
Efforts in redesigning the antileukemic drug 6-thiopurine: decreasing toxic side effects while maintaining efficacy†
Arnaldo X.
Torres Hernandez‡
ab,
Chamitha J.
Weeramange‡
b,
Prathibha
Desman
b,
Anthony
Fatino
b,
Olivia
Haney
b and
Ryan J.
Rafferty
 *b
*b
aDepartment of Chemistry, Pontifical Catholic University of Puerto Rico, 2250 Boulevard Luis A. Ferré Aguayo, Suite 626, Ponce, PR 00717-0777, Puerto Rico
bDepartment of Chemistry, Kansas State University, 1212 Mid-Campus Drive North, Manhattan, KS 66506, USA. E-mail: rjraff@ksu.edu
First published on 20th December 2018
Abstract
6-Thiopurine (6TP) is a currently prescribed drug in the treatment of diseases ranging from Crohn's disease to acute lymphocytic leukemia. While its potent mode of action is through incorporation into DNA as a thiol mimic of deoxyguanosine, severe toxicities are associated with its administration which hinder the potential therapeutic application. We have previously reported in vitro that the oxidative metabolites of 6TP, specifically 6-thiouric acid (6TU, Ki 7 μM), are potent inhibitors of UDP-glucose dehydrogenase (UDPGDH), an enzyme that is responsible for the formation of UDP-glucuronic acid (UDPGA), an essential substrate that is used in detoxification processes in the liver. An in vivo investigation was undertaken to probe if 6TU inhibits UDPGDH in rat hepatocytes, and it was observed that 6TU does greatly suppress the conjugation of bilirubin with UDPGA. The failed excretion of bilirubin is linked to a majority of the reported toxicities associated with 6TP administration. Efforts were undertaken for the construction of 6TP analogs, substituted at the C8 position, to reduce inhibition of UDPGDH while retaining therapeutic efficacy. Three new 6TP analogs bearing a halogen (Br, Cl, and F) at the C8 position have been achieved over five-synthetic steps in overall yields of 16 to 32%. Each of these analogs were shown to have reduced inhibition towards UDPGDH, with Ki values of 192, 163, 215 μM, respectively. In addition, the bromine, chlorine, and fluorine analogs were shown to possess cytotoxicity towards the REH cell line (acute lymphocytic leukemia) having IC50 values of 9.54 μM (±0.97), 3.95 μM (±1.94), and 4.71 μM (±1.40), respectively. These three new 6TP analogs represent the first steps in the redesign of this potent anticancer agent into a better drug that possesses reduced toxic side effects while retaining therapeutic potency.
1. Introduction
6-Thiopurine (6TP, 1) has been a continuously prescribed therapeutic since its FDA approval in 1952, shortly after its discovery in 1950 by Gertrude Elion and George Hitchings at Burroughs Wellcome.1 This potent therapeutic serves as a treatment option for numerous diseases, such as, but not limited to: acute lymphocytic leukemia (ALL),2,3 non-Hodgkin's leukemia,4,5 Crohn's disease,6–8 and inflammatory bowel disease.9,10 6TP has no therapeutic properties itself, however, it is metabolized into a therapeutic form, a deoxyguanosine nucleotide mimic, commencing with the phosphoribosylation by hypoxanthine–guanine phosphoribosyltransferase (HGPRT) forming the nucleotide mimic 6-thioinosinic acid monophosphate (TIMP, 2). HGPRT is an enzyme that usually catalyzes phosphoribosylation of hypoxanthine (C6 hydroxyl) to inosinic acid (IMP).11 Oxidation of the C2 position of 6TP is accomplished by inosine-5′-monophosphate dehydrogenase (IMPD), an enzyme that is overexpressed in the presence of 6TP, followed by C2 amine installation by guanine monophosphate synthase (GMPS) giving rise to either thioguanosine mono phosphate (TGMP or dTGMP, 3).11–13 Multiple kinase transformations then afford dTGTP (4), which gets incorporated into DNA in place of dGTP. However, incorporation of dTGTP does not immediately result in apoptosis, via base pair mismatching, but requires multiple passages through the S-phase allowing for sufficient dTGTP incorporation into DNA.14 It has also been shown that 6-thioguanine (6TG, 5) can be used in conjunction with 6TP and is acted upon by HGPRT accessing the monophosophate nucleotide formed via 6TP metabolism.Checkpoint activation is required for recognition of the mismatched base pairing within DNA post 4 incorporation; a process commonly suppressed within cancer cells.15–17 Mutually 6TP and 6TG can be methylated to form the corresponding thiol ethers by thiopurine methyltransferase (TPMT), neither possessing any cytotoxic properties, but do induce checkpoint activation.18 Additionally, TIMP (2) can be transformed into its corresponding methyl thiol ether, but rather than activating checkpoints this species has been proven to inhibit de novo purine biosynthesis.19 This inhibition allows for the cellular uptake of purines to increase, including 6TP, which corresponds to a higher probability of dTGTP incorporation into DNA that can then lead to apoptosis, via base pair mismatching (Fig. 1).
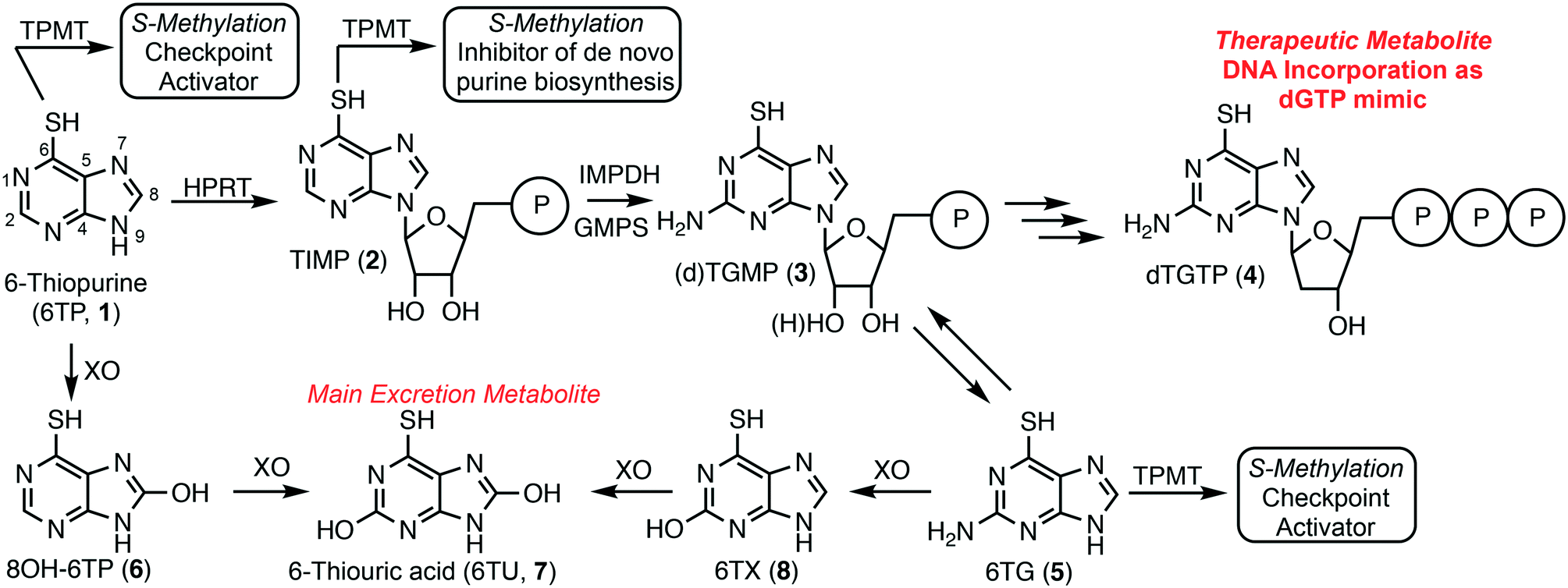 | ||
| Fig. 1 Metabolism of 6-thiopurine: 1) therapeutic pathway forming dTGTP, a dGTP mimic, which incorporates into DNA resulting in apoptosis, and 2) the excretion pathway allowing for the removal of 6-thiopurine and its biosynthetic metabolites. | ||
In parallel to 6TP's therapeutic metabolism, efficient oxidative pathways remove 6TP from the body, thereby decreasing its therapeutic potential. Xanthine oxidase (XO) can directly oxidize the C8 position of 6TP forming 8-OH-6TP (6), a species that is not a substrate for HGPRT, thus, eliminating any therapeutic potential.20 Moreover, 6 can be further oxidized at the C2 position forming 6-thiouric acid (6TU, 7), which is the terminal excretion metabolite. This highly efficient oxidation by XO limits the bioavailability of 6TP. In route to dTGTP (4), dTGMP (3) can be formed into 6TG, which is rapidly oxidized to 6-thioxanthine (6TX, 8), a species that is further oxidized by XO to 6TU. Once 6TU is formed, it is well retained by the body beyond 24 hours post-6TP administration.20
As with nearly all therapeutic drugs, the benefits come with side effects that limit effectiveness and general applications. 6TP is not immune to this trend, with multiple toxicities associated with its use including, but not limited to: pulmonary lesions, myocarditis, reticulocytopenia, depletion of cellularity in bone marrow, intestinal ulcerations, jaundice, hepatotoxicity, and even death.13,15 Of these, the most predominate toxic side effects are jaundice and hepatotoxicity, corresponding to the reported increase in bilirubin levels within patients taking 6TP. In addition to the toxicities listed above, increased bilirubin levels within the body can cause hyperbilirubinemia, kernicterus, Crigler–Najjar syndrome, Gilbert's syndrome, liver failure, and death.21–23 While 6TP is a currently prescribed anticancer agent, its toxic side effects are so potent and prevalent that the administration of 6TP is given in an on/off strategy, allowing time for the toxic species to be cleared by the body. Unfortunately, this greatly reduces its therapeutic efficacy, restricts the quality of life of the patients taking the drug, and has ultimately limited its use as an anticancer therapy. Our laboratory is interested in repurposing dismissed or under-utilized anticancer therapies, such as 6TP. Noting the common connection between the predominate toxic side effects possibly originating in the bilirubin pathway, this prompted us to begin a systematic study to explore the effects of 6TP and its oxidative metabolites.
The bilirubin pathway refers to the excretion of bilirubin from the body through detoxification in the liver.24–26 Heme groups are released when red blood cells undergo senescence, which in the presence of oxygen and NADPH, reduces the heme to bilirubin (11, also referred to as unconjugated bilirubin, UCB).24 Bilirubin is a relatively large non-polar compound that is not easily excreted by the body (Fig. 2).27 The excretion of bilirubin is achieved through conjugation by UDP-glucuronyl transferase (UGT-1A) with two UDP-glucuronic acids (UDPGA, 10) forming bilirubin diglucuronide (BDG, 12), a water-soluble and excretable compound.28,29 Physiologically, UDPGA (10) is formed from the oxidation of UDP-glucose (UDPG, 9) via UDP-glucose dehydrogenase (UDPGDH).30 We have reported that 6TP and its excretion/oxidative metabolites inhibit UDPGDH, but not UGT-1A (Fig. 2).31 Furthermore, in vitro investigations revealed that 6TP and its excretion metabolites resulted in an increase in bilirubin and a decrease in conjugated bilirubin levels, relative to controls. While 6TP was shown to have low inhibition towards UDPGDH, its main excretion metabolite 6TU has a 41-fold increase in inhibition. Noting that the formation of 6TU via XO is rapid, we were led to investigate the other oxidative metabolites. Investigations of 6-thioxanthine (8, C2 hydroxyl) revealed a five-fold increase in inhibition towards UDPGDH and 8-OH-6TP a 20-fold increase in inhibition, relative to 6TP. From this, it is suggested that the C8 position is critical in the inhibition properties towards UDPGDH.31 As such, we propose that analogs that block the C8 position of 6TP, thereby preventing oxidation by XO to the C8–OH, will limit, if not eliminate, inhibition of UDPGDH. We speculate that the reduced inhibition of UDPGDH will result in a higher concentration of the UDPGA liable pool to allow for conjugation of bilirubin and its excretion, thereby reducing one of the predominate toxic side-effects of 6TP administration.
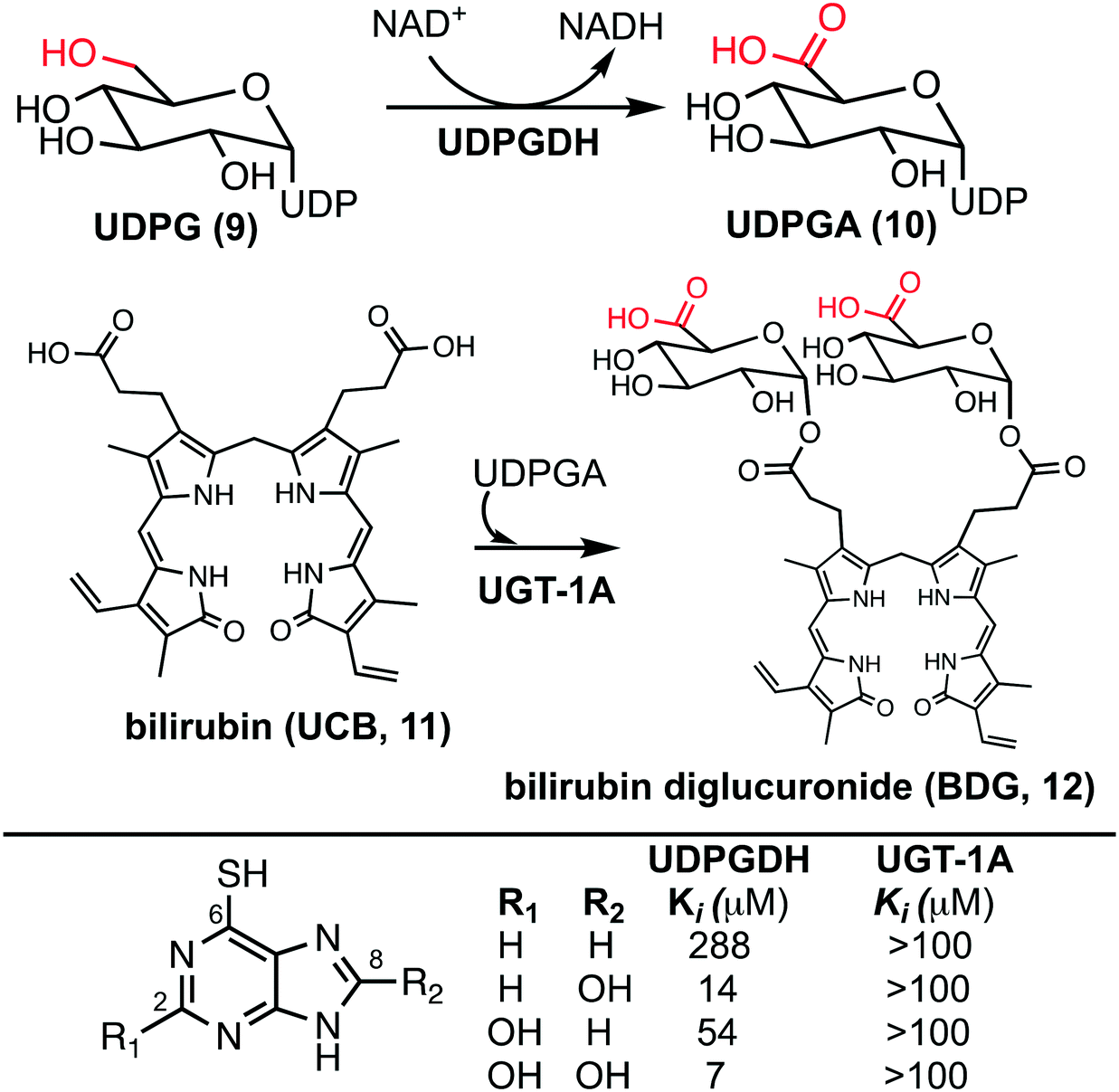 | ||
| Fig. 2 (Upper) Biosynthesis of UDPGA from the oxidation of UDPG by UDPGDH and conjugation of bilirubin with UDPGA by UGT-1A forming the excretable bilirubin diglucuronide. (Lower) Inhibition profiles of 6TP and its excretion metabolites towards UDPGDH and UGT-1A. | ||
2. Results and discussion
2.1. Hepatotoxicity evaluation of 6-thiopurine and 6-thiouric acid
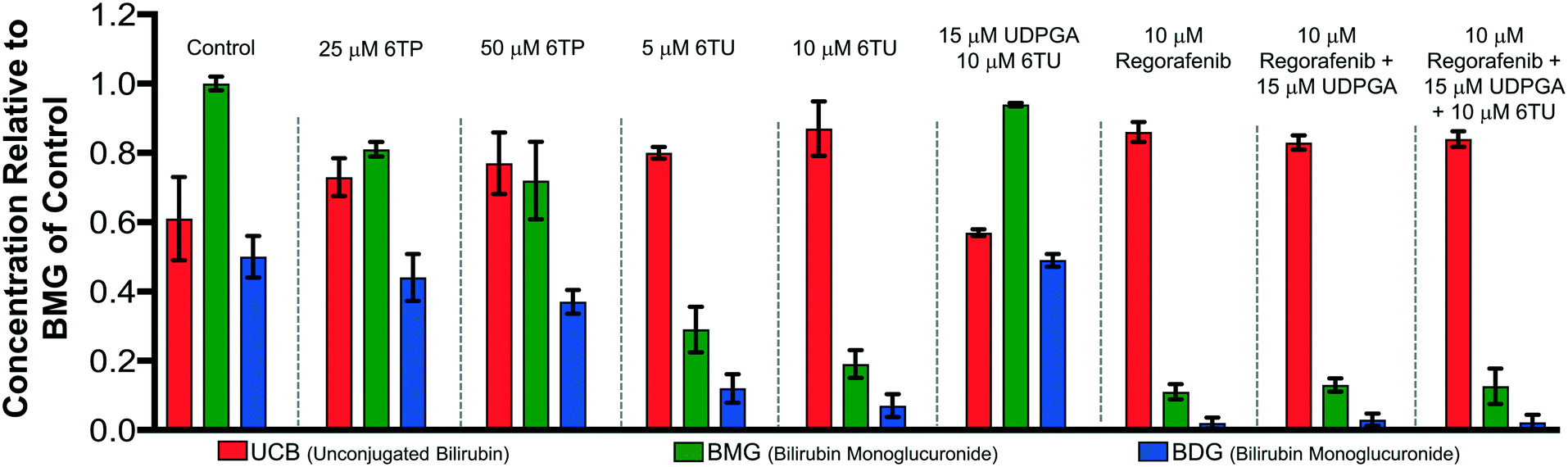
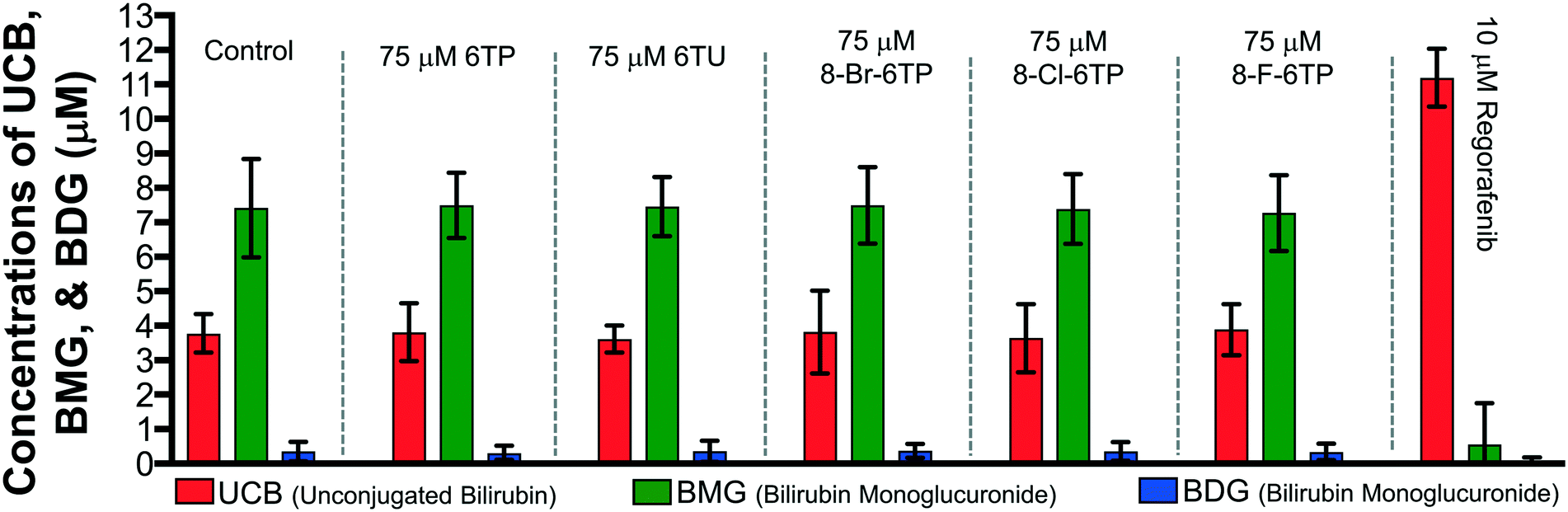
Previously, our lab has shown that the excretion metabolites of 6TP, specifically those hydroxylated at both, or either, C2/8, possess inhibition towards UDPGDH but not UGT-1A, when both enzymes were screened independently in vitro.31 To further corroborate these findings and to give a potential correlation to in vivo effects, we set forth to investigate our initial claim that 6TP and 6TU inhibit UDPGDH in a bio-mimic in vivo assay, via isolated perfused livers of rat hepatocytes. While it is difficult to correlate the exact enzyme being inhibited by a small molecule in a whole cell experiment, we decided to explore UDPGDH inhibition with regards to these two steps through quantification of the glucuronidated bilirubin species. Full glucuronidation of UCB leads to the formation of BDG (12, Fig. 2), however monoglucuronidation results in two different monoglucuronidated bilirubin species (BMG1 and 2); in this work, both BMG1 and 2 were quantified together (herein refered to as BMG). Rat liver hepatocytes were used for the assessment of formation of BMG and BDG in the presence of 6TP and 6TU at high and low concentrations. Table 1 shows the quantification of UCB, BMG, and BDG in hepatocytes. Quantification of the bilirubin species throughout this study is normalized to the BMG levels of the control set.
|
Subjecting the harvested hepatocytes to 25 μM 6TP showed an overall decrease in the conjugation of UCB and formation of BDG; relative to the control set, UCB levels increased by 19%, and both BMG and BDG levels were decreased by 19% and 12%, respectively. Based upon our previous conducted in vitro assays, this finding was not unexpected and further supports that inhibition of UDPGDH is responsible for the decrease in bilirubin conjugation and subsequent toxicity. Increasing the dosage of 6TP to 50 μM revealed greater decrease in BMG and BDG levels, 28% and 26%, respectively, relative to controls. The observed decrease in BMG and BDG levels is assumed to arise from the increased conversion of 6TP to 6TU via XO, thereby inhibiting UDPGDH. Treatment with 5 and 10 μM of 6TU revealed potent inhibition of BMG and BDG formation, far greater than when 6TP was administered. When 5 μM of 6TU was employed an increase of 31% in UCB levels was observed, relative to the controls. Furthermore, BMG and BDG levels decreased by 71% and 76, respectively. The levels of BMG formation decreased by 81% and BDG levels by 86% when 10 μM of 6TU was used, with an increase in UCB levels by 42%, all relative to the controls. If the reduced BDG formation does originate from the decrease in the liable pool of UDPGA, addition of UDPGA to the system should allow recovery of the system similar to the controls. The addition of 15 μM of UDPGA along with 10 μM of 6TU resulted in the production of BDG levels similar to that of the control group, suggesting that 6TU does suppresses the formation of UDPGA via inhibition of UDPGDH in vivo but does not inhibit UGT-1A, but does require further investigations into UGT-1A itself is required to substantiate this claim.
Treatment with 10 μM regorafenib, a known inhibitor of UGT-1A and proven non-inhibitor of UDPGDH by our group (see ESI†), resulted in strong inhibition in the formation of both BMG (89% decrease) and BDG (96% decrease) is observed, both greater than 6TU. When treated with 10 μM regorafenib and 15 μM of UDPGA a similar pattern of reduced BMG and BDG formation is observed. Furthermore, treatment with 10 μM of regorafenib, 10 μM 6TU, and 15 μM of UDPGA also showed the same profile of BMG and BDG levels. Thus, the decrease in BMG and BDG levels from regorafenib are from UGT-1A inhibition, which can be translated into confirming that the effects of 6TP and 6TU are upon UDPGDH and not UGT-1A.
With the confirmation of in vivo inhibition of UDPGDH by 6TP and 6TU, the latter being more potent with regards to suppressing BDG formation, the construction of 6TP analogs was considered the next logical step. From previously conducted in vitro investigations, it was observed that hydroxylation upon the C2 and C8 positions does translate into inhibition of UDPGDH.31 The C2 position of 6TP cannot be modified as amine installation post phosphoribosylation is required to form its therapeutically active form; therefore, modification of the C8 position is considered the only viable strategy for analog construction.
2.2. Chemistry
Construction of C8-substituted purine systems is reported in the literature,32,33 however the inclusion of the C6 thiol with C8 substitution, restricted to alkyl and phenyl analogs, is limited. In the efforts to construct analogs that block the C8 position from oxidation by XO, we set forth to construct analogs that bear halogens about the C8 position in hopes of retaining the therapeutic character of 6TP. Halogens were chosen for the C8 substitution, rather than alkyl or aryl substituents, to decrease the probability of failed DNA incorporation due to sterics about the C8 position. Starting from the commercially available 6-chloropurine (13), protection of the N9 nitrogen was successfully accomplished with potassium carbonate and para-methoxybenzyl chloride (PMBCl) to access 14 in 58% yield (Scheme 1), matching reported characterization found in the literature.34–36 In addition to obtaining pure 14, these conditions also allow access to the N7 PMB protected compound (15) in 34% yield. Further synthetic elaborations were conducted upon 14 but not 15. While 15 can be employed in the efforts of accessing C8 halogen 6TP analogs in the same route as 14, there is a decrease in yields using this material and will be discussed below. With 14 in hand, efforts towards the installation of halogens (bromine,37,38 chlorine,34 and fluorine39) at the C8 position were undertaken through modifications of reported procedures. Deprotonation of the C8 hydrogen of 14 was accomplished with freshly prepared LDA; the lithiated species was then treated with 1,2-dibromotetrachloroethane to access the C8-bromo compound (16a) in 75% yield. The substitution of the 6-chloro group upon purines has precedent in the literature,40,41 following these reported procedures, thiol installation with thiourea and ethanol reflux, afforded 17a in 92% yield over two-steps. Deprotection of the PMB protecting group was performed by refluxing sulfuric acid, thus, gaining access to the 8-bromo-6TP (18a) in 81% yield, with an overall 32% yield over five synthetic transformations from 13. Additional 8-Br-6TP (18a) was accessed from 15 by subjecting it to the same conditions with comparable yields, except about the thiol installation step. From the N7-PMB variant of 16a, subjection to thiol installation provided a 58% yield. It is thought that the additional steric congestion brought from the N7 PMB is the cause for the decrease in yields, as nearly 34% of starting material is recovered.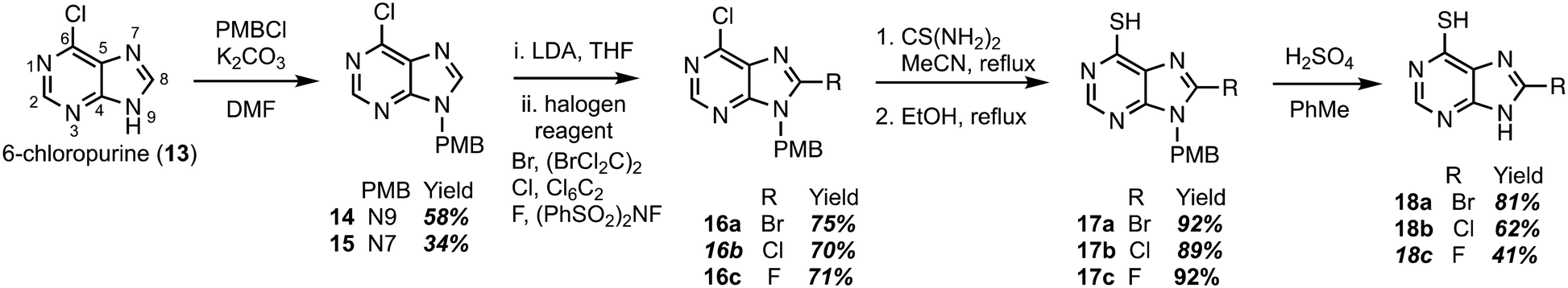 | ||
| Scheme 1 Synthetic route accessing the bromo-, chloro-, and fluoro-C8-substituted 6TP analogs from 6-chloropurine. | ||
Following the same approach described in Scheme 1, accessing the C8 chloride and fluoride analogs were undertaken. Deprotonation of the C8 hydrogen of 14 was accomplished with LDA and subsequent treatment with hexachloroethane afforded 6,8-dichloro-N-paramethoxbenzylpurine (16b) in 70% yield. In an analogous fashion, treatment with N-fluorobenzenesulfonimide (NFSI) afforded 16c in 71% yield, with 19% of starting material recovered. Thiol installation was performed upon 16b and 16c accessing 17b and 17c in 89% and 92% yields, respectively. When the same transformation was attempted upon the N7 variants, a suppression in yields were obtained similar to the bromine analog described previously. Removal of the PMB group was performed under refluxing sulfuric acid to give access to 18b and 18c in 62% and 41% yields. The chloro- and fluoro-analogs were constructed over five-steps in overall yields of 22% and 16%, respectively.
2.3. Biological evaluation of 6TP analogs
Inhibition assessment of the synthesized C8-halogenated 6TP analogs was performed independently against the two enzymatic steps of the bilirubin pathway, UPDGDH and UGT-1A. A cytotoxicity assessment was performed against the acute lymphocytic leukemia cell line REH as well.
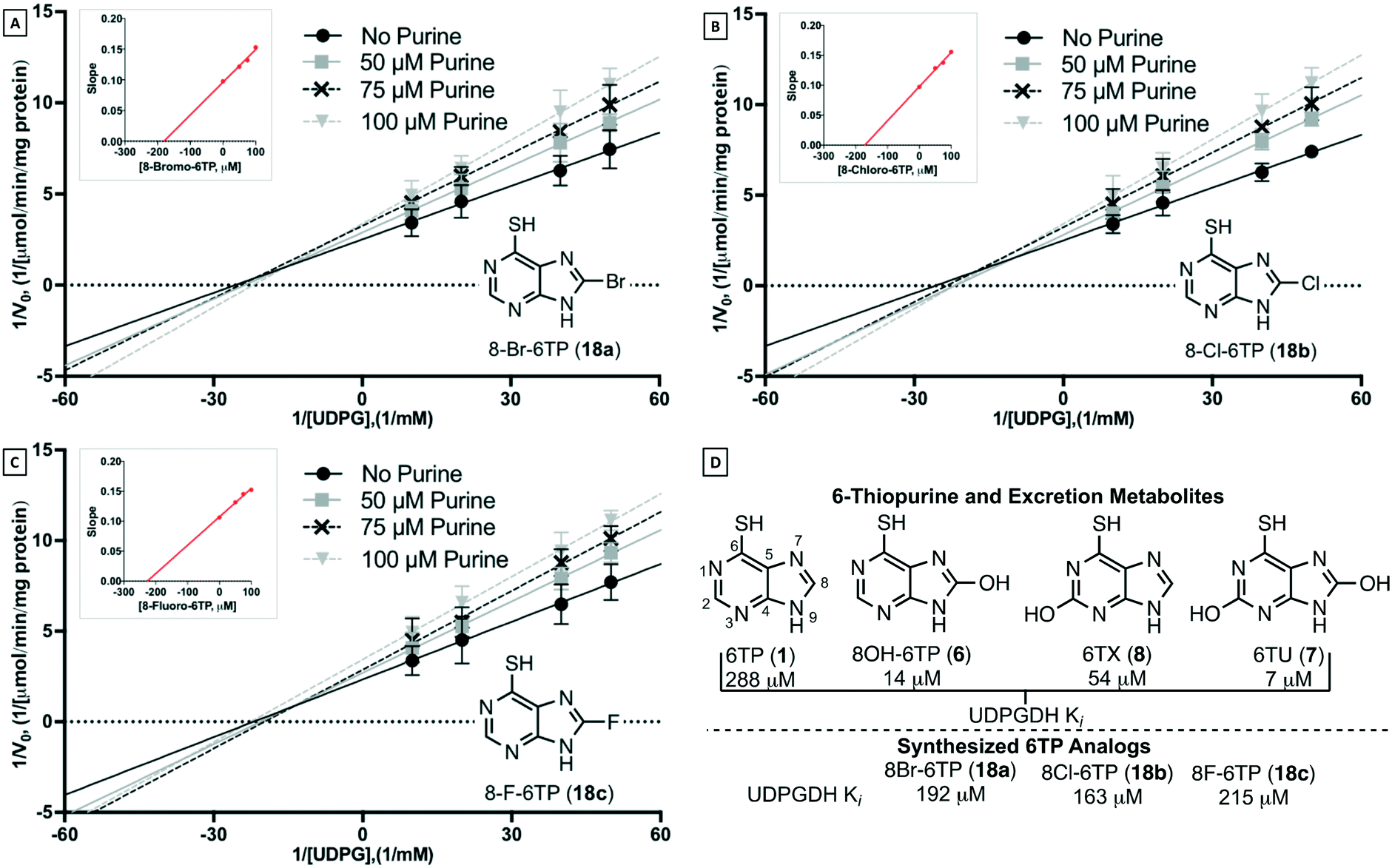
|
|
| Compound | IC50 (μM) REH |
|---|---|
| 6TP | 2.94 (±0.48) |
| 6TU | >10 |
| 6TX | >10 |
| 8-OH-6TP | 9.91 (±2.36) |
| 18a (Br) | 9.54 (±0.97) |
| 18b (Cl) | 3.95 (±1.94) |
| 18c (F) | 4.71 (±1.40) |
| Doxorubicin | 0.285 (±0.08) |
It is assumed that the cytotoxicity of these analogs arise from their incorporation into DNA, in a similar pathway and sequence to that of 6TP, but this has not been proven in this study. Assuming the same mode of action as 6TP, the observed lower potency of the bromine compared to the chlorine and fluorine analogs is thought to arise from its size. The size of bromine upon the C8 position of the purine within the deoxyguanosine mimic could hinder DNA incorporation. Similarly, the increase bulk of the bromine upon the C8 position could prevent enzymatic recognition by the HGPRT, as well as, the other enzymatic steps leading to the formation of 4. The increased cytotoxicity observed by both the chlorine and fluorine analogs lend further support to the possible role of size in the observed trend of cytotoxicity. However, the greater cytotoxicity exhibited by the chlorine analog versus the fluorine analog contradicts the argument of size as a function of activity, but could also lead to an electronic argument for activity. Further investigations are required to fully understand the observed trend. Most importantly, through these analogs it was shown that substitution of the C8 position is not only tolerated, but does give access to 6TP analogs that retain cytotoxicity.
3. Conclusions
Through this study, further investigations into the mode of toxicity associated with the therapeutic application of 6-thiopurine has been accomplished. It has been found that inhibition of UDPGDH, one of two critical steps in the bilirubin pathway, is highly inhibited by 6TU, the primary excretion metabolite of 6TP. Based upon previous structure–activity relationships of the excretion metabolites of 6TP, it was postulated that substitution about the C8 position could decrease inhibition towards UDPGDH while retaining its cytotoxic character. The synthesis of the C8 substituted bromine, chlorine, and fluorine analogs were successfully accomplished from 6-chloropurine in 32%, 22%, and 16% yields, respectively, all over five synthetic steps. Each of the analogs were shown to have low inhibition (192, 163, 215 μM, respectively) towards UDPGDH, similar to 6TP (288 μM) and no inhibition towards UGT-1A. The C8-bromo analog had limited cytotoxicity towards the acute lymphocytic leukemia cell line REH, IC50 of 9.54 μM, however the chloro- and fluoro-6TP analogs were shown to retain cytotoxicity with IC50 values of 3.95 and 4.71 μM, respectively. These results have effectively shown that structural modifications of 6-thiopurine will not only retain cytotoxicity, but also decrease inhibition towards UDPGDH, a critical step found to be responsible for the most predominate toxic side effects of 6TP administration. It is anticipated that with a decrease in toxicity of new 6TP analogs, an increase in dosage can be administered, resulting in further increase of therapeutic properties of 6TP treatment. As such, newly constructed analogs of 6TP will lead to a more potent anti-leukemic drug that can help maintain the quality of life for patients through decreased associated toxicities.4. Experimental section
4.1. Chemistry
All reagents were commercially available and used without purification unless otherwise stated. NMR spectra were recorded with a Varian 400 MHz instrument. The chemical shifts are given in parts per million (ppm) relative to residual CHCl3 at δ 7.26 ppm or DMSO δ 2.50 ppm for proton spectra and relative to CDCl3 at δ 77.23 ppm or DMSO δ 39.52 ppm for carbon spectra, unless otherwise noted. Low-resolution mass spectra were obtained using a Waters Xevo-TQD via direct injection; samples were dissolved in methanol, filtered, and the supernatant injected. Flash column chromatography was performed with silica gel grade 60 (230–400 mesh). Dichloromethane (CH2Cl2), tetrahydrofuran (THF), toluene (PhMe), N,N-dimethylformamide (DMF), and acetonitrile (CH3CN) were all degassed with argon and passed through a solvent purification system containing alumina or molecular sieves. All commercially available reagents were used as received. All procedures including anhydrous solvents were performed with rigorously dried glassware under inert atmosphere.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane of 1:2 to 9:1) to afford 14 (1.55 g, 58% yield) and 15 (0.9 g, 34% yield). 1H NMR (DMSO, 400 MHz) δ (ppm): 8.81 (s, 1H), 8.78 (s, 1H), 7.33 (d, J = 8.0 Hz, 2H), 6.89 (d, J = 8.1 Hz, 2H), 5.43 (s, 2H), 3.70 (s, 3H). 13C NMR (DMSO, 101 MHz) δ (ppm): 159.8, 152.3, 149.8, 148.0, 145.3, 131.5, 130.1, 128.6, 114.8, 55.8, 47.3. LRMS (ESI): m/z calcd for C13H11ClN4NaO: [M + Na]+ 297.05, found 297.12.
:hexane of 1:2 to 9:1) to afford 14 (1.55 g, 58% yield) and 15 (0.9 g, 34% yield). 1H NMR (DMSO, 400 MHz) δ (ppm): 8.81 (s, 1H), 8.78 (s, 1H), 7.33 (d, J = 8.0 Hz, 2H), 6.89 (d, J = 8.1 Hz, 2H), 5.43 (s, 2H), 3.70 (s, 3H). 13C NMR (DMSO, 101 MHz) δ (ppm): 159.8, 152.3, 149.8, 148.0, 145.3, 131.5, 130.1, 128.6, 114.8, 55.8, 47.3. LRMS (ESI): m/z calcd for C13H11ClN4NaO: [M + Na]+ 297.05, found 297.12.
4.1.2.1. 8-Bromo-6-chloro-9-(4-methoxybenzyl)-9H-purine (16a). The crude material was purified via flash silica gel chromatography (Et2O
:hexane 1:1) to afford 16a in 75% as a yellowish amorphous solid (mp: 115–117). 1H NMR (DMSO, 400 MHz) δ (ppm): 8.81 (s, 1H), 7.23 (d, J = 8.4 Hz, 2H), 6.88 (d, J = 8.4 Hz, 2H), 5.41 (s, 2H), 3.70 (s, 3H). 13C NMR (DMSO, 101 MHz) δ (ppm): 159.7, 153.6, 152.7, 148.3, 136.2, 131.8, 129.6, 127.6, 114.9, 55.8, 47.9. LRMS (ESI): m/z calcd for C13H10ClBrN4NaO+: [M + Na+] 374.96, found 375.08.
4.1.2.2. 6,8-Dichloro-9-(4-methoxybenzyl)-9H-purine (16b). The crude material was purified via flash silica gel chromatography (Et2O
:hexane 3:1) to afford 16b in 70% yield as a white amorphous solid (mp: 111–113 °C). 1H NMR (DMSO, 400 MHz) δ (ppm): 8.84 (s, 1H), 7.26 (d, J = 8.5 Hz, 2H), 6.88 (d, J = 8.5 Hz, 2H), 5.42 (s, 2H), 3.70 (s, 3H). 13C NMR (DMSO, 101 MHz) δ (ppm): 159.5, 152.2, 152.1, 149.6, 147.8, 131.3, 129.9, 128.4, 114.6, 55.6, 47.1. LRMS (ESI): m/z calcd for C13H10Cl2N4NaO+: [M + Na+] 331.01, found 331.04.
4.1.2.3. 8-Fluoro-6-chloro-9-(4-methoxybenzyl)-9H-purine (16c). The crude material was triturated in diethyl ether and filtered (×2) to afford 16c in 71% yield as a white apmorphous solid (mp: 110–112). 1H NMR (DMSO, 400 MHz) δ (ppm): 8.99 (s, 1H), 7.23 (d, J = 8.5 Hz, 2H), 6.72 (d, J = 8.5 Hz, 2H), 6.06 (s, 2H), 3.63 (s, 3H). 13C NMR (DMSO, 101 MHz) δ (ppm):159.7, 152.7, 146.5, 139.2 & 136.26 (C8), 131.8, 127.6, 114.9, 105.0, 55.8, 47.9. LRMS (ESI): m/z calcd for C13H10ClFN4NaO+: [M + Na+] 315.04, found 315.03.
4.1.3.1. 8-Bromo-9-(4-methoxybenzyl)-9H-purine-6-thiol (17a). Once at room temperature, the resulting yellow crystals were filtered via vacuum filtration, washed with diethyl ether, and left to dry over night to afford 17a (181 mg 92% yield) as a white amorphous solid. 1H NMR (DMSO, 400 MHz) δ (ppm): 13.52 (bs, 1H), 8.23 (s, 1H), 7.31 (d, J = 8.4 Hz, 2H), 6.85 (d, J = 8.5 Hz, 2H), 5.26 (s, 2H), 3.70 (s, 3H). 13C NMR (DMSO, 101 MHz) δ (ppm): 169.5, 164.5, 159.4, 147.1, 144.0, 130.1, 128.5, 125.9, 114.5, 55.8, 45.6. LRMS (ESI): m/z calcd for C13H11BrN4NaOS+: [M + Na+] 372.97, found 372.95.
4.1.3.2. 8-Chloro-9-(4-methoxybenzyl)-9H-purine-6-thiol (17b). Once at room temperature, the resulting yellow crystals were filtered via vacuum filtration, washed with diethyl ether, and left to dry over night to afford 17b (177 mg 89% yield) as a white amorphous solid. 1H NMR (DMSO, 400 MHz) δ (ppm): 13.51 (s, 1H), 8.23 (s, 1H), 7.31 (d, J = 8.5 Hz, 2H), 6.85 (d, J = 8.5 Hz, 2H), 5.26 (s, 2H), 3.69 (s, 3H). 13C NMR (DMSO, 101 MHz) δ (ppm): 169.3, 164.2, 159.2, 147.0, 143.8, 129.8, 128.3, 125.6, 114.3, 55.5, 45.4. LRMS (ESI): m/z calcd for C13H11ClN4NaOS+: [M + Na+] 329.02, found 328.99.
4.1.3.3. 8-Fluoro-9-(4-methoxybenzyl)-9H-purine-6-thiol (17c). Once at room temperature, the resulting yellow crystals were filtered via vacuum filtration, washed with methanol, and left to dry over night to afford 17c (181 mg 92% yield) as a white amorphous solid. 1H NMR (DMSO, 400 MHz) δ (ppm): 13.51 (s, 1H), 8.27 (s, 1H), 7.25 (d, J = 8.4 Hz, 2H), 6.82 (d, J = 8.4 Hz, 2H), 5.26 (s, 2H), 3.61 (s, 3H). 13C NMR (DMSO, 101 MHz) δ (ppm): 176.5, 159.1, 149.9, 145.9, 140.5 & 134.9 (C8), 130.1, 128.9, 114.1, 110.0, 55.4, 47.2. LRMS (ESI): m/z calcd for C13H11FN4NaOS+: [M + Na+] 313.05, found 313.05.
4.1.4.1. 8-Bromo-9H-purine-6-thiol (18a). The PhMe was decanted off, and the residue was triturated with Et2O and filtered via vacuum filtration to obtain red/orange crude crystals. The water-soluble impurities were removed by partially dissolving the material in water, gently heating the mixture, and then allowed to cool to room temperature. The resulting yellow crystals that formed were collected by vacuum filtration and washing with Et2O to obtain 18a in 81% yield. 1H NMR (DMSO, 400 MHz) δ (ppm): 13.70 (bs, 1H), 8.50 (s, 1H). 13C NMR (DMSO, 101 MHz) δ (ppm): 176.2, 165.0, 148.7, 137.4, 133.1. LRMS (ESI): m/z calcd for C5H3BrN4NaS+: [M + Na]+ 252.92, found 252.92.
4.1.4.2. 8-Chloro-9H-purine-6-thiol (18b). The PhMe was decanted off, and the residue was triturated with Et2O and filtered via vacuum filtration to obtain the crude crystals. The water-soluble impurities were removed by partially dissolving the material in water, gently heating the mixture, and then allowed to cool to room temperature. The resulting white crystals that formed were collected by vacuum filtration and washing with Et2O to obtain 18b in 62% yield. 1H NMR (DMSO, 400 MHz) δ (ppm): 13.71 (bs, 1H), 13.51 (bs, 1H), 8.52 (s, 1H). 13C NMR (DMSO, 101 MHz) δ (ppm): 177.5, 165.3, 148.5, 135.1, 131.9. LCMS (ESI): m/z calcd for C5H3ClN4NaS+: [M + Na]+ 208.97, found 208.91.
4.1.4.3. 8-Fluoro-9H-purine-6-thiol (18c). The PhMe was decanted off, and the residue was triturated with Et2O and filtered via vacuum filtration to obtain the crude crystals. The resulting solid was then dissolved in a minimum amount of warm ethanol, followed by a slow addition of NaHCO3(aq) forming a pale-yellow cloudy solution. Dropwise addition of 1 M HCl(aq) to a pH of 2.5 gave a transparent solution that upon cooling to 0 °C afforded yellow/orange crystals, which were filtered and rinsed with cold ethanol (×3) and left to dry to obtain 18c in 41% yield. 1H NMR (DMSO3, 400 MHz) δ (ppm): 13.68 (bs, 1H), 12.42 (bs, 1H), 8.28 (s, 1H). 13C NMR (DMSO, 101 MHz) δ (ppm): 177.1, 168.9, 159.2, 153.1 & 149.9 (C8), 133.7. LCMS (ESI): m/z calcd for C5H3FN4NaS+: [M + Na]+ 193.00, found 192.97.
4.2. Biological
4.2.1.1. Inhibitor assessment – saturating NAD+ varying UDPG concentration. For each analysis, the cuvette was prepared in the same fashion as outlined above. The final concentration of the components of the mixture were 150 mM Gly-Gly, 0.1 unit per mL UDPGDH, 3 mM NAD+ and varied concentrations of 0.1, 0.05, 0.025 and 0.02 mM of UDPG, obtained from stock solution addition. Nanopure water was used as a variable component to ensure that a final volume of 1 mL was obtained. Inhibitor analysis of the four purines was performed at two concentrations: 50 and 100 μM for 6TP, 20 and 50 μM for 6TX and 8OH-6TP, and 5 and 10 μM for 6TU, obtained from their corresponding stock solutions. Each assessment was performed in triplicate. The average of the three were plotted and the slopes were used to determined inhibition values.
4.2.2.1. Quantification of bilirubin, and mono/di-glucuronide levels. Bilirubin was quantified directly from the generated standard curve (see ESI†). A total of ten peaks for the glucuronide species, including their isomers were detected in the incubation samples. Peak assignment and identification of UCB, BMG1, BMG2, BDG and their isomers were based on their lipophilicity and polarity, as well as the elution pattern, chromatographic peak position and relative retention time from previous reports.21–23,42 The calibration curves for bilirubin were used to determine the concentration of the mono- and di-glucuronide species employing the gradient HPLC bilirubin method described above. Quantification of UDPGA levels was determined through the use of the constructed standard curve within the isocratic HPLC method developed for UDGPA.
Bilirubin glucuronide formation: bilirubin glucuronidation was performed at 37 °C in a shaking water bath. All steps taken were performed in the lowest light conditions possible; the glucuronide formed was found to be unstable to ambient lighting. The following was added to an Eppendorf tube to achieve the final concentrations indicated, final volume 200 μL: potassium phosphate buffer (50 mM, pH 7.4), bilirubin (10 μM), MgCl2·6H2O (0.88 mM), rat liver microsomes (RLM, 100 μg of protein per mL), alamethicin (22 μg mL−1), and allowed to pre-incubated for 2 min. Addition of UDPGA (3.5 mM), referred to as the zero-time point, initiated the reaction. The mixture was allowed to shake at 37 °C for each of the time course experiments. To each reaction 600 μL of ice-cold methanol containing 200 mM ascorbic acid was added to terminate the enzymatic reaction, vortexed for 2 min, and then centrifuged at 12000 rpm for 10 min. The supernatant was then analyzed by the developed gradient HPLC protocol for separation and quantification of UCB, BMG1, BMG2, and BDGs.
Validation of bilirubin glucuronide formation: quantification of UCB, BMG1&2 and BDGs were performed post the quenching of UGT1A1, which was performed by immersing the Eppendorf tube with the reaction mixture in a cold-water bath for two min. No ascorbic acid was used, as the residual material would quench the glucuronidase enzyme to be added. To this sample 0.1 mg mL−1 glucuronidase enzyme was added, inverted (×3), and then analyzed by the HPLC protocol developed to quantify the levels of bilirubin and BMG1&2 and BDGs for formation confirmation.
4.2.2.2. Inhibitor assessment of bilirubin glucuronide formation. Employing the same protocol delineated above for the formation of the bilirubin glucuronide species, inhibitor assessment was performed. To the Eppendorf tube, the purine (50 and 75 μM final concentrations) was added alongside a control (no purine added) and allowed to pre-incubate for 2 min. Addition of UDPGA initiated the reaction for each of the time course experiments. The gradient HPLC method was employed for the 45 min time course experiments for the quantification of the glucuronide species.
General procedure: compounds were solubilized in DMSO (10 μM stock solutions) and added to a 96-well plate over a range of concentrations (31.6 nM to 200 μM) with media, and 40 μL was added to the 384-well plate in triplicate for each concentration of compound. Cells were then added to the plate (2000 cells per well) in 10 μL of media. After 69 h of continuous exposure, 5 μL of Alamar Blue was added to each well, and the cells were allowed to incubate for an additional 3 h. The plates were then read for fluorescence intensity with an excitation of 560 nm and emission of 590 nm on a BioTek Synergy H1 plate reader. Doxorubin and etoposide were both used as positive death controls, and wells with no compounds added as negative death controls. IC50 values were determined from three or more independent experiments using GraphPad Prism 7.0 (LaJolla, CA, USA).
000 cells per well, in 1 mL of WME media in a 12-well plate and incubated at 37 °C in a 5% CO2, 95% humidity atmosphere. Following the procedure established by Ito,44 cell media was exchanged and the hepatocytes were left to grow for 5 days.
Assessment: hepatocytes were transferred to a 24-well plate (20000 cells per well) in 500 of HWE media and allowed to adhere for 24 h. The media was removed and replaced with 1 mL of a buffer solution comprised of: HWE, bilirubin (10 μM), MgCl2·6H2O (0.88 mM), alamethicin (22 μg mL−1), and 15 μM UDPG and left to incubate. To this buffered solution was added the purines of interest, as well 15 μM UDPGA and 10 μM of regorafenib. The addition of the buffered solution was considered the 0 min time point. After 24 h, the liquid mixture was transferred to labelled Eppendorf tubes and 2 mL of trypsin was added to well and left to incubate for 20 min at which 4 mL of WME media was added. The 6 mL of the cell mixture was then centrifuged (10000 g, 2 min), the supernatant removed, washed three times with PBS buffer via gentle resuspension and centrifugation. The pellet was treated with 0.7 mL of a lysis solution (ETA, SDS, Tris HCl at pH 8.00) and 600 μL of ice-cold methanol containing 200 mM ascorbic acid was added to terminate the enzymatic reaction, vortexed for 2 min, centrifuge at 200000×g for 15 min (pellet large cellular debris), the supernatant was added to the original liquid and assessed for UCB, BMG, and BDG levels via the HPLC previously described.
Ethics statement
All animal experiments were conducted under the supervision of Kansas State University Institutional Animal Care and Use Committee (IACUC) and were conducted according to approved protocols in accordance with all applicable federal, state, local and institutional laws or guidelines governing animal research.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work could not have been undertaken without the gracious financial support from the Johnson Cancer Research Center of Kansas State University and Startup Capital from Kansas State University. Funding for this research was provided by the NSF REU program under grant number CHE-1460898. A. X. T. H. was supported by the NSF-REU program.References
- G. H. Hitchings and G. B. Elion, Pharmacol. Rev., 1963, 15, 365–405 CAS.
- J. M. Torpy, C. Lynm and R. M. Glass, JAMA, J. Am. Med. Assoc., 2009, 301, 452–452 CrossRef CAS PubMed.
- T. Moriyama, R. Nishii, T.-N. Lin, K. Kihira, H. Toyoda, N. Jacob, M. Kato, K. Koh, H. Inaba and A. Manabe, Pharmacogenet. Genomics, 2017, 27, 236–239 CrossRef CAS PubMed.
- O. Eden, J. Clin. Pathol., 2000, 53, 55–59 CrossRef CAS PubMed.
- C. Vendrik, J. Bergers, W. De Jong and P. Steerenberg, Cancer Chemother. Pharmacol., 1992, 29, 413–429 CrossRef CAS PubMed.
- C. Cuffari, E. Seidman, S. Latour and Y. Theoret, Can. J. Physiol. Pharmacol., 1996, 74, 580–585 CrossRef CAS PubMed.
- D. S. Rampton, BMJ [Br. Med. J.], 1999, 319, 1480 CrossRef CAS.
- M. L. Seinen, D. P. van Asseldonk, N. K. de Boer, N. Losekoot, K. Smid, C. J. Mulder, G. Bouma, G. J. Peters and A. A. van Bodegraven, Optimalisation of conventional therapies in inflammatory bowel disease, J. Crohn's Colitis, 2016, 7, 812–819 CrossRef PubMed.
- X. Roblin, L. P. Biroulet, J. M. Phelip, S. Nancey and B. Flourie, Am. J. Gastroenterol., 2008, 103, 3115 CrossRef CAS PubMed.
- R. B. Gearry and M. L. Barclay, J. Gastroenterol. Hepatol., 2005, 20, 1149–1157 CrossRef CAS PubMed.
- J. A. Nelson, J. W. Carpenter, L. M. Rose and D. J. Adamson, Cancer Res., 1975, 35, 2872–2878 CAS.
- S. Haglund, J. Taipalensuu, C. Peterson and S. Almer, Br. J. Clin. Pharmacol., 2008, 65, 69–77 CrossRef CAS PubMed.
- K. Rowland, L. Lennard and J. S. Lilleyman, Xenobiotica, 1999, 29, 615–628 CrossRef CAS PubMed.
- D. M. Tidd and A. R. P. Paterson, Cancer Res., 1974, 34, 738–746 CAS.
- P. Karran and N. Attard, Nat. Rev. Cancer, 2008, 8, 24–36 CrossRef CAS PubMed.
- P. Karran, Br. Med. Bull., 2006, 79–80, 153–170 CrossRef PubMed.
- L. Chouchana, A. A. Fernández-Ramos, F. Dumont, C. Marchetti, I. Ceballos-Picot, P. Beaune, D. Gurwitz and M.-A. Loriot, Genome Med., 2015, 7, 37 CrossRef PubMed.
- T. Dervieux, J. G. Blanco, E. Y. Krynetski, E. F. Vanin, M. F. Roussel and M. V. Relling, Cancer Res., 2001, 61, 5810–5816 CAS.
- T. Dervieux, T. L. Brenner, Y. Y. Hon, Y. Zhou, M. L. Hancock, J. T. Sandlund, G. K. Rivera, R. C. Ribeiro, J. M. Boyett, C.-H. Pui, M. V. Relling and W. E. Evans, Blood, 2002, 100, 1240–1247 CrossRef CAS PubMed.
- J. M. Carethers, M. T. Hawn, D. P. Chauhan, M. C. Luce, G. Marra, M. Koi and C. R. Boland, J. Clin. Invest., 1996, 98, 199–206 CrossRef CAS.
- G. Ma, J. Lin, W. Cai, B. Tan, X. Xiang, Y. Zhang and P. Zhang, J. Pharm. Biomed. Anal., 2014, 92, 149–159 CrossRef CAS PubMed.
- D. Zhang, T. J. Chando, D. W. Everett, C. J. Patten, S. S. Dehal and W. G. Humphreys, Drug Metab. Dispos., 2005, 33, 1729–1739 CrossRef CAS PubMed.
- J. Zhou, T. S. Tracy and R. P. Remmel, Drug Metab. Dispos., 2011, 39, 322–329 CrossRef CAS PubMed.
- R. Troxler and S. Brown, in Bilirubin, CRC Press, 2018, pp. 1–38 Search PubMed.
- L. Weaver, A.-R. A. Hamoud, D. E. Stec and T. D. Hinds, Am. J. Physiol., 2018, 4, G668–G676 Search PubMed.
- B. H. Billing, in Bilirubin, CRC Press, 2018, pp. 85–102 Search PubMed.
- M. Vögelin, L. Biedermann, P. Frei, S. R. Vavricka, S. Scharl, J. Zeitz, M. C. Sulz, M. Fried, G. Rogler and M. Scharl, PLoS One, 2016, 11, e0155218 CrossRef PubMed.
- E. A. Hullah, P. A. Blaker, A. M. Marinaki, M. P. Escudier and J. D. Sanderson, J. Oral Pathol. Med., 2015, 44, 761–768 CrossRef CAS PubMed.
- P. P. Siva, K. M. Murali, A. Deepak, S. Murali, S. Michael and M. Sandhya, Drug Metab. Lett., 2016, 10, 264–269 Search PubMed.
- N. R. Beattie, N. D. Keul, A. M. Sidlo and Z. A. Wood, Biochemistry, 2017, 56, 202–211 CrossRef CAS PubMed.
- C. J. Weeramange, C. M. Binns, C. Chen and R. J. Rafferty, J. Pharm. Biomed. Anal., 2018, 151, 106–115 CrossRef CAS PubMed.
- G. B. Elion, E. Burgi and G. H. Hitchings, J. Am. Chem. Soc., 1952, 74, 411–414 CrossRef CAS.
- R. Sariri and G. Khalili, Russ. J. Org. Chem., 2002, 38, 1053–1055 CrossRef CAS.
- Z. Luo, Z. Jiang, W. Jiang and D. Lin, J. Org. Chem., 2018, 83, 3710–3718 CrossRef CAS PubMed.
- M. Brændvang and L.-L. Gundersen, Bioorg. Med. Chem., 2007, 15, 7144–7165 CrossRef PubMed.
- L.-L. Gundersen, J. Nissen-Meyer and B. Spilsberg, J. Med. Chem., 2002, 45, 1383–1386 CrossRef CAS.
- S. H. Jung, G.-S. Hwang, S. I. Lee and D. H. Ryu, J. Org. Chem., 2012, 77, 2513–2518 CrossRef CAS PubMed.
- J. Bergman and L. Venemalm, J. Org. Chem., 1992, 57, 2495–2497 CrossRef CAS.
- A. K. Ghosh, P. Lagisetty and B. Zajc, J. Org. Chem., 2007, 72, 8222–8226 CrossRef CAS PubMed.
- S. Prekupec, D. Svedružić, T. Gazivoda, D. Mrvoš-Sermek, A. Nagl, M. Grdiša, K. Pavelić, J. Balzarini, E. De Clercq, G. Folkers, L. Scapozza, M. Mintas and S. Raić-Malić, J. Med. Chem., 2003, 46, 5763–5772 CrossRef CAS PubMed.
- S. Ciro, C. Rubio, K. Estieu-Gionnet, L. Latxague, G. Déléris, R. Bareille, J. Amédée and C. Baquey, ChemBioChem, 2002, 3, 341–347 CrossRef CAS PubMed.
- M. G. Bartlett and G. R. Gourley, Semin. Perinatol., 2011, 35, 127–133 CrossRef PubMed.
- E. Ogimura, S. Sekine and T. Horie, Biochem. Biophys. Res. Commun., 2011, 416, 313–317 CrossRef CAS PubMed.
- C. Liu, S. Sekine and K. Ito, Toxicol. Appl. Pharmacol., 2016, 302, 23–30 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8md00463c |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2019 |