
Controlled polymers: accessing new platforms for material synthesis
Keri L.
Kaligian
and
Melissa M.
Sprachman
 *
*
Charles Stark Draper Laboratory Inc, Cambridge, Massachusetts, USA. E-mail: MSprachman@draper.com
First published on 14th January 2019
Abstract
Advances in polymer synthesis and characterization have laid the foundation for expanding the scope of synthetic polymer applications. Precisely controlled polymer synthesis could enable a generation of polymers with unique and predictable properties, akin to the syntheses of nucleic acids and peptides. Applications of synthetic polymers include information storage and assembly of 2-D and 3-D organic and composite nanomaterials. Herein, methods to achieve sequence control are discussed, including DNA-templated chemistries, iterative additions, and controlled living polymerization. The possibility of incorporating a verification method at each step is also presented.
Design, System, ApplicationThis article is a review of methods in controlled polymer synthesis. Design strategies include templated synthesis, reagent control/photo-control, and use of protecting groups. The article is intended as a resource for readers seeking applications such as information storage and synthesis of novel materials. |
Introduction
The exquisite capacity for biological polymers to store and transfer information has long been recognized as a tool with uses beyond life science applications. One key area is information storage, where multidisciplinary efforts are underway to realize the transition from hard-drives in warehouses (106 sq. ft. per exabyte) to arrays of encoded polymers (1000 sq. ft. per exabyte).1 The use of DNA to encode information has been demonstrated, with developments such as encoding digital movies into the genomes of bacteria.2More recently, researchers at the University of Washington in collaboration with Microsoft demonstrated the ability to encode over 400 MB of data and recover distinct files using a random access approach.3 DNA has been leveraged because it is well understood in terms of sequence control (writing), amplification, and sequencing (reading). Well-characterized DNA sequences are also used in materials chemistry as tools for patterning (e.g., use of DNA origami for “single molecule” printing)4 or for controlled assembly of nanomaterials into hierarchical structures.5
As much as DNA is lauded for programmability and accessibility, there are limitations including restricted reaction conditions and limited chemical properties of the polymers (e.g., limited stability outside aqueous environments). One only need look at another biopolymer family, peptides and proteins, to understand the chemical and material space that can be accessed through different combinations of monomers. Examples include assembly of proteins into hierarchical structures with unique physical and mechanical properties.6,7
Synthetic polymers have also been well-studied in terms of their potential to form well-ordered materials and self-assemble.8,9 More attention has been given to general architecture (length, polarity, shape) versus sequence control. If polymers could be synthesized in a predictable manner, one could envision new methods for generating materials with specific and tunable properties. Although data storage remains a key application, one could also envision using synthetic polymers for programmable design and assembly of novel materials.
This article contains a summary of current methods being developed for sequence control in oligomer and polymer synthesis. The possibility of using distinct monomers as coding modules is discussed, among other applications, and the merits and limitations of various synthetic methods are evaluated in the context of possible applications. This article is not meant to be comprehensive but rather inspire researchers to think about using polymers for data storage and construction of new materials.
Sequence controlled methods
Templated synthesis
The oldest form of polymer-based information storage is DNA, where information is stored in the prescribed base sequences. A canonical template-mediated synthesis is transcription, where RNA is formed from a template DNA strand. Researchers have sought means to replicate the same level of sequence control in non-natural polymer synthesis by using a pre-designed template that can interact with incoming monomers.In DNA-Templated synthesis (DTS), monomers are placed in desired positions through use of a DNA template. An example of DTS is the Wittig transfer reaction to produce a decamer (Fig. 1).10 The polymers have a pre-determined initial and final monomers with interchangeable monomers between them. Specifically, the intermediate monomers consist of peptides B and C (Fig. 1A), with monomer C bearing an alkyne for further click chemistry modifications.
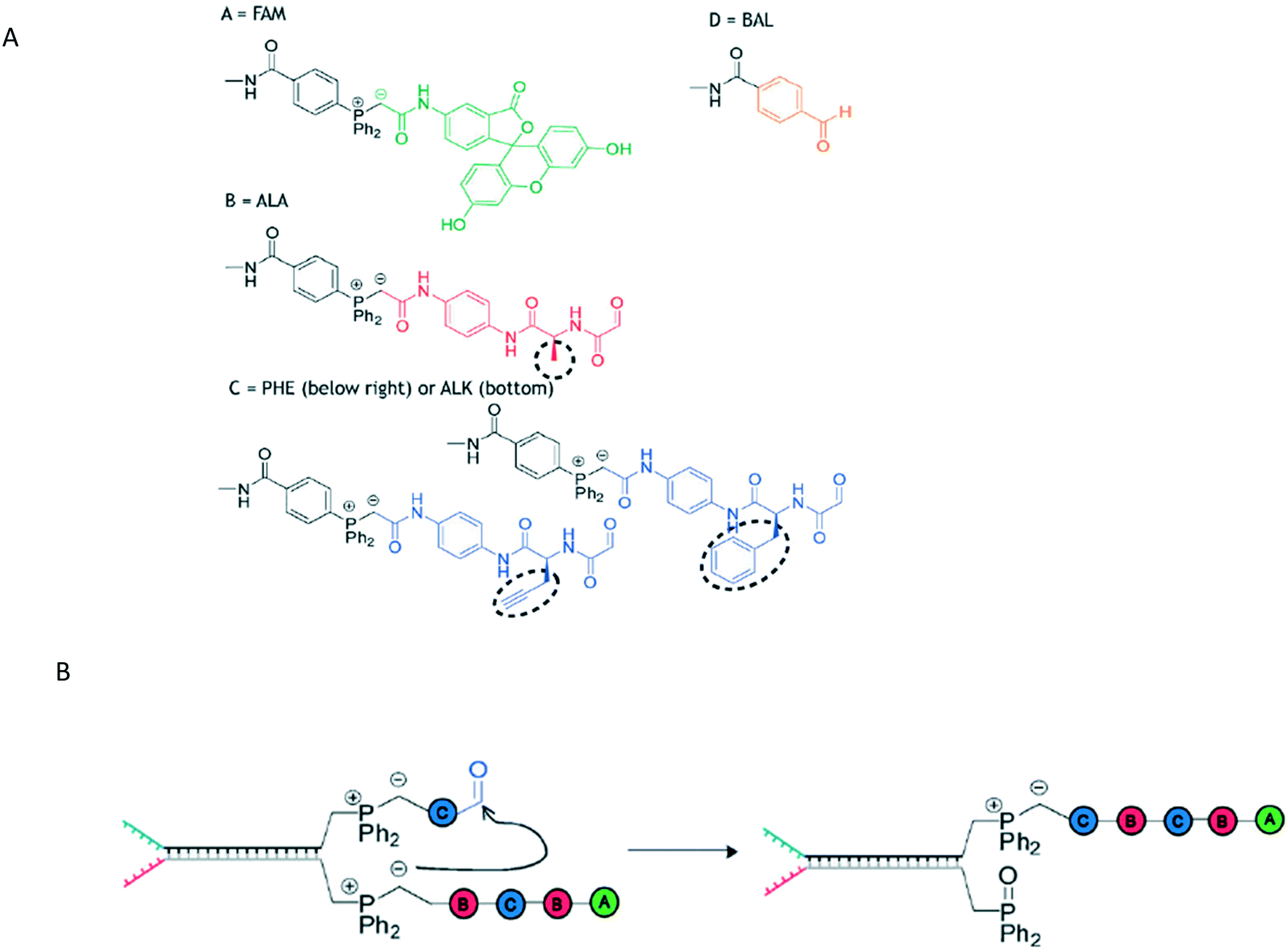 | ||
| Fig. 1 A) The structures of monomers A, B, C, and D used in the Wittig reaction and B) the propagation step of the Wittig reaction. Reproduced from ref. 10 with permission from The Royal Society of Chemistry.10 | ||
In this Wittig reaction, a template strand terminating in a phosphonium ylide is hybridized to a complement with an aldehyde. When the Wittig reaction takes place, the oxophosphonium intermediate becomes the Wittig product and the phosphine oxide. This effectively adds a building block onto a growing oligomer chain; the strand containing the phosphine oxide is a removable byproduct.
The phosphine-oxide containing strand can undergo strand displacement with a strand containing the next monomer unit, continuing the chain growth. Because the oligomers are formed by sequential additions/displacement steps, the DNA adapter sequence can be reused. The overall yield from this approach is limited by final purity – typical purities of 73% and 63% resulted in ca. 31% and 23% yields respectively, giving an average yield of 88% and 85% yield per step.10
Liu and coworkers have developed several DTS methods for generating synthetic polymer libraries. In dilute solutions of DNA tagged reactants, the effective concentration of reactive partners is increased upon DNA hybridization of complementary strands. Several reactions can be run in parallel as long as each monomer is tagged with a unique DNA barcode.11 DTS has been used to construct macrocycle libraries of greater than 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 members with at least four sequential monomer unit additions. The process was refined further by identifying the smallest number of defective codons that could be removed from the monomer pool.11
000 members with at least four sequential monomer unit additions. The process was refined further by identifying the smallest number of defective codons that could be removed from the monomer pool.11
Liu and coworkers have also developed a templated synthesis that involves using DNA oligonucleotides as templates for direct oligomerization of polymer building blocks tethered to peptide nuclei acid adapters (Fig. 2).12 Each building block has an adapter containing a PNA pentamer with a bifunctional polymer building block, for example a polyethylene glycol chain terminated with an alkyne at one end and an azide at the other.
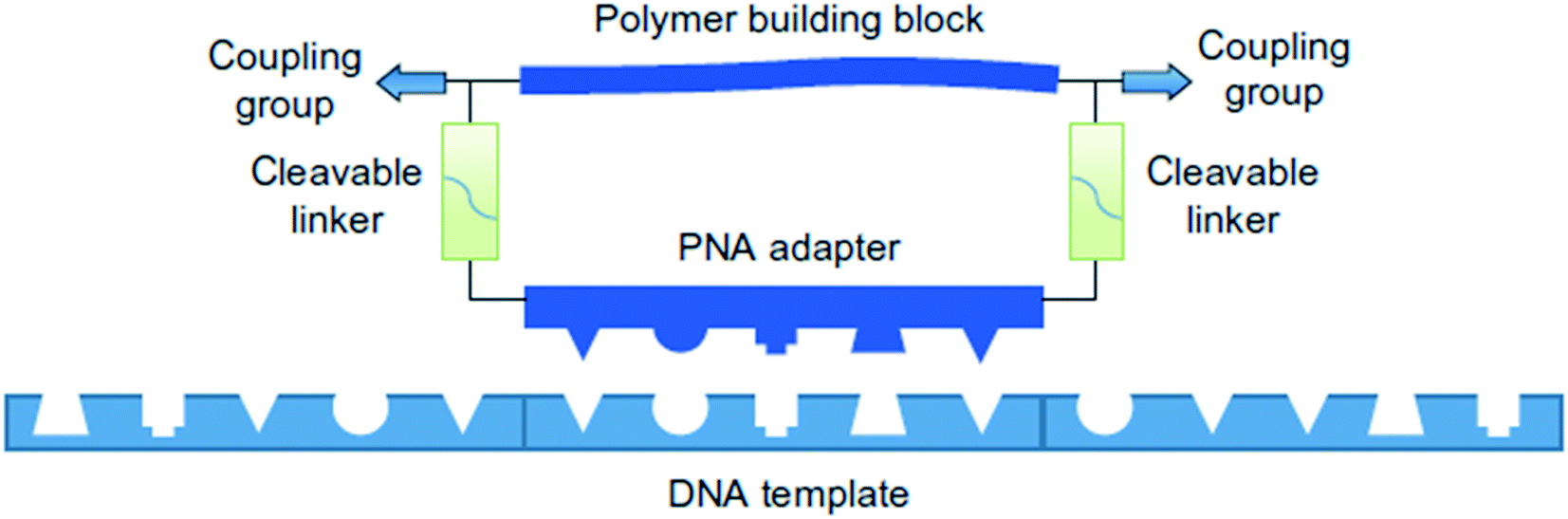 | ||
| Fig. 2 A simplified example of the PNA adapter method. Reprinted with permission from ref. 12. Copyright 2013 Springer Nature.12 | ||
Sequence control is achieved by assigning each polymer building block an appropriate PNA adapter to hybridize with the DNA template. CuAAC (copper(I)-catalyzed alkyne-azide cycloaddition) is used to connect the building blocks. The system is compatible with polymer backbones including polyethylene glycol (PEG), β-peptides, and α-(D)-peptides, reaching molecular weights up to 26 kDa formed from 16 consecutive substrate couplings.13
Liu and coworkers also took advantage of enzymatic methods by synthesizing densely functionalized nucleic acids via a T4 DNA ligase mediated DNA-templated polymerization of 5′-phosphorylated trinucleotides having various substituents (Fig. 3).14 The process mimics the coding done during ribosomal translation of mRNA into proteins (using a tricodon system). A template DNA is flanked by initial and terminating binding sites to specify a reading frame.
 | ||
| Fig. 3 The translation strategy to turn DNA into a functionalized DNA alongside the amine-linked bases that were incorporated into the chain. Reprinted with permission from ref. 14. Copyright 2013 ACS Publications. https://pubs.acs.org/doi/abs/10.1021%2Fja311331m%20 further permissions related to use of this material should be directed to ACS publications.14 | ||
The reading frame contains trinucleotide codons that correspond with the trinucleotide substrates; upon templating, DNA-T4 ligase adds the trinucleotide to the templated strand. In this example, up to eight different functional groups were incorporated throughout a polymer product, and it is possible to incorporate up to 64 different functions. Overall, this process can be used to generate polymers of at least 50 building blocks having a molecular weight of approximately 60 kDa.14
Yet another form of DTS involves DNA Walkers, wherein building blocks are autonomously added to a growing polymer chain (Fig. 4).15 The walker itself is a single strand of DNA that attaches to a track. As the polymer chain grows from each walker strand on the track, the growing chain is transferred by strand displacement. The primary advantage of this method is its autonomous nature; external intervention is not needed to propagate the monomer addition.
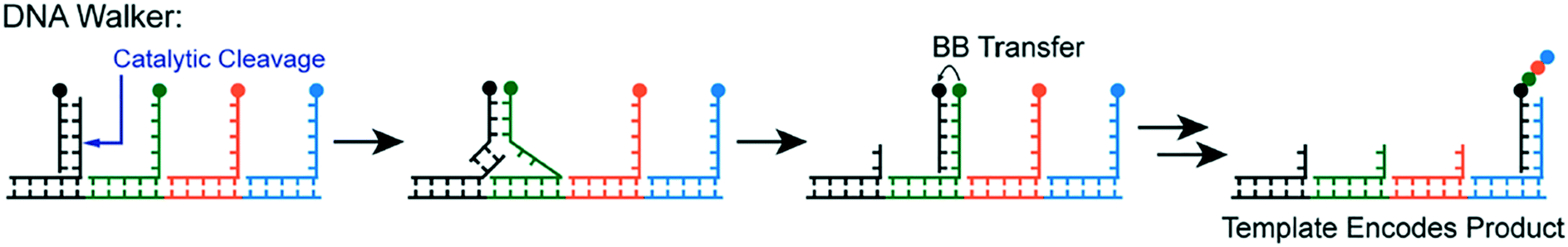 | ||
| Fig. 4 The DNA walker method—building blocks (circles) are transferred sequentially by “walking” to complementary strands containing the next building block in the sequence. The building block is transferred from one chain to the next, propagating growth on a primary strand. Reprinted with permission from ref. 16. Copyright 2017 ACS publications: https://pubs.acs.org/doi/abs/10.1021%2Facs.accounts.7b00280 further permission related to use of this material should be directed to ACS publications.16 | ||
Templated synthesis benefits from translating the superb sequence control of DNA to a synthetic polymer sequence. The methods are still restricted to chemistries that are compatible with DNA; moreover, the process of synthesizing oligonucleotide-small molecule conjugates can be a limiting factor. Templates other than DNA have also been used; examples include templates generated from methacrylate monomers substituted with nucleobases.17,18
Depending on the application, the templated methods could suffer from mass balance/atom economy issues as the templates are not incorporated into the final product. Template-free methods for producing well-defined sequences could prove advantageous.
Controlled iterative additions
Controlled iterative addition refers to the stepwise addition of monomers to a growing oligomer or polymer chain, giving the user control over each sequential addition. This method often relies on orthogonal protection/deprotection methods to maintain control, and polymer lengths and yields will be limited based on the efficiencies of these steps.Classic examples of iterative oligomer growth are solid-phase peptide synthesis (SPPS, Merrifield) and oligonucleotide synthesis. Merrifield synthesis relies on iterative additions of orthogonally protected amino acids—classically, N-fluorenylmethyloxycarbonyl (Fmoc) protected amino acids with a free carboxyl group coupled to a resin-bound peptide chain.19 After each addition the Fmoc group is removed to allow reaction with the free carboxyl of another N-protected amino acid.
SPPS has facilitated the synthesis of large peptide libraries, and the platform can be extended to non-peptide amide-containing oligomers.19,20 SPPS has utility due to its improved yields from liquid phase synthesis as well as process automation;20 automated synthesizers can produce a large amount of peptides up to the kg scale.21 Longer protein-like peptide chains can be produced by coupling protected segments or by efficient ligations.22
Oligonucleotide synthesis, also performed on solid support, is a fast and efficient means to produce small quantities of material.23 These oligomers are produced using the phosphoramidite method (Fig. 5). The method is now automated with several companies (e.g., IDT, Sigma-Aldrich) providing custom oligonucleotide synthesis, and bench-top oligonucleotide synthesizers are available for purchase.
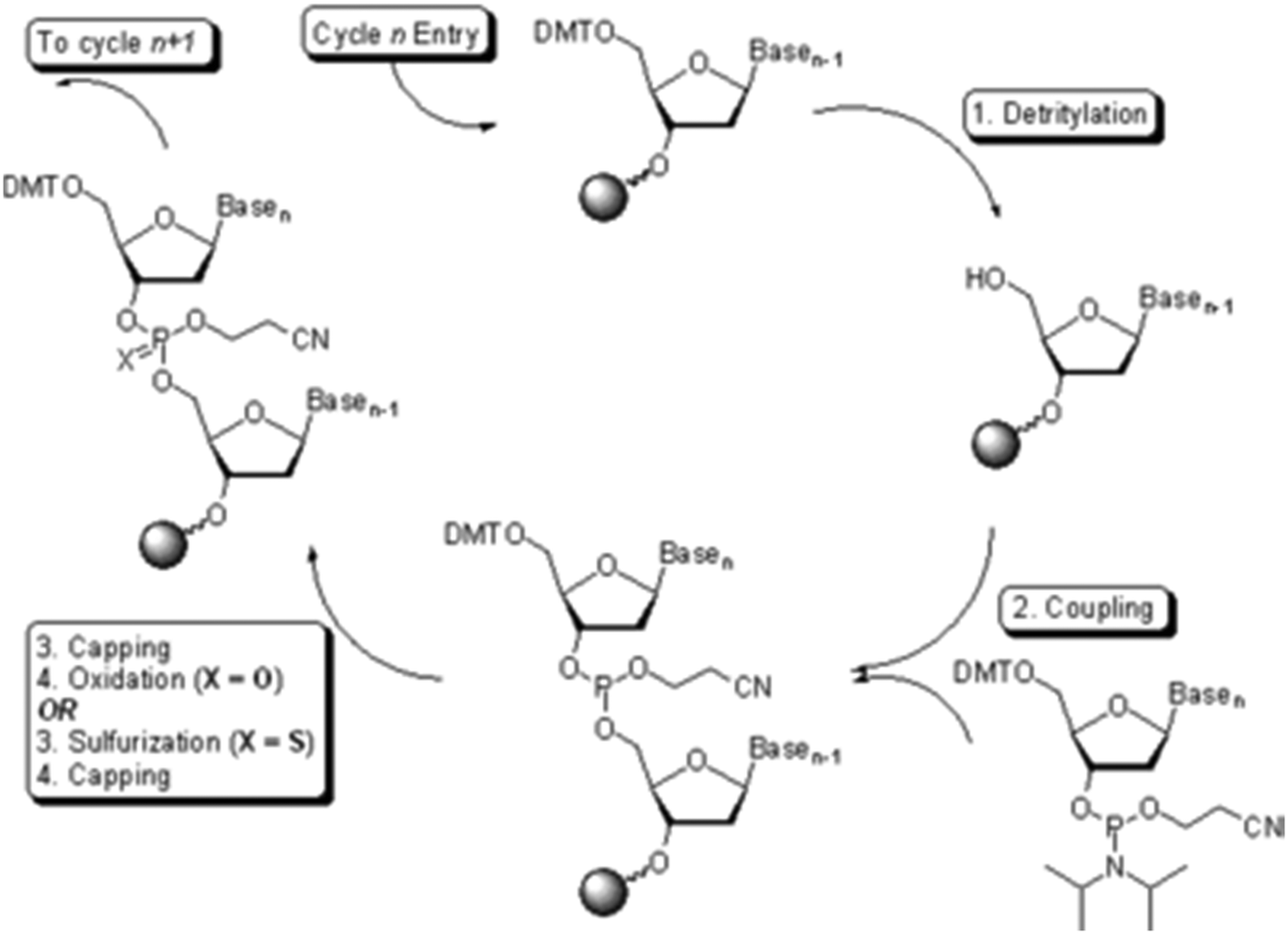 | ||
| Fig. 5 The phosphoramidite approach to solid phase oligonucleotide synthesis. Source: Wikipedia.24 | ||
Carbon–carbon bonds can also be achieved using iterative strategies. The Burke group has designed bifunctional haloboronic acid building blocks where the boronic acid reactivity is attenuated by complexation with MIDA (N-methylimidodiacetic acid) ligands (Fig. 6). The user can trigger removal of the MIDA complex under basic conditions (e.g., NaOH in THF/water), allowing effective deprotection of the boronic acid moiety for sequential reactions as shown in Fig. 7.25 Burke surmises that this cross-coupling can be similar to the iterative nature of SPPS (Fig. 7).
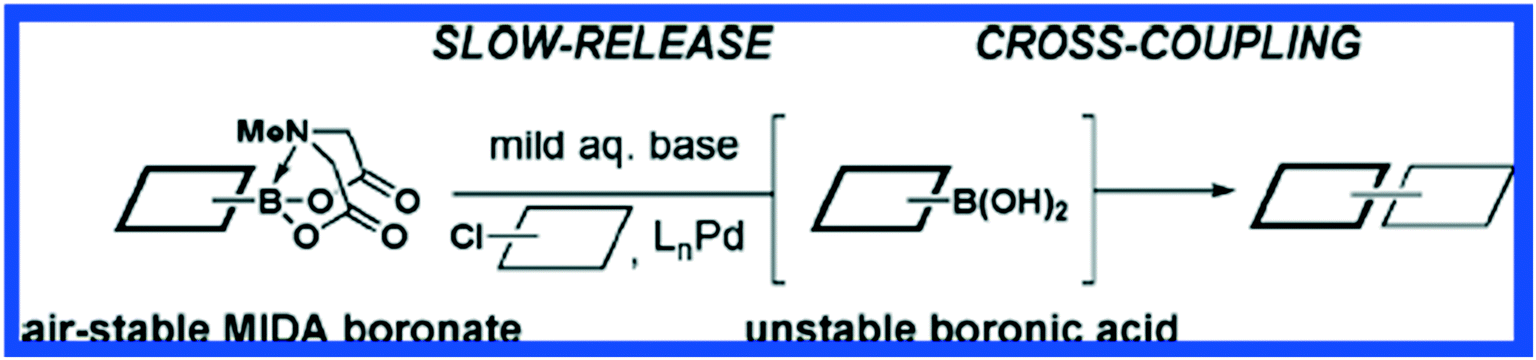 | ||
| Fig. 6 The MIDA Boronate is stable in air and is effectively a protected boronic acid. Upon treatment with mild base, the boronic acid is revealed and undergoes Pd-mediated cross coupling reactions. Reprinted with permission from ref. 26. Copyright 2009 ACS Publications.26 | ||
 | ||
| Fig. 7 Comparison of Suzuki–Miyaura chemistry to peptide chemistry. Both methods can be used for sequential additions of building blocks. Reprinted with permission from ref. 25. Copyright 2015 ACS Publications.25 | ||
The haloboronic acids have enhanced bench-top stability because the MIDA ligand effectively removes the Lewis acid p-orbital. Over 200 possible MIDA boronates are commercially available having a variety of different functional groups, oxidation states, and pre-installed stereochemistries.25
The variety and stability of the haloboronic acid building blocks paired with the user-enabled deprotection methodologies makes the MIDA boronate chemistry an attractive candidate for iterative oligomer growth. Although efforts have been made to translate this methodology to solid-phase approaches, synthetic efforts have focused mostly on building complex molecules of, at most, 12 MIDA boronate blocks.25
Copper-catalyzed azide/alkyne cycloadditions (CuAAC) can also be used for sequential addition of monomers—an example is shown in Fig. 8A.27 Iterations of CuAAC using macromers with molecular weights ranging from 3 to 5.5 kDA are used to generate polymers.27 The polymer formed can reach a molecular weight up to 50000 DA in the form of a spherical miktoarm star structure. Much like MIDA boronates, iterative CuAAc cannot currently be used for long chains with a great deal of control; one solution to this problem is Iterative Exponential Growth (IEG), which will be discussed in the following section.
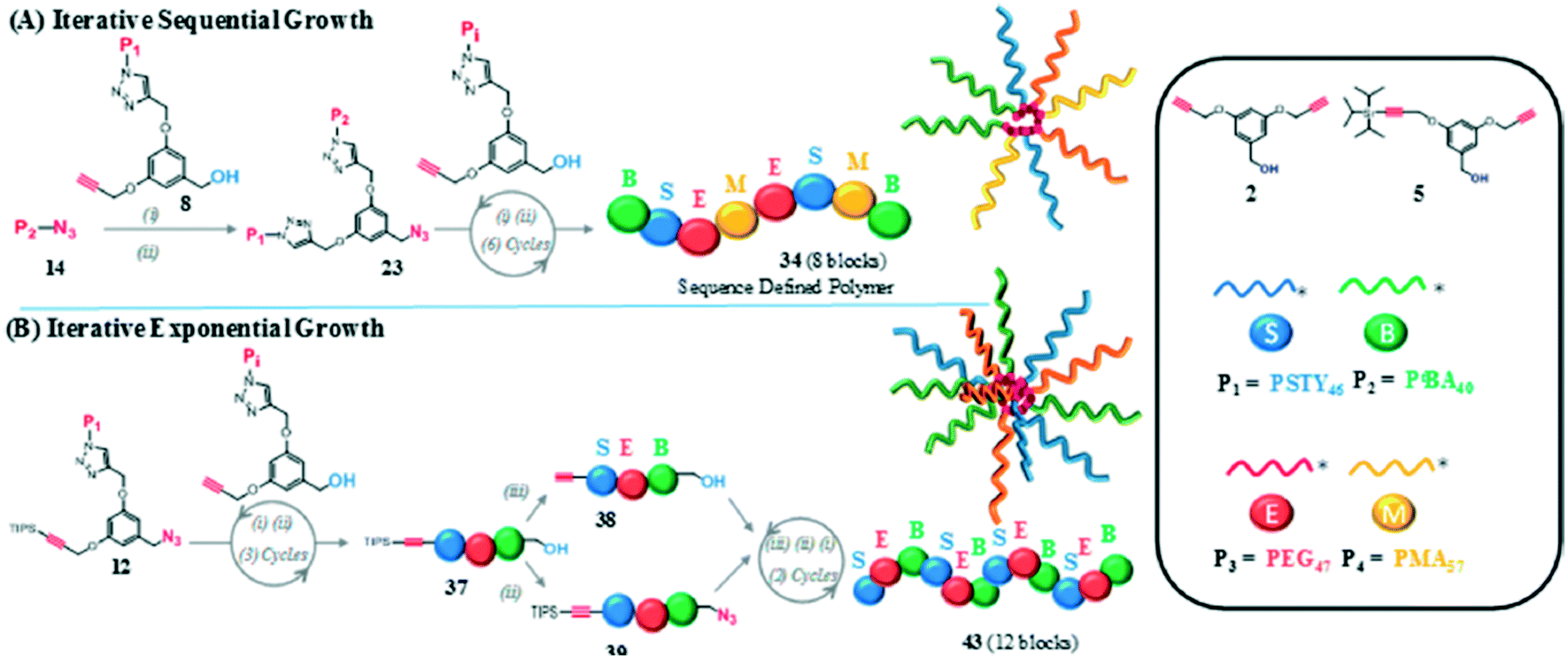 | ||
| Fig. 8 A generalized visual displaying the different mechanisms for A) iterative sequential growth and B) iterative exponential growth (IEG) in the formation of Miktoarm star polymers. Reprinted with permission from ref. 27. Copyright 2016 ACS Publications.27 | ||
The controlled iterative additions discussed thus far involve orthogonally protected monomer groups (drawing on the analogy to orthogonally protected amino acids for SPPS). Another class of iterative addition involves radical chemistries – one example being reversible addition-fragmentation chain transfer (RAFT). RAFT chemistry is initiated by redox pairs at room temperature, reducing side reactions that commonly occur with acrylates.28
Some RAFT methodologies avoid the use of metals, and no intermediate purification steps are needed. In one case, use of TBHP/AsAc as the redox pair enabled full monomer conversion to occur. The method has limitations when attempting to achieve additions of high molecular weight blocks due to reduced livingness causing termination reactions to occur during the polymerization, as shown through attempts to create (PHEA100)6 and only being able to produce (PHEA100)4.28
Bulky tertiary ester methacrylates have been used to create sequence controlled vinyl polymers (Fig. 9).29 The bulky polymers prevent sequential propagation, making it easier to perform acidolysis and esterification after each radical addition, but the overall method does suffer from low efficiency.
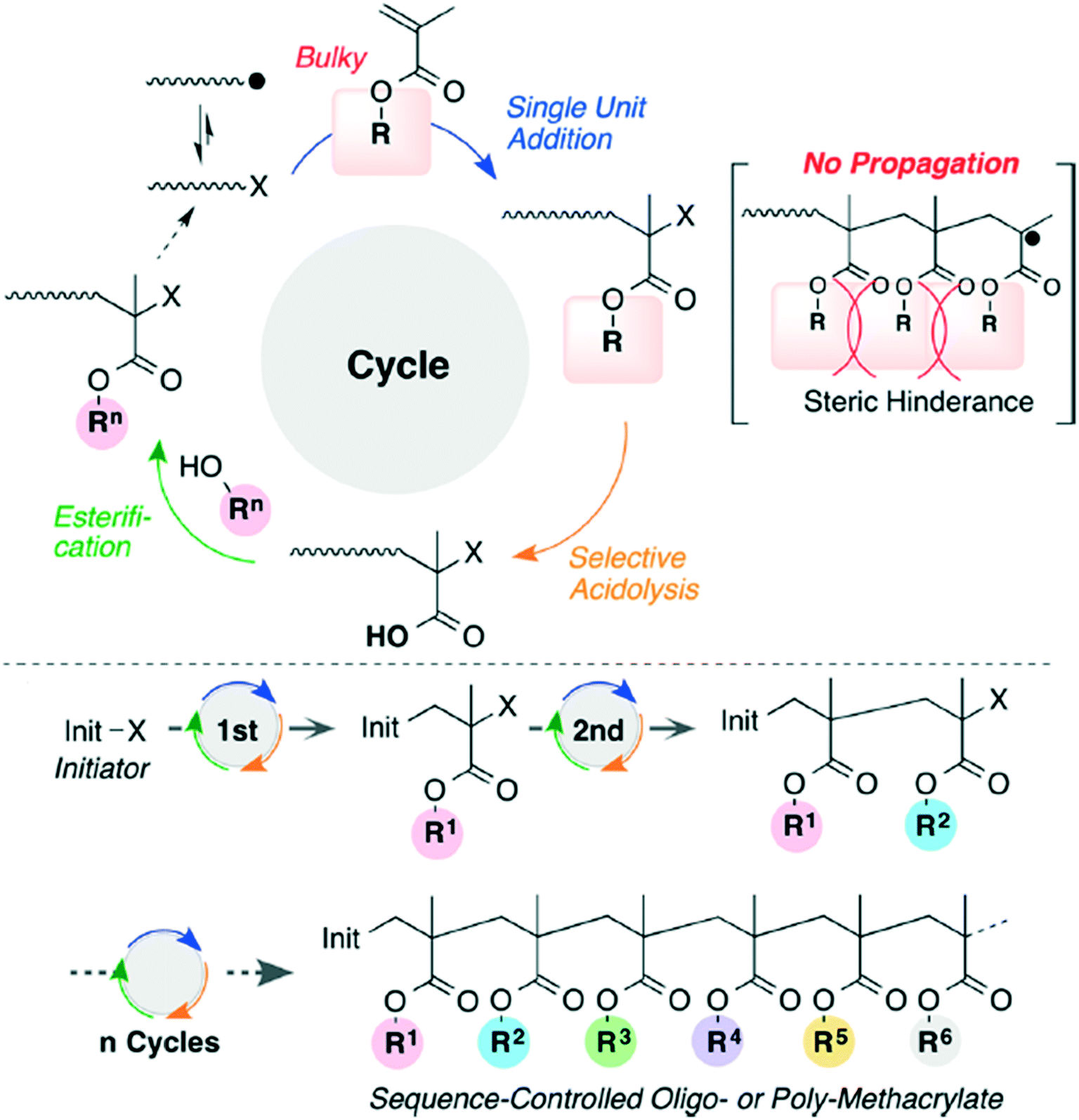 | ||
| Fig. 9 The reaction scheme for iterative radical addition of methacrylates to create vinyl polymers. Reprinted with permission from ref. 29. Copyright 2016 ACS Publications.29 | ||
Iterative exponential growth
Iterative exponential growth (IEG) refers to a strategy where orthogonally protected molecules of length l are repeatedly activated/coupled so that oligomers/polymers with length l-2#cycles are formed.30,31 One limitation of IEG is that the sequences will consist of repeating units. To further the utility of IEG, Johnson and coworkers have incorporated side chain functionalization in a method coined IEG+ (Fig. 10).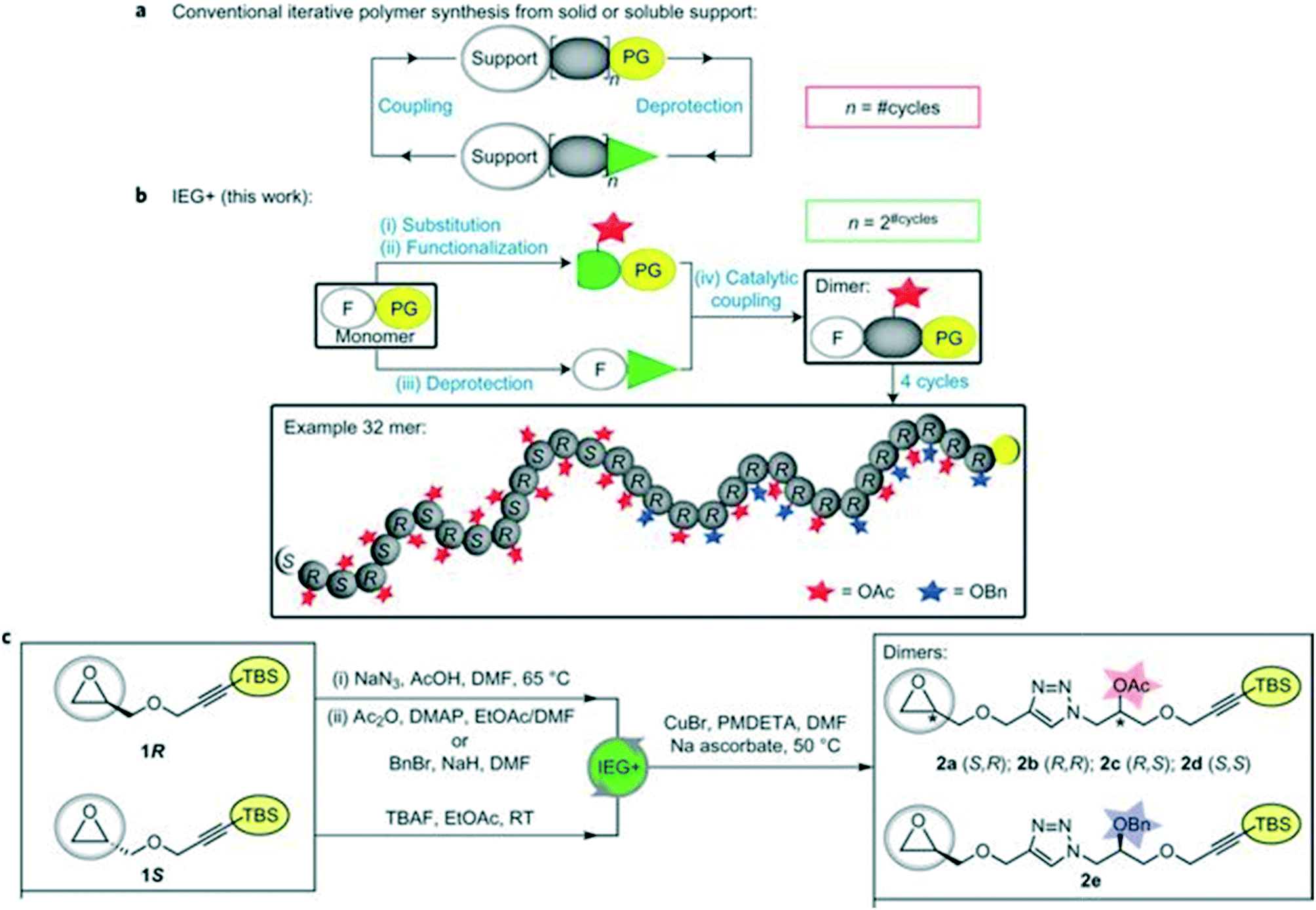 | ||
| Fig. 10 Schematic comparing traditional iterative polymer synthesis with IEG+. Part a illustrates conventional iterative addition, where PG = protecting group. In part b, the concept of side-chain manipulation is demonstrated. In part c, representative conditions for epoxide opening, purposeful functional group protection (either acetyl or benzyl) followed by silane removal for click chemistry is shown. Reprinted with permission from ref. 32. Copyright 2015 Springer Nature.32 | ||
Iterative exponential growth plus side chain functionalization (IEG+) is a specialized form of IEG designed to exert control over the sequence and stereochemistry of a polymer.32 IEG+ uses either chiral uniform oligomers or polymers with variable sequences of acetyl (Ac) and benzyl (Bn) protected alcohols.30,32 The main reactions used are opening epoxides with azides, fluoride-mediated desilylation of alkynes, and CuAAC.
An extension of this work involves incorporation of allyl groups that then undergo thiol–ene reactions to impart added functionality. IEG+ relies on extremely efficient reactions to produce macromolecules with excellent yields and high atom economy. By incorporating functional group handles, additional properties are imparted.
Flow-IEG, an extension of IEG chemistries, was developed by Jamison, Leibfarth, and Johnson in 2015.33 Flow-IEG benefits from throughput, reproducibility, and the ability to telescope reactions, simplifying the redundant aspects of IEG.34 One advantage for this new method is the ability to program the thermal and mechanical properties of the finished oligomers in a semi-automated manner.
Controlled iterative methods benefit from direct control over each insertion. Because most require sequential protection/deprotection steps, the processes are often time-consuming and suffer from diminished yields as chain lengths are extended. An alternative but less controlled approach is living polymerization.
Controlled living polymerization
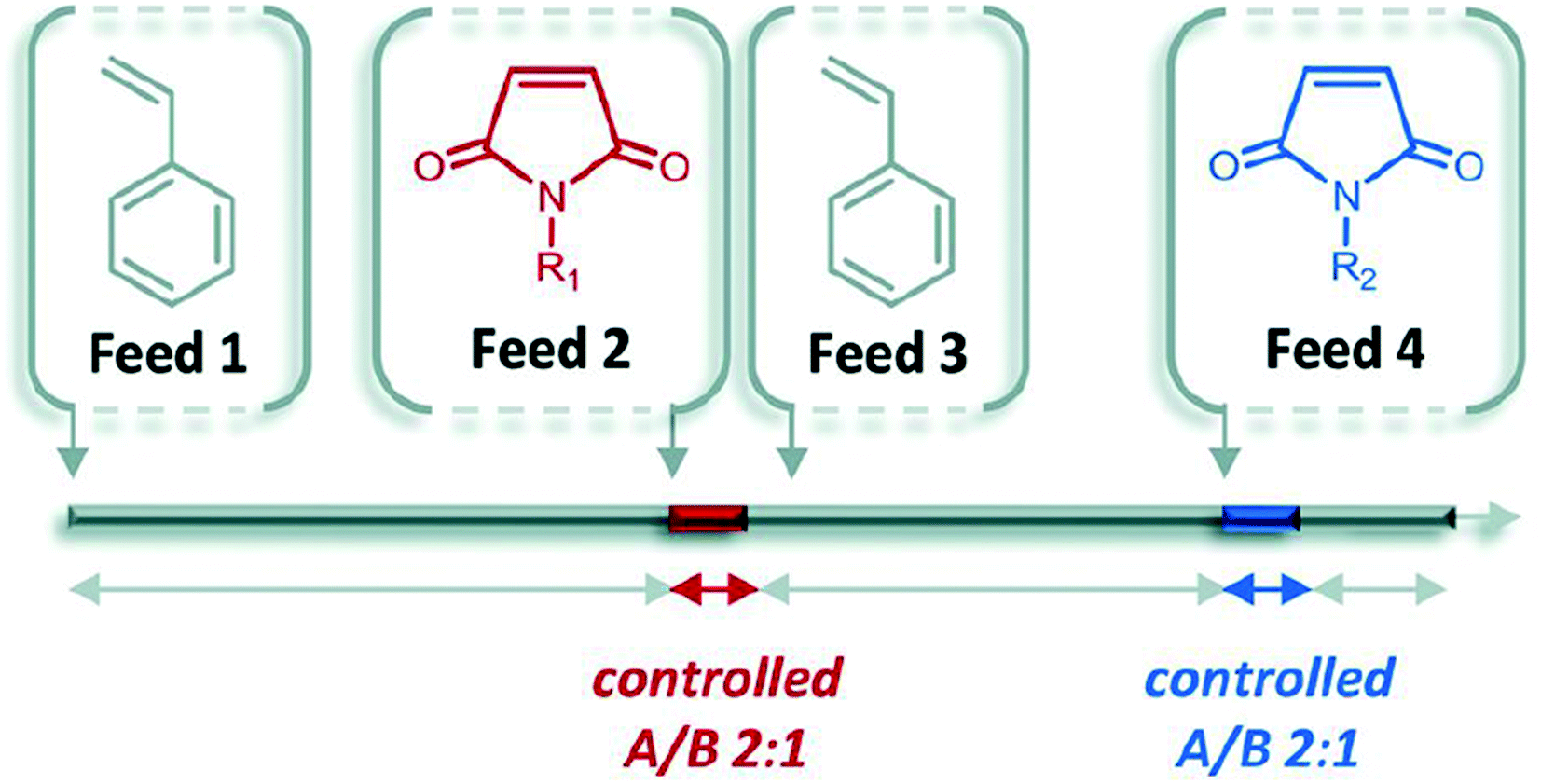
In contrast to the step-by-step nature of iterative addition, chain growth polymerization is typically used to prepare long chains with little care for sequence control. Living polymerization is a sub-category of chain growth polymerization where the forming chain is unable to terminate the reaction. The polymerization, therefore, continues until all monomers are consumed and acts as though the polymer itself is alive. There are opportunities to control sequence through careful selection of polymer backbone and timed monomer additions.One exploration of this method is the control of living polymerization through the use of a donator backbone and an acceptor that is precisely inserted into the backbone.35 For example, styrene and select styrene derivatives serve as a homopolymer backbone that has a low homopolymerization rate in comparison to the copolymerization rates of the donor and acceptor monomers. Functional groups (N-substituted maleimides) can be precisely inserted based on timing of introduction into the reaction mixture (Fig. 11).
 | ||
| Fig. 11 The general concept of ultra-precise insertion of functionalized monomers. Reprinted with permission from ref. 36. Copyright 2014 ACS Publications.36 | ||
This method has been termed the “ultra-precise insertion” method and requires that the initial donor monomer reaches a certain conversion (around 60%) before inserting the acceptor. Once the acceptor is fully incorporated, more donor is added to maintain the livingness of the chain for further additions.
The main difficulty with this method is the uncertainty of the acceptor monomer distribution. Even with precise monomer insertion, uncertainty occurs because the monomers are distributed inconsistently across the multiple chains that are forming, mainly due to the occurrence of side reactions such as chain transfer and pre-mature termination.37
Another strategy for controlling maleimide insertion into polystyrene backbones is to use a furan-protected maleimide as a latent monomer.38 The maleimide is deprotected through retro-Diels–Alder (rDA) which can be triggered by strategic thermodynamic control to begin its incorporation into the polystyrene. This method differs from the ultra-precise insertion (relying on adding the monomers at requisite time points) in that the maleimides' incorporation is consistent across all polymer chains, making this method more advantageous for achieving sequence control.38
An alternative method involves living polymerization with photo-induced sequence control.39,40 This method requires a photo-initiator to add each copolymer block. An example is shown in Fig. 12, wherein homopolymerization forms blocks from the initial monomers—polyacrylate blocks initially formed through homopolymerization are used as effective monomers. These blocks are then combined via photo-induced methods, resulting in a copolymer with a varied number of polymer blocks defining its architecture.39 Sequence control in this method is achieved by selective actuation of a UV source.
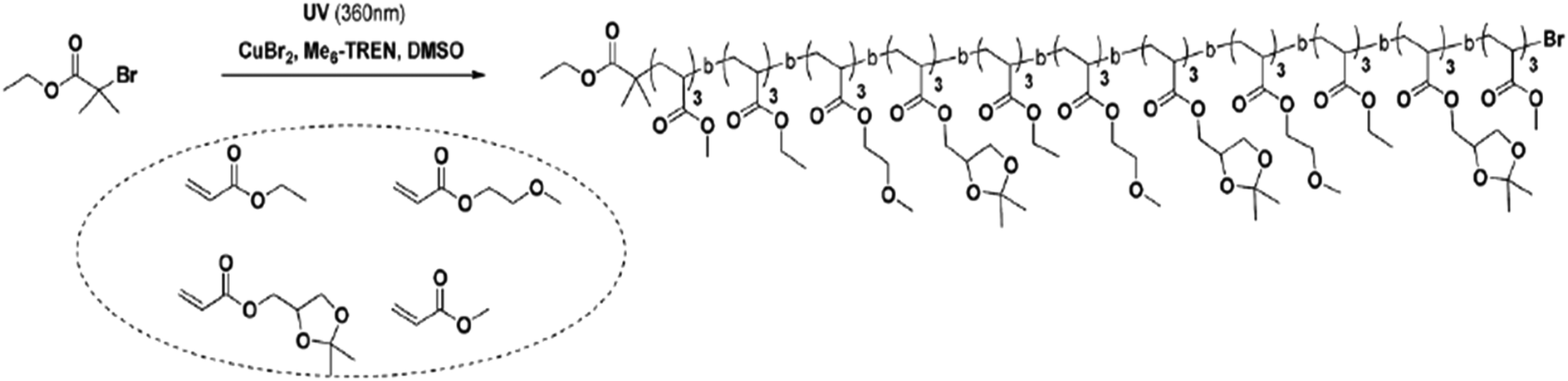 | ||
| Fig. 12 The reaction scheme for one-pot photo-induced living polymerization, wherein the polymer blocks are added to initial bromide. Reproduced from ref. 39 with permission from The Royal Society of Chemistry.39 | ||
Although photo-induced polymerization allows for greater sequence control than the “ultra-precise” N-substituted maleimides insertions, there are limitations to the number of block additions when using high molecular weight polyacrylate blocks. For example, Anastasaki et al. reported generating only hexablock copolymers from 100-mer polyacrylate blocks while producing undecablock copolymers from 3-mer polyacrylate copolymers.39
Single-electron transfer living radical polymerization (SET-LRP) can also be used to create sequence controlled multi-block copolymers.41 The SET-LRP method is performed in water at 0 °C, with the reaction performed at low temperature to prevent bromide hydrolysis. In this process, disproportionation of Cu (Me6TREN) Br generates the catalyst system in situ, and unprecedented reaction rates and control are achieved (Fig. 13). The method does suffer from early terminations and the potential for a loss of ω-Br chain ends. Due to the use of aqueous media, this method has the possibility of being compatible with biological systems.
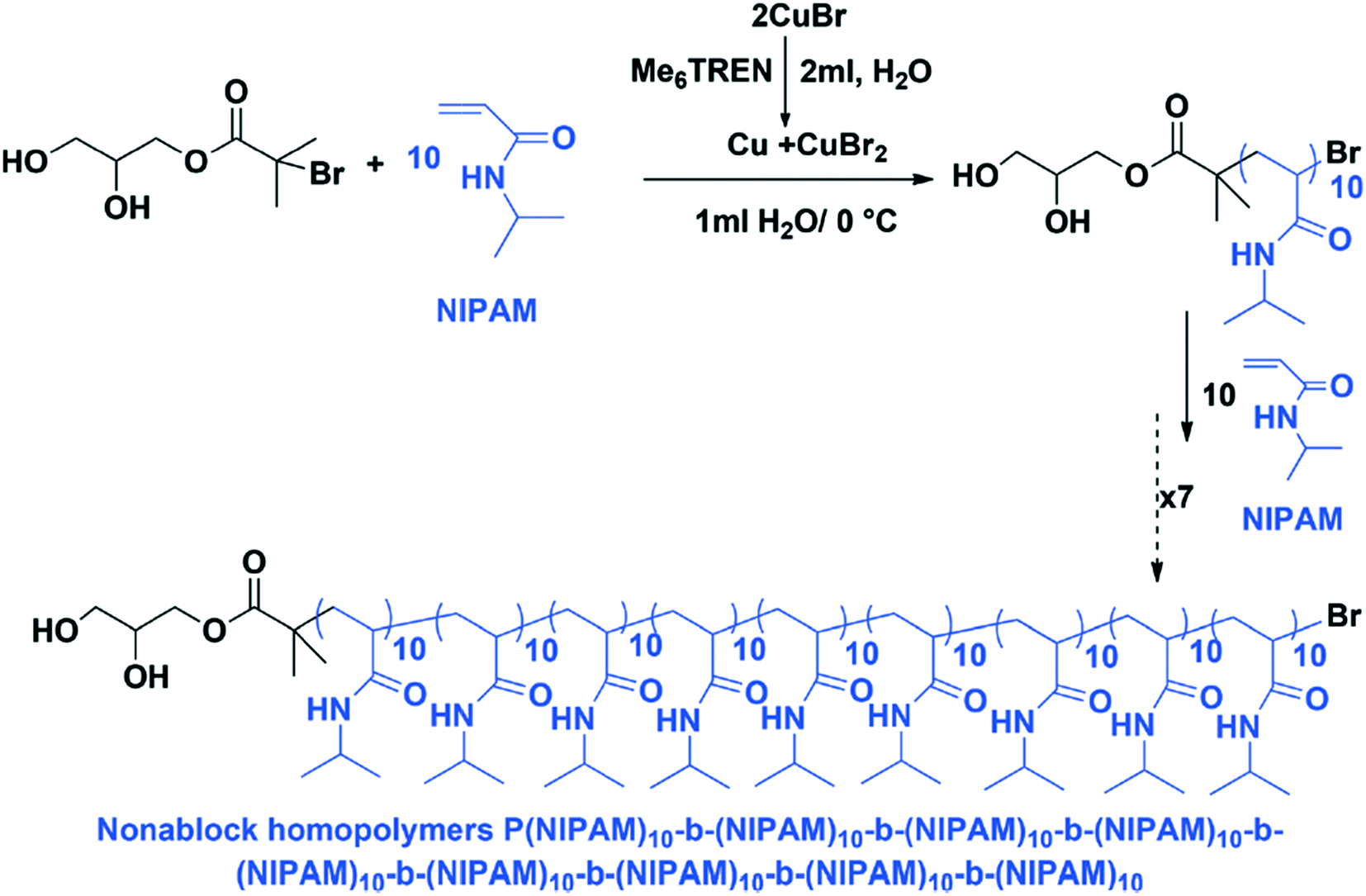 | ||
| Fig. 13 A reaction scheme for SET-LRP at 0 °C. Reproduced from ref. 41 with permission from The Royal Society of Chemistry.41 | ||
Sequence control of synthetic polymers remains a challenge, but the chemistries described herein constitute just a few examples of the advances that have been made. Templated synthesis and iterative chemistries achieve sequence control at the expense of throughput and speed. These chemistries might be more suited for applications in materials science where precision supersedes the need for fast-paced development. On the other hand, applications such as information storage necessitate high speed and low costs, but errors can be tolerated. The next sections explore some applications of sequence-controlled polymers.
Applications and adaptations
A natural extension of generating sequence controlled polymers is to use them in similar applications as the canonical sequence-controlled polymer, DNA. In an era where significant efforts are underway to use DNA for data storage,3,42–44 the possibility of using synthetic polymers is tantalizing. Lutz and coworkers have coined “binary coded oligomers,” which have repeating functional groups that are marked by chemical tags as either a 1 byte or a 0 byte.45 To generate the oligomers, iterative additions are carried out using phosphoramidite chemistry, CuAAc, or anhydride-amine/nitroxide radical couplings (Fig. 14).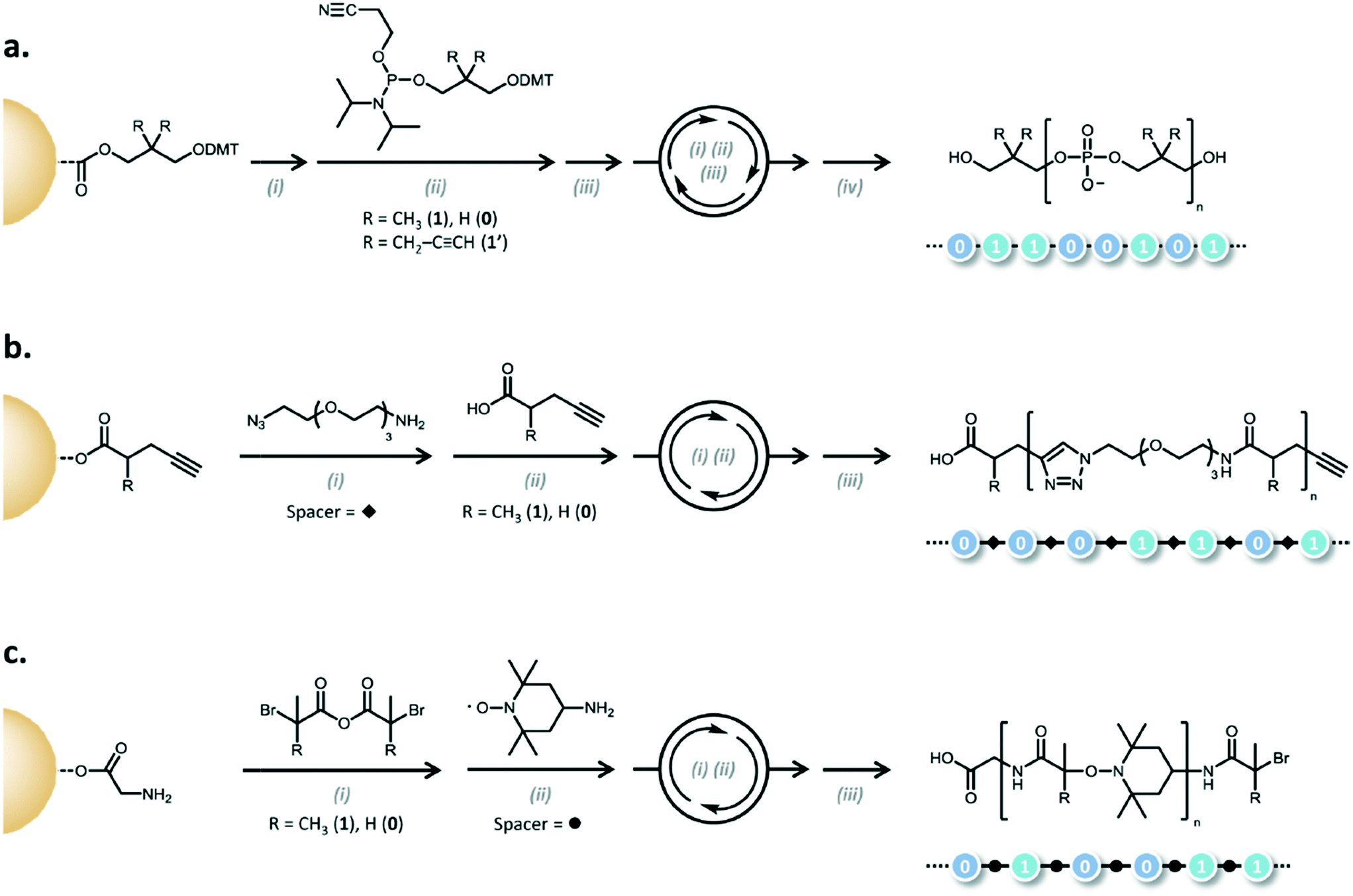 | ||
| Fig. 14 Examples of the different mechanisms for binary coding. a) Phosphoramidite (i) DMT deprotection: CCl3–COOH, CH2Cl2; (ii) coupling step: RT, MeCN, tetrazole; (iii) oxidation: RT, I2, H2O/pyridine/THF; (iv) cyanoethyl deprotection and cleavage: piperidine, MeCN then NH3, H2O, dioxane. b) AB/CD spacer (carboxylic acid/alkyne) c) AB/CD spacer (anhydride/alkyl bromide) (i) anhydride–amine coupling: THF, i-Pr2EtN or K2CO3; (ii) radical–radical coupling: CuBr, Me6TREN, DMSO; (iii) cleavage: TFA,CH2Cl2). Reprinted with permission from ref. 45. Copyright 2015 ACS Publications https://pubs.acs.org/doi/abs/10.1021%2Facs.macromol.5b00890. Further permissions related to use of this material should be directed to ACS publications.45 | ||
The phosphoramidite chemistry exploits the same methods used in oligonucleotide synthesis to generate non-natural polymers (Fig. 5). In the case of the examples shown in Fig. 14, bifunctional monomers undergo iterative additions where so-called AB blocks bearing functional tags are connected by CD spacers (in the case of CuAAc and anhydride-amine/nitroxide radical couplings).45
An AB block is composed of either carboxylic acid (A) and alkyne (B) moieties or acyclic symmetric anhydride acid (A) and alkyl bromides (B).45 The CD building block is typically composed of azide moieties (C) and primary amines (D) for the carboxylic acid alkyne pair or a nitroxide (C) and a primary amine (D). A spacer is used to couple between two AB blocks and serves no part in the actual coding. The coding is done primarily through the R group tag on the AB blocks.
The phosphoramidite coupling method can be automated using DNA synthesizers; this capability has been demonstrated by the generation of polymers with a degree of polymerization (DP) above 100.46 Conversely, the CuAAc and anhydride-amine/nitroxide chemistries have not been translated to automated processes. The time it takes to couple each monomer to a growing chain greatly reduces the output, as it would take roughly 15 coupling steps to create an oligomer with a byte worth of information.47
In an attempt to streamline these processes, a library of dyads was created to reduce the number of coupling steps from 15 to 7 to aid in faster synthesis of AB/CD binary oligomers.47 Once more processes are optimized and automated, this technology can be explored further.
In addition to the chemistries developed by Lutz with the explicit intent of information storage, several methods detailed herein could have merit in this field. For example, the DNA-templated Wittig chemistry developed by O'Reilly and coworkers10 has an explicit end and beginning monomer. These well-defined monomers could be set as terminal coding regions. If an appropriate translation method were developed, the oligomers could be used for storing information, where retrieval (“reading”) would rely on recognition of these elements.
Hierarchical assembly of organic nanomaterials and structural control of nanocomposites
Using polymeric materials as precursors or building blocks for hierarchical, structured materials is an exciting challenge. Because DNA is readily available, highly programmable, and well-understood, complex DNA origami structures have been constructed.48 Advances in the understanding of DNA origami have been a boon to researchers in many fields; for example, immobilized DNA origami can be used for nanometer-scale spatial control of ligand arrays. Generation of nanoscale DNA “boxes” that selectively open and shut have applications in drug delivery and selective release of enzymes.48Although synthetic polymers have not been fabricated into the complex 3-D structures achieved using DNA origami, some synthetic polymers have been assembled into two-dimensional nanosheets.49 A key example is the use of peptoids, which are peptidomimetic polymers consisting of repeated N-substituted glycine monomer units, where the side chains are appended to the nitrogen atoms instead of the α-carbon.
Peptoids are synthesized with excellent sequence-control using SPPS, and they are engineered to have hydrophobic and polar components placed at designated areas with precision; this engineered control enables peptoids to be arranged into nanosheets. The growing understanding of these materials makes them amenable to applications such as biomolecular recognition, catalysis, and templating the growth of organic or inorganic structures. For example, Zuckermann and coworkers have demonstrated use of peptoid nanosheets for mineralization.49
Using peptoids for construction of nanosheets is one example where sequence-defined polymers are engineered for a particular application. The possible structures, however, are still limited to structures rising from polymers generated through traditional SPPS means. The IEG and IEG+ work demonstrated by Johnson and co-workers may also lend itself well to applications where purposeful introduction of functional side-chains could dictate 2-D (or potentially 3-D) organic nanomaterial formations.30,32
Organic polymers can also control assembly of inorganic nanocomposites—an example being the extensive work in the field of DNA-mediated assembly of inorganic (mainly gold) nanoparticles.5 Controlled assembly of nanomaterials has myriad applications, one being the generation of plasmonic nanocomposites for optoelectronics and sensing applications.50 While assembly of nanocomposites is well-understood using DNA, there are limitations including reagent/solvent compatibility and long-term stability.
To address these limitations, nanocomposite tectons—inorganic nanoparticles functionalized with polymer chains that have molecular recognition elements at the termini—have been developed by Macfarlane and co-workers.51 The nanocomposite tectons can be assembled into ordered bcc superlattices. This work presents an intriguing application for sequence controlled polymers; one could envision tuning the physical properties of the polymer backbone or side-chains to mediate nanocomposite assemblies.
In the context of nanomaterial assemblies, the IEG+ work could be of interest for structure–function studies of how side-chain modifications affect nanocomposite structure. Alternatively, work involving placement of maleimides along a polystyrene backbone,35,38 could find application in this field, especially because exact sequence may not be as important as the order or frequency of side-chain modifications.
Photo-induced polymerization
Combining the speed of living polymerization with the control of iterative additions would be a huge boon to controlled polymer synthesis. To introduce control to living polymerization, selective actuation of catalysts has been explored, namely through photo-activation. A typical photo-induced method uses UV light to initiate the reaction; removing the light source stops reaction.39 Using different wavelengths to activate chain growth at different times could be used for developing distinctive structures and formations.52One such example is a method using single unit monomer addition (SUMI) via photoinduced electron/energy transfer-reversible addition-fragmentation chain transfer (PET-RAFT).52 Selective activation of different RAFT agents by photoredox catalysts stimulates synthesis depending on the color of light used. For example, the RAFT agent 4-cyanopentanoic acid dithiobenzoate (CPADB) is activated by Pheophorbide a (PheoA) using red light. Meanwhile, 2-(n-butyltrithiocarbonate) propanoic acid (BTPA) is activated by Zinc tetraphenylporphine (ZnTPP) using green light (Fig. 15).
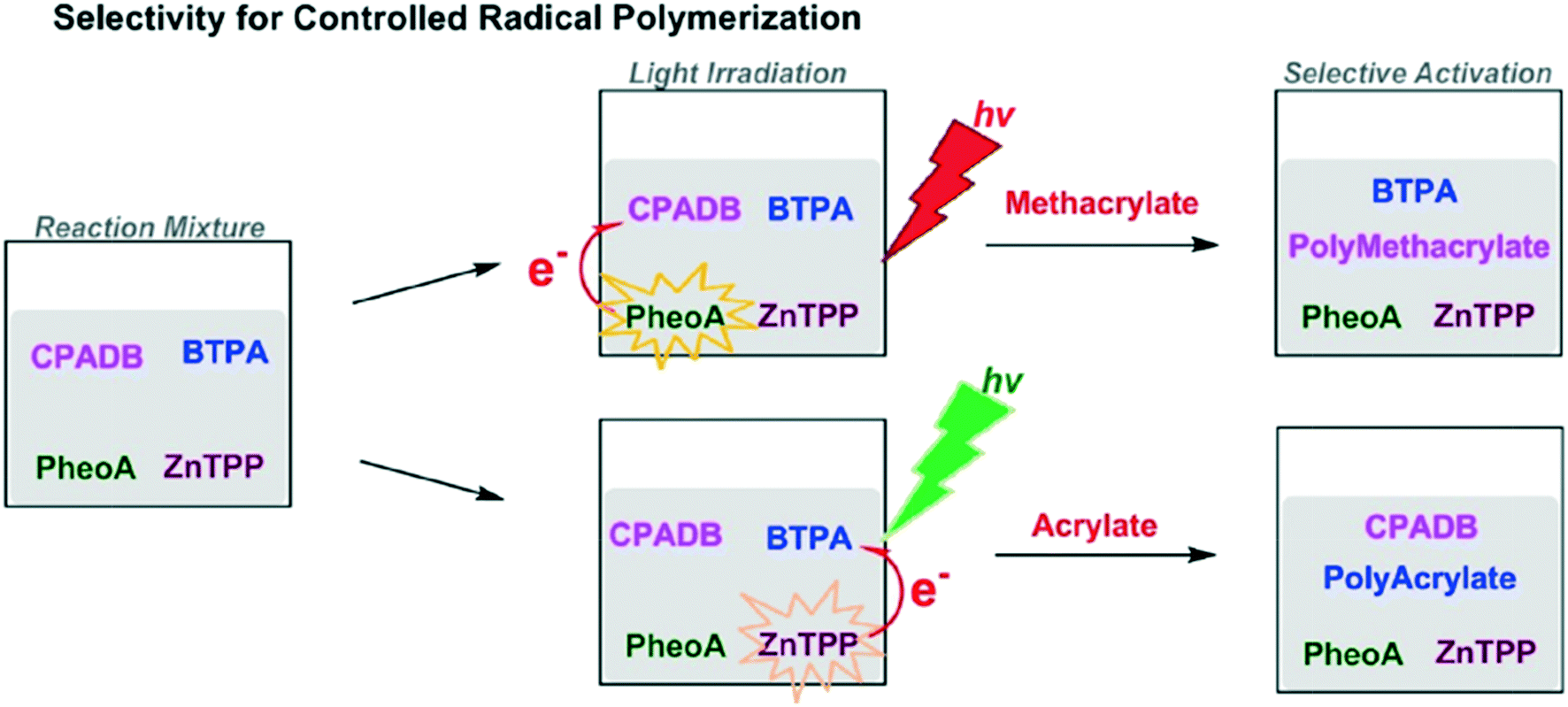 | ||
| Fig. 15 The selective activation of RAFT agents depends on light. Reprinted with permission from ref. 52. Copyright 2016 ACS Publications.52 | ||
This selective activation allowed for the generation of complex graft copolymers.52 Using red light produced a methacrylate chain whereas the green light activation generates a polyacrylate side chain graft. The complex structure of the copolymers generated could potentially be used in conjunction with functional tags to produce encoded structures for information storage or to generate polymers optimized for 2-D and 3-D nanomaterial assembly.
Another photoinduced method involves using blue light to activate an iridium catalyst to perform PET-RAFT (Fig. 16).53 Acrylate and acrylamide monomers alongside vinyl acetate, vinyl pivalate (VP), among others, are polymerized with high control of the molecular weight (MW) and polydispersity index (PDI). This method could be orthogonal to the processes catalyzed by PheoA and ZnTPP as only blue light activates the synthesis; use of green to red light (500–700 nm) resulted in no polymerization.
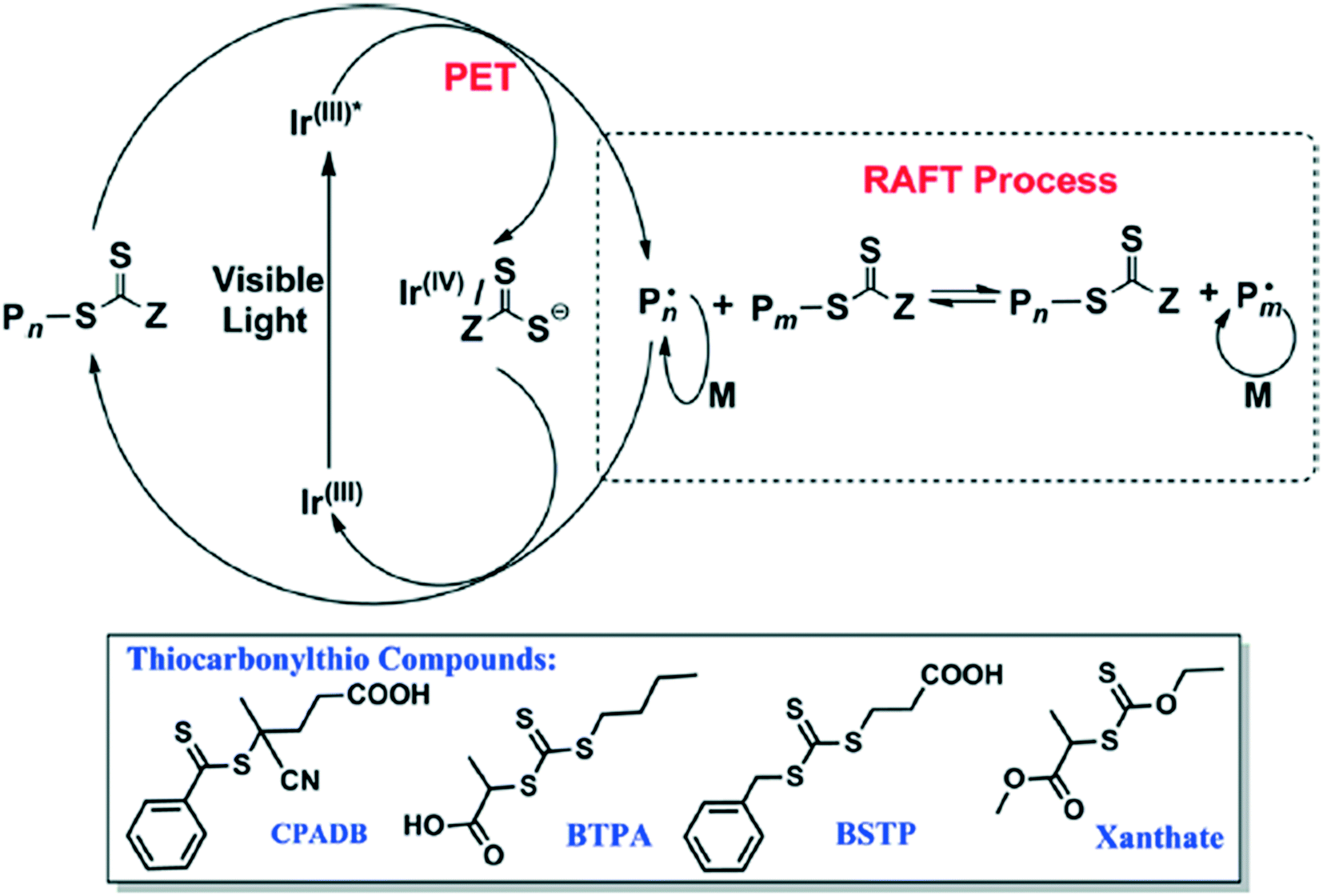 | ||
| Fig. 16 The reaction scheme for the blue light induced polymerization along with the RAFT agents used. Reprinted with permission from ref. 53. Copyright 2014 ACS Publications.53 | ||
The blue-light mediated PET RAFT process can occur at low catalyst concentration and low wattage.53 Using ultra-low catalyst concentration (1 ppm) resulted in an increase in reaction time while still maintaining a large degree of synthetic control. Low wattage (1–2.4 W) resulted in an increase in the inhibition period before the synthesis and a lower polymerization rate. Despite this drawback, the lower wattage prevented side reactions and degradation of the polymer while still maintaining the control over the MW and PDI.
Use of light is a promising means for controlling polymer growth. Approaches where light sources (e.g., LEDs) are integrated with automated or microfluidic platforms could be used to generate controlled polymers in an efficient manner. Careful consideration to the photolytic stability of the growing polymer chains and lifetime of the photoredox catalysts must be taken into account. One advantage of using light obviates the need for continuous washing/introduction of new reagents in these types of processes.
Perspective on methods and applications
This review highlights methods toward achieving sequence-controlled polymers with a focus on developing applications such as information storage or generation of organic nanomaterials and nanocomposite structures. If an exacting, precise sequence is needed, the iterative methods relying on controlled additions/deprotections (akin to SPPS) should be used. The work disclosed by Johnson and coworkers on IEG+ involves synthesis of reasonably long polymers with the ability to incorporate a diverse range of side chains—this method could be used to engineer structured materials (such as 2-D or 3-D organic nanomaterials) with engineered properties; of particular interest is the ability to incorporate chirality into the materials.In applications where the order of components is important but not necessarily the spacing, controlled living polymerizations can be used. A good example would be the “ultra-precise” addition of maleimides to a polystyrene backbone. Photoinduced polymerizations (where sequence control is dictated by photo-activated catalysts) and the SET-LRP methods would also lend themselves well to such applications.
Lastly, templated synthesis has applications including rapid library screening and the ability to precisely place monomers for reactions, limiting the need for protecting groups. The disadvantage of DTS is in the nature of the approach itself—the requirement of a template.
One outstanding goal might be an oligomer synthesis that has the precision of either iterated, protecting-group mediated or DNA-templated approaches without the need for a template or a protecting group. The photoinduced method, where monomer placement is controlled by light,52,53 or the thermodynamically controlled additions of maleimides into polystyrene backbones38 may be the closest methods toward achieving this goal.
Analysis and reading methods
Peptide, peptoids, and other sequence controlled oligomers can be sequenced—a key property exploited when using these compounds to store information. Mass spectral based methods for sequencing peptides and oligomers can be applied to non-natural oligomers and polymers. For example, an open access decoding program was recently released which uses tandem MS with an ESI-MS device.54The tandem MS/MS method decodes binary coded polymers in milliseconds.54 It uses an electrospray ionization mass spectrometer (ESI-MS) to read polyurethane, poly (alkoxyamine phosphodiester), and poly (alkoxyamine amide) type polymers. By utilizing the weak O–(CO) or C–ON bonds, the polymer is divided into easily identifiable segments whose molar mass can be classified into the 0 and 1 byte necessary for the binary code, illustrated in Fig. 17.
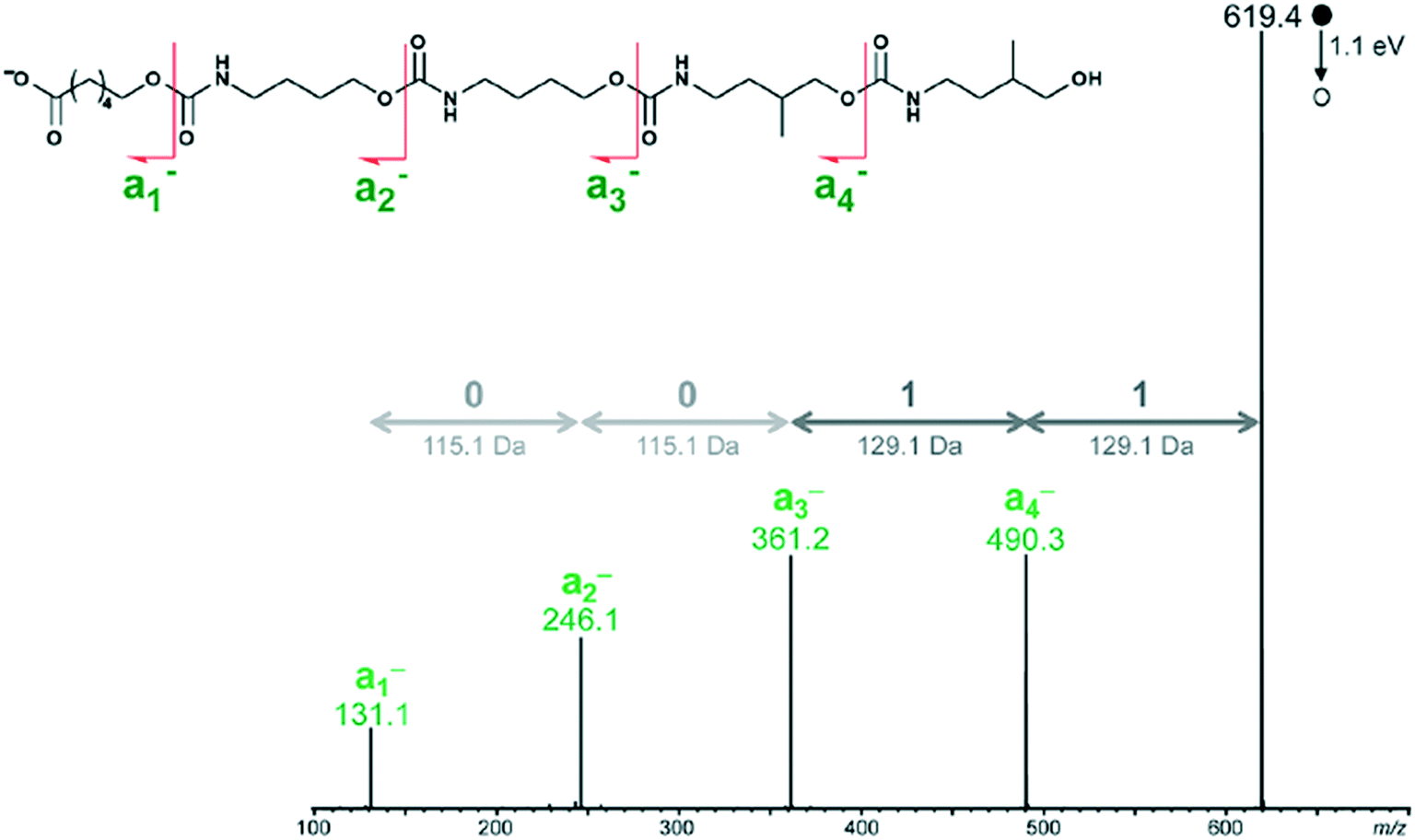 | ||
| Fig. 17 How the tandem MS decoder reads polymer chains. Reprinted with permission from ref. 54. Copyright 2017 ACS Publications.54 | ||
The program then automates the decoding process by using the relative molar mass of the block to determine whether it has a 0 byte or 1 byte tag. The decoding process finishes within milliseconds of starting. Mass spectroscopy is one method to read polymer codes, but the system necessitates design of polymers with distinctive fragmentation patterns. Another tool that could be used for polymer reading is nanopore sequencing—a technique developed for sequencing nucleotides.
Nanopores utilize changes in current across a membrane to detect different nucleotides.55 Different nucleotides create different resistances in the electrical current, allowing for identification of the nucleotide crossing the membrane. This technique has also been demonstrated with metal ions and small molecules alongside proteins and nucleotides.55
Use of the nanopore method does require that each monomer or small molecule be characterized, but suitable polymers could be characterized and used with a nanopore for synthetic polymer sequencing. Although identification of synthetic polymers has not been the focus of nanopore research, it is conceivable that the principles could be applied to such an endeavor.
Limitations in reading homopolymers with accuracy does pose a challenge when translating nanopore technology to synthetic polymer sequencing. The N-maleimide homopolymer method, for instance, would be incompatible with the nanopore reading technique. This is due in part to the long homopolymer producing a constant potential difference for the duration of its time in the nanopore.55 This long segment would interfere with the nanopore's ability to differentiate between the different small molecules.
In the case of other sequence-defined polymers, traditional analytical methods are used. Table 1 shows the variety analytical tools of each method. Most methods that have the potential to store information in their chains currently lack a means to read the information stored within.
| Chemistry | Primary analytical methods | Secondary analytical methods | Yield |
|---|---|---|---|
| MIDA Boronates | H NMR | X-ray | 49–96% |
| IEG+ | GPC | SAXS | 89% |
| IEG/iterative additions | MALDI-ToF | H NMR, SEC | 68% < x ≤ 87% |
| Binary coding | ESI-MS/MS | H NMR, C NMR, MALDI-ToF-MS, HPLC | 87–98% |
| N-Substituted maleimides | SEC | H NMR | N/A |
| Photo-induced | H NMR | HR-ESI_MS, FT-IR | 1% due to purification |
| Wittig reagent | LC-MS | HPLC | Up to 80% |
Limitations of known methods
A general problem in generation of sequence-defined polymers is the lack of automation. The speed of the synthesis in the binary coded method is slow due to manually cross-coupling each of the monomer units.46 Until the process is automated or adapted to do so, it will be time consuming to produce lengthy chains.Methods that generate long oligomers/polymers within a suitable time-frame often suffer from lack of precision, as is the case for the ultra precise insertion method.35 Although a long homopolymer with functional monomers inserted at specific points has been demonstrated, there is a limit to how precise the insertions can be. As discussed by Harrisson et al., the distribution of the functional monomer can be modeled using a Poisson or beta distribution.37 One problem is that side reactions can occur, resulting in terminations and chain transfers that upset the distribution. More importantly, even under ideal conditions, the resulting polymer will lack the precision necessary to control monomer distribution. By extension, such polymers cannot be used to store information properly. An example given is the precise placement of a monomer B in position 11 would only occur in 12.5% of the chains created, whereas its placement before 8 or after 14 occurs in 26.6% of the chains (Fig. 18).37
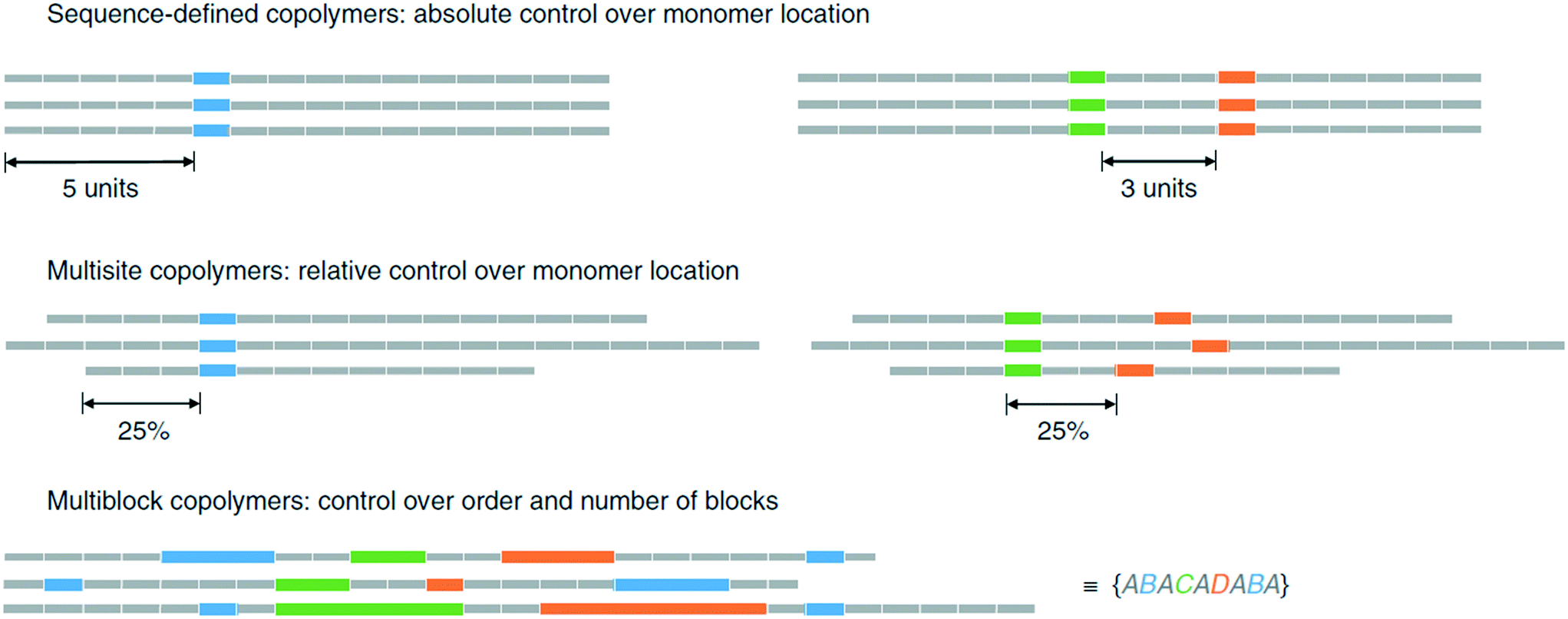 | ||
| Fig. 18 Visual explaination of polymer chain distributions, reprinted from ref. 37. | ||
The advantage of controlled iterative methods is the ability to regulate the monomer addition. The precision comes at the cost of requiring multiple steps, which will lead to diminishing yields, limiting the lengths that can be achieved. For example, SPPS is typically limited to 50 amino acid-length peptides; oligonucleotides can be scaled to lengths of ca. 100 nucleotides.56,57
One possible improvement to all methods presented is the incorporation of molecular evolution.58 Molecular evolution enables one to sample a large chemical space without requiring all possible molecules to be synthesized prior to sampling. Repeated translation and selection are achieved from an initial set of instructions. Each repeated selection is amplified to be improved from its previous iteration until an evolved product is produced with optimized properties, circumventing investigation of each possible sample.
Fig. 19 shows how each iteration would work, starting with the initial iteration, which is produced from a library of DNA templated instructions. After a set of enriched products is selected from the initial batch, they are amplified into a new set of instructions; the cycle is then repeated several times until a final optimized product is produced.
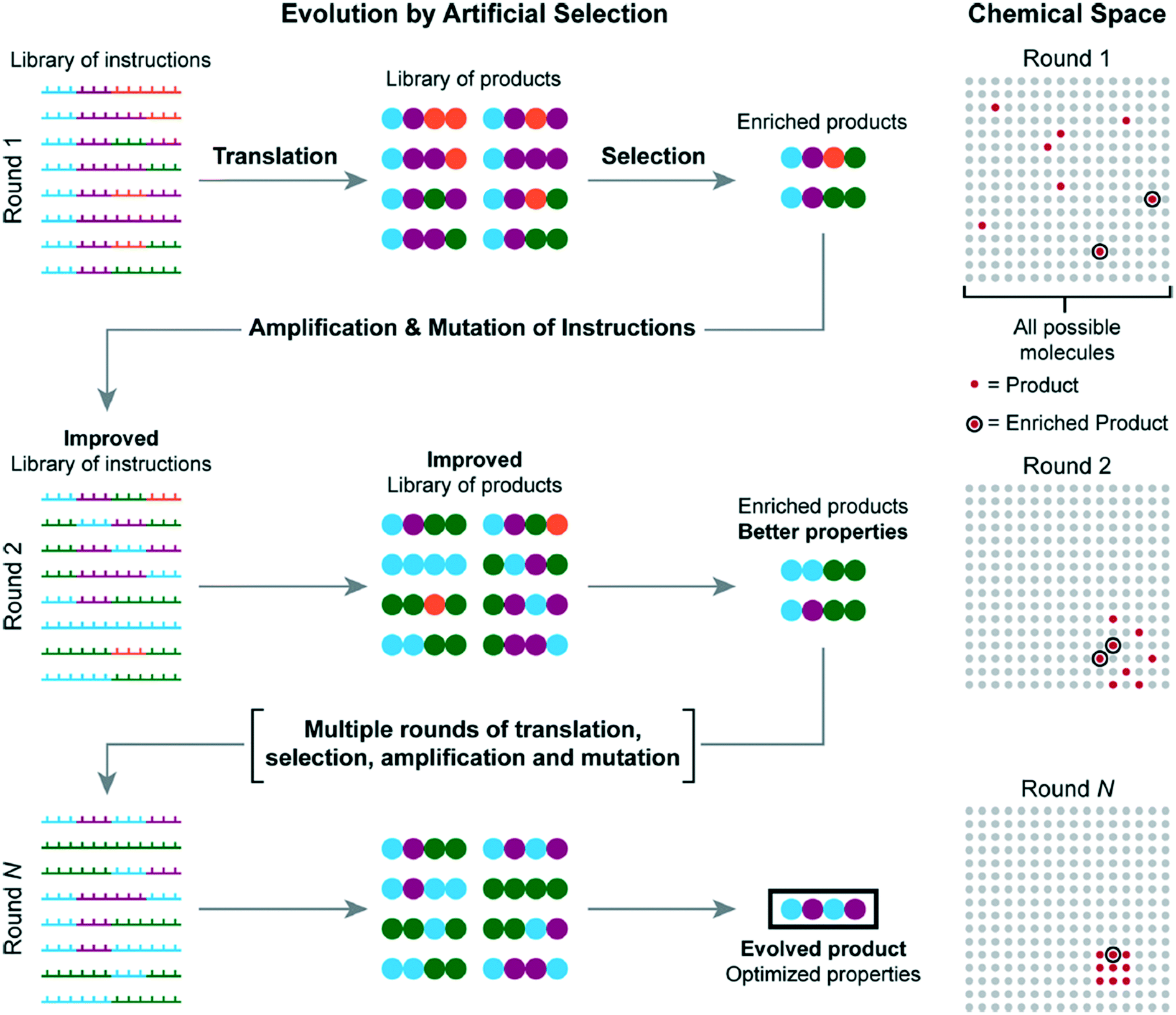 | ||
| Fig. 19 A visual explanation of molecular evolution. Reprinted with permission from ref. 16. Copyright 2017 ACS publications: https://pubs.acs.org/doi/abs/10.1021%2Facs.accounts.7b00280 further permission related to use of this material should be directed to ACS publications.16 | ||
Where to go from here
Key challenges in forming sequence-controlled polymers include speed and precision. Some applications value precision over rapid polymer generation. For example, designing new materials with a desired function may require a great deal of rigor with ample time to produce the desired specimen. For these applications, iterative methods will suffice as long as the required lengths do not exceed those feasible for the chosen chemistry.For applications of data storage, speed supersedes precision because information storage applications can tolerate error rates. For example, writing a terabyte/day would require a write speed of 1.2 × 107 byte per s. Although it may seem simple to envision installing a code into polymers generated through iterative approaches (such as the “1, 0” substituents employed by Lutz), the translation of methods to a feasible information writing approach will necessitate chemistries that occur at higher speeds. Automation of the approaches would also be a necessary advancement. Using DNA synthesizers and adapting them to work with synthetic polymers has shown a large increase in the number of monomers one can add in the same amount of time as manual methods.46
In addition to this automation, cleavable indicators that signal when each monomer is added would help improve polymer precision. For example, one could envision adapting the MIDA boronate chemistry with CuAAc to have a fluorescent tag on the protecting group. Notional cases are depicted in Fig. 20.59 In one instance, the N-methyl group of the MIDA ligand is replaced with an alkyl linker/fluorophore.
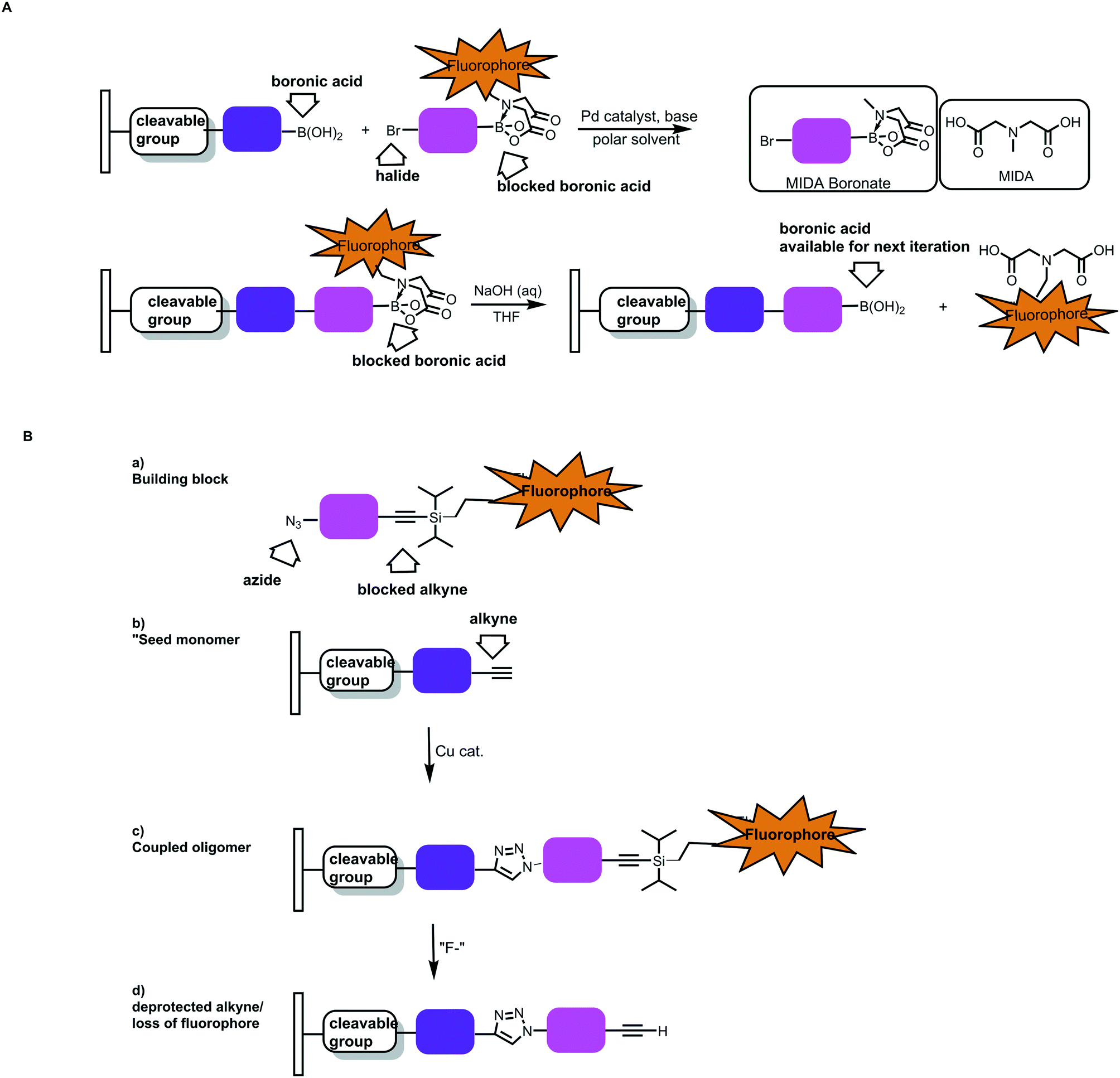 | ||
| Fig. 20 Notional cases for building arrays using sequential, optically verified chemistries. A) Sequential Suzuki–Miyaura couplings. A starting boronic acid would be immobilized in a reversible fashion (via a cleavable linker). Variants of the MIDA boronates wherein the boronic acid protecting group contains a fluorophore would be used. B) Sequential CuAAC chemistries wherein bifunctional molecules containing an azide and a protected alkyne are used. If the alkyne protecting group bears a fluorophore, the monomer incorporation events could be monitored optically.59 | ||
Alternatively, CuAAc chemistry could be used if fluorescent silyl groups were used to cap terminal alkynes. As the polymer grows from an immobilized platform, the incorporation of the monomer unit could be verified optically. The deprotection step could also be monitored. One could envision performing the syntheses in array format and selecting for populations where the length/incorporation events were monitored throughout the process.
Chain growth polymerization methods would be more challenging to improve as the main limitations reside in statistical distributions of the monomer insertions. If the concept could be adapted into a more precisely controlled chemistry, it would be possible to use a library of functionalized monomers to write a translatable code similar to DNA. The thermo-responsive furan-protected maleimide method shows promise in developing a chain growth methodology with higher precision.38
Finally, an alternative way to improve the precision of chain growth polymerizations could be to perform the synthesis in a microfluidic platform, which can operate with a high switching speed.8 In such a platform, the concentration of monomer could be introduced and flushed out at rates that could overcome the current obstacles of statistical distributions. As with the other methods, a means to read the code should also be created. If the method has similar weak O–(CO) bonds as binary coded polymers do, it may be as simple as adapting the Tandem MS program.54
Precision polymerization offers exciting possibilities for applications in materials science and information storage. A wide array of techniques and building blocks exist, meaning that one can match a precision polymerization method with a particular application. Key advances may include enhancing the processes through automation and in situ verification methods. Moreover, streamlining characterization processes and even “reading” newly synthesized polymers would be a boon to the field. Coupling robust polymer chemistries with microfluidic platforms or optical assays would be examples of ways to improve the viability of precision polymer uses.
The detailed understanding of DNA has led to advances well outside applications in the life sciences. Because of its programmable nature, DNA has found application in information storage and in materials science. If one could develop an understanding of synthetic polymers at the level to which we understand DNA, a wide array of possibilities could abound. Means to control polymer sequence are a crucial and key first step to attaining this level of knowledge.
Abbreviations
| Ac | Acetyl |
| Bn | Benzyl |
| DMSO | Dimethylsulfoxide |
| DMT | Dimethoxytrityl |
| LED | Light-emitting diode |
| PHEA | Poly(2-hydroxyethyl acrylate) |
| TBHP | tert-Butyl hydroperoxide |
| THF | Tetrahydrofuran |
| TFA | Trifluoroacetic acid |
| TREN | tris(2-Aminoethyl)amine |
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by DARPA (N66001-17-C-4034). The authors are thankful to Dr. A. Magyar (Draper) for helpful insights and to Dr. B. C. Hoefler (Draper) and Professor J. A. Johnson (MIT) for critical review of the manuscript.Notes and references
- V. Zhirnov, R. M. Zadegan, G. S. Sandhu, G. M. Church and W. L. Hughes, Nat. Mater., 2016, 15, 366–370 CrossRef CAS PubMed.
- S. L. Shipman, J. Nivala, J. D. Macklis and G. M. Church, Nature, 2017, 547, 345–349 CrossRef CAS PubMed.
- L. Organick, S. D. Ang, Y.-J. Chen, R. Lopez, S. Yekhanin, K. Makarychev, M. Z. Racz, G. Kamath, P. Gopalan, B. Nguyen, C. N. Takahashi, S. Newman, H.-Y. Parker, C. Rashtchian, K. Stewart, G. Gupta, R. Carlson, J. Mulligan, D. Carmean, G. Seelig, L. Ceze and K. Strauss, Nat. Biotechnol., 2018, 36, 242–248 CrossRef CAS PubMed.
- A. Gopinath, E. Miyazono, A. Faraon and P. W. K. Rothemund, Nature, 2016, 535, 401 CrossRef CAS PubMed.
- M. R. Jones, N. C. Seeman and C. A. Mirkin, Science, 2015, 347, 840 CrossRef CAS PubMed.
- N. A. Carter and T. Z. Grove, J. Am. Chem. Soc., 2018, 140, 7144–7151 CrossRef CAS PubMed.
- Q. Luo, T. Pan, Y. Liu and J. Liu, in Advances in Bioinspired and Biomedical Materials Volume 1, American Chemical Society, 2017, ch. 7, vol. 1252, pp. 129–148 Search PubMed.
- P. M. Valencia, E. M. Pridgen, M. Rhee, R. Langer, O. C. Farokhzad and R. Karnik, ACS Nano, 2013, 7, 10671–10680 CrossRef CAS PubMed.
- J. De Neve, J. J. Haven, L. Maes and T. Junkers, Polym. Chem., 2018, 9, 4692–4705 RSC.
- P. J. Milnes, M. L. McKee, J. Bath, L. Song, E. Stulz, A. J. Turberfield and R. K. O'Reilly, Chem. Commun., 2012, 48, 5614–5616 RSC.
- D. L. Usanov, A. I. Chan, J. P. Maianti and D. R. Liu, Nat. Chem., 2018, 10, 704–714 CrossRef CAS PubMed.
- J. Niu, R. Hili and D. R. Liu, Nat. Chem., 2013, 5, 282–292 CrossRef CAS PubMed.
- R. E. Kleiner, Y. Brudno, M. E. Birnbaum and D. R. Liu, J. Am. Chem. Soc., 2008, 130, 4646–4659 CrossRef CAS PubMed.
- R. Hili, J. Niu and D. R. Liu, J. Am. Chem. Soc., 2013, 135, 98–101 CrossRef CAS PubMed.
- Y. He and D. R. Liu, Nat. Nanotechnol., 2010, 5, 778–782 CrossRef CAS PubMed.
- R. K. O'Reilly, A. J. Turberfield and T. R. Wilks, Acc. Chem. Res., 2017, 50, 2496–2509 CrossRef PubMed.
- M. Garcia, K. Kempe, D. M. Haddleton, A. Khan and A. Marsh, Polym. Chem., 2015, 6, 1944–1951 RSC.
- Y. Kang, A. Lu, A. Ellington, M. C. Jewett and R. K. O'Reilly, ACS Macro Lett., 2013, 2, 581–586 CrossRef CAS.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- J. Jin, T. L. Graybill, M. A. Wang, L. D. Davis and M. L. Moore, J. Comb. Chem., 2001, 3, 97–101 CrossRef CAS PubMed.
- aappTec, Equipment for Solid Phase Peptide Synthesis, https://www.peptide.com/equipment-solid-phase-peptide-synthesis-i-249.html, (accessed 6/6/18, 2018) Search PubMed.
- I. Coin, M. Beyermann and M. Bienert, Nat. Protoc., 2007, 2, 3247–3256 CrossRef CAS PubMed.
- C. B. Reese, Org. Biomol. Chem., 2005, 3, 3851–3868 RSC.
- Wikipedia, 2009, Synthetic cycle in preparation of oligonucleotides.
- J. Li, A. S. Grillo and M. D. Burke, Acc. Chem. Res., 2015, 48, 2297–2307 CrossRef CAS PubMed.
- D. M. Knapp, E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2009, 131, 6961–6963 CrossRef CAS PubMed.
- F. Amir, Z. Jia and M. J. Monteiro, J. Am. Chem. Soc., 2016, 138, 16600–16603 CrossRef CAS PubMed.
- L. Martin, G. Gody and S. Perrier, Polym. Chem., 2015, 6, 4875–4886 RSC.
- D. Oh, M. Ouchi, T. Nakanishi, H. Ono and M. Sawamoto, ACS Macro Lett., 2016, 5, 745–749 CrossRef CAS.
- Y. Jiang, M. R. Golder, H. V.-T. Nguyen, Y. Wang, M. Zhong, J. C. Barnes, D. J. C. Ehrlich and J. A. Johnson, J. Am. Chem. Soc., 2016, 138, 9369–9372 CrossRef CAS PubMed.
- S. Binauld, D. Damiron, L. A. Connal, C. J. Hawker and E. Drockenmuller, Macromol. Rapid Commun., 2011, 32, 147–168 CrossRef CAS PubMed.
- J. C. Barnes, D. J. C. Ehrlich, A. X. Gao, F. A. Leibfarth, Y. Jiang, E. Zhou, T. F. Jamison and J. A. Johnson, Nat. Chem., 2015, 7, 810–815 CrossRef CAS PubMed.
- F. A. Leibfarth, J. A. Johnson and T. F. Jamison, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10617–10622 CrossRef CAS PubMed.
- A. C. Wicker, F. A. Leibfarth and T. F. Jamison, Polym. Chem., 2017, 8, 5786–5794 RSC.
- M. Zamfir and J.-F. Lutz, Nat. Commun., 2012, 3, 1–8 RSC.
- N. Baradel, O. Shishkan, S. Srichan and J.-F. Lutz, ACS Symp. Ser., 2014, 1170, 119–131 CrossRef CAS.
- G. Gody, P. B. Zetterlund, S. B. Perrier and S. Harrisson, Nat. Commun., 2016, 7, 1–8 CrossRef PubMed.
- Y. Ji, L. Zhang, X. Gu, W. Zhang, N. Zhou, Z. Zhang and X. Zhu, Angew. Chem., Int. Ed., 2017, 56, 2328–2333 CrossRef CAS PubMed.
- A. Anastasaki, V. Nikolaou, G. S. Pappas, Q. Zhang, C. Wan, P. Wilson, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Sci., 2014, 5, 3536–3542 RSC.
- Y.-M. Chuang, A. Ethirajan and T. Junkers, ACS Macro Lett., 2014, 3, 732–737 CrossRef CAS.
- F. Alsubaie, A. Anastasaki, P. Wilson and D. M. Haddleton, Polym. Chem., 2015, 6, 406–417 RSC.
- E. Strickland, IEEE Spectrum, 2016, 53, 9–11 Search PubMed.
- N. Goldman, P. Bertone, S. Chen, C. Dessimoz, E. M. LeProust, B. Sipos and E. Birney, Nature, 2013, 494, 77–80 CrossRef CAS PubMed.
- R. Heckel, I. Shomorony, K. Ramchandran and D. N. C. Tse, Conference Proceeding from 2017 IEEE International Symposium on Information Theory (ISIT), 2017 Search PubMed.
- J.-F. Lutz, Macromolecules, 2015, 48, 4759–4767 CrossRef CAS.
- A. A. Ouahabi, M. Kotera, L. Charles and J.-F. Lutz, ACS Macro Lett., 2015, 4, 1077–1080 CrossRef.
- T. T. Trinh, L. Oswald, D. Chan-Seng, L. Charles and J.-F. Lutz, Chem. – Eur. J., 2015, 21, 11961–11965 CrossRef CAS PubMed.
- A. A. Arora and C. de Silva, Nano Rev. Exp., 2018, 9, 1430976 CrossRef PubMed.
- E. J. Robertson, A. Battigelli, C. Proulx, R. V. Mannige, T. K. Haxton, L. Yun, S. Whitelam and R. N. Zuckermann, Acc. Chem. Res., 2016, 49, 379–389 CrossRef CAS PubMed.
- S.-W. Hsu, A. L. Rodarte, M. Som, G. Arya and A. R. Tao, Chem. Rev., 2018, 118, 3100–3120 CrossRef CAS PubMed.
- J. Zhang, P. J. Santos, P. A. Gabrys, S. Lee, C. Liu and R. J. Macfarlane, J. Am. Chem. Soc., 2016, 138, 16228–16231 CrossRef CAS PubMed.
- J. Xu, S. Shanmugam, C. Fu, K.-F. Aguey-Zinsou and C. Boyer, J. Am. Chem. Soc., 2016, 138, 3094–3106 CrossRef CAS.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed.
- A. Burel, C. Carapito, J.-F. Lutz and L. Charles, Macromolecules, 2017, 50, 8290–8296 CrossRef CAS.
- W. Shi, A. K. Friedman and L. A. Baker, Anal. Chem., 2017, 89, 157–188 CrossRef CAS PubMed.
- L. Raibaut, N. Ollivier and O. Melnyk, Chem. Soc. Rev., 2012, 41, 7001–7015 RSC.
- R. Behrendt, P. White and J. Offer, J. Pept. Sci., 2016, 22, 4–27 CrossRef CAS PubMed.
- Y. Brudno and D. R. Liu, Chem. Biol., 2009, 16, 265–276 CrossRef CAS.
- A. P. Magyar and M. M. Sprachman, WO2018129328A1, 2018.
| This journal is © The Royal Society of Chemistry 2019 |