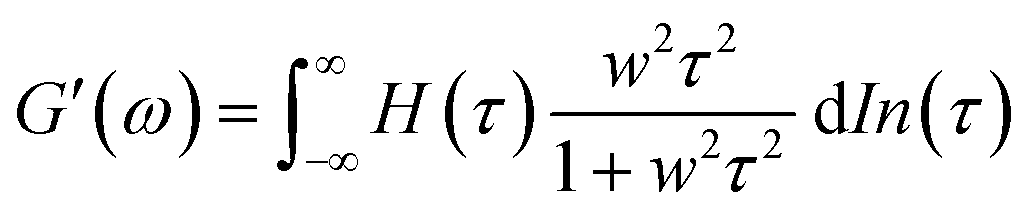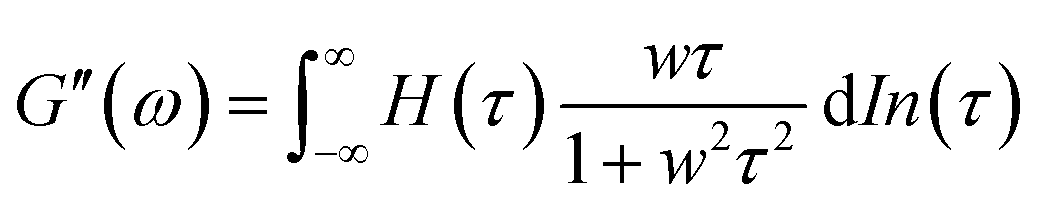Synthesis of bio-inspired viscoelastic molecular networks by metal-induced protein assembly†
Received
20th February 2019
, Accepted 10th May 2019
First published on 10th May 2019
Abstract
An inducible protein assembly system is desirable for developing high-order biomolecular architectures with dynamic properties. Here we demonstrate the creation of molecular networks with distinct stress-relaxation behavior using metal-induced protein assembly—a process that involves the folding and reconstitution of a pair of split IgG-binding GB1 proteins. In addition, metal–ligand coordination within the protein networks exerted great influence over their viscoelastic properties. The resulting protein networks are self-healable, amenable to biochemical decoration via SpyTag/SpyCatcher chemistry, and compatible with 3D culture of fibroblasts. This study points to a simple and robust strategy for designing recombinant protein hydrogels with tunable biochemical and mechanical properties.
 Fei Sun | Dr. Fei Sun is currently an Assistant Professor at the Department of Chemical and Biological Engineering, the Hong Kong University of Science and Technology. Before joining HKUST in 2014, Dr. Fei Sun worked as a postdoctoral researcher with Prof. Frances H. Arnold at the California Institute of Technology. He obtained his PhD in Chemistry from the University of Chicago under the supervision of Prof. Chuan He in 2012 and BS degree from Peking University in 2007. His primary research interests include protein materials, protein topology engineering and synthetic biology. |
Design, System, Application
Assembling recombinant proteins into molecular networks has proven to be a powerful approach for creating bioactive materials with genetically programmable properties. In order to achieve dynamic control over these protein-based molecular systems, we leveraged a Cu2+-induced protein assembly system involving the folding and reconstitution of a pair of split IgG-binding GB1 proteins upon oxidation and created entirely protein-based molecular networks with varied stress-relaxation behaviour. The resulting protein networks were versatile and possessed several important features, including the abilities of shear-thinning and self-healing, the amenability toward covalent decoration with complex globular proteins via SpyTag/SpyCatcher chemistry, and the suitability for cell encapsulation, which provide new opportunities for injectable therapeutic/cell delivery.
|
1. Introduction
Construction of macromolecular systems with precise control over their structural and functional properties constitutes a fundamental challenge for molecular engineering. A plausible solution is to assemble genetically engineered proteins into advanced architectures under mild conditions while preserving their biochemical activities.1,2 Inspired by an isopeptide bond-containing bacterial adhesin domain, genetically encoded SpyTag/SpyCatcher chemistry has recently emerged as a powerful tool for covalently stitching together protein molecules,3,4 which has thus led to numerous applications such as protein topology engineering5–8 and design of synthetic vaccines9–12 and functional materials.13–24 As SpyTag/SpyCatcher chemistry proceeds spontaneously under physiological conditions and requires no external stimuli for activation, a stimuli-responsive protein assembly system would be complementary and may further enable the creation of protein materials with delicately controlled dynamic properties.
Mussel foot proteins (Mfp's) can form self-healing and tough fibers largely through phase transition involving metal–ligand coordination, which allows mussels to adhere to underwater substrates.25 Inspired by those natural materials, several synthetic polymer materials including tough hydrogels and self-healing solid materials have been developed using dynamic metal–ligand coordination.26–29 Using multiple, kinetically distinct metal–ligand interactions, Grindy and coworkers recently demonstrated the assembly of 4-arm histidine-modified poly(ethylene glycol) (4A-PEG-His) into hydrogels with mechanical properties decoupled from their spatial structure.30 Using a similar polymer precursor, 4A-PEG-His, Wegner and coworkers developed cobalt cross-linked redox-responsive hydrogels that can transition from self-healing viscoelastic liquids to stable elastic solids upon oxidation of Co2+ to Co3+.31 In addition to synthetic polymers, de novo designed proteins can undergo metal-directed self-assembly to form advanced nano-architectures including 2D- and 3D- cage-like structures.32
In this study, using metal-directed protein assembly, we developed an entirely engineered protein-based hydrogel system with several useful features like genetic programmability, tunable viscoelasticity and self-healing abilities. To assemble such a polymeric protein network, we leveraged a pair of structural motifs, GB1N and GB1C, derived from split IgG-binding GB1 proteins that can self-assemble upon oxidation.33,34 The resulting materials exhibited marked self-healing behavior and cytocompatibility, thus showing great potential in biomedical applications.
2. Experimental section
2.1. Plasmid construction
The gene encoding the MEP protein has been described previously.33,34 It was purchased as a synthetic gBlocks gene fragment from Integrated DNA Technologies (IDT). It was inserted into pQE80l-AAA using the restriction sites, SacI and SpeI. All mutants used in this study were generated using QuikChange mutagenesis based on the manufacturer's recommended protocol (Agilent Technologies, Inc.). The sequences of all genes were verified by Sanger sequencing (Beijing Genomics Institute).
2.2. Protein expression and purification
E-MEP-E and its mutants were expressed using Escherichia coli strain BL21 (DE3). The bacterial cells were cultured in Luria broth containing 100 mg per L ampicillin at 37 °C until the OD600 reached 0.6–0.8. Isopropyl-β-D-1-thiogalactopyranoside (IPTG, 500 μM) was added to induce the protein expression at 30 °C for 4 h. Cells were harvested at 4200 × g centrifugation. After being stored at −80 °C overnight, the pellets were resuspended in Buffer A (NaCl 300 mM, Tris-HCl 10 mM, pH 8.0), and sonicated on ice with 20 s × 4 pulse to lyse the cells. The cell lysates were centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 30 min at 4 °C to obtain the supernatants. The target protein was purified from the supernatants using Ni-NTA affinity column (HisTrap HP, 5 mL, GE Healthcare Life sciences) with ATKA FPLC system (GE Healthcare) and eluted by buffer B (NaCl 300 mM, Tris-HCl 10 mM, imidazole 500 mM, pH 8.0). The proteins were dialyzed against MilliQ H2O to remove the salts and lyophilized at −80 °C.
000 × g for 30 min at 4 °C to obtain the supernatants. The target protein was purified from the supernatants using Ni-NTA affinity column (HisTrap HP, 5 mL, GE Healthcare Life sciences) with ATKA FPLC system (GE Healthcare) and eluted by buffer B (NaCl 300 mM, Tris-HCl 10 mM, imidazole 500 mM, pH 8.0). The proteins were dialyzed against MilliQ H2O to remove the salts and lyophilized at −80 °C.
2.3. Hydrogel preparation
The protein powder was dissolved in phosphate buffered saline (PBS) containing 2.67 mM metal (Cu2+, Ni2+ or Co2+) to make a 10 wt% protein solution (the molar ratio of E-MEP-E to metal, 1:1). The gel was cured at 4 °C with sufficient humidity overnight before further analysis.
2.4. Size exclusion chromatography
The reactions of proteins (50 μM) and Cu2+ (50 μM) were incubated at 4 °C for 12 h. The resulting products were analyzed by size exclusion chromatography with a Superdex 12 10/300GL column in an ATKA FPLC system (GE Healthcare). The running buffer was PBS (pH 7.5). The flow rate was 0.5 mL min−1.
2.5. Rheological measurements
Rheological measurements were performed in a TA Instruments ARES-RFS strain-controlled rheometer with a standard steel parallel-plate geometry (8 mm in diameter). For dynamic time-sweep tests, the frequency was held at 1 rad s−1 and the strain at 5% at 23 °C. For frequency- and strain-sweep tests, hydrogels were prepared one day before. For frequency-sweep tests, the strain was held constantly at 10% at 23 °C while varying the oscillatory frequency from 100 to 0.01 rad s−1. Strain-sweep measurements were performed by holding the frequency at 30 rad s−1 at 23 °C and varying the strain amplitude from 0.1 to 200%.
2.6. Encapsulation of fibroblasts
NIH/3T3 mouse fibroblasts were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM, Gibco) containing 10% fetal calf serum (FCS, Gibco), 1% penicillin/streptomycin (Sangon). Cells between passages P10 and P15 were used. The cells were suspended in the medium containing E-MEP-E (10 wt%) and 2 mM Cu2+. The gel was cured at 37 °C, 5% CO2 for 30 min before adding culture medium to cover the gel. The cells were maintained at 37 °C, 5% CO2 for 24 h. Cell viability was determined by LIVE/DEAD staining assay (Invitrogen). Cell morphology was determined by Hoechst 33342 dye (Life Technologies) for nucleus and Alexa Fluor® 546 phalloidin (Life Technologies) for actin. Zeiss laser scanning confocal microscope [LSM7 DUO (710+LIVE)] was used to visualize the stained cells to determine cell viability and morphology.
2.7. Fabrication of E-MEP-E/Cu2+ hydrogel microspheres
A PDMS microfluidic device with Y-shaped inlet microchannel was used to generate the hydrogel microspheres. Mineral oil with 3% EM90 and 0.05% Triton-X 100 served as the carrier fluid, and the solution of 6.7 wt% E-MEP-E and Cu2+ (1.7 mM) in PBS served as the aqueous flow. The oil and aqueous streams ran into a mixture at constant flow rates of 21 and 10 μL min−1, respectively. The droplets were cured for 2 days.
2.8. Post-gelation modification
Hydrogel microspheres were washed by PBS three times followed by treatment with 5% BSA for 1 h. The microspheres were then immersed in 50 μM A-YFP-A or B-YFP-B for 2 h. After wash by PBS three times, the microspheres were imaged with a fluorescence microscope. To quantify the fluorescence intensity, the microspheres were immersed in PBS containing 0.1 M EDTA and were completely dissolved within 12 h. Their relative fluorescence was measured using a microplate reader (Thermo Scientific Varioskan LUX multimode).
3. Results and discussion
3.1. Protein construct design
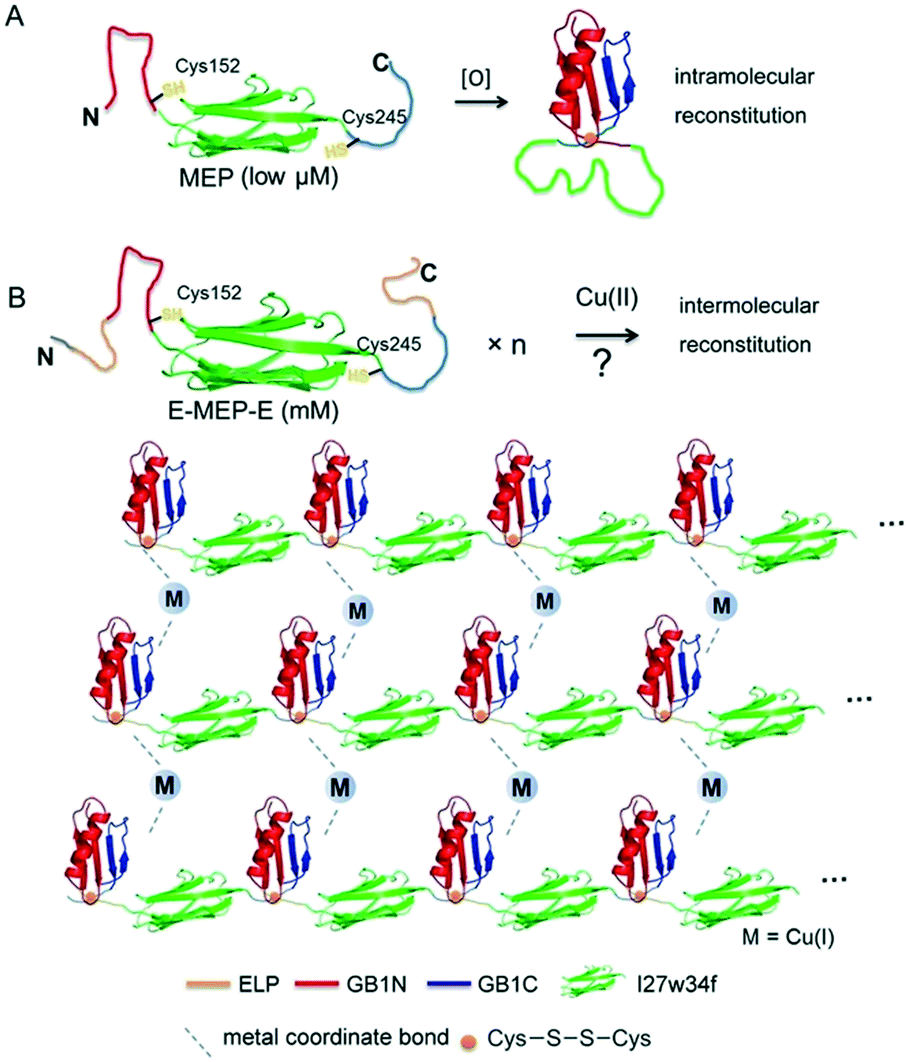
Careful examination of literatures revealed an interesting mutually exclusive protein (MEP) based redox-potential switch, GB1N-I27w34f-GB1C, that consists of the I27w34f domain derived from the giant muscle protein titin and two flanking split fragments (GB1N and GB1C) derived from the mutant of GB1.33,34 Under reducing conditions, GB1N and GB1C were unstructured and separated apart by a folded I27w34f domain (Fig. 1A). Cys152 in GB1N and Cys245 in GB1C formed an intramolecular disulfide bond on oxidation, which was accompanied with the reconstitution of a folded GB1 domain and the unfolding of I27w34f.
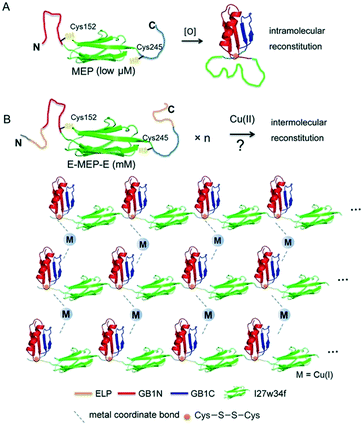 |
| | Fig. 1 Schematic illustration of induced protein assembly. (A) Redox-dependent protein switch—a mutually exclusive protein (MEP) consisting of the I27w34f domain derived from the giant muscle protein titin and the flanking split GB1 proteins (GB1N and GB1C). At a low protein concentration (μM), the two Cys residues, Cys152 and Cys245, form an intramolecular disulfide bond, which leads to the reconstitution and folding of GB1 and the unfolding of I27w34f.33 GB1 (PDB code: 1PGB). I27w34f (PDB code: 1TIT). (B) Oxidation of high-concentration ELP-MEP-ELP (E-MEP-E) may lead to high-order polymerization by forming intermolecular disulfide bonds between Cys152 and Cys245. In addition to the covalent polymerization, metal coordination with side chains of the residues like Cys and His could contribute to interchain interactions within the molecular network. For clarity, only the MEP domains are shown in the molecular network. | |
It is noteworthy that a low concentration of protein (low μM) was used in all the experiments previously reported.33,34 We envisioned that GB1N/GB1C might constitute an ideal pair for oxidation-induced protein assembly at a high protein concentration, whereas intermolecular reconstitution between GB1N and GB1C would be kinetically favored (Fig. 1B). To test this hypothesis, we designed a genetic construct, ELP-GB1N-I27w34f-GB1C-ELP (or E-MEP-E), that fuses the MEP with two elastin-like polypeptides (ELPs) (Fig. S1†). The intermolecular GB1N/GB1C reconstitution is likely to circumvent the energy barrier imposed by the unfolding of I27w34f. The ELP we chose is composed of repetitive pentapeptides (VPGXG)15, where X can be either valine or glutamate at a ratio of 4:1. The two ELP domains were introduced to improve the expression and solubility of the recombinant E-MEP-E protein.35 In addition, this particular ELP composition exhibited a LCST that falls within the range of 45–60 °C.36 Since all experiments in this study were performed at either room temperature or 37 °C, far below this LCST, any influence of the phase transition of the ELP on the gel's mechanics would be negligible.
3.2. Cu2+-induced protein polymerization
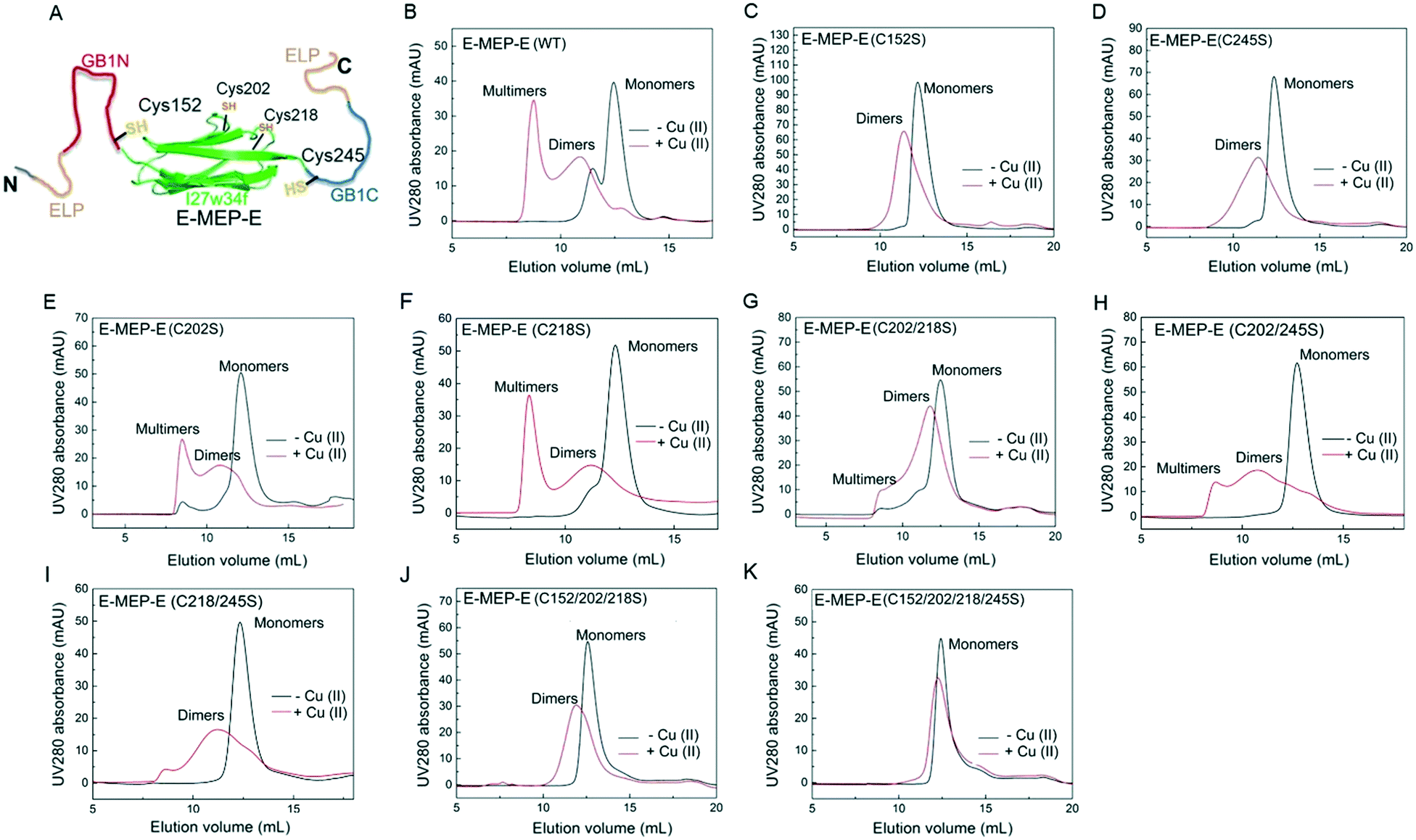
The His6-tagged E-MEP-E protein was produced using an E. coli expression system and purified in a considerable yield (∼20 mg per liter bacterial culture) using a Ni-NTA chromatography. Cu2+ has proven to be effective in oxidizing the thiols of cysteine residues into disulfide bonds within proteins.37 To examine whether Cu2+ is able to induce the intermolecular assembly of GB1N and GB1C, the lyophilized E-MEP-E protein powder was dissolved in phosphate buffered saline (PBS) to make a 50 μM solution, followed by treatment with a stoichiometric amount of Cu2+. As a result, E-MEP-E indeed polymerized into mostly multimers with an elution volume approaching the void volume of the size-exclusion column, in addition to a small portion of dimerization (Fig. 2B). Besides the putative oxidation-sensitive Cys152 and Cys245, E-MEP-E possesses two more Cys residues, Cys202 and Cys218 (Fig. 1A). It turned out that mutating either Cys152 or Cys245 to Ser significantly reduced multimerization and resulted in predominantly dimerization according to the SEC analysis, suggesting that Cys152 and Cys245 are mainly responsible for the observed Cu2+ induced polymerization (Fig. 2C and D). Protein multimers, while varying in amounts relative to dimers, still formed on oxidation after the single mutation of Cys202 or Cys218 to Ser or the double mutation of both Cys202 and Cys218 to Ser, further confirming that it is Cys152/245, but not Cys202/218, that are key to the oxidation-induced polymerization (Fig. 2E and F). We also observed a low level of polymerization of both mutants, C218/245S and C202/245S, suggesting that Cys202 or Cys218, in the presence of Cys152, could also contribute to the polymerization (Fig. 2H and I). With the only remaining Cys245, the Cys152/202/218Ser mutant was still able to dimerize upon oxidation, showing the proneness of Cys245 to oxidation (Fig. 2J). The Cys152/202/218/245Ser mutant containing no Cys residue exhibited neither multimerization nor dimerization in the presence of Cu2+, showing that the oxidation of Cys is essential for the observed protein oligomerization (Fig. 2H). Taken together, these results show that Cu2+ is able to induce the polymerization of E-MEP-E presumably via the oxidation of Cys152 in GB1N and Cys245 in GB1C.
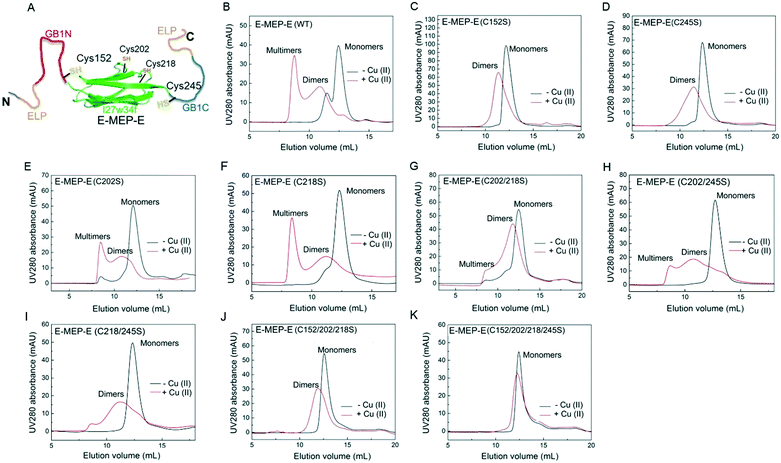 |
| | Fig. 2 Cys152 and Cys245 are mainly responsible for oxidation-induced polymerization of E-MEP-E. (A) All four cysteine residues within E-MEP-E, including Cys152 and Cys245 in the unfolded GB1N and GB1C and Cys202 and Cys218 in the folded I27w34f. For clarity, SpyTags are not shown. (B–K) Size exclusion chromatography (SEC) analyses showing the influence of cysteine mutations on the oligomerization of proteins upon Cu2+ oxidation. | |
3.3. Assembly of E-MEP-E networks by Cu2+
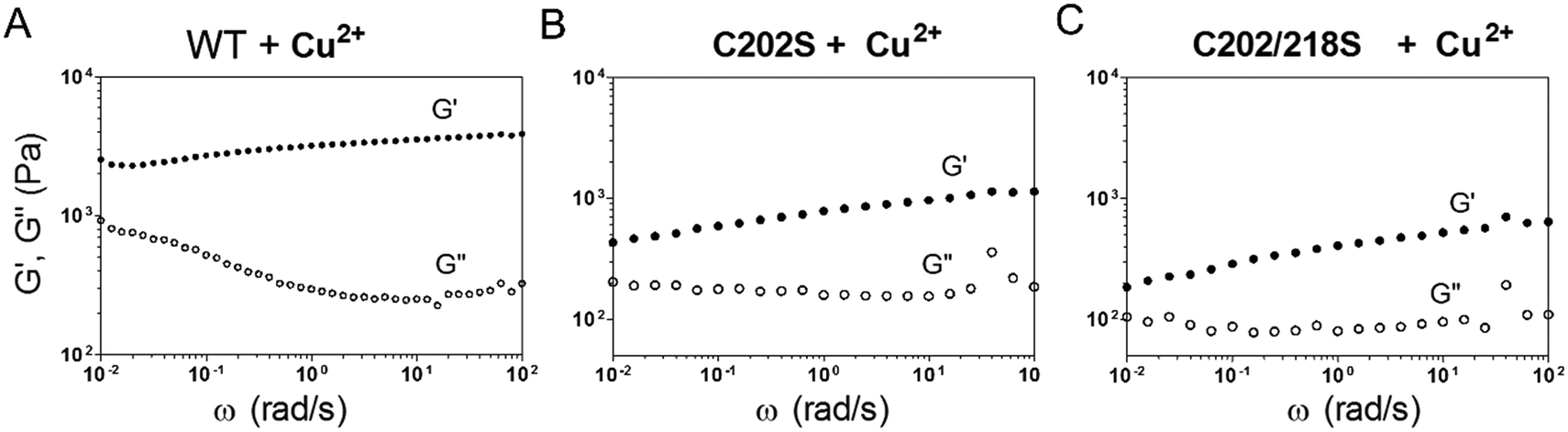
Mixing E-MEP-E (10 wt% in PBS) with Cu2+ at an equimolar ratio led to the formation of a gel-like material, of which the storage modulus G' reached plateau after 12 h according to time-sweep tests (Fig. S2†). According to the dynamic shear frequency-sweep test, this material exhibited storage modulus G′ of ∼2.2–3.9 kPa over a frequency range of 0.01–100 rad s−1, substantially higher than loss modulus G′′ (∼0.9–0.3 kPa), suggesting the formation of a viscoelastic solid (Fig. 3A). Among the four Cys residues within E-MEP-E, Cys152 and Cys245 have proven to be important for the protein linear polymerization (Fig. 2) presumably through the formation of intermolecular disulfide bond upon oxidation, whereas Cu2+ would be reduced to Cu+. Interestingly, oxidation of the C202S mutant and the C202/218S mutant by Cu2+ also led to the formation of gels, both exhibiting dramatically decreased G′ (0.4–1.1 and 0.2–0.6 kPa) (Fig. 3B and C). Despite their seemingly marginal roles in the protein polymerization (Fig. 2), the rheological measurements on these mutant proteins suggested that Cys202 and Cys218 could be crucial for the interchain interactions within the protein network, presumably through S-Cu+ coordination. Since E-MEP-E is a His6-tagged protein, the metal/His6-tag coordination may also contribute to the mechanics of the resulting gels. Indeed, the double mutant protein, C202/218S, crosslinked by Cu exhibited a solid-like property (G′ > G′′)—indicative of effective interchain interactions likely due to His-6 tag, though its G′ ∼ 0.2–0.6 kPa was substantially lower than that of WT + Cu ∼ 2.2–3.9 kPa, suggesting that the metal/His6-tag coordination contributed only moderately to the gels' mechanics.
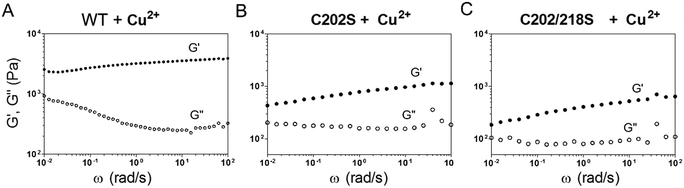 |
| | Fig. 3 Genetically programming mechanical properties of the MEP networks. Frequency sweep tests on the products of wild-type E-MEP-E (A) and its mutants, C202S (B) and C202/218S (C), oxidized by Cu2+. The strain was held at 5%. | |
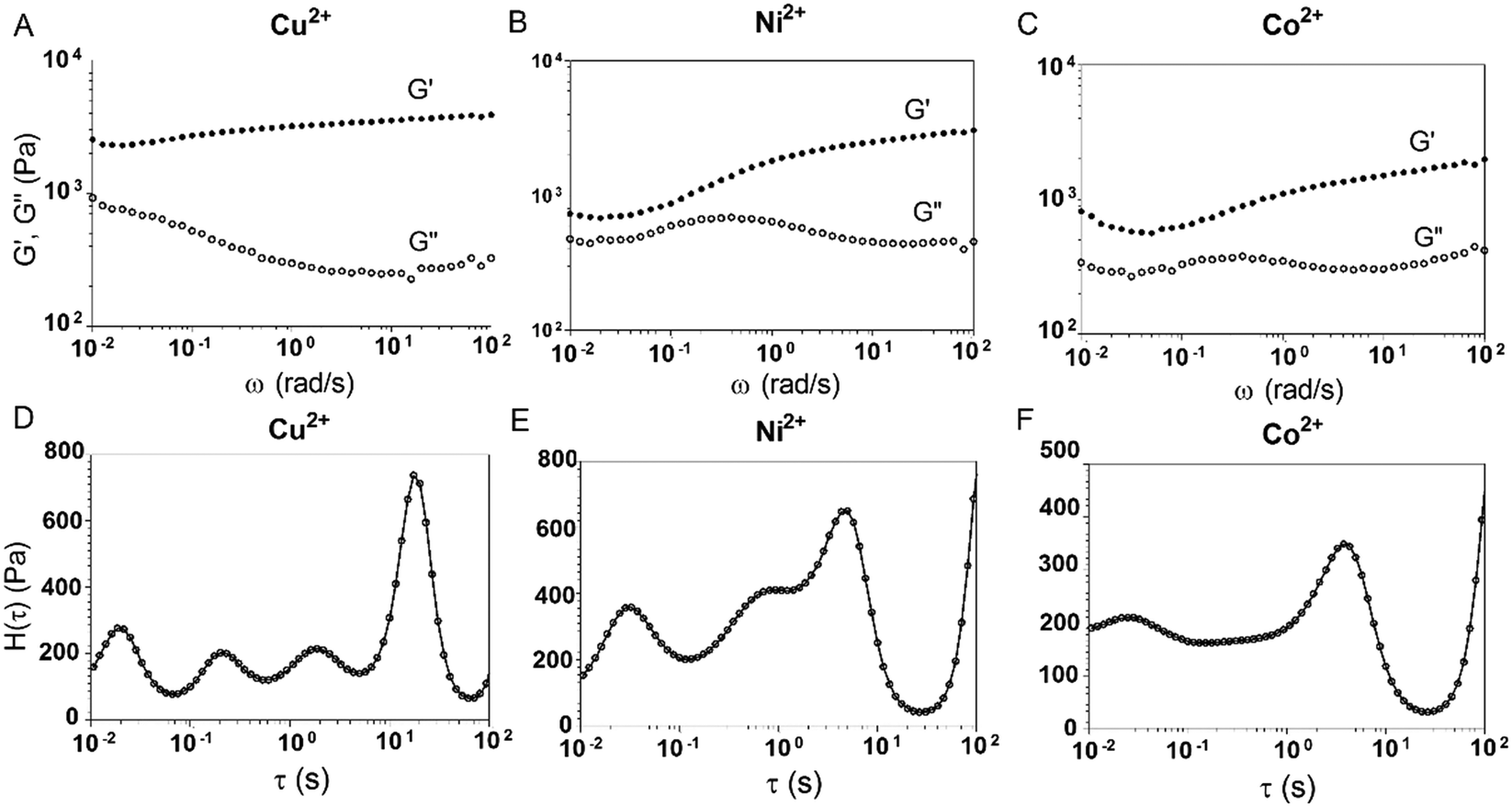
3.4. Assembly of E-MEP-E networks by other divalent metals
A previous study showed that not only oxidation but divalent metal ions like Ni2+ could also trigger the GB1N/GB1C assembly and folding within the MEP.38 It turned out that addition of a stoichiometric amount of the metal ions including Co2+ and Ni2+ both induced the gelation of E-MEP-E. Frequency-sweep tests showed that these resulting materials all exhibited G′ larger than G′′ over a frequency range of 0.01–100 rad s−1, suggesting the formation of viscoelastic solids. The values of G′ at high frequency, which reflect the combined contributions of stable and transient interchain crosslinks, follow the order of Co < Ni < Cu, which coincides with the Irving-Williams order.39 The G's of these materials exhibited more pronounced frequency-dependence than those assembled by Cu, indicating the more dynamic nature of these metal–ligand interactions (Fig. 4B and C). In order to better elucidate the influence of metal–ligand coordination on stress-relaxation behavior, the frequency-sweep data were converted into the continuous relaxation spectra H (τ) using eqn (1) and (2) shown below:30| | 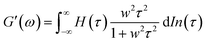 | (1) |
| | 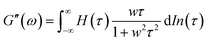 | (2) |
 |
| | Fig. 4 Influence of different divalent metal ions on viscoelastic properties of the E-MEP-E networks. (A–C) Frequency sweep tests on E-MEP-E (10 wt%) with divalent metal ions at an equimolar ratio. (D–F) The continuous relaxation spectra H(τ) were calculated using eqn (1) and (2). The strain was held at 5%. | |
The relaxation spectrum of the Cu network exhibited a major energy dissipation peak at τ ∼ 10–102 s, while the other (Ni and Co) showed major dissipation at τ ∼ 100–101 s, significantly faster than the Cu network (Fig. 4). The disparity in their energy dissipation rates well reflected the distinct kinetics of metal–ligand interactions within these networks. The results together demonstrated the feasibility of using divalent metal ions to fine-tune the mechanics of these protein networks.
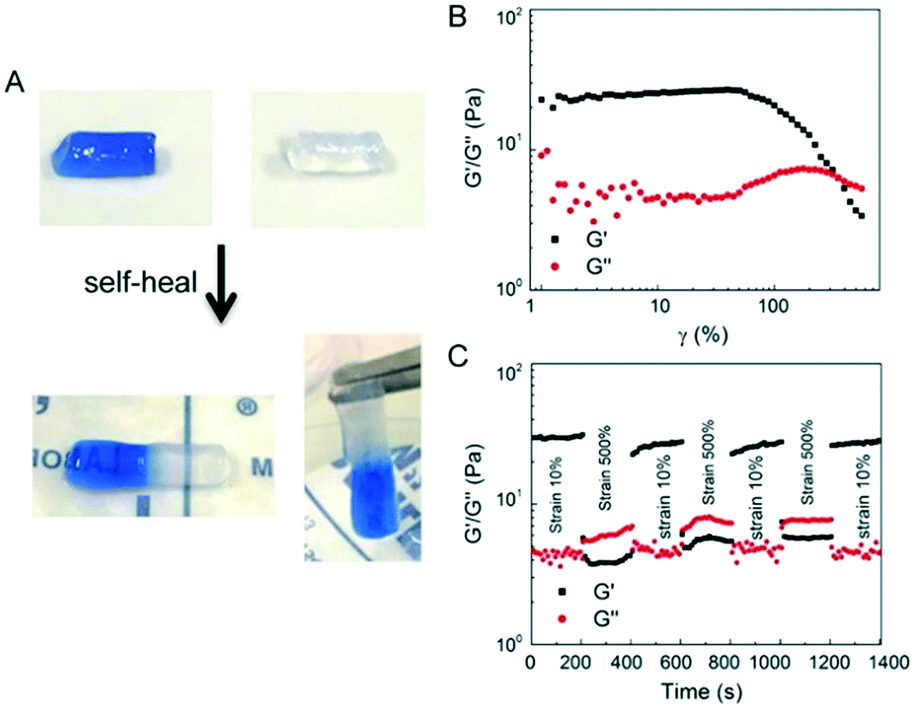
3.5. Shear-thinning and self-healing of E-MEP-E/Cu hydrogels
The E-MEP-E/Cu hydrogel displayed the capability of self-healing at room temperature (Fig. 5A). Strain-sweep tests on these hydrogels revealed a broad linear viscoelastic region that ends at the yield point (γy, ∼60%), in addition to a crossover point G′ = G′′, also known as the flow point (γf, ∼120%) (Fig. 5B), highly indicative of shear-thinning behavior. The material exhibits solid-like and liquid-like properties at low (<γf) and high strain (>γf), respectively, showing its potential for injectable delivery. Continuous step-strain measurements were then performed to assess the quick recovery of the hydrogel after network failure at a high-amplitude strain. A high strain (500%) was applied for 100 s to rupture the hydrogel network, thus leading to a liquid-like status (G′′ > G′), followed by a low-amplitude strain (10%) for 100 s, which immediately restored the solid-like status (G′ > G′′). After three cycles of network rupture, G′ could still be restored once the low-amplitude strain was applied, showing fast and complete recovery of the mechanical properties. The ability of rapid and complete self-healing over repeated cycles of breaking and reforming highlights the reversible and robust metal–ligand coordinate crosslinks within the networks.
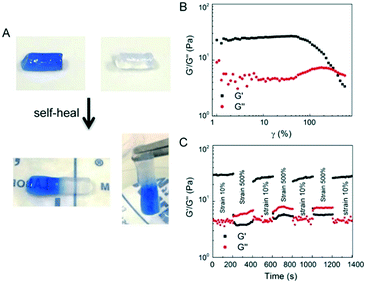 |
| | Fig. 5 Self-healing and shear-thinning behavior of the E-MEP-E/Cu hydrogels. (A) Self-healing of these gels at room temperature. One piece of gel is dyed blue to make the interface distinguishable. (B) Strain sweep test showing the yield point, γy = 60%, and the flow point, γf = 120%, of the hydrogel. (C) Continuous step-strain measurements. | |
3.6. Post-gelation modification via SpyTag/SpyCatcher chemistry
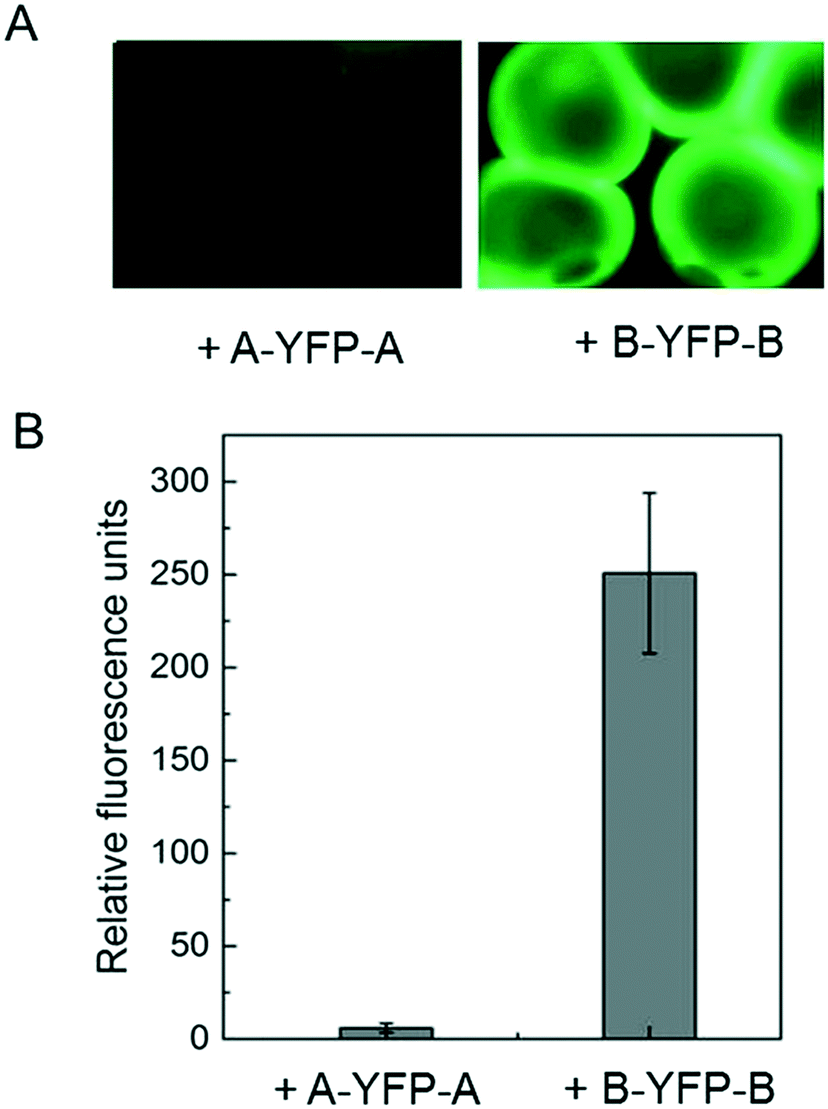
Immobilization of biomolecules, especially recombinant proteins, onto hydrogels may offer great opportunities for therapeutic delivery as well as studying cell–matrix interactions. E-MEP-E was designed to contain SpyTag—a short peptide consisting of AHIVMVDAYKPTK—on both N- and C- termini. To investigate the feasibility of using SpyTag/SpyCatcher chemistry to immobilize protein molecules onto the E-MEP-E/Cu hydrogel, the hydrogel microspheres comprising E-MEP-E + Cu were created using a Y-shaped two-channel microfluidic device,40 followed by treatment with recombinant fluorescent proteins including SpyTag-YFP-SpyTag (A-YFP-A) and SpyCatcher-YFP-SpyCatcher (B-YFP-B). After intense wash by PBS, only B-YFP-B, but not A-YFP-A, remained bound to the microspheres in a significant amount, highlighting the specificity and robustness of the covalent bond-forming SpyTag/SpyCatcher chemistry (Fig. 6).
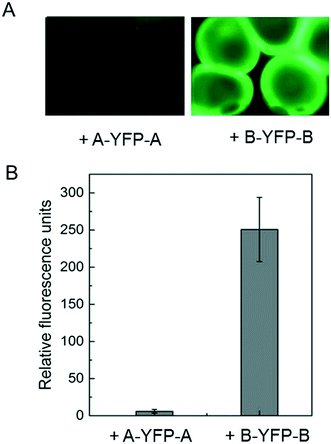 |
| | Fig. 6 Post-gelation modification by SpyTag/SpyCatcher chemistry. (A) Immobilization of YFP onto the E-MEP-E/Cu hydrogel microspheres containing SpyTag. (B) Quantitation of fluorescent labeling of hydrogel microspheres. Since E-MEP-E possesses SpyTag on both N- and C- termini, the resulting hydrogels were decorated with SpyCatcher-YFP-SpyCatcher (B-YFP-B) but not the control sample SpyTag-YFP-SpyTag (A-YFP-A). Data are presented as mean ± SD (n = 3). | |
3.7. Cell encapsulation
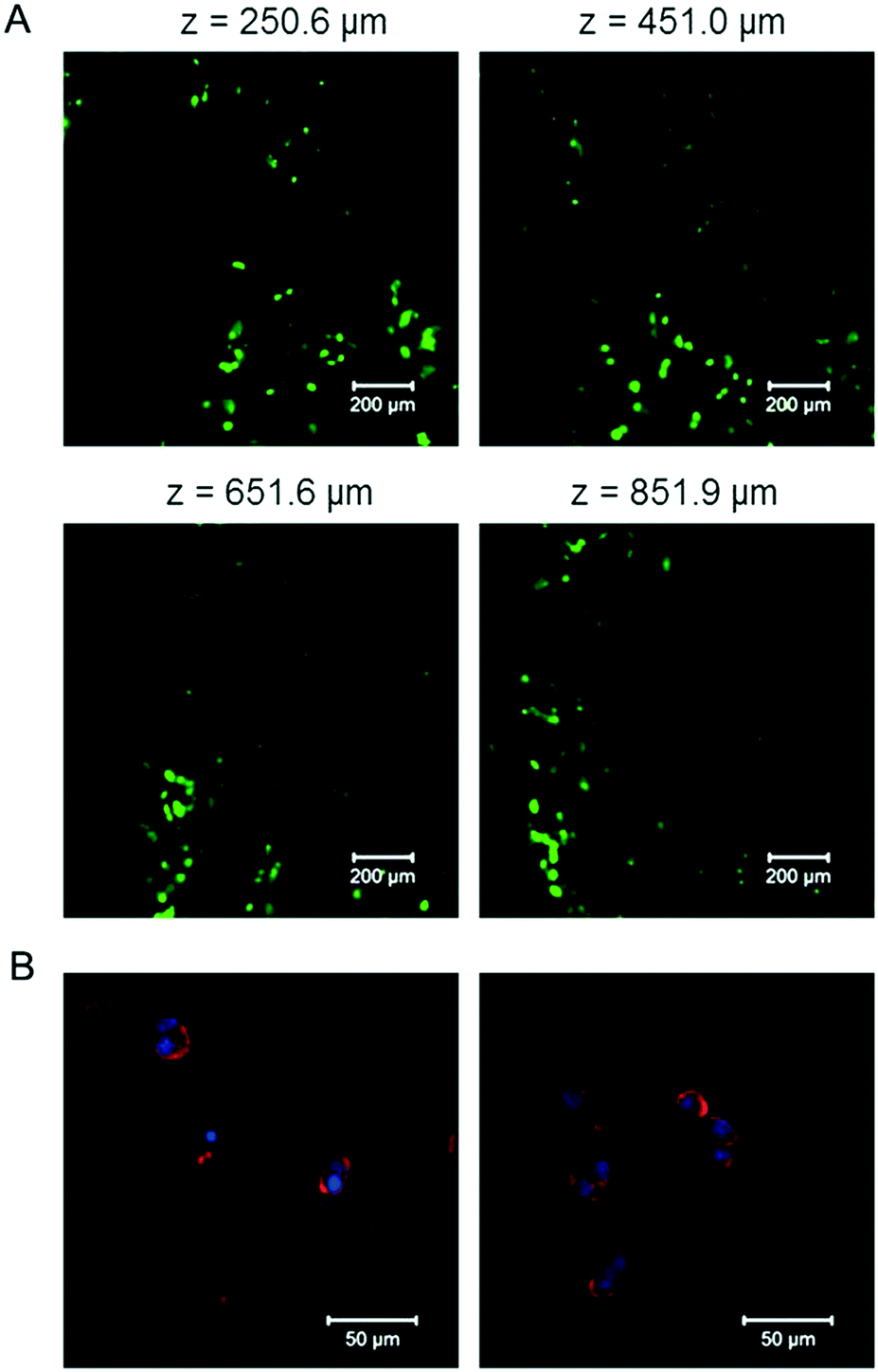
To test the cytocompatibility of the E-MEP-E/Cu hydrogel, cell encapsulation experiments were performed by suspending mouse 3T3 fibroblasts in the mixture of E-MEP-E and Cu2+. After curing at 37 °C for 30 min, the gel was covered with the culture medium and then was placed under a standard cell culture condition (37 °C, 5% CO2). After 24 h, Live/Dead staining assay and laser scanning confocal microscopy were performed. High cell viability (>95%) was observed with z-stack imaging (Fig. 7A), which was further corroborated by the natural structure of the cytoskeleton of these encapsulated cells according to actin–nucleus staining (Fig. 7B). Although free-floating transition metal ions like Cu2+ are often considered to be cytotoxic, a stoichiometric amount of metal ions (molar ratio of Cu to E-MEP-E, 1:1) were used in the gelation. They were predominantly bound to the side chains of the residues such as His and Cys in a reduced form (Cu+), which may explain the marked cytocompatibility of this E-MEP-E/Cu hydrogel.
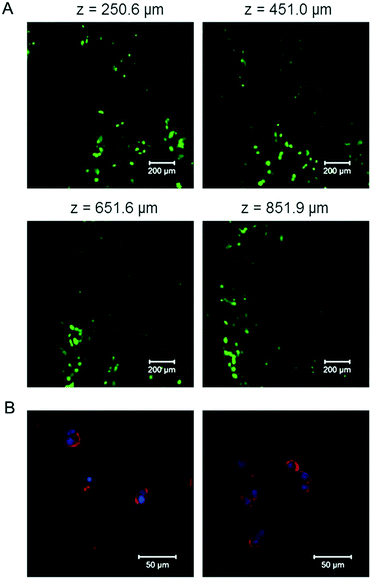 |
| | Fig. 7 Encapsulation of NIH/3T3 fibroblasts by the E-MEP-E/Cu hydrogels. (A) Confocal fluorescence z-slice micrographs of live (green, calcein AM) and dead (red, ethidium homodimer) cells. Scale bars, 200 μm. (B) Actin (red) and nuclei (blue) staining of encapsulated cells. Scale bars, 50 μm. Representative micrographs are shown. | |
4. Conclusions
In this study, the induced assembly of split GB1 protein fragments has proven to be feasible for creating protein-based materials with dynamic properties. The affinity and kinetics of metal–ligand coordination can exert great influence over the viscoelastic properties of bulk gels, thus offering a simple way to tune the gel mechanics. Moreover, the protein networks assembled by Cu2+ have exhibited several important features, including pronounced shear-thinning and self-healing behavior, amenability to biochemical decoration by SpyTag/SpyCatcher chemistry, and compatibility with encapsulated fibroblasts. Metal-induced protein assembly—a prevalent mechanism that underlies numerous molecular interactions in biology—has opened up a new dimension for designing soft materials for biomedical applications.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The funding supports from Science, Technology and Innovation Commission of Shenzhen Municipality to F. S. (Basic Research Program Grant # JCYJ20170818114000727), from PKU-HKUST ShenZhen-HongKong Institution to F. S. (SZIER18EG02), from the Research Grants Council of Hong Kong SAR Government to F. S. (RGC-ECS Grant #26103915, CRF Grant # C6004-17GF and AoE/M-09/12) are acknowledged. F. S. is grateful to the Department of Chemical and Biological Engineering, HKUST for the faculty start-up fund.
References
- S. A. Maskarinec and D. A. Tirrell, Protein engineering approaches to biomaterials design, Curr. Opin. Biotechnol., 2005, 16, 422–426 CrossRef CAS PubMed.
- R. Langer and D. A. Tirrell, Designing materials for biology and medicine, Nature, 2004, 428, 487–492 CrossRef CAS PubMed.
- B. Zakeri, J. O. Fierer, E. Celik, E. C. Chittock, U. Schwarz-Linek, V. T. Moy and M. Howarth, Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E690–E697 CrossRef CAS PubMed.
- L. Li, J. O. Fierer, T. A. Rapoport and M. Howarth, Structural Analysis and Optimization of the Covalent Association between SpyCatcher and a Peptide Tag, J. Mol. Biol., 2014, 426, 309–317 CrossRef CAS PubMed.
- W.-B. Zhang, F. Sun, D. A. Tirrell and F. H. Arnold, Controlling Macromolecular Topology with Genetically Encoded SpyTag–SpyCatcher Chemistry, J. Am. Chem. Soc., 2013, 135, 13988–13997 CrossRef CAS PubMed.
- D. Liu, W.-H. Wu, Y.-J. Liu, X.-L. Wu, Y. Cao, B. Song, X. Li and W.-B. Zhang, Topology Engineering of Proteins in Vivo Using Genetically Encoded, Mechanically Interlocking SpyX Modules for Enhanced Stability, ACS Cent. Sci., 2017, 3, 473–481 CrossRef CAS PubMed.
- X.-W. Wang and W.-B. Zhang, Cellular Synthesis of Protein Catenanes, Angew. Chem., Int. Ed., 2016, 55, 3442–3446 CrossRef CAS.
- L. J. Xu and W. B. Zhang, Topology: a unique dimension in protein engineering, Sci. China: Chem., 2018, 61, 3–16 CrossRef CAS.
- K. D. Brune and M. Howarth, New Routes and Opportunities for Modular Construction of Particulate Vaccines: Stick, Click, and Glue, Front. Immunol., 2018, 9, 1432 CrossRef.
- Z. Liu, H. Zhou, W. Wang, W. Tan, Y.-X. Fu and M. Zhu, A novel method for synthetic vaccine construction based on protein assembly, Sci. Rep., 2014, 4, 7266 CrossRef CAS.
- M. Fairhead, G. Veggiani, M. Lever, J. Yan, D. Mesner, C. V. Robinson, O. Dushek, P. A. van der Merwe and M. Howarth, SpyAvidin Hubs Enable Precise and Ultrastable Orthogonal Nanoassembly, J. Am. Chem. Soc., 2014, 136, 12355–12363 CrossRef CAS PubMed.
- T. U. J. Bruun, A.-M. C. Andersson, S. J. Draper and M. Howarth, Engineering a Rugged Nanoscaffold To Enhance Plug-and-Display Vaccination, ACS Nano, 2018, 12, 8855–8866 CrossRef CAS.
- F. Sun, W. B. Zhang, A. Mahdavi, F. H. Arnold and D. A. Tirrell, Synthesis of bioactive protein hydrogels by genetically encoded SpyTag-SpyCatcher chemistry, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11269–11274 CrossRef CAS.
- P. Q. Nguyen, Z. Botyanszki, P. K. R. Tay and N. S. Joshi, Programmable biofilm-based materials from engineered curli nanofibres, Nat. Commun., 2014, 5, 4945 CrossRef CAS.
- A. Y. Chen, Z. Deng, A. N. Billings, U. O. S. Seker, Michelle Y. Lu, R. J. Citorik, B. Zakeri and T. K. Lu, Synthesis and patterning of tunable multiscale materials with engineered cells, Nat. Mater., 2014, 13, 515–523 CrossRef CAS.
- B. An, X. Wang, M. Cui, X. Gui, X. Mao, Y. Liu, K. Li, C. Chu, J. Pu, S. Ren, Y. Wang, G. Zhong, T. K. Lu, C. Liu and C. Zhong, Diverse Supramolecular Nanofiber Networks Assembled by Functional Low-Complexity Domains, ACS Nano, 2017, 11, 6985–6995 CrossRef CAS PubMed.
- R. Wang, Z. Yang, J. Luo, I. M. Hsing and F. Sun, B12-dependent photoresponsive protein hydrogels
for controlled stem cell/protein release, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5912–5917 CrossRef CAS PubMed.
- M. Obana, B. R. Silverman and D. A. Tirrell, Protein-Mediated Colloidal Assembly, J. Am. Chem. Soc., 2017, 139, 14251–14256 CrossRef CAS.
- J. Wei, W.-H. Wu, R. Wang, Z. Yang, F. Sun and W.-B. Zhang, B12-Dependent Protein Oligomerization Facilitates Layer-by-Layer Growth of Photo/Thermal Responsive Nanofilms, ACS Macro Lett., 2018, 7, 514–518 CrossRef CAS.
- G. Veggiani, T. Nakamura, M. D. Brenner, R. V. Gayet, J. Yan, C. V. Robinson and M. Howarth, Programmable polyproteams built using twin peptide superglues, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1202–1207 CrossRef CAS.
- C. Zhang, J. Huang, J. Zhang, S. Liu, M. Cui, B. An, X. Wang, J. Pu, T. Zhao, C. Fan, T. K. Lu and C. Zhong, Engineered Bacillus subtilis biofilms as living glues, Mater. Today, 2019, 4, 3389 Search PubMed.
- J. Huang, S. Liu, C. Zhang, X. Wang, J. Pu, F. Ba, S. Xue, H. Ye, T. Zhao, K. Li, Y. Wang, J. Zhang, L. Wang, C. Fan, T. K. Lu and C. Zhong, Programmable and printable Bacillus subtilis biofilms as engineered living materials, Nat. Chem. Biol., 2018, 15, 34–41 CrossRef.
- X. Gao, S. Lyu and H. Li, Decorating a Blank Slate Protein Hydrogel: A General and Robust Approach for Functionalizing Protein Hydrogels, Biomacromolecules, 2017, 18, 3726–3732 CrossRef CAS.
- X. Gao, J. Fang, B. Xue, L. Fu and H. Li, Engineering Protein Hydrogels Using SpyCatcher-SpyTag Chemistry, Biomacromolecules, 2016, 17, 2812–2819 CrossRef CAS.
- B. P. Lee, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Mussel-Inspired Adhesives and Coatings, Annu. Rev. Mater. Res., 2011, 41, 99–132 CrossRef CAS.
- J. Y. Sun, X. H. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. G. Suo, Highly stretchable and tough hydrogels, Nature, 2012, 489, 133–136 CrossRef CAS.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Double-network hydrogels with extremely high mechanical strength, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- K. J. Henderson, T. C. Zhou, K. J. Otim and K. R. Shull, Ionically Cross-Linked Triblock Copolymer Hydrogels with High Strength, Macromolecules, 2010, 43, 6193–6201 CrossRef CAS.
- D. Mozhdehi, S. Ayala, O. R. Cromwell and Z. B. Guan, Self-Healing Multiphase Polymers via Dynamic Metal-Ligand Interactions, J. Am. Chem. Soc., 2014, 136, 16128–16131 CrossRef CAS.
- S. C. Grindy, R. Learsch, D. Mozhdehi, J. Cheng, D. G. Barrett, Z. B. Guan, P. B. Messersmith and N. Holten-Andersen, Control of hierarchical polymer mechanics with bioinspired metal-coordination dynamics, Nat. Mater., 2015, 14, 1210–1216 CrossRef CAS PubMed.
- S. V. Wegner, F. C. Schenk, S. Witzel, F. Bialas and J. P. Spatz, Cobalt Cross-Linked Redox-Responsive PEG Hydrogels: From Viscoelastic Liquids to Elastic Solids, Macromolecules, 2016, 49, 4229–4235 CrossRef CAS.
- E. N. Salgado, R. J. Radford and F. A. Tezcan, Metal-Directed Protein Self-Assembly, Acc. Chem. Res., 2010, 43, 661–672 CrossRef CAS PubMed.
- Q. Peng, N. Kong, H. C. E. Wang and H. B. Li, Designing redox potential-controlled protein switches based on mutually exclusive proteins, Protein Sci., 2012, 21, 1222–1230 CrossRef CAS PubMed.
- Q. Peng and H. Li, Direct observation of tug-of-war during the folding of a mutually exclusive protein, J. Am. Chem. Soc., 2009, 131, 13347–13354 CrossRef CAS PubMed.
- D. W. Urry, Physical chemistry of biological free energy transduction as demonstrated by elastic protein-based polymers, J. Phys. Chem. B, 1997, 101, 11007–11028 CrossRef CAS.
- Z. G. Yang, S. Z. Kou, X. Wei, F. J. Zhang, F. Li, X. W. Wang, Y. Lin, C. Wan, W. B. Zhang and F. Sun, Genetically Programming Stress-Relaxation Behavior in Entirely Protein-Based Molecular Networks, ACS Macro Lett., 2018, 7, 1468–1474 CrossRef CAS.
- Z. Y. Hao, H. B. Lou, R. F. Zhu, J. H. Zhu, D. M. Zhang, B. X. S. Zhao, S. Z. Zeng, X. Chen, J. Chan, C. He and P. R. Chen, The multiple antibiotic resistance regulator MarR is a copper sensor in Escherichia coli, Nat. Chem. Biol., 2014, 10, 21–U48 CrossRef CAS.
- N. Kong, L. Fu, Q. Peng and H. Li, Metal Chelation Dynamically Regulates the Mechanical Properties of Engineered Protein Hydrogels, ACS Biomater. Sci. Eng., 2016, 3, 742–749 CrossRef.
- H. Irving and R. J. P. Williams, 637. The stability of transition-metal complexes, J. Chem. Soc., 1953, 3192 RSC.
- S. Kou, Z. Yang and F. Sun, Protein Hydrogel Microbeads for Selective Uranium Mining from Seawater, ACS Appl. Mater. Interfaces, 2017, 9, 2035–2039 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures. See DOI: 10.1039/c9me00027e |
|
| This journal is © The Royal Society of Chemistry 2020 |

 b and
Fei
Sun
b and
Fei
Sun