficiency was observed for E. coli. This minor difference in the killing efficiency could be explained by the difference in the membrane structures of Gram positive and negative bacteria.57 The DNA-mediated etching formed smaller AgNPs with a greater surface area to volume ratio and also free Ag+ ions. After being internalized by the cancer or bacterial cells, these etched species were more toxic by interacting with important enzymes and production of reactive oxygen species.58,59
Conclusions
In summary, we communicated the first example of controlled etching of AgNPs using poly-C DNA, while most previous studies focused on DNA-directed growth or assembly of nanomaterials. This work expands the scope of interactions between DNA and nanomaterials to a new dimension. The reactions consist of two stages: etching or dissolution of AgNPs at a low concentration of DNA, followed by ripening with a further increased DNA concentration. Poly-C DNA-dependent etching was performed on Ag nanomaterials of different shapes and sizes, displaying interesting color changes. At the same time, etching enhanced the cytotoxicity of the AgNPs to both cancer cells and bacterial cells. Our work will likely impact DNA-directed materials synthesis, biosensor development, and anti-microbial and anti-cancer research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this work was from the National Natural Science Foundation of China (21575166, 21705166 and 21876208) and the Natural Sciences and Engineering Research Council of Canada (NSERC).Notes and references
- M. R. Jones, N. C. Seeman and C. A. Mirkin, Science, 2015, 347, 1260901 CrossRef PubMed.
- F. A. Aldaye, A. L. Palmer and H. F. Sleiman, Science, 2008, 321, 1795–1799 CrossRef CAS PubMed.
- Q. Hu, H. Li, L. Wang, H. Gu and C. Fan, Chem. Rev., 2018 DOI:10.1021/acs.chemrev.7b00663.
- Z. Chen, C. Liu, F. Cao, J. Ren and X. Qu, Chem. Soc. Rev., 2018, 47, 4017–4072 RSC.
- F. Pu, J. Ren and X. Qu, Chem. Soc. Rev., 2018, 47, 1285–1306 RSC.
- W. Zhao, M. M. Ali, M. A. Brook and Y. Li, Angew. Chem., Int. Ed., 2008, 47, 6330–6337 CrossRef CAS PubMed.
- R. J. Macfarlane, B. Lee, M. R. Jones, N. Harris, G. C. Schatz and C. A. Mirkin, Science, 2011, 334, 204–208 CrossRef CAS PubMed.
- L. H. Tan, H. Xing and Y. Lu, Acc. Chem. Res., 2014, 47, 1881–1890 CrossRef CAS PubMed.
- W. Sun, E. Boulais, Y. Hakobyan, W. L. Wang, A. Guan, M. Bathe and P. Yin, Science, 2014, 346, 1258361 CrossRef PubMed.
- X. Lan, Z. Su, Y. Zhou, T. Meyer, Y. Ke, Q. Wang, W. Chiu, N. Liu, S. Zou, H. Yan and Y. Liu, Angew. Chem., Int. Ed., 2017, 56, 14632–14636 CrossRef CAS PubMed.
- L. H. Tan, Y. Yue, N. S. Satyavolu, A. S. Ali, Z. Wang, Y. Wu and Y. Lu, J. Am. Chem. Soc., 2015, 137, 14456–14464 CrossRef CAS PubMed.
- J. Wu, L. H. Tan, K. Hwang, H. Xing, P. Wu, W. Li and Y. Lu, J. Am. Chem. Soc., 2014, 136, 15195–15202 CrossRef CAS PubMed.
- J. Richter, R. Seidel, R. Kirsch, M. Mertig, W. Pompe, J. Plaschke and H. K. Schaket, Adv. Mater., 2000, 12, 507–510 CrossRef CAS.
- J. T. Petty, J. Zheng, N. V. Hud and R. M. Dickson, J. Am. Chem. Soc., 2004, 126, 5207–5212 CrossRef CAS PubMed.
- T. Song, L. Tang, L. H. Tan, X. Wang, N. S. R. Satyavolu, H. Xing, Z. Wang, J. Li, H. Liang and Y. Lu, Angew. Chem., Int. Ed., 2015, 54, 8114–8118 CrossRef CAS PubMed.
- W.-Y. Chen, G.-Y. Lan and H.-T. Chang, Anal. Chem., 2011, 83, 9450–9455 CrossRef CAS PubMed.
- S. P. Surwade, F. Zhou, B. Wei, W. Sun, A. Powell, C. O'Donnell, P. Yin and H. Liu, J. Am. Chem. Soc., 2013, 135, 6778–6781 CrossRef CAS PubMed.
- D. Nykypanchuk, M. M. Maye, D. van der Lelie and O. Gang, Nature, 2008, 451, 549 CrossRef CAS PubMed.
- S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, Nature, 2008, 451, 553 CrossRef CAS PubMed.
- J. Sharma, R. Chhabra, A. Cheng, J. Brownell, Y. Liu and H. Yan, Science, 2009, 323, 112–116 CrossRef CAS PubMed.
- B. D. Smith and J. Liu, J. Am. Chem. Soc., 2010, 132, 6300–6301 CrossRef CAS PubMed.
- J. Shen, Q. Tang, L. Li, J. Li, X. Zuo, X. Qu, H. Pei, L. Wang and C. Fan, Angew. Chem., Int. Ed., 2017, 56, 16077–16081 CrossRef CAS PubMed.
- C. Lu, Z. Huang, B. Liu, Y. Liu, Y. Ying and J. Liu, Angew. Chem., Int. Ed., 2017, 56, 6208–6212 CrossRef CAS PubMed.
- A. Kumar, S. Kim and J. M. Nam, J. Am. Chem. Soc., 2016, 138, 14509–14525 CrossRef CAS PubMed.
- A. Samanta and I. L. Medintz, Nanoscale, 2016, 8, 9037–9095 RSC.
- B. Liu, Z. Huang and J. Liu, Angew. Chem., Int. Ed., 2018, 57, 9439–9442 CrossRef CAS PubMed.
- W. Zhou, J. Ding and J. Liu, Chem. Commun., 2015, 51, 12084–12087 RSC.
- H. Pei, F. Li, Y. Wan, M. Wei, H. Liu, Y. Su, N. Chen, Q. Huang and C. Fan, J. Am. Chem. Soc., 2012, 134, 11876–11879 CrossRef CAS PubMed.
- L. Ma, B. Liu, P. J. Huang, X. Zhang and J. Liu, Langmuir, 2016, 32, 5672–5680 CrossRef CAS PubMed.
- C. M. Cobley, M. Rycenga, F. Zhou, Z.-Y. Li and Y. Xia, J. Phys. Chem. C, 2009, 113, 16975–16982 CrossRef CAS.
- X. Lin, S. Lin, Y. Liu, M. Gao, H. Zhao, B. Liu, W. Hasi and L. Wang, Langmuir, 2018, 34, 6077–6084 CrossRef CAS PubMed.
- L. Chen, X. Fu, W. Lu and L. Chen, ACS Appl. Mater. Interfaces, 2013, 5, 284–290 CrossRef CAS PubMed.
- J. B. Zeng, Y. Y. Cao, J. J. Chen, X. D. Wang, J. F. Yu, B. B. Yu, Z. F. Yan and X. Chen, Nanoscale, 2014, 6, 9939–9943 RSC.
- J.-S. Lee, H. Kim and W. R. Algar, J. Phys. Chem. C, 2017, 121, 28566–28575 CrossRef CAS.
- Y. Zhou, W. Huang and Y. He, Sens. Actuators, B, 2018, 270, 187–191 CrossRef CAS.
- Z. Chen, J. Li, X. Chen, J. Cao, J. Zhang, Q. Min and J. J. Zhu, J. Am. Chem. Soc., 2015, 137, 1903–1908 CrossRef CAS PubMed.
- X. Yang, Y. Yu and Z. Gao, ACS Nano, 2014, 8, 4902–4907 CrossRef CAS PubMed.
- A. Ono, H. Torigoe, Y. Tanaka and I. Okamoto, Chem. Soc. Rev., 2011, 40, 5855–5866 RSC.
- D. Zhu, J. Chao, H. Pei, X. Zuo, Q. Huang, L. Wang, W. Huang and C. Fan, ACS Appl. Mater. Interfaces, 2015, 7, 11047–11052 CrossRef CAS PubMed.
- S. Basu, S. Jana, S. Pande and T. Pal, J. Colloid Interface Sci., 2008, 321, 288–293 CrossRef CAS PubMed.
- X. Zhang, M. R. Servos and J. Liu, Langmuir, 2012, 28, 3896–3902 CrossRef CAS PubMed.
- X. Zhang, M. R. Servos and J. Liu, Chem. Commun., 2012, 48, 10114–10116 RSC.
- W. Cheng, S. Dong and E. Wang, Angew. Chem., Int. Ed., 2003, 42, 449–452 CrossRef CAS PubMed.
- A. J. Frank, N. Cathcart, K. E. Maly and V. Kitaev, J. Am. Chem. Soc., 2010, 87, 1098–1101 CAS.
- M. McEachran and V. Kitaev, Chem. Commun., 2008, 5737–5739 RSC.
- Y. Sun and Y. Xia, Adv. Mater., 2003, 15, 695–699 CrossRef CAS.
- Q. Zhang, W. Li, C. Moran, J. Zeng, J. Chen, L.-P. Wen and Y. Xia, J. Am. Chem. Soc., 2010, 132, 11372–11378 CrossRef CAS PubMed.
- L. J. Sherry, R. Jin, C. A. Mirkin, G. C. Schatz and R. P. Van Duyne, Nano Lett., 2006, 6, 2060–2065 CrossRef CAS PubMed.
- L. Fang, Y. Wang, M. Liu, M. Gong, A. Xu and Z. Deng, Angew. Chem., 2016, 128, 14508–14512 CrossRef.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280–3294 CrossRef CAS PubMed.
- Y. Yang, J. Liu, X. Sun, L. Feng, W. Zhu, Z. Liu and M. Chen, Nano Res., 2016, 9, 139–148 CrossRef CAS.
- G. D. Hamblin, K. M. M. Carneiro, J. F. Fakhoury, K. E. Bujold and H. F. Sleiman, J. Am. Chem. Soc., 2012, 134, 2888–2891 CrossRef CAS PubMed.
- X. Qu, J. O. Trent, I. Fokt, W. Priebe and J. B. Chaires, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 12032–12037 CrossRef CAS PubMed.
- V. Voliani, G. Signore, O. Vittorio, P. Faraci, S. Luin, J. Peréz-Prieto and F. Beltram, J. Mater. Chem. B, 2013, 1, 4225–4230 RSC.
- S. S. Agasti, A. Chompoosor, C.-C. You, P. Ghosh, C. K. Kim and V. M. Rotello, J. Am. Chem. Soc., 2009, 131, 5728–5729 CrossRef CAS PubMed.
- C. Riccardi and I. Nicoletti, Nat. Protoc., 2006, 1, 1458–1461 CrossRef CAS PubMed.
- C. Liu, K. W. Chan, J. Shen, H. M. Wong, K. W. Kwok Yeung and S. C. Tjong, RSC Adv., 2015, 5, 72288–72299 RSC.
- S. Prabhu and E. K. Poulose, Int. Nano Lett., 2012, 2, 32 CrossRef.
- S. Hu, B. Ye, H. Tang, F. Wu, X. Yi, T. Yi, D. Wu, L. Wu and J. Wang, J. Mater. Chem. B, 2018, 6, 1187–1194 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8mh01126e |
| This journal is © The Royal Society of Chemistry 2019 |