
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePaper-based microfluidic aluminum–air batteries: toward next-generation miniaturized power supply†
Liu-Liu
Shen
a,
Gui-Rong
Zhang
 *a,
Markus
Biesalski
b and
Bastian J. M.
Etzold
*a
*a,
Markus
Biesalski
b and
Bastian J. M.
Etzold
*a
aErnst-Berl-Institut für Technische und Makromolekulare Chemie, Department of Chemistry, Technische Universität Darmstadt, Alarich-Weiss-Straße 8, 64287 Darmstadt, Germany. E-mail: g.zhang@tc1.tu-darmstadt.de; etzold@tc1.tu-darmstadt.de
bLaboratory of Macromolecular Chemistry and Paper Chemistry, Department of Chemistry, Technische Universität Darmstadt, Alarich-Weiss-Straße 8, 64287 Darmstadt, Germany
First published on 26th September 2019
Abstract
Paper-based microfluidics (lab on paper) emerges as an innovative platform for building small-scale devices for sensing, diagnosis, and energy storage/conversions due to the power-free fluidic transport capability of paper via capillary action. Herein, we report for the first time that paper-based microfluidic concept can be employed to fabricate high-performing aluminum–air batteries, which entails the use of a thin sheet of fibrous capillary paper sandwiched between an aluminum foil anode and a catalyst coated graphite foil cathode without using any costly air electrode or external pump device for fluid transport. The unique microfluidic configuration can help overcome the major drawbacks of conventional aluminum–air batteries including battery self-discharge, product-induced electrode passivation, and expensive and complex air electrodes which have long been considered as grand obstacles to aluminum–air batteries penetrating the market. The paper-based microfluidic aluminum–air batteries are not only miniaturized in size, easy to fabricate and cost-effective, but they are also capable of high electrochemical performance. With a specific capacity of 2750 A h kg−1 (@20 mA cm−2) and an energy density of 2900 W h kg−1, they are 8.3 and 12.6 times higher than those of the non-fluidic counterpart and significantly outperform many other miniaturized energy sources, respectively. The superior performance of microfluidic aluminum–air batteries originates from the remarkable efficiency of paper capillarity in transporting electrolyte along with O2 to electrodes.
1. Introduction
The rapid advance of information technology and future demands for internet of things (IoT) devices have stimulated explosive growth in the market for small, portable and disposable electronics, which are envisioned to provide versatile functions such as sensing, diagnostics, display, or medical therapy in our daily life.1–3 This raises concerns about how to mitigate the power needs of the dramatically rising number of small-sized devices. Lithium-ion batteries (LIBs) have been the dominant technology. However, the low abundance of lithium and significant environmental impacts during lithium mining and battery manufacturing have raised serious doubt about their feasibility for large-scale sustainable applications. It is also anticipated that a huge number of batteries, most of which are still almost fully charged after a single use, might be disposed in an uncontrolled manner. This will lead to energy waste and serious environmental impact.3,4 Alternative energy sources are urgently required to meet the power requirement of small electronic devices at minimal environmental cost. Aluminum–air (Al–air) batteries have great potential because of their numerous practical advantages, such as remarkably high theoretical specific energy density (8.1 kW h kg−1), high theoretical voltage (2.7 V), abundance of raw materials (aluminum is the most abundant metal element in the Earth's crust at 8.1 wt%), low-cost, environmentally friendly nature of aluminum, and safety for use in residential and commercial environments.5–10Despite the great potential of Al–air batteries to be employed as next-generation power supplies, several scientific and technical issues need to be tackled before their commercial-scale implementation: 1) self-discharge: the parasitic corrosion reaction of aluminum and electrolyte causes self-discharge and shortens the battery shelf life; 2) passivation of anode by discharge products: formation of gel-like aluminum hydroxide and/or hydrated alumina passivates the anode surface and prevents further discharging; 3) expensive and complex air electrode: careful design of the air electrode, with an active catalyst, gas diffusion layer and current collector, is essential to promote oxygen diffusion/reduction5–7,11,12 and prevent pore clogging by carbonate formation and deposition at air electrodes.13 Due to its structural complexity, the air electrode usually represents the most expensive part in an Al–air battery.
A lot of research has been done on these problems. For example, intensive works are dedicated to modifying aluminum with other metals to form an alloy; using a non-aqueous electrolyte to alleviate the anode corrosion and passivation;14–22 developing novel electrolytes, catalysts and air electrodes with sophisticated pore structures to enhance oxygen reduction reaction (ORR) activity and increase the oxygen mass transfer rate.23–37 However, these advancements can lead to increased system costs, and complicated and costly procedures for battery component recycling. Al–Air batteries with flowing electrolytes are an alternative way to mitigating the oxygen mass transfer challenge, but external pressure sources (e.g., pumps) are needed to maintain electrolyte flow. This would also add to system costs, making miniaturization of the flow battery system for small devices unrealistic.
In the current work, a paper microfluidic concept has been implemented for the first time in constructing Al–air batteries, and the fabricated paper-based microfluidic Al–air battery (denoted as μAl–air) can be employed as a miniaturized power supply for those small-sized disposable electronics. Moreover, those scientific and technical problems faced by conventional Al–air batteries can be largely relieved or even eliminated on these miniaturized energy devices. As a matter of fact, paper materials have already been employed as a key component in making low-cost, eco-friendly, portable and flexible power sources, and take function as electrode separator, electrolyte/redox species reservoir, or electrodes (e.g., electric conductive paper).38–44 Different from these reports, here in the current work, paper is employed as microfluidic channel to continuously and spontaneously transport electrolyte solution with steady flow rate, largely benefiting from the capillary action on paper with cellulosic fibrous structure. The μAl–air batteries are composed of an aluminum foil anode, a catalyst loaded graphite foil cathode, and a sheet of porous paper, which serves as both separator and microfluidic channel for carrying electrolytes to electrode surfaces (Fig. 1a). This unique paper-based microfluidic design is expected to bring about the following advantages to an Al–air battery: first, Al anode is stored separately from the electrolyte before use, which can completely prevent parasitic corrosion induced self-discharge and prolong battery shelf life. Second, continuous flow of the electrolyte can minimize the accumulation of non-reactive discharge products (e.g., Al(OH)3) at anode/cathode surfaces, preventing electrodes from surface passivation. Third, the capillary driven flow within the porous paper channels enables sufficient mass transfer of O2 to cathode and completely omits the use of expensive air electrodes and bypasses the pore clogging issue of air electrodes, which represents one of the major causes of battery failure. Compared with traditional (micro) flow batteries, no external equipment, e.g., pumps, is needed in the paper-based μAl–air battery to circulate the electrolyte. The latter is unique to paper-based capillary transport systems.45–48 The resultant structure, together with cost-effective and eco-friendly battery components, makes μAl–air batteries feasible as the next-generation of power supply for portable and disposable electronic applications, particularly in single-use devices in diagnosing and sensing fields.
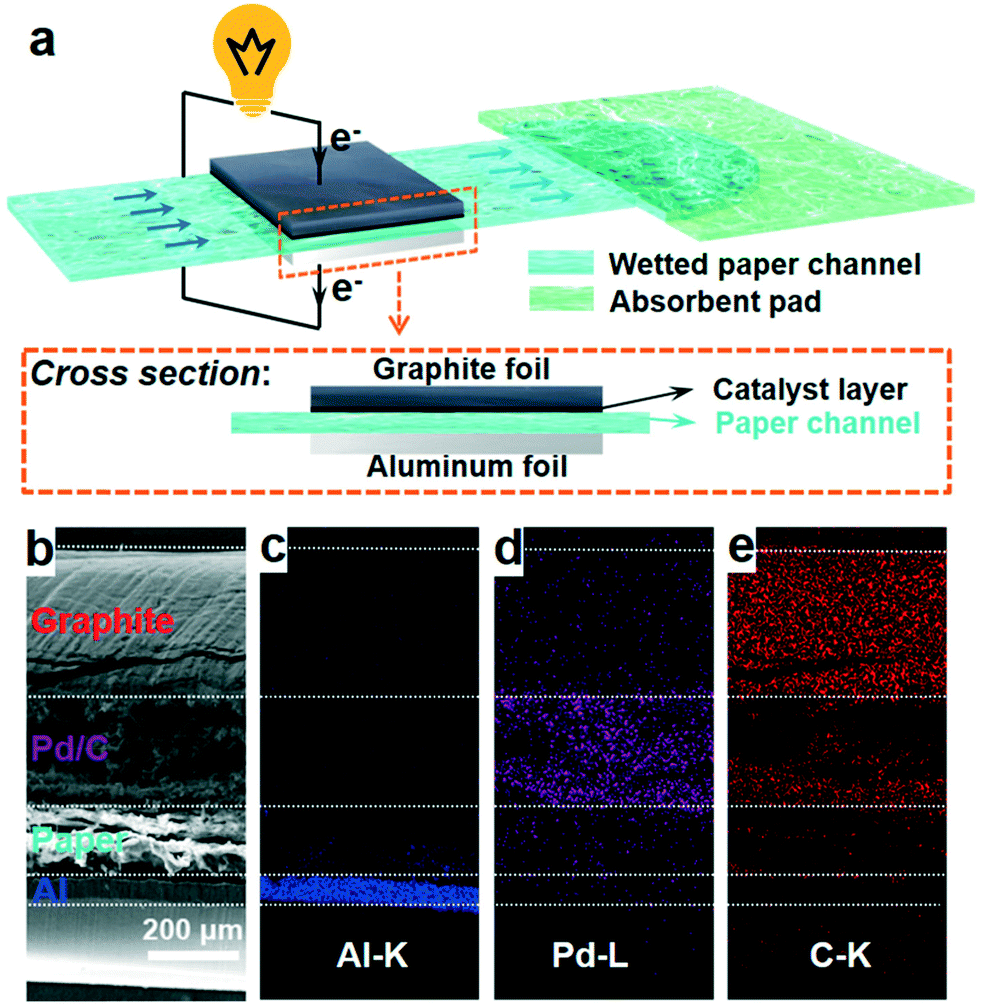 | ||
| Fig. 1 (a) Schematic illustration of a paper-based μAl–air battery; (b) cross sectional SEM image; and (c–e) EDS elemental mapping images of a paper-based μAl–air battery. | ||
2. Experimental
Material and chemicals
Palladium 10% on carbon (type 487, Alfa Aesar), potassium hydroxide (Alfa Aesar), ethanol (VWR), Nafion solution (5 wt%, Sigma-Aldrich), conductive silver printing ink (resistivity 5–6 μΩ cm, Sigma-Aldrich) were all analytical-grade reagents and used as received without any further purification. Aluminum foil (thickness: 0.05 mm) was purchased from Sigma-Aldrich. Graphite foil (thickness: 0.35 mm) was purchased from Sigraflex. Filter paper (grade 417, thickness: 0.15 mm, mean pore diameter: 50 μm, mean porosity: 50%) was purchased from VWR. All solutions were prepared with ultra-pure water. Gas diffusion layer (GDL, 28BC) was purchased from Ionpower. An electric fan (A105) with a power rating of 10 mW was purchased from H-TEC Education GmbH.Preparation of the battery prototype
Cathode electrode is Pd/C on graphite foil, with Pd/C as catalyst and graphite foil as current collector. Graphite foil was first cut into rectangular shape pieces with 25 mm in length, and then cleaned with ethanol and ultrapure water under ultrasonication. PMMA substrate (60 × 40 mm) was cleaned under the same protocols. A catalyst ink (20 mg mL−1) was prepared by sonicating Pd/C in water. Typically, 8 μL of catalyst ink (Pd loading: 0.04 mg cm−2) was carefully applied onto one end of the graphite foil by drop casting, covering a surface area of 40 mm2 (Fig. S1a†). Catalyst coated electrodes were then left air dried at room temperature for 30 min prior to use. In order to prevent catalyst detaching from the graphite foil, a certain amount of Nafion solution was dropped onto the catalyst layer and then dried at room temperature for another 30 min. The mass ratio of Pd to Nafion is fixed at 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Aluminum foil was cut into rectangular shape pieces with 25 mm in length and used as anode directly, as shown in Fig. S1a.† The electrodes are fixed on the PMMA substrate by tape for easy operation, as detailed in Fig. S1b.† The anode and cathode electrodes were places face-to-face and separated by a piece of filter paper (VWR grade 417, 80 × 10 mm2). Note, we used this particular paper, as it consists of cotton linters fibers which are composed of almost pure alpha cellulose with very little lignin and further polysaccharides, respectively. The width of the geometric electrode active area (the overlapping area of the electrode and electrolyte) was 8 mm. A rectangular shaped absorbent pad (40 × 30 mm2) was placed at the end of the paper strip, and the other end of the paper strip was immersed in electrolyte during battery discharge (Fig. S1c†).
:1. Aluminum foil was cut into rectangular shape pieces with 25 mm in length and used as anode directly, as shown in Fig. S1a.† The electrodes are fixed on the PMMA substrate by tape for easy operation, as detailed in Fig. S1b.† The anode and cathode electrodes were places face-to-face and separated by a piece of filter paper (VWR grade 417, 80 × 10 mm2). Note, we used this particular paper, as it consists of cotton linters fibers which are composed of almost pure alpha cellulose with very little lignin and further polysaccharides, respectively. The width of the geometric electrode active area (the overlapping area of the electrode and electrolyte) was 8 mm. A rectangular shaped absorbent pad (40 × 30 mm2) was placed at the end of the paper strip, and the other end of the paper strip was immersed in electrolyte during battery discharge (Fig. S1c†).
Flow rate measurements
Flow rate of electrolyte (1.5 M KOH) within paper channel (VWR grade 417, 10 mm in width) was measured by monitoring the mass gain of the absorbent pads (VWR grade 417, 40 × 30 mm2) over time using an analytical balance. A digital webcam (Logitech C920 PRO HD) was used to record the readings of the balance. As shown in Fig. S2a,† one end of the paper channel was immersed in the reservoir out of the balance, and the absorbent pad was placed on the other end of the paper channel on the balance. The reservoir was filled with 1.5 M KOH solution (1.056 g mL−1 at 20 °C). The solution density was obtained by weighting 1.000 mL solution. The balance was tared immediately when the liquid front reached the absorbent pad. A steady weight increase can be observed until the absorbent pad was fully saturated by the electrolyte. The volume (mL) of the electrolyte in absorbent pad was converted from the mass (g) of the electrolyte, and then plotted against time (min), and linear regression was used to determine the volumetric flow rate (Fig. S2b†).Fabrication of the pouch battery
The paper-based μAl–air pouch battery is shown in Fig. S3a† and 6. The anode and cathode electrode, together with the filter paper strip and absorbent pad, were attached to a piece of laminating pouch, and their relative positions were the same as the prototypical battery on a PMMA substrate mentioned previously. The punch battery was sealed by using an office laminator (Fellowes Lunar A4). Each battery underwent the laminating press twice, once from each direction: 1) along the paper channel (from absorbent pad to the electrodes), 2) across the paper channel (from cathode electrode to anode electrode). A sponge (around 1 cm3 in volume) saturated with electrolyte was placed at the end of the paper channel to serve as an electrolyte reservoir. When two batteries were connected in series (Fig. S3b†), they were placed at a proper angle; the end of the paper channel overlaps with each other, so that one piece of sponge is able to trigger two cells at the same time. Conductive silver printing ink was used to connect batteries to reduce the ohmic resistance.Electrochemical measurements
Electrochemical measurements were carried out on a PMC-1000 multichannel potentiostat (AMETEK) controlled by Versastudio software at room temperature. The electrochemical measurements were carried out shortly after the electrolyte flowed along the paper channel through the electrodes and reached the absorbent pads. The cell resistance between anode and cathode electrodes was determined by conducting the AC impedance analysis (100 kHz, 5 mV), with a cell resistance of under 10 Ω implying a good connection. The open circuit voltage (OCV) and power of the battery were measured by LSV from OCV to 10 mV with a step height of 20 mV at 2 s intervals. Galvanostatic discharge of batteries was performed using chronopotentiometry technique. For comparison, the projected geometric electrode area (overlapping area between the Al foil and catalyst coated cathode at the reaction zone) was used to calculate current density and power density during normalization. For each condition, the measurements were conducted at least four times, using different batteries to generate the error bars (defined as the relative standard deviation).Structural analysis
The thickness of the aluminum foil, graphite foil and paper materials were measured using a high precision dial thickness gauge with 0.001 mm reading (F-1101/30, Käfer Messuhren GmbH). The morphology and elemental mapping of battery cross-section and electrodes were probed using a scanning electron microscopy (Philips XL30 FEG) equipped with an EDAX X-ray detector (CDU Leap XL-30) operated at an accelerating voltage of 30 kV.3. Results and discussion
Structure and design of paper-based μAl–air batteries
A three-layer sandwich structure consisting of cathode, separator and anode is employed to construct the reaction zone of μAl–air batteries, as shown in Fig. 1a and S1,† respectively. The microfluidic paper channel (separator) is sandwiched between Al foil (anode) and Pd/C coated graphite foil (cathode), and the reaction zone lies in between the electrolyte reservoir and absorbent pad. Instead of using a gas diffusion air electrode, poreless graphite foil is used at the cathode as it acts as both current collector and substrate for coating Pd/C to catalyze the ORR process. The microscopic structure of the paper-based μAl–air battery was investigated using cross-sectional scanning electron microscopy (SEM) and energy dispersive X-ray (EDX) elemental mapping analyses (Fig. 1b–e), with which the layered structure can be easily distinguished. The signals from carbon and aluminum roughly follow the shape of the graphite foil and aluminum foil, respectively, while the catalyst layer features a strong signal from Pd. The thicknesses of each layer can also be determined: 50 μm – Al anode, 165 μm – paper channel, 230 μm – catalyst layer (Pd loading 0.04 mg cm−2), and 350 μm – graphite foil.Successful implementation of the paper microfluidic Al–air battery concept relies heavily on the feasibility of sustainable capillary flow within the paper channel. In a paper-based μAl–air battery, the electrolyte flows along the paper channel in a laminar way, driven by the capillary force; no external auxiliaries (e.g., pumps) are used to maintain the laminar flow. From a macroscopic point of view, the single-phase flow of a Newtonian liquid within a porous medium (e.g., paper) is governed by Darcy's law:
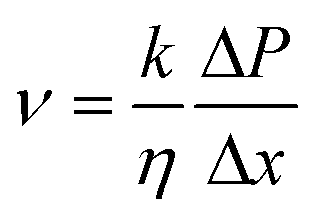 | (1) |
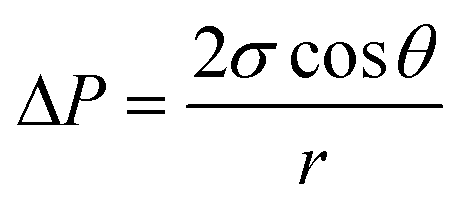 | (2) |
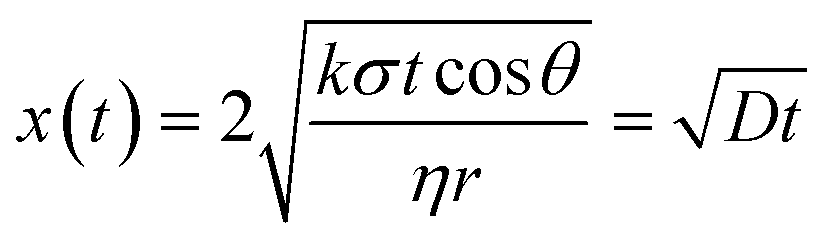 | (3) |
Electrochemical performance of paper-based μAl–air batteries
The possible influences of two critical chemical components, the Pd/C catalyst and the KOH electrolyte, were studied and optimized before analyzing the electrochemical performance of the paper-based μAl–air battery (Fig. 2). Pd/C was employed as the cathode catalyst because of its fairly high catalytic activity toward ORR.54,55 The Pd loading on the cathode was varied from 0.025 to 0.075 mg cm−2 to investigate its influence on battery performance. As shown in Fig. 2a, the peak power density increases linearly with the Pd loading, from 0.025 to 0.04 mg cm−2, and then reaches a plateau with increasing Pd loading up to 0.075 mg cm−2, which is probably due to low catalyst utilization associated with the thick catalyst layer. Therefore, a Pd loading of 0.04 mg cm−2 was chosen in the following investigations. Experiments were also performed to investigate the influence of electrolyte concentration (Fig. 2b). The KOH concentration was varied in the range of 0.2 to 2.5 M. The peak power density increases by almost 3 times when the KOH concentration increases from 0.2 to 1.5 M, while a deviation from this trend is observed by further increasing the KOH concentration up to 2.0 and 2.5 M. This unique dependence behavior is a consequence of the interplay between facilitated Al oxidation reaction at the anode, and disfavored ORR at the cathode, with more concentrated KOH electrolyte.56–58 The battery performance becomes unstable and power density starts declining when the KOH concentration reaches 2.0 M (Fig. S4†), which may originate from degradation of the cathode catalyst and/or cellulose paper in the presence of a strong alkaline medium.59 In view of these results, a KOH concentration of 1.5 M is adopted as a tradeoff between high peak power density and good battery operation stability. | ||
| Fig. 2 (a) Influence of the Pd loading on peak power density. KOH concentration: 1.0 M; the geometric electrode active area: 8 × 5 mm2. (b) Influence of the KOH concentration on the peak power density. Pd loading: 0.04 mg cm−2; the geometric electrode active area: 8 × 5 mm2. | ||
The electrochemical performance of the paper-based μAl–air batteries was then characterized by recording their polarization (V–I) and power density curves (Fig. 3). At the same time, a non-fluidic counterpart Al–air battery with a similar layout but using electrolyte-wetted paper between anode and cathode (Fig. S5†) was also constructed and tested as a reference. The linear sweep voltammetry (LSV) from open circuit voltage (OCV) to 10 mV was recorded 10 times. Fig. 3a shows the polarization (V–I) and power density curves recorded on a paper-based μAl–air battery under continuous LSV. Both the maximum current density and peak power density of the μAl–air battery first increase and then level off after the 3rd voltammetric scan. The OCV of the battery approaches ca. 1.55 V, and the maximum current density reaches 66.5 mA cm−2 (absolute current: 26.6 mA). The battery can also deliver a peak power density of 22.5 mW cm−2 (absolute power: 9.0 mW), which is significantly superior to many other miniaturized power supplies (compared in Fig. 3d and Table S1†). In contrast to the μAl–air battery, the performance of the non-fluidic counterpart drops quickly with the scan number (Fig. 3b). Its peak power density decreases by 40% and 75% after the 3rd and the 10th voltammetric scans, respectively, indicating that the non-fluidic Al–air battery suffers severe degradation during the polarization tests.
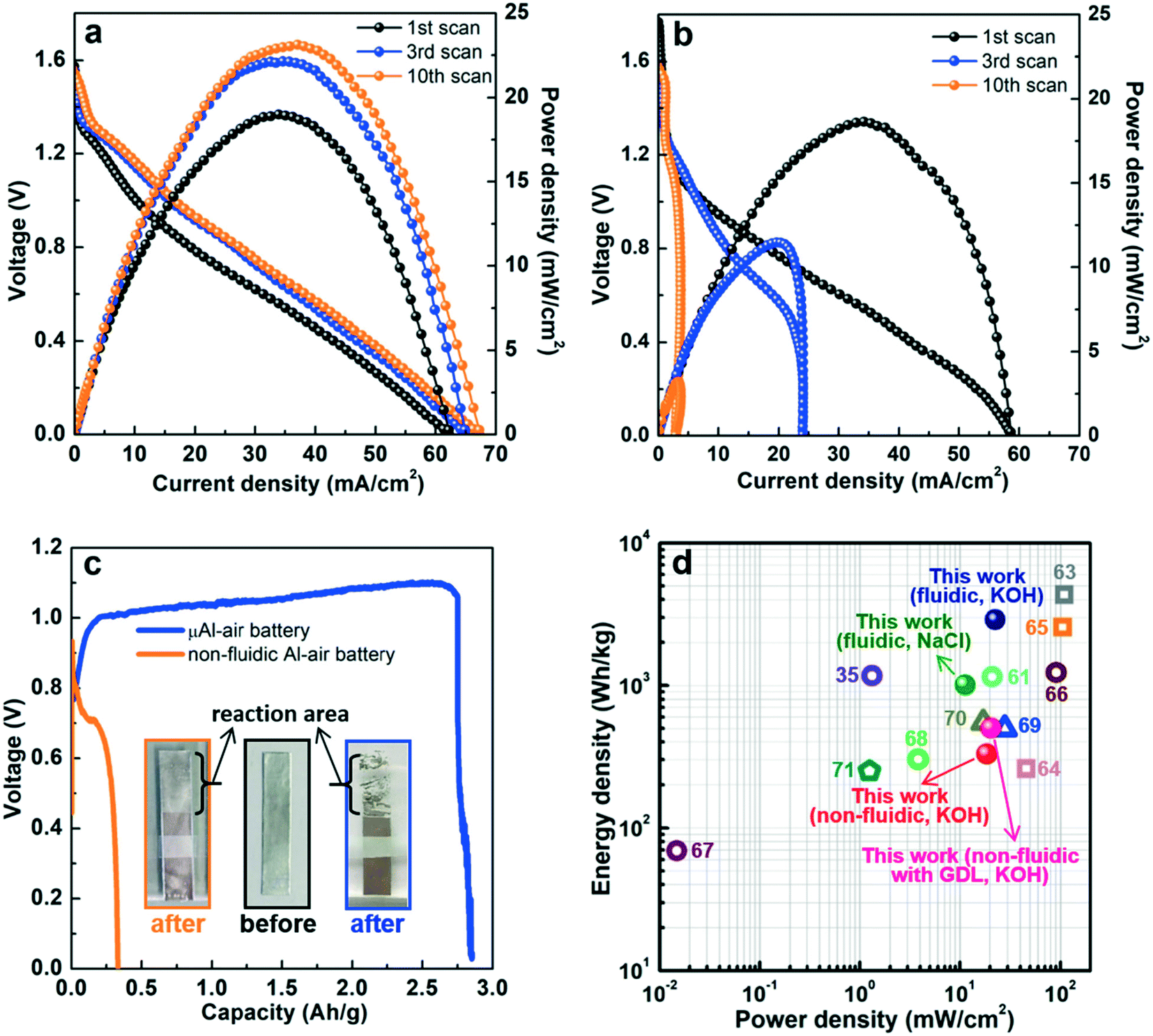 | ||
| Fig. 3 Polarization (V–I) and power density curves recorded at different scan numbers measured by continuous LSV: (a) paper-based μAl–air battery; and (b) paper-based Al–air battery with stagnant (non-fluidic) configuration; (c) discharge curves at current density of 20 mA cm−2. The insets in (c) are the optical images of the anode aluminum foil before (middle) and after discharge (left: non-fluidic Al–air battery; right: μAl–air battery); (d) comparison of power density vs. energy density of μAl–air batteries with other energy storage devices. Square: primary Al–air battery; circle: miniaturized Al–air battery; triangle: miniaturized Zn–air battery; pentagon: miniaturized eutectic gallium–indium–air battery.35,61,63–71 | ||
Continuous battery operation is studied by recording the galvanostatic discharge curves at various current densities, ranging from 5 to 20 mA cm−2 (Fig. 3c and S6†). The μAl–air battery can exhibit a relatively steady discharge voltage, and all of the Al anodes were almost fully consumed regardless of the discharge current, as shown in the inset of Fig. 3c and S6a.† At a discharge current density of 20 mA cm−2, the specific capacity (based on the total mass of Al in the anode) is estimated to be 2750 mAh g−1 (corresponding to a coulombic efficiency of 92%), implying that the parasitic self-corrosion of Al is largely suppressed in the μAl–air battery. Most recently, Leung et al. reported the fabrications of paper-based Al–air battery in which a piece of paper was sandwiched between an air electrode (consisting of ORR catalyst, Ag mesh current collector and a gas diffusion layer) and an Al anode, and they also found that porous and tortuous microstructure of paper can greatly impede the diffusive transport of OH−, consequently suppressing Al corrosion despite a non-fluidic configuration is employed during the battery discharge.60,61
The specific energy density (based on the total mass of Al foil) is calculated to be 2900 W h kg−1, which represents one of the highest values among miniaturized/flexible Al–air batteries. A relatively lower discharge current density is always accompanied by a lower specific capacity, which may stem from the parasitic self-corrosion of Al (Al + 3H2O + OH− → 3/2H2 + Al(OH)4−).5,62 As shown in Fig. S7,† it takes 57 min for a piece of Al foil (8 × 5 mm2) to be fully dissolved in 1.5 M KOH solution. Lower discharge current along with longer discharge time could have led to severer parasitic Al corrosion, which results in lower overall specific capacity of the battery.5,62 A different picture emerges for the non-fluidic Al–air battery: voltage drops quickly regardless of the discharge current densities (Fig. S6b†); this result is in line with the rapid degradation of the non-fluidic Al–air battery during polarization tests (Fig. 3b). The Al anodes stay intact after discharging, although the surface of the Al foil becomes coarse, and some white deposit covers the Al foil surface (Fig. 3c and S6b†). When the battery is discharging at 20 mA cm−2, the specific capacity and energy density of the non-fluidic battery are calculated as 330 mA h g−1 and 230 W h kg−1, respectively, which is 8.3 and 12.6 times lower than the values obtained using the μAl–air battery. Attempt were also made to construct a non-fluidic Al–air battery using a gas diffusion layer air electrode at the cathode, which is widely adapted in conventional Al–air batteries. The performance is shown in Fig. S8:† the peak power density declines significantly with the voltammetric scan numbers, which is similar to the non-fluidic Al–air battery without air electrode. Both the peak power density (20.5 mW cm−2, read at the 1st V–I scan) and specific capacity (500 mA h g−1) are superior to those of the non-fluidic counterpart without air electrode, which is not surprising since the mass transfer of O2 is facilitated using the air electrode, while at the same time the performance is still much worse than that of the μAl–air battery, demonstrating the great advantage of the microfluidic configuration in building high-performing Al–air batteries.
The performance of the paper-based μAl–air battery was also tested using neutral electrolyte (2 M NaCl; Fig. S9†). The μAl–air battery can exhibit an OCV of 0.70 V, and a maximum power density of 11.3 mW cm−2. It can also be observed that the battery can discharge steadily at 20 mA cm−2 until most of the anode aluminum foil is consumed, and the battery exhibits a specific capacity of 2200 mA h g−1, which corresponds to 80% that of the battery using KOH electrolyte. These results indicate that the possible anode passivation by insoluble reaction product (e.g., Al(OH)3) is less likely taking place on these paper-based μAl–air batteries regardless of the nature of electrolyte. It is hypothesized that insoluble products might be flushed away by the flow of electrolyte, which helps minimize the accumulation of those insoluble products and prevent electrode surface from passivation. Moreover, as shown in Fig. 3d and Table S1,† paper-based μAl–air batteries, whether using an alkaline or neutral electrolyte, exhibit superior power and energy densities to recently reported miniaturized metal–air batteries, and are even comparable to some of the primary Al–air batteries. The success in using NaCl electrolyte also raises the possibility of activating μAl–air batteries by directly using naturally abundant seawater, which sheds new light on developing fully sustainable energy storage/conversion devices in the future.
Rechargeability of the μAl–air battery was also investigated. Although Al–air batteries with aqueous electrolyte are in principle not electrically rechargeable, the mature Al refining/recycling technology and market make it feasible to “mechanically” recharge Al–air batteries by simply replacing entire Al anodes. The μAl–air battery can exhibit excellent cycle performance during a mechanical recharging process (Fig. S10†), which is done by replacing the Al foil after a preceding full discharge operation at 20 mA cm−2.The discharging curves are in a comparable range after 3 cycles, implying that the cathode electrode is robust and can be used repeatedly in the μAl–air battery, which would be desirable for practical applications.
Insight into the advantage of paper-based μAl–air batteries
Despite their simple structure, paper-based μAl–air batteries exhibit superior performance in both power density and energy density, largely due to the unique microfluidic configuration. The most intriguing question is about the advantage of microfluidic design. Before this question can be answered, the rapid battery drain of non-fluidic Al–air batteries must be assessed.As shown in Fig. 3c, the non-fluidic Al–air battery can barely sustain steady voltage during discharge and rapidly shuts down due to severe degradation. To probe the degradation mechanism of non-fluidic Al–air batteries, attempts were made to recover the degraded non-fluidic Al–air battery, using either a new anode or cathode, along with newly prepared electrolyte. The discharge capacity of the non-fluidic Al–air battery can only be recovered when a new cathode is used (Fig. S11†), indicating that the cathode is mainly responsible for the degradation of non-fluidic Al–air batteries. In a conventional proton exchange membrane (PEM) fuel cell, complete flooding of the cathode by an aqueous electrolyte would result in a significant loss of cell performance due to the limited diffusion coefficient of O2 in aqueous phase and associated restricted mass transfer of O2 to ORR catalysts.72–75 To find out whether degradation of the non-fluidic Al–air battery is caused by analogous cathode flooding, the cathode was dried out for 2 h, then re-used in the non-fluidic Al–air battery for the next discharge. Battery performance, in terms of discharge voltage and capacity, can be fully regained using the recovered cathode (Fig. S11c†). This confirms that the fully flooded cathode is the main cause of battery degradation.
Based on this, there is a possible rationale for the dramatic difference in performance between the μAl–air battery and its non-fluidic counterpart. In this work, oxygen molecules for ORR mainly come from naturally dissolved O2 from air. In a non-fluidic battery, the channels and pores in the catalyst layer are fully flooded by the electrolyte. The continuous consumption of oxygen at the cathode and slow diffusion of O2 from bulk electrolyte to catalyst pores results in a build-up of the depletion layer for the dissolved O2 near the catalyst surface; the gradually decreasing O2 concentration leads to a decline in power density (Fig. 3b) and eventually battery shut down (Fig. 3c) when the O2 within the catalyst layer is depleted. In contrast, the power density of a μAl–air battery slightly increases from the 1st to the 3rd LSV, which is probably related to the electrolyte penetrating into the catalyst layer. After this, fairly stable power output can be obtained (Fig. 3a). As shown in the galvanostatic discharge measurements (Fig. 3c and S10†), the μAl–air battery can discharge steadily until the Al anode is completely consumed. After mechanical recharge by replacing the Al foil, the μAl–air battery has almost unchanged capacity, indicating that no limitation from O2 transportation has come into effect during the discharge process.
To gain further insight into the superiority of the microfluidic configuration in facilitating the mass transfer of O2 to the cathode, an additional experiment was performed by placing a piece of porous paper onto the microfluidic paper channel (Fig. 4a). The additional porous paper was used to mimic the catalyst layer in direct contact with the microfluidic paper channel. Then one end of the paper channel was alternately dipped into a dye solution and a clear water solution. The time lapse images of solution flow patterns within the overlay paper were recorded (Fig. 4a). The flow pattern within the overlay paper always follows that of the paper channel underneath when switching the inlet solution from dye to water or vice versa. It seems that a portion of the solution diverts into the overlay paper. The diverted solution flows through the overlay paper and then converge into the main stream in the underneath paper channel. These results imply that the liquid phase within the pores of the overlay paper is not stagnant, and the pores keep being replenished by the new solution via convection. By applying this knowledge to the μAl–air battery, O2-rich fresh electrolyte can be “convectively” transported to the cathode catalyst layer in a continuous manner, which could prevent the build-up of the O2 depletion layer within the cathode (Fig. 4b). This is not the case in a non-fluidic Al–air battery as mass transfer of O2 can only be achieved by slow diffusion in an aqueous solution, which would lead to rapid degradation of non-fluidic Al–air battery due to fast depletion of O2 at near surfaces of the cathode (Fig. 4c). Even though using an air electrode in a non-fluidic Al–air battery can postpone battery shutdown by directly feeding air to the cathode, its overall capacity is still limited by the cathode, as reflected by the declining discharge curve accompanied by a well-reserved Al anode after discharge (Fig. S8b†). The much lower overall capacity compared to the μAl–air battery again demonstrates the great structural advantage of the microfluidic configuration in facilitating mass transfer of O2.
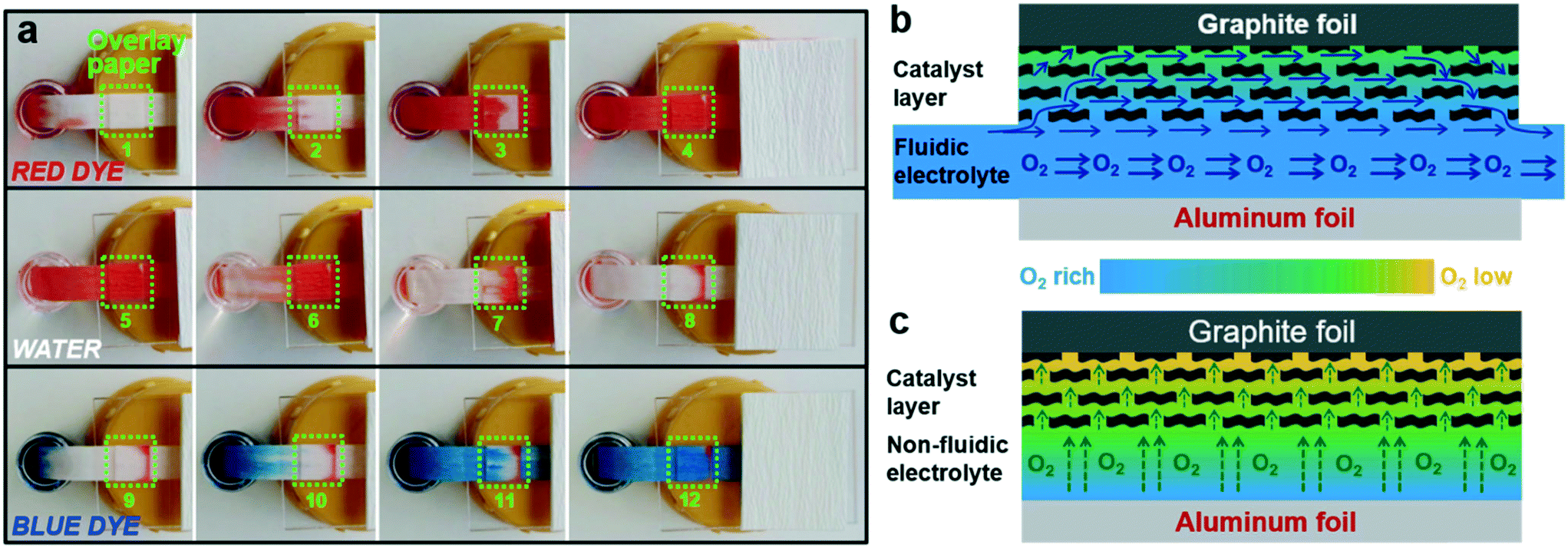 | ||
| Fig. 4 (a) Time-lapse photos showing flow patterns of solutions within the overlay paper, where the green squares mark the overlay paper. Schematic illustrations of O2-rich/low electrolyte distribution in (b) μAl–air battery, and (c) non-fluidic Al–air battery in cross-section. | ||
Practical applications
To meet the power requirements of various electronic devices, the power scalability of paper-based μAl–air batteries was first investigated by either enlarging the electrodes (geometric electrode active area) or connecting several batteries in series. The peak power of the μAl–air batteries can be increased by 37.5% and 85.2% when the electrode width is enlarged from 5 to 10 and 15 mm, respectively (Fig. 5a and S12†). The areal power density (power normalized to the geometric electrode active area) is reduced on larger electrodes (Fig. 5a), which probably originates from the severer depletion of O2 at the electrode along the direction of flow, as has been recently reported.53 Nevertheless, both OCV (3.0 V) and power density (17.9 mW) are nearly doubled when two batteries are connected in series (Fig. 5b), indicating good voltage and power scalability of μAl–air batteries in series configuration.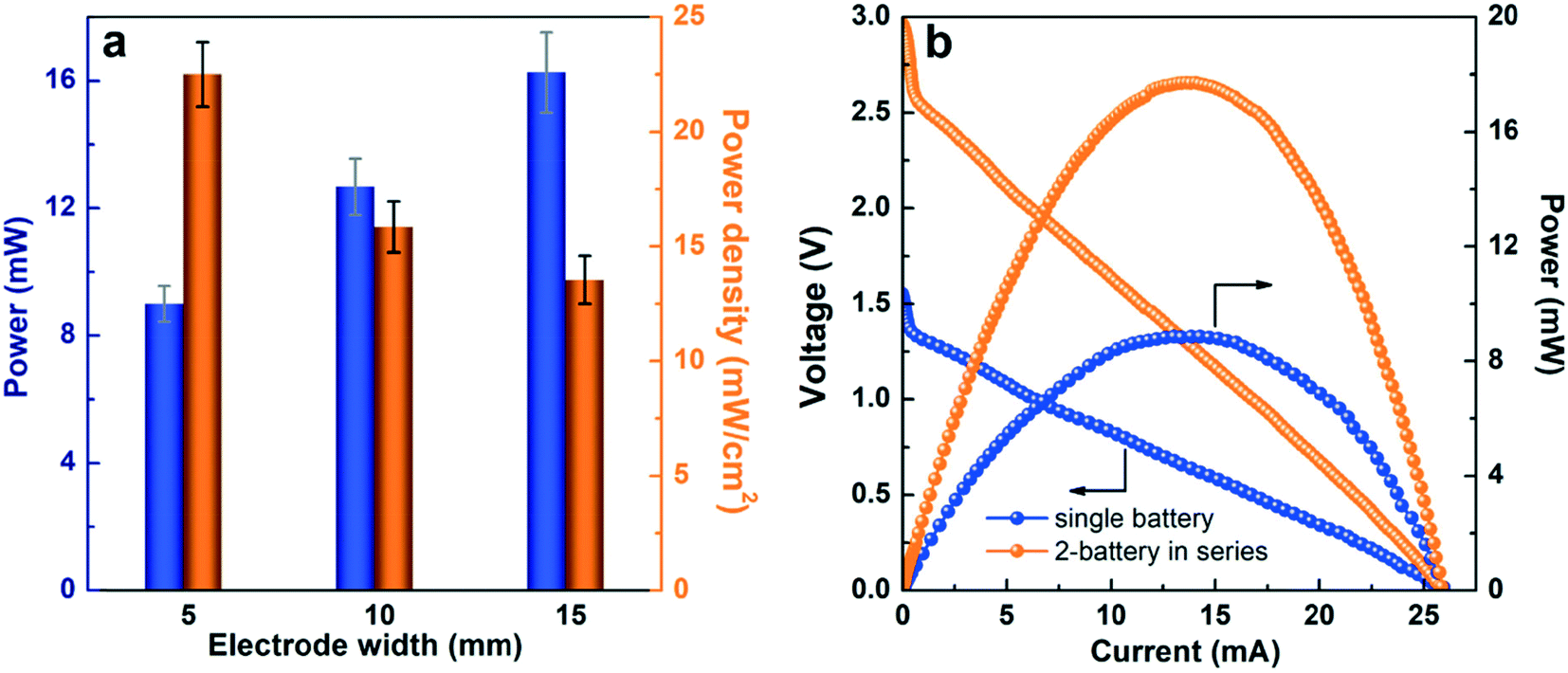 | ||
| Fig. 5 (a) Comparison of power/power density in paper-based μAl–air batteries with three electrode sizes (geometric electrode active area: 8 × 5, 8 × 10, 8 × 15 mm2). (b) Polarization (V–I) and power density curves of a single battery and two paper-based μAl–air batteries connected in a series. | ||
The initial lab scale testing setup (Fig. S1†) of the paper-based μAl–air batteries was expanded to a pouch cell configuration (Fig. 6a and b and S3†) which is more user-friendly. The pouch batteries can be activated by simply contacting the paper with an electrolyte storage device (in this case a piece of sponge pre-wetted with 1.5 M KOH electrolyte), as shown in Fig. S3.† Polarization curves were then recorded by performing LSV measurements on the pouch batteries. The voltage–current characteristic is identical to the prototypical μAl–air battery (Fig. S3c†). Therefore, the pouching process has little impact on the performance of the paper-based μAl–air battery. The pouch batteries were then used to power some small electronic devices. A single battery (geometric electrode active area: 8 × 5 mm2) was able to fully power a hand-held calculator with a LCD display (Fig. 6c). A single battery (geometric electrode active area: 8 × 15 mm2) is capable to keep a mini-fan running for over 70 min, as shown in Fig. 6d and S13.† Attempts were also made to use two connected batteries to power a LED lamp (Fig. 6e); more LED lamps can be on at the same time due to the high overall power output (Fig. 6f). These results successfully demonstrate the great potential of μAl–air batteries to meet the power demand of small portable and disposable electronic devices. Further optimization of the battery geometry and miniaturization of the battery is now required, which could be achieved by integrating solid KOH or NaCl into the inlet of the paper channel. The μAl–air battery can then be conveniently activated by adding water, which will bring the μAl–air battery closer to being an ideal power supply option for small devices. Moreover, cellulose fibers with high mechanical properties and well-defined porous structures, such as TEMPO-oxidized cellulose nanofibers, hold great potential to further boost the performance of paper-based microfluidic batteries.59,76,77
 | ||
| Fig. 6 A paper-based μAl–air pouch battery from (a) top and (b) side view. (c) A single battery (geometric electrode active area: 8 × 5 mm2) powering a calculator. (d) A single battery (geometric electrode active area: 8 × 15 mm2) powering a mini-fan. Two series-connected μAl–air pouch batteries (geometric electrode active area: 8 × 5 mm2), powering (e) one and (f) six LED lamp(s). | ||
4. Conclusion
In addition to developing novel electrocatalysts and structural engineering of complex air electrodes, adoption of a microfluidic configuration turns out to be a highly efficient and inexpensive way to construct high-performing Al–air batteries. In comparison to conventional Al–air batteries, where the electrolyte is pre-loaded, in a μAl–air battery a porous paper channel enables spontaneous capillary flow of electrolyte, which can efficiently shuttle O2 to the cathode, overcoming many shortcomings of conventional Al–air batteries, e.g., self-discharge, short shelf life, and spontaneous electrode passivation by reaction products. The microfluidic paper-based design eliminates expensive air electrode from the Al–air battery, which helps minimize overall system cost and battery encapsulation effort since the bothersome open cell structure of air electrodes can be omitted, and at the same time bypasses the pore clogging issues. The μAl–air battery exhibits impressive electrochemical performance in specific capacity (2750 A h kg−1) and energy density (2900 W h kg−1), both of which are significantly superior to Al–air batteries with non-fluidic configuration, irrespective of whether an air electrode was used. The excellent performance of μAl–air batteries is also reflected by their capacity to power electronic devices with different power requirements. We believe that the paper-based microfluidic concept demonstrated here has the potential to significantly advance the design of metal–air battery systems, creating next-generation high-efficiency, low-cost and eco-friendly energy sources, and can potentially have strong implications for the construction of other high-performing energy storage/conversion systems.Author contributions
L.-L. S. performed investigation, writing-original draft, methodology, and formal analysis; G.-R. Z. performed conceptualization, supervision, investigation, and writing-review and editing; M. B. performed writing-review and editing, and resources; B. E. performed supervision, writing-review and editing, project administration, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Ms. Laura Riedler for her kind help on preparing paper-based materials. The authors acknowledge the funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement No. 681719). L.-L. S. acknowledges the financial support of the China Scholarship Council (CSC, No. 201506210077).References
- S. Chou, W. Yang, K. Chua, J. Li and K. Zhang, Appl. Energy, 2011, 88, 1–16 CrossRef CAS.
- M. Safdar, J. Jänis and S. Sanchez, Lab Chip, 2016, 16, 2754–2758 RSC.
- J. Esquivel, F. Del Campo, J. G. de la Fuente, S. Rojas and N. Sabate, Energy Environ. Sci., 2014, 7, 1744–1749 RSC.
- O. A. Ibrahim, P. Alday, N. Sabaté, J. P. Esquivel and E. Kjeang, J. Electrochem. Soc., 2017, 164, A2448–A2456 CrossRef CAS.
- Y. Liu, Q. Sun, W. Li, K. R. Adair, J. Li and X. Sun, Green Energy & Environ., 2017, 2, 246–277 Search PubMed.
- L. Fan, H. Lu and J. Leng, Electrochim. Acta, 2015, 165, 22–28 CrossRef CAS.
- Q. Li and N. J. Bjerrum, J. Power Sources, 2002, 110, 1–10 CrossRef CAS.
- S. Yang and H. Knickle, J. Power Sources, 2002, 112, 162–173 CrossRef CAS.
- N. Jayaprakash, S. K. Das and L. A. Archer, Chem. Commun., 2011, 47, 12610–12612 RSC.
- D. J. Kim, D.-J. Yoo, M. T. Otley, A. Prokofjevs, C. Pezzato, M. Owczarek, S. J. Lee, J. W. Choi and J. F. Stoddart, Nat. Energy, 2018, 1 Search PubMed.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- M. Mokhtar, M. Z. M. Talib, E. H. Majlan, S. M. Tasirin, W. M. F. W. Ramli, W. R. W. Daud and J. Sahari, J. Ind. Eng. Chem., 2015, 32, 1–20 CrossRef CAS.
- Z. P. Cano, D. Banham, S. Ye, A. Hintennach, J. Lu, M. Fowler and Z. Chen, Nat. Energy, 2018, 3, 279 CrossRef.
- H. El Shayeb, F. A. El Wahab and S. Z. El Abedin, Corros. Sci., 2001, 43, 655–669 CrossRef CAS.
- M. Nestoridi, D. Pletcher, J. A. Wharton and R. J. Wood, J. Power Sources, 2009, 193, 895–898 CrossRef CAS.
- I. Smoljko, S. Gudić, N. Kuzmanić and M. Kliškić, J. Appl. Electrochem., 2012, 42, 969–977 CrossRef CAS.
- D. Egan, C. P. De León, R. Wood, R. Jones, K. Stokes and F. Walsh, J. Power Sources, 2013, 236, 293–310 CrossRef CAS.
- J. Ma, J. Wen, J. Gao and Q. Li, J. Electrochem. Soc., 2014, 161, A376–A380 CrossRef CAS.
- D. Gelman, B. Shvartsev and Y. Ein-Eli, J. Mater. Chem. A, 2014, 2, 20237–20242 RSC.
- M. Kar, T. J. Simons, M. Forsyth and D. R. MacFarlane, Phys. Chem. Chem. Phys., 2014, 16, 18658–18674 RSC.
- R. Revel, T. Audichon and S. Gonzalez, J. Power Sources, 2014, 272, 415–421 CrossRef CAS.
- H. Wang, S. Gu, Y. Bai, S. Chen, F. Wu and C. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 27444–27448 CrossRef CAS PubMed.
- G.-R. Zhang and S. Wöllner, Appl. Catal., B, 2018, 222, 26–34 CrossRef CAS.
- L.-L. Shen, G.-R. Zhang, S. Miao, J. Liu and B.-Q. Xu, ACS Catal., 2016, 6, 1680–1690 CrossRef CAS.
- Y. Wang, M. Zhao, Q. Zhao, Q. Li and H. Pang, Nanoscale, 2018, 10, 15755–15762 RSC.
- X. Li, Y. Fang, X. Lin, M. Tian, X. An, Y. Fu, R. Li, J. Jin and J. Ma, J. Mater. Chem. A, 2015, 3, 17392–17402 RSC.
- G.-L. Chai, K. Qiu, M. Qiao, M.-M. Titirici, C. Shang and Z. Guo, Energy Environ. Sci., 2017, 10, 1186–1195 RSC.
- S. Liu, Z. Wang, S. Zhou, F. Yu, M. Yu, C. Y. Chiang, W. Zhou, J. Zhao and J. Qiu, Adv. Mater., 2017, 29, 1700874 CrossRef PubMed.
- H. Peng, F. Liu, X. Liu, S. Liao, C. You, X. Tian, H. Nan, F. Luo, H. Song, Z. Fu and P. Huang, ACS Catal., 2014, 4, 3797–3805 CrossRef CAS.
- X.-H. Yan and B.-Q. Xu, J. Mater. Chem. A, 2014, 2, 8617–8622 RSC.
- T. Y. Ma, J. Ran, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 4646–4650 CrossRef CAS PubMed.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444 CrossRef CAS PubMed.
- S. Li, C. Cheng, H. W. Liang, X. Feng and A. Thomas, Adv. Mater., 2017, 29, 1700707 CrossRef PubMed.
- C. Tang, B. Wang, H. F. Wang and Q. Zhang, Adv. Mater., 2017, 29, 1703185 CrossRef PubMed.
- Y. Xu, Y. Zhao, J. Ren, Y. Zhang and H. Peng, Angew. Chem., 2016, 128, 8111–8114 CrossRef.
- J. Li, Z. Zhou, K. Liu, F. Li, Z. Peng, Y. Tang and H. Wang, J. Power Sources, 2017, 343, 30–38 CrossRef CAS.
- M. Zhang, Q. Dai, H. Zheng, M. Chen and L. Dai, Adv. Mater., 2018, 30, 1705431 CrossRef PubMed.
- K. B. Lee, J. Micromech. Microeng., 2005, 15, S210–S214 CrossRef CAS.
- K. B. Lee, J. Micromech. Microeng., 2006, 16, 2312–2317 CrossRef CAS.
- G. Nyström, A. Razaq, M. Strømme, L. Nyholm and A. Mihranyan, Nano Lett., 2009, 9, 3635–3639 CrossRef PubMed.
- H. Liu and R. M. Crooks, Anal. Chem., 2012, 84, 2528–2532 CrossRef CAS PubMed.
- N. K. Thom, K. Yeung, M. B. Pillion and S. T. Phillips, Lab Chip, 2012, 12, 1768–1770 RSC.
- Y. Koo, J. Sankar and Y. Yun, Biomicrofluidics, 2014, 8, 054104 CrossRef PubMed.
- M. Mohammadifar and S. Choi, Adv. Mater. Technol., 2017, 2, 1700127 CrossRef.
- A. Böhm, F. Carstens, C. Trieb, S. Schabel and M. Biesalski, Microfluid. Nanofluid., 2014, 16, 789–799 CrossRef.
- F. Sharifi, S. Ghobadian, F. R. Cavalcanti and N. Hashemi, Renewable Sustainable Energy Rev., 2015, 52, 1453–1472 CrossRef.
- A. Böhm and M. Biesalski, MRS Bull., 2017, 42, 356–364 CrossRef.
- M. M. Gong and D. Sinton, Chem. Rev., 2017, 117, 8447–8480 CrossRef CAS PubMed.
- M. Liu, J. Wu, Y. Gan, D. A. Hanaor and C. Chen, Langmuir, 2016, 32, 9899–9904 CrossRef CAS PubMed.
- E. W. Washburn, Phys. Rev., 1921, 17, 273 CrossRef.
- S. Mendez, E. M. Fenton, G. R. Gallegos, D. N. Petsev, S. S. Sibbett, H. A. Stone, Y. Zhang and G. P. López, Langmuir, 2009, 26, 1380–1385 CrossRef PubMed.
- L.-L. Shen, G.-R. Zhang, W. Li, M. Biesalski and B. J. Etzold, ACS Omega, 2017, 2, 4593–4603 CrossRef CAS PubMed.
- L.-L. Shen, G.-R. Zhang, T. Venter, M. Biesalski and B. J. Etzold, Electrochim. Acta, 2019, 298, 389–399 CrossRef CAS.
- F. Lima, J. Zhang, M. Shao, K. Sasaki, M. Vukmirovic, E. Ticianelli and R. Adzic, J. Phys. Chem. C, 2007, 111, 404–410 CrossRef CAS.
- M. H. Seo, S. M. Choi, H. J. Kim and W. B. Kim, Electrochem. Commun., 2011, 13, 182–185 CrossRef CAS.
- D. Chu and R. F. Savinell, Electrochim. Acta, 1991, 36, 1631–1638 CrossRef CAS.
- R. Davis, G. Horvath and C. Tobias, Electrochim. Acta, 1967, 12, 287–297 CrossRef CAS.
- H. Li, H. Liu, Z. Jong, W. Qu, D. Geng, X. Sun and H. Wang, Int. J. Hydrogen Energy, 2011, 36, 2258–2265 CrossRef CAS.
- N. Mittal, F. Ansari, K. Gowda, V. C. Brouzet, P. Chen, P. T. Larsson, S. V. Roth, F. Lundell, L. Wagberg and N. A. Kotov, ACS Nano, 2018, 12, 6378–6388 CrossRef CAS PubMed.
- Y. Wang, H. Kwok, W. Pan, H. Zhang and D. Y. Leung, J. Power Sources, 2019, 414, 278–282 CrossRef CAS.
- Y. Wang, H. Y. Kwok, W. Pan, H. Zhang, X. Lu and D. Y. Leung, Appl. Energy, 2019, 251, 113342 CrossRef CAS.
- H. Wang, D. Y. Leung, M. K. Leung and M. Ni, Energy Fuels, 2010, 24, 3748–3753 CrossRef CAS.
- Q. Hong and H. Lu, Sci. Rep., 2017, 7, 3378 CrossRef PubMed.
- L. Xu, H. Fan, L. Huang, J. Xia, S. Li, M. Li, H. Ding and K. Huang, Electrochim. Acta, 2017, 239, 1–9 CrossRef CAS.
- J. Ryu, H. Jang, J. Park, Y. Yoo, M. Park and J. Cho, Nat. Commun., 2018, 9, 3715 CrossRef PubMed.
- Z. Zhang, C. Zuo, Z. Liu, Y. Yu, Y. Zuo and Y. Song, J. Power Sources, 2014, 251, 470–475 CrossRef CAS.
- S. Choi, D. Lee, G. Kim, Y. Y. Lee, B. Kim, J. Moon and W. Shim, Adv. Funct. Mater., 2017, 27 Search PubMed.
- Y. Liu, Q. Sun, X. Yang, J. Liang, B. Wang, A. Koo, R. Li, J. Li and X. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 19730–19738 CrossRef CAS PubMed.
- J. Fu, D. U. Lee, F. M. Hassan, L. Yang, Z. Bai, M. G. Park and Z. Chen, Adv. Mater., 2015, 27, 5617–5622 CrossRef CAS PubMed.
- X. Chen, B. Liu, C. Zhong, Z. Liu, J. Liu, L. Ma, Y. Deng, X. Han, T. Wu and W. Hu, Adv. Energy Mater., 2017, 7, 1700779 CrossRef.
- G. Liu, J. Y. Kim, M. Wang, J. Y. Woo, L. Wang, D. Zou and J. K. Lee, Adv. Energy Mater., 2018, 1703652 CrossRef.
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC.
- G.-R. Zhang, M. Munoz and B. J. Etzold, ACS Appl. Mater. Interfaces, 2015, 7, 3562–3570 CrossRef CAS PubMed.
- G.-R. Zhang, M. Munoz and B. J. Etzold, Angew. Chem., Int. Ed., 2016, 55, 2257–2261 CrossRef CAS PubMed.
- H. Li, Y. Tang, Z. Wang, Z. Shi, S. Wu, D. Song, J. Zhang, K. Fatih, J. Zhang and H. Wang, J. Power Sources, 2008, 178, 103–117 CrossRef CAS.
- C. J. Brett, N. Mittal, W. Ohm, M. Gensch, L. P. Kreuzer, V. Körstgens, M. Månsson, H. Frielinghaus, P. Müller-Buschbaum and L. D. Söderberg, Macromolecules, 2019, 52, 4721–4728 CrossRef CAS.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9lc00574a |
| This journal is © The Royal Society of Chemistry 2019 |