
Multiplexed immunoassay using post-synthesis functionalized hydrogel microparticles†
Hyun Jee
Lee
 a,
Yoon Ho
Roh
a,
Hyeon Ung
Kim
a,
Sun Min
Kim
*b and
Ki Wan
Bong
*a
a,
Yoon Ho
Roh
a,
Hyeon Ung
Kim
a,
Sun Min
Kim
*b and
Ki Wan
Bong
*a
aDepartment of Chemical and Biological Engineering, Korea University, Seoul, Republic of Korea. E-mail: bong98@korea.ac.kr
bDepartment of Obstetrics and Gynecology, Seoul Metropolitan Government-Seoul National University Boramae Medical Center, Seoul, Republic of Korea. E-mail: obgysun@gmail.com
First published on 23rd November 2018
Abstract
In response to a growing demand for simultaneous detection of multiple proteins in a single sample, multiplex immunoassay platforms have emerged at the forefront of proteomic analysis. In particular, detections using graphically encoded hydrogel microparticles synthesized via flow lithography have received attention for integrating a hydrogel, a substrate that can provide enhanced kinetics and high loading capacity, into the bead-based multiplex platform. Currently, the method of microparticle functionalization involves copolymerization of antibodies with the gel during particle synthesis. However, its practical operation is too precarious to be adopted because antibodies are susceptible to aggregation due to incompatibility with hydrophobic photoinitiators used in the photo-induced gel polymerization. In this work, we present a multiplex immunoassay platform that uses encoded hydrogel microparticles that are functionalized after particle synthesis by conjugating antibodies with remnant active groups readily available in the hydrogels. The method not only precludes antibody aggregation but also augments the loading density of the antibodies, which translates into enhanced detection performance. In addition to multiplexing, our platform demonstrates high sensitivity, a broad assay range, and a fast detection rate that outperform the enzyme linked immunosorbent assay (ELISA).
Introduction
Protein detection and quantification is increasingly becoming an indispensable analytical practice in various applications, including food science,1 biopharmacy,2 toxicology,3 and medical diagnostics.4,5 For decades, protein detection has been performed most heavily using enzyme-linked immunosorbent assay (ELISA), which uses highly specific antibody–antigen interactions to immobilize and label a target protein on the surface of a microwell.6 In the sandwich ELISA, which is the most robust and commonly used format, the target protein binds to a capture antibody attached to the plate and is labeled with a secondary antibody functionalized for colorimetric or fluorescent readout. Although the method is well-established with a wide range of validated antibody pairs, one of its critical limitations has been its difficulty of detecting multiple targets simultaneously.7 Because only a single analyte can be measured per well, multiplexing in the ELISA requires arduous procedures and large sample volumes.This limitation has become more pronounced with the advances in medical diagnostics that recognized the complexity of diseases and interdependence of protein biomarkers.8 As opposed to monitoring a single protein, measuring a large panel of protein biomarkers is likely to provide more accurate clinical information.9 Moreover, the requirement of large sample volumes has been a major drawback for analysis using limited sample volumes, such as in pediatrics10 and obstetrics.11 Consequently, many efforts have been made to adapt ELISA's fundamental mechanism in multiplex platforms to enable simultaneous detection of multiple targets in a single sample.12 Current multiplex immunoassay techniques can be classified into two main categories: planar and suspension microarrays. In planar microarrays, capture antibodies are immobilized in micro-spots on a solid support, enabling spatial encoding of hundreds of different target proteins.13 On the other hand, suspension (bead-based) microarrays use particles conjugated with capture antibodies to encode targets based on the optical, graphical, or electronic properties of the particles.14 Suspension microarrays feature several advantages over planar microarrays that have rendered them favorable under many assay conditions; they offer solution-phase reaction kinetics in contrast to solid-phase kinetics in planar arrays, customizable and flexible target panels, and higher precision due to the large number of particles per analyte.15
Being the pioneer in commercial suspension arrays, Luminex technology is well-recognized as the representative technology of its kind. It uses polystyrene microspheres that contain a distinct combination of red and near-infrared fluorophores to provide spectral encoding. Because of spectral overlaps, the maximum number of codes has been theoretically limited to 100.12 While Luminex is reported to demonstrate an assay performance comparable to the ELISA,14,16 there has been an ongoing research interest to develop suspension assay platforms with enhanced performance in regard to multiplexing capacity, sensitivity, specificity, dynamic range, and detection time.17,18
One of the notable approaches that combine an advanced encoding scheme with a promising bead substrate is the use of barcoded hydrogel microparticles.19 In this technique, microparticles are encoded graphically, which allows for easy implementation of much higher encoding capacities (i.e. of the order of 106 codes on a 100 μm particle) compared to spectral encoding.17 Graphical encoding was realized by synthesizing the particles using a microfluidics-based photolithography technique called stop flow lithography.20 A shape-defined UV light is selectively exposed to a layer of prepolymer stream, polymerizing microparticles in 2-D extrusions of virtually any graphical shape. Furthermore, the technique adopted hydrophilic gels as the constituting material for the microparticles. Owing to their permeable 3-dimensional scaffolds, gels have emerged as a promising material that can enhance the binding kinetics, alleviate steric hindrance caused by rigid immobilization, and improve the loading capacity for the capture antibodies.21,22 However, despite the technique's novel concept, there exists an inherent problem in its antibody immobilization condition. Antibodies are copolymerized during the photo-induced particle polymerization in an incompatible prepolymer solution containing hydrophobic photoinitiators.23 The immiscibility causes antibodies to form aggregates, which are significantly limited in binding affinity with the target protein. The procedure necessitates vigorous mixing followed by centrifugation to reduce and partially remove the aggregates from the prepolymer.24 Nevertheless, such measures do not address the fundamental issue and lead to a considerable loss of available antibodies. This problem, which is aggravated at high antibody concentrations, results in a low loading density of functional capture antibodies that translates to poor sensitivity and a narrow assay range.
In this work, we present a hydrogel microparticle based immunoassay platform with a novel method of particle functionalization that allows for immobilization of antibodies at high concentrations without aggregation. The antibody aggregation is completely eliminated by conjugating the antibodies after particle synthesis with remnant reactive groups abundantly available in the hydrogel network. The binding reaction, which takes place in an aqueous medium in the absence of photoinitiators, enables immobilization of antibodies in high density and enhances the detection performance. We compare our assay's detection sensitivity, detection rate, and dynamic range with those of ELISA in the detection of three clinically relevant proteins. We also validate our technique's suitability for multiplex detection in a 3-plex immunoassay.
Experimental
Fabrication of the microfluidic device
The first step of the device fabrication is soft lithography of PDMS on a silicon master wafer that had been patterned using an SU-8,25 photoresist (MicroChem) to form positive relief microchannels of 50 μm height and 250 μm width. PDMS (SYLGARD 184, Dow Corning) mixed at a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 base to curing agent ratio was poured over the master and cured for 4 h at 70 °C in an oven. Then, the cured PDMS was detached from the master and perforated with 1.0 mm and 10.0 mm punches (Miltex) to create inlet and outlet reservoirs, respectively. A microscope slide was thinly coated with PDMS and partially cured at 70 °C for 30 min. Then, the perforated PDMS block, with the negative relief pattern facing down, was attached onto the partially cured surface of the glass slide and fully cured overnight.
:1 base to curing agent ratio was poured over the master and cured for 4 h at 70 °C in an oven. Then, the cured PDMS was detached from the master and perforated with 1.0 mm and 10.0 mm punches (Miltex) to create inlet and outlet reservoirs, respectively. A microscope slide was thinly coated with PDMS and partially cured at 70 °C for 30 min. Then, the perforated PDMS block, with the negative relief pattern facing down, was attached onto the partially cured surface of the glass slide and fully cured overnight.
Stop flow lithography set-up
The actuation of stop flow lithography involved using a UV-pressure synchronization system controlled by a customized circuit board and a LabView (National Instrument) script. With the microfluidic device mounted on an inverted microscope (Axiovert 200, Zeiss), the prepolymer solution was loaded into the device inlet in a pipette tip reservoir. With a Tygon tube, the reservoir was connected to a compressed air tank, bridged by a pressure regulator (ITV0031-3BL, SMC Pneumatics). A photomask, designed using AutoCAD and printed using a high-resolution printer (50000 dpi), was inserted into the field-stop of the microscope. An LED lamp (M365LP1, Thorlabs) was used as the UV photo-polymerization source. In this work, the UV intensity was fixed at 2200 mW cm−2 as measured through a 20× objective using a UV power meter (GT-510, Giltron).
Immunoassay reagents
For direct comparison with the ELISA, proteins and antibody pairs from ELISA kits were used: IL-6 (R&D Systems, cat. no. DY206), VEGF (R&D Systems, cat. no. DY293B), PIGF (R&D Systems, cat. no. DY264), and CG beta (R&D Systems, cat. no. DY9034-05). Reagents were reconstituted with consideration of their working concentrations specified in the product certificate of analysis. The post-synthesis functionalized (PSF) particle assay was designed to use approximately 1 well equivalent of reagents per particle. Capture antibodies were reconstituted with 1× phosphate buffered saline (PBS; Welgene). Secondary antibodies, which were biotinylated as provided, were reconstituted with 5% bovine serum albumin (BSA) in 1× PBS. Target proteins were reconstituted and serially diluted in 2% BSA in 1× PBS.Copolymerization functionalized particle fabrication
The particle synthesis closely followed the previously reported protocol.24 IL-6 capture antibodies were functionalized by combining 25 μg μL−1 antibody with 50 μg μL−1 heterobifunctional poly(ethylene glycol) (PEG) linker (ACRL-PEG2000-NHS, Nanocs) in a 4:1 (vol/vol) ratio. The mixture was incubated in a shaker for 3 h at 1000 r.p.m. at 25 °C. Then, the functionalized antibodies were combined in a 1:9 ratio with a prepolymer solution consisting of 20% (v/v) poly(ethylene glycol) diacrylate (PEGDA700, Mn = 700 Da, Sigma-Aldrich), 40% (v/v) poly(ethylene glycol) (PEG200; Mn = 200 Da, Sigma-Aldrich), 35% (v/v) 3× Tris-EDTA (TE) buffer (Sigma-Aldrich), and 5% (v/v) 2-hydroxy-2-methylpropiophenone (photoinitiator, Sigma-Aldrich). The mixture was vortexed for 5 min and then centrifuged for 5 min at 2000g. The supernatant was loaded onto the microchannel for particle synthesis. One cycle of particle synthesis via stop flow lithography consisted of 400 ms of flow, 200 ms of stop, 75 ms of UV exposure, and 200 ms of hold time. 30 min synthesis produced around 2000 particles using a 1-particle photomask. After synthesis, the particles were collected in a microtube and washed three times in 1× PBST (1× PBS with 0.05% TWEEN®20 (Sigma-Aldrich)).
Post-synthesis functionalized particle fabrication
Particle synthesis and functionalization were carried out in two discrete steps. The prepolymer solution for particle synthesis consisted of 20% (v/v) PEGDA700, 40% (v/v) PEG600, 35% (v/v) deionized (DI) water, and 5% (v/v) photoinitiator. One cycle of particle synthesis via stop flow lithography consisted of 400 ms of flow, 200 ms of stop, 75 ms of UV exposure, and 200 ms of hold time. For each protein assay, 1 h synthesis produced around 4000 particles using a 1-particle photomask. Different photomasks were used to encode each protein group. After synthesis, particles were separately collected in a microtube and washed three times in 1× PBST. For particle functionalization, 16.5 μL of particles (55 particles μL−1) were incubated with 12 μL of the reconstituted capture antibody (6 μg μL−1 for IL-6, 3 μg μL−1 for VEGF, 12 μg μL−1 for PIGF, and 3 μg μL−1 for CG beta) and 1.5 μL of heterobifunctional PEG linker (Thiol-PEG2000-NHS, Nanocs) for 48 h at 1500 r.p.m. at 25 °C. Based on our finding that the isolation of steps did not produce any discernible advantage, functionalization of antibodies and their immobilization on the particles were performed in a single step to reduce the number of steps as well as the preparation time. After functionalization, the particles were washed three times in 1× PBST.Protein detection
For each assay condition, 50 functionalized particles in 25 μL 1× PBST were placed in a microtube and mixed with 25 μL of 2× target protein in 2% BSA in 1× PBS. The particles were incubated with the target protein for 2 h (or at various time intervals for detection time analysis) at 1500 r.p.m at 25 °C. After rinsing three times in 1× PBST, the protein-bound particles were suspended in 40 μL 1× PBST. 10 μL of the reconstituted secondary antibody (12.5 ng μL−1 for IL-6, 25 ng μL−1 for VEGF, 15 ng μL−1 for PIGF, and 125 ng μL−1 for CG beta) was added to give a final volume of 50 μL. The mixture was incubated for 1 h at 1500 r.p.m. at 25 °C. After three washes in 1× PBST, the particles were again suspended in 40 μL 1× PBST. The secondary antibodies bound to protein targets were fluorescently labeled by adding 10 μL of streptavidin–phycoerythrin (SA–PE; Life Technologies) diluted in 5% BSA in 1× PBST by a factor of 50. The particles were washed five times with 1× PBST before imaging.Particle imaging and data analysis
Monochrome fluorescence images of the particles were taken on an inverted fluorescence microscope (Axiovert 200, Zeiss) equipped with a scientific complementary metal-oxide-semiconductor (sCMOS) camera (Prime, Photometrics, USA) and a fluorescence light source (Illuminator HXP 120 V, Zeiss). For detecting fluorescence SA–PE signals, the light was filtered using a red fluorescence filter set (λex/λem = 546/590 nm). The images were taken using the 20× objective with a camera exposure time of 50 ms. The intensity of the fluorescence light source was fixed at 1100 mW cm−2. Images were retrieved in TIFF format and the signal brightness was measured using an image processing program (ImageJ).The ELISA
The procedure closely followed the general ELISA protocol provided by the manufacturer. A 96-well microplate (R&D Systems) was coated with 100 μL per well of diluted VEGF capture antibody (1 μg mL−1) and incubated overnight at room temperature. The wells were washed three times with 400 μL of 1× PBST. To block free sites, the plate was incubated for 1 h with 300 μL of 1% BSA in 1× PBS per well. After two washes, the plate was incubated for 2 h with 100 μL of the target protein diluted in 1% BSA in 1× PBS per well. The wells were washed three times before 100 μL of the diluted detection antibody (100 ng mL−1) was added. The plate was incubated for 2 h (or at various time intervals for detection time analysis) at room temperature. After three washes, 100 μL of 40-fold diluted Streptavidin-HRP (R&D Systems) was added to each well and incubated for 20 min at room temperature. The plate was washed three times, and 100 μL of substrate solution (R&D Systems, cat. no. DY999) was added to each well and incubated for 20 min at room temperature. After adding 50 μL of stop solution (R&D Systems, cat. no. DY994), the optical density of each well was determined using a microplate reader (SpectraMax iD5, Molecular Devices) set to 450 nm and 570 nm. The readings at 570 nm were subtracted from the readings at 450 nm to correct for optical imperfections in the plate.Multiplex detection
For each of the 8 combinations, a total of 150 functionalized particles (50 per protein) in 100 μL 1× PBST were placed in a microtube. Then, 100 μL of pre-mixed 2× target proteins in 2% BSA in 1× PBS was added. The pre-mixture was prepared by mixing 25 μL of 2% BSA in 1× PBS with 25 μL 8× target protein in 2% BSA in 1× PBS for target presence and 25 μL of 2% BSA in 1× PBS for target absence. The particles in 8 different target combinations were incubated for 2 h at 1500 r.p.m at 25 °C. The particles were washed three times in 1× PBST and suspended in 20 μL 1× PBST. 10 μL of each of the three secondary antibodies (25 ng μL−1 for VEGF, 15 ng μL−1 for PIGF, and 125 ng μL−1 for CG beta) were pre-mixed and added to the particles to give a final volume of 50 μL. The mixtures were incubated for 1 h at 1500 r.p.m. at 25 °C. After three washes in 1× PBST, the particles were suspended in 120 μL 1× PBST. The secondary antibodies were fluorescently labeled by adding 30 μL of streptavidin–phycoerythrin (SA–PE; Life Technologies) diluted in 5% BSA in 1× PBST by a factor of 50. The particles were washed five times in 1× PBST before imaging.Results and discussion
Design and synthesis of hydrogel microparticles
The most fundamental difference of the hydrogel microparticle based assay from ELISA is its ability to perform multiplex detections. Multiplexing was made possible through graphical encoding of the microparticles using stop flow lithography (Fig. 1). Unlike previously reported barcoded hydrogel microparticles,19,25 which had internal divisions within the particles for target identification and binding, our particle design integrated the two functions for simplicity, high synthesis throughput, and a low fraction of defectives. Because the entire particle is responsible for target binding, particles can be easily synthesized in a single prepolymer flow without structural defects that occur in misalignment of multiple prepolymer flows. Also, more than one particle can be synthesized perpendicular, as well as parallel, to the direction of the flow, increasing the batch size with a 2 dimensional particle array.26 Non-superimposable codes were created with combinations of shape holes on particles shaped as a square prism with a protruding bar (Fig. 1). Changing the shape and number of the holes can easily result in an encoding capacity of over 500. | ||
| Fig. 1 Elimination of capture antibody aggregation in particle functionalization. (a) In copolymerization functionalization, in which capture antibodies are conjugated during particle synthesis, the antibodies are prone to aggregate due to their immiscibility with water-insoluble photoinitiators required for particle synthesis. The irregular fluorescence signals for IL-6 detection at a high concentration (30000 pg mL−1) show the aggregated antibodies, which render the particles inadequate for immunoassay. Note that the image brightness has been increased for improved visualization. (b) In post-synthesis functionalization, thiolated antibodies are conjugated after particle synthesis via thiol–ene “click” reaction with remnant double bonds in the hydrogel. This method allows antibodies to be conjugated without aggregation as evidenced by uniform fluorescence signals at the same target concentration. Scale bars are 50 μm. | ||
Elimination of capture antibody aggregation
The susceptibility of antibodies to aggregation during copolymerization with the hydrogel and the nature of aggregation were examined. Interleukin-6 (IL-6), one of the most frequently assayed cytokines in diverse disciplines,27,28 was assayed with hydrogel particles functionalized with antibodies during particle synthesis. Although the previously reported protocols19,24 were strictly adhered to, microemulsions were observed during particle synthesis, and the aggregation was clearly visible in fluorescence images of the particles after protein detection (Fig. 1a). The protein detection was carried out as outlined in Fig. 2. The resulting detection signals were too low to detect the protein in the ELISA assay range (9.4–600 pg mL−1), and the aggregated spots were only made visible in a high target concentration (30000 pg mL−1). Microemulsions developed upon mixing of the functionalized capture antibodies with the prepolymer solution, and sequential addition of the prepolymer constituents revealed that emulsions are formed only when the photoinitiator is introduced. The water-soluble antibodies were incompatible with the phenyl-containing water-insoluble photoinitiator. This demonstrated that the aggregation is nearly unavoidable in design, and a reliable detection performance is highly challenging to reproduce.
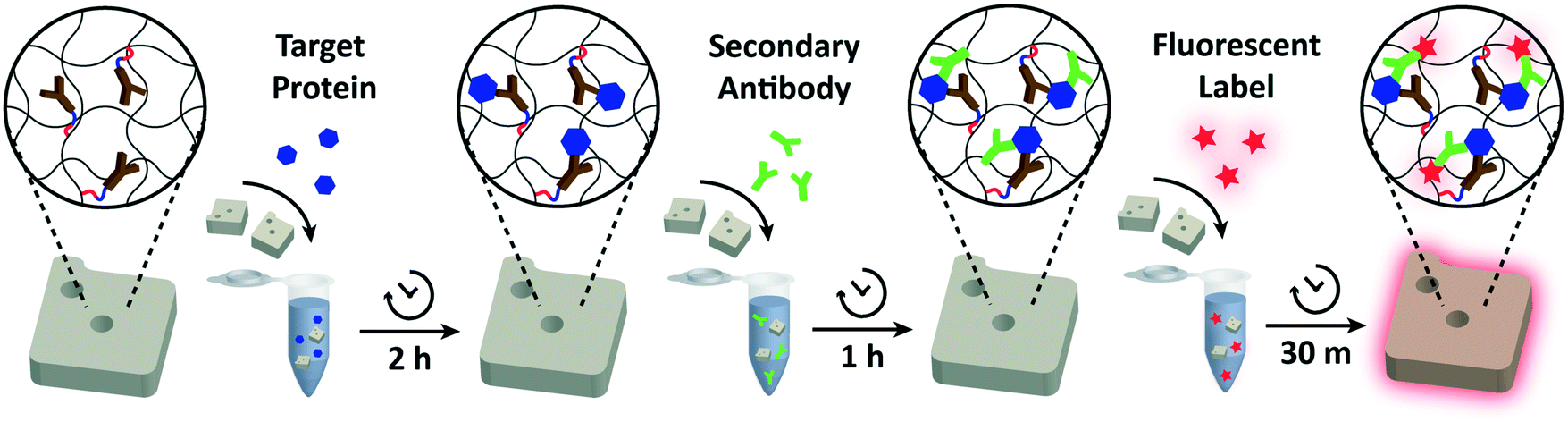 | ||
| Fig. 2 Schematic overview of the assay. The general binding scheme correlates with the sandwich ELISA. Antibody functionalized particles are mixed into the target sample and proteins (blue) are allowed to bind to the capture antibodies. After washing the particles to remove unbound proteins, secondary antibodies (green) are added. These biotinylated antibodies attach to a non-overlapping epitope of the target protein, forming a sandwich unit. Another wash step is performed to remove excess secondary antibodies. Then, streptavidin–phycoerythrin (SA–PE; red) is added to fluorescently label the particle-bound secondary antibodies. Unbound fluorescent labels are washed in a final wash step and the particles are imaged using fluorescence microscopy. | ||
Since the aggregation is caused by the immiscibility of antibodies with the photoinitiator required for hydrogel particle synthesis, the most definitive solution would be to spatially isolate the two. Our approach was to functionalize the particles in a protein-stabilizing solution after particle synthesis in the absence of the photoinitiator, enabling a uniform dispersion of antibodies for interaction with the particles. Intuitively, antibodies can be conjugated after particle synthesis by adding antibody-reactive species in the prepolymer mixture prior to particle synthesis.29–32 However, the compatibility of the additional species with the prepolymer is not guaranteed, and their copolymerization can undesirably alter the particles' physical and chemical properties. For instance, the incorporation of charged species can result in non-specific binding of targets of the opposite charge33,34 and degrade the particles' structural integrity by altering the swelling behaviour.35 Also, copolymerization of additional species is particularly disadvantageous for particle synthesis via flow lithography because of its low copolymerization yield (∼10%) inevitable due to its limiting polymerization conditions.15,36 The low yield directly restricts the availability of capture antibodies that are responsible for the assay. To circumvent these issues, we directed our focus to the existence of unreacted monomer active sites remaining in the hydrogel network after particle synthesis. The unreacted active sites, which are predominantly acrylate groups for poly(ethylene glycol) (PEG) based hydrogels, exist as a result of incomplete free radical polymerization reaction in flow lithography due to insufficient radical initiation and steric hindrance.37 Using these remnant active sites for antibody conjugation removes the complications of introducing the aforementioned additional species. Our work aimed to fully exploit the abundance of the unreacted active sites, which have neither received much attention nor been used for reaction.
To make use of the remnant active groups for functionalization, we employed the thiol–ene chemistry (hydrothiolation of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond), which is recognized as a “click” reaction given its simplicity and versatility, the absence of by-products, and its high yield without catalysts.38 The covalent conjugation of capture antibodies using the thiol–ene chemistry was implemented using a heterobifunctional PEG linker (SH-PEG-NHS). The N-hydroxysuccinimide (NHS) ester group reacts with primary amines found in antibodies, and the thiol group, bridged by PEG, binds to the double bond of remnant acrylate groups in the particles. Antibody conjugation may also be aided by direct thiol–ene reactions with sulfhydryl groups on the antibody.39 IL-6 detection using post-synthesis functionalized (PSF) particles demonstrated the complete elimination of antibody aggregation evidenced by uniform fluorescence detection signals (Fig. 1b). Compared to particles copolymerized with capture antibodies during synthesis, post-synthesis functionalized (PSF) particles produced uniform and much stronger detection signals that were readily distinguishable from the experimental control.
C bond), which is recognized as a “click” reaction given its simplicity and versatility, the absence of by-products, and its high yield without catalysts.38 The covalent conjugation of capture antibodies using the thiol–ene chemistry was implemented using a heterobifunctional PEG linker (SH-PEG-NHS). The N-hydroxysuccinimide (NHS) ester group reacts with primary amines found in antibodies, and the thiol group, bridged by PEG, binds to the double bond of remnant acrylate groups in the particles. Antibody conjugation may also be aided by direct thiol–ene reactions with sulfhydryl groups on the antibody.39 IL-6 detection using post-synthesis functionalized (PSF) particles demonstrated the complete elimination of antibody aggregation evidenced by uniform fluorescence detection signals (Fig. 1b). Compared to particles copolymerized with capture antibodies during synthesis, post-synthesis functionalized (PSF) particles produced uniform and much stronger detection signals that were readily distinguishable from the experimental control.
Assay performance of the PSF assay
Post-synthesis functionalization not only eliminated antibody aggregation, but it also resulted in improved assay performance. For performance evaluation, ELISA, to which most multiplex assay platforms are compared, served as the basis of comparison because of its acceptance as the “gold standard” for protein measurement in clinical tests.14 Three of the most important criteria for protein assays are discussed: detection sensitivity, assay range, and detection time.The performance of an immunoassay is largely dependent on the antibody affinity, the density of capture antibodies, and the reaction environment.19,40 Since the antibody affinities vary even among those that target the same protein, identical antibodies should be used for valid comparisons between different platforms.41 Because we used identical antibodies supplied in ELISA kits of verified performance, our result reflects the performance differences derived from intrinsic characteristics of our platform. Because a single PSF particle has the function of one ELISA well, each particle was designed to react with 1 well-equivalent of capture and secondary antibodies. The target proteins were selected in consideration of our future goal to develop a detection technique for preeclampsia, which is a serious pregnancy complication whose unmet need for early diagnosis has been emphasized.42,43 We detected three proteins that are known to be dysregulated in preeclampsia: vascular endothelial growth factor (VEGF), placental growth factor (PlGF), and chorionic gonadotropin beta (CG beta).44,45 VEGF and PIGF are growth factors involved in angiogenesis, the development of new blood vessels, which takes place during placental development.46 CG beta, commonly tested during pregnancy, is a glycoprotein hormone secreted in pregnancy by trophoblastic tissues, and its abnormal level has been suggested as a predictor of preeclampsia.47,48
The detection performances of ELISA and PSF particle assay were compared by creating standard calibration curves for VEGF (Fig. 3a). ELISA was performed as directed by the provided protocol with the following reaction times: 2 h target conjugation, 2 h secondary antibody conjugation, 20 min enzyme conjugation, and 20 min substrate incubation. The PSF particle assay followed the previously reported reaction times:24 2 h target conjugation, 1 h secondary antibody conjugation, and 30 min fluorescent labeling (Fig. 2). As expected, ELISA's assay range was in agreement with the kit specification of 31.2–2000 pg mL−1 (1.8 log). The assay using PSF particles, however, was able to detect VEGF up to 60000 pg mL−1, which was the practical limit of target dilution. The lower bound of the range was determined by the limit of detection (LOD) of 17.7 pg mL−1, resulting in a 3.5 log range. The lower bound of the assay was not limited by the non-linearity of the curve but its LOD, which is defined as the concentration that corresponds to the control signal plus three standard deviations (three signal to noise ratios). Also, the rates of detection were compared by examining the increase in detection signal over a 240 min target incubation period (Fig. 3b). The signals were normalized with respect to the signals at 240 min, which were taken as the saturation points. The PSF particle assay exhibited a higher rate of detection, evidenced by the higher slope up until 60 min of incubation. A little less than 1 h incubation in the PSF particle assay produced a signal that is comparable to 2 h incubation in the ELISA. This implies a reduction of the total assay time from 4 h 40 min in ELISA to 2 h 30 m in the PSF particle assay. From the results, it was apparent that the PSF particle assay demonstrated higher detection sensitivity, a broader assay range, and faster detection compared to the ELISA. While the assay was performed in phosphate buffered saline containing bovine serum albumin as in the general ELISA, detection of VEGF in animal serum did not show a significant deviation in the calibration curve (ESI† Fig. S1). Similar results were observed in the standard calibration curves of two other preeclampsia-relevant proteins, PIGF and CG beta (Fig. 4). PSF particle assays of both proteins exhibited an improved LOD and a greater assay range compared to the ELISA (Table 1). The fluorescence images of the PIGF and CG beta particles are available in the ESI (Fig. S2 and S3†). A particle's fluorescence pattern is dependent on the diffusion rate of the specific target and its antibody pairs. Reactions of large-sized targets and antibodies are characterized by a large Damköhler number due to the limited diffusion into the inner matrix, resulting in concentrated fluorescence on the edges of the particle, as seen in the detection of VEGF and PIGF. For small-sized targets and antibodies, however, the rate of diffusion is greater than the rate of reaction. In this case, diffusion reaches an equilibrium before the reaction, so the fluorescence intensity is uniform throughout the particle, as observed in the detection of CG beta. For signal measurement, the average of the values of the entire particle area was recorded. It should be noted that the particles' orientation, facing front or back, did not result in a difference in signal intensity (Fig. S4†).
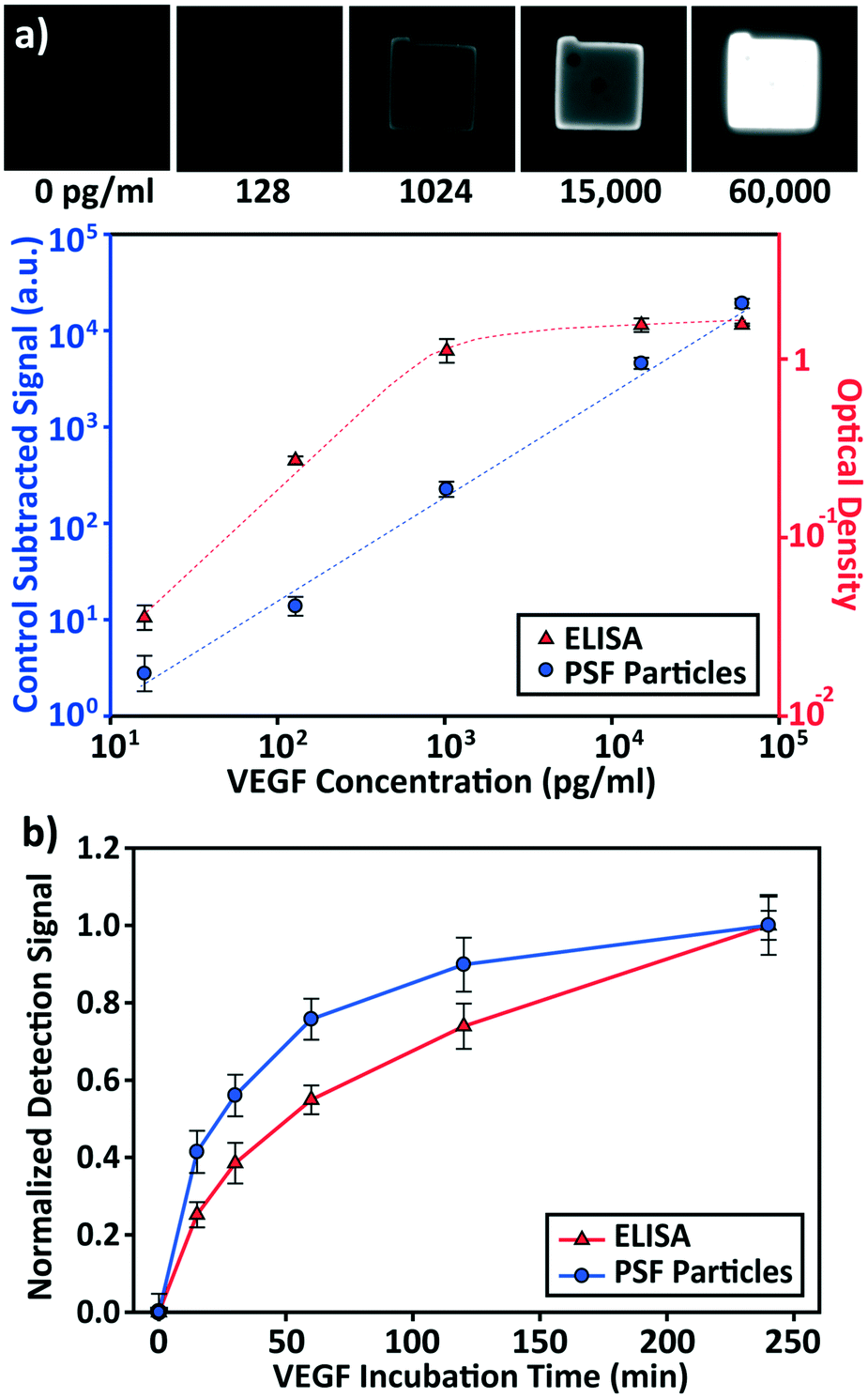 | ||
| Fig. 3 Assay performance evaluation and its comparison with the ELISA. (a) VEGF standard calibration curves of ELISA and post-synthesis functionalized (PSF) particle based assay. ELISA's performance is in accordance with the specified assay range of the kit (31.2–2000 pg mL−1). PSF particle assay features a broader assay range of 17.7–60000 pg mL−1, with the lower bound as the limit of detection. (b) Detection signal vs. VEGF incubation time. The signals were normalized with respect to the signals at 240 min. For ELISA, each data point and the vertical error bar represent the average signal and standard deviation of 3 wells. For PSF particle assay, each data point and the vertical error bars represent the average signal and standard deviation of >10 particles. | ||
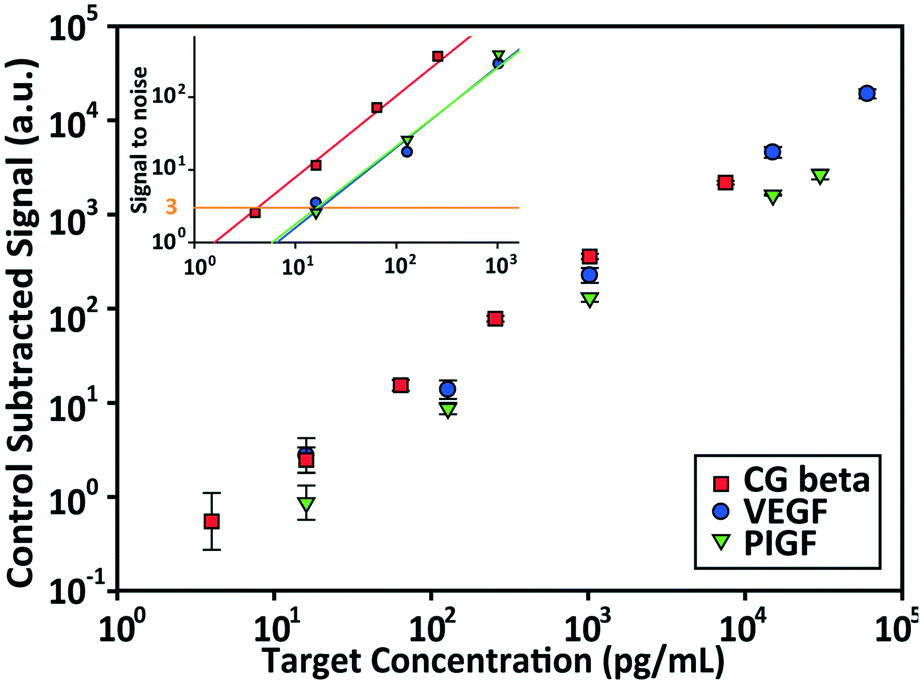 | ||
| Fig. 4 Standard calibration curves for VEGF, PIGF, and CG beta and their signal to noise plots. The limit of detection (LOD) is defined as the concentration that produces three signal to noise ratios (control subtracted signal divided by the standard deviation of the control). The linear lines are the least squares regression lines of the signal to noise ratios. Each data point and the vertical error bars represent the average signal and standard deviation of >10 particles. | ||
| Target | Assay | Range [LOD-max.]d (log10) |
|---|---|---|
| a Product information R&D Systems Human VEGF Duoset ELISA (DY293B). b Product information R&D Systems Human PIGF Duoset ELISA (DY264). c Product information R&D Systems Human CG beta (HCG beta) Duoset ELISA (DY9034-05). d The lower bound of the assay range is determined to be the limit of detection (LOD), which lies on the linear plot. | ||
| VEGF | ELISAa | [31.2–2000 pg mL−1] (1.8) |
| PSF particle assay | [17.7–60000 pg mL−1] (3.5) |
|
| PIGF | ELISAb | [31.2–2000 pg mL−1] (1.8) |
| PSF particle assay | [17.5–30000 pg mL−1] (3.2) |
|
| CG beta | ELISAc | [7.8–500 pg mL−1] (1.8) |
| PSF particle assay | [4.2–7500 pg mL−1] (3.3) | |
The enhanced performance is attributable to the characteristics of our hydrogel microparticles. Unlike two dimensional antibody immobilization in the ELISA, the hydrogel microparticles' hydrophilic three dimensional scaffolds enable a higher loading capacity of capture antibodies.49,50 This is further coupled with the high efficiency of post-synthesis functionalization to achieve a higher antibody density per projected area. Assuming first-order Langmuir kinetics for the antibody–antigen interaction, a higher antibody density would result in a greater number of bound targets at equilibrium and display a heightened signal, increasing the detection sensitivity and decreasing the detection time.51,52 Also, a higher antibody density allows for the quantification of targets in a high concentration by increasing the saturation capacity for the target protein. This widens the assay range, which is beneficial in avoiding unnecessary dilutions of a sample of an unknown concentration, providing substantial improvement in assay time and cost.53 It should be noted that a higher antibody density is unrealizable in two dimensional surface immobilizations due to steric limitations as well as the limitation of a single layer surface. There exists a maximum antibody density (∼104 molecules μm−2) that can be achieved with surface immobilization, and increasing the antibody concentration does not result in a higher density beyond this point.50
Multiplex detection
Independent of sensitivity, an assay must demonstrate high specificity for target proteins in order to detect multiple targets simultaneously. The specificity of an assay is determined by the compatibility of the platform with the antibody–antigen interactions. The specificity of the PSF particle assay was investigated with simultaneous detections of the three preeclampsia-relevant proteins, VEGF, PIGF, and CG beta. These proteins were confirmed to exhibit no cross-reactivity or interference in the ELISA. The assays were performed in a total of eight possible combinations of the three proteins in order to uncouple and identify potential causes of cross reactivity (Fig. 5). The three proteins were spiked at different concentrations to mimic different concentrations of proteins present in clinical samples. Protein-specific detection signals were interpreted by the particle code and the fluorescence intensities. The recovery of all three proteins were within 17% of the actual spike concentration and showed no significant signs of cross reactivity (Table 2). Such recoveries demonstrate the multiplex capability of the PSF particle assay that does not compromise the specificity of the antibody–antigen interactions.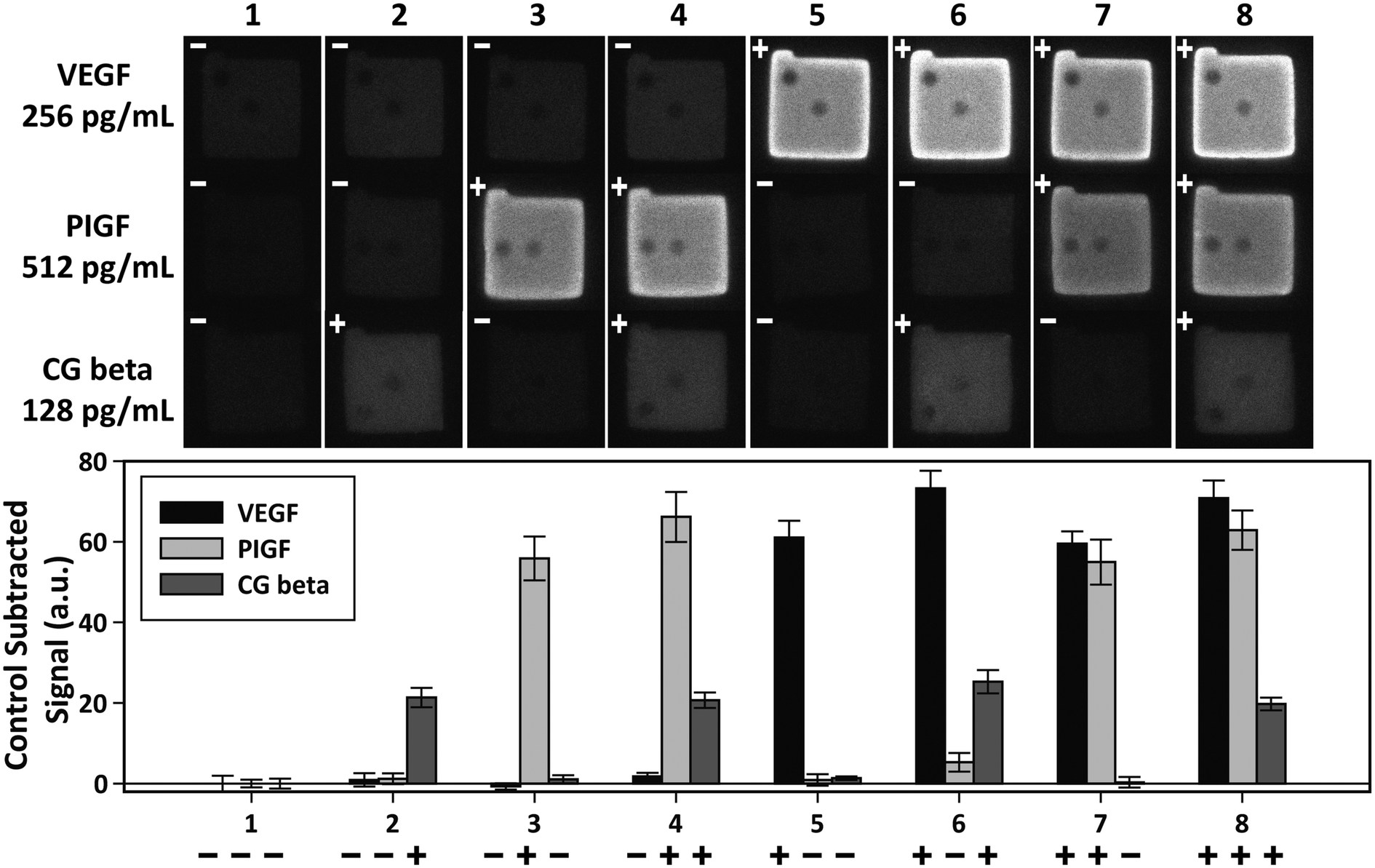 | ||
| Fig. 5 Multiplex detection of VEGF, PIGF, and CG beta. The three proteins were simultaneously assayed in eight possible combinations to check for cross-reactivity. The plus and minus signs indicate the presence (+) and absence (−) of the target. VEGF, PIGF, and CG beta were spiked at 256 pg mL−1, 512 pg mL−1, and 128 pg mL−1, respectively. The bar graph shows the average of control subtracted signals and the standard deviation of >10 particles (provided in Table 2). | ||
| Case | Control subtracted signala (a.u.) | |||||
|---|---|---|---|---|---|---|
| VEGF | PIGF | CG beta | ||||
| a All detection signals for each target were subtracted with the control signals (case 1) to exclude non-specific signals. Each signal is the mean ± standard deviation of >10 particles. The signs in front of the signals indicate the presence (+) and absence (−) of the target. VEGF, PIGF, and CG beta were spiked at 256 pg mL−1, 512 pg mL−1, and 128 pg mL−1, respectively. b The four signals of target presence were averaged for each target. c The recovery of each target was calculated as the concentration determined from the standard calibration curve divided by the actual spike-in concentration. | ||||||
| 1 | − | 0 ± 2.0 | − | 0 ± 0.9 | − | 0 ± 1.2 |
| 2 | − | 0.9 ± 1.7 | − | 1.2 ± 1.3 | + | 21.4 ± 2.4 |
| 3 | − | −0.7 ± 0.8 | + | 55.8 ± 5.5 | − | 1.1 ± 1.0 |
| 4 | − | 1.8 ± 0.9 | + | 66.2 ± 6.2 | + | 20.7 ± 1.9 |
| 5 | + | 61.1 ± 7.1 | − | 0.9 ± 1.4 | − | 1.3 ± 0.4 |
| 6 | + | 73.2 ± 4.3 | − | 5.3 ± 2.3 | + | 25.3 ± 2.9 |
| 7 | + | 59.5 ± 3.1 | + | 54.9 ± 5.6 | − | 0.3 ± 1.3 |
| 8 | + | 70.8 ± 4.4 | + | 62.9 ± 4.9 | + | 19.7 ± 1.6 |
| Avg.b | 66.2 ± 6.9 | 60.0 ± 5.4 | 21.8 ± 2.4 | |||
| Recov.c (%) | 117.3 | 110.4 | 100.0 | |||
Conclusions
In this paper, we introduced a new method of functionalizing hydrogel microparticles for multiplex protein detection. Unlike the current method of functionalizing hydrogels with antibodies during particle synthesis, our method functionalizes them after particle synthesis by exploiting the reactivity of remnant active groups in the hydrogels. Our antibody immobilization method was able to effectively eliminate the antibody aggregation problem that has been associated with the current method. Moreover, PSF particles demonstrated an enhanced assay performance, outperforming ELISA in terms of detection sensitivity, range, and time. We were able to detect three proteins in multiplex assays with no significant signs of cross reactivity, which demonstrated the compatibility of our immunoassay platform for multiplex detection. As we have only presented the feasibility of using PSF hydrogel microparticles for multiplex immunoassays, we plan to further develop the platform for application in the diagnosis of preeclampsia by expanding the target protein panels and detecting in clinical samples. We also anticipate the utilization of the PSF particles in point-of-care applications through combination with microfluidic chips that enable semi to full automation of the assay. Although we have focused our attention on the application of post-synthesis functionalization in proteomics, the functionalization method itself may well be employed in functionalizing hydrogel particles for other various practices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Engineering Research Center of Excellence Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2016R1A5A1010148) and the Next-Generation Biogreen 21 Program funded by the Rural Development Administration of the Republic of Korea (No. PJ013158). This research was also supported by the Basic Science Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2018R1D1A1B07046577 and NRF-2017R1D1A1B03029883).References
- L. Asensio, I. González, T. García and R. Martín, Food Control, 2008, 19, 1 CrossRef CAS.
- G. Hopfgartner, A. Lesur and E. Varesio, TrAC, Trends Anal. Chem., 2013, 48, 52 CrossRef CAS.
- T. Kupiec, L. DeCicco, V. Spiehler, G. Sneed and P. Kemp, J. Anal. Toxicol., 2002, 26, 513 CrossRef CAS PubMed.
- W. E. Benitz, M. Y. Han, A. Madan and P. Ramachandra, Pediatrics, 1998, 102, e41 CrossRef CAS PubMed.
- K. Blennow, NeuroRx, 2004, 1, 213 CrossRef PubMed.
- E. Engvall and P. Perlmann, Immunochemistry, 1971, 8, 871 CrossRef CAS PubMed.
- B. Schweitzer and S. F. Kingsmore, Curr. Opin. Biotechnol., 2002, 13, 14 CrossRef CAS PubMed.
- E. F. Petricoin III, V. E. Bichsel, V. S. Calvert, V. Espina, M. Winters, L. Young, C. Belluco, B. J. Trock, M. Lippman and D. A. Fishman, J. Clin. Oncol., 2005, 23, 3614 CrossRef PubMed.
- S. T. Chang, J. M. Zahn, J. Horecka, P. L. Kunz, J. M. Ford, G. A. Fisher, Q. T. Le, D. T. Chang, H. Ji and A. C. Koong, J. Transl. Med., 2009, 7, 105 CrossRef PubMed.
- E. Burgos-Ramos, G. Á. Martos-Moreno, J. Argente and V. Barrios, Adv. Immunoassay Technol., 2012, 165 CAS.
- R. Gitau, N. M. Fisk, J. M. Teixeira, A. Cameron and V. Glover, J. Clin. Endocrinol. Metab., 2001, 86, 104 CAS.
- Q. Fu, J. Zhu and J. E. Van Eyk, Clin. Chem., 2010, 56, 314 CrossRef CAS PubMed.
- M. F. Templin, D. Stoll, M. Schrenk, P. C. Traub, C. F. Vöhringer and T. O. Joos, Drug Discovery Today, 2002, 7, 815 CrossRef CAS PubMed.
- M. F. Elshal and J. P. McCoy, Methods, 2006, 38, 317 CrossRef CAS PubMed.
- G. C. Le Goff, R. L. Srinivas, W. A. Hill and P. S. Doyle, Eur. Polym. J., 2015, 72, 386 CrossRef CAS PubMed.
- S. Pang, J. Smith, D. Onley, J. Reeve, M. Walker and C. Foy, J. Immunol. Methods, 2005, 302, 1 CrossRef CAS PubMed.
- S. Birtwell and H. Morgan, Integr. Biol., 2009, 1, 345 RSC.
- S. F. Kingsmore, Nat. Rev. Drug Discovery, 2006, 5, 310 CrossRef CAS PubMed.
- D. C. Appleyard, S. C. Chapin and P. S. Doyle, Anal. Chem., 2010, 83, 193 CrossRef PubMed.
- D. Dendukuri, S. S. Gu, D. C. Pregibon, T. A. Hatton and P. S. Doyle, Lab Chip, 2007, 7, 818 RSC.
- J. Moorthy, R. Burgess, A. Yethiraj and D. Beebe, Anal. Chem., 2007, 79, 5322 CrossRef CAS PubMed.
- G. Nallur, R. Marrero, C. Luo, R. M. Krishna, P. E. Bechtel, W. Shao, M. Ray, S. Wiltshire, L. Fang and H. Huang, Biomed. Microdevices, 2003, 5, 115 CrossRef CAS.
- A. Y. Rubina, A. Kolchinsky, A. A. Makarov and A. S. Zasedatelev, Proteomics, 2008, 8, 817 CrossRef CAS PubMed.
- D. C. Appleyard, S. C. Chapin, R. L. Srinivas and P. S. Doyle, Nat. Protoc., 2011, 6, 1761 CrossRef CAS PubMed.
- S. C. Chapin, D. C. Appleyard, D. C. Pregibon and P. S. Doyle, Angew. Chem., Int. Ed., 2011, 50, 2289 CrossRef CAS PubMed.
- K. W. Bong, K. T. Bong, D. C. Pregibon and P. S. Doyle, Angew. Chem., 2010, 122, 91 CrossRef.
- M. Helle, L. Boeije, E. de Groot, A. de Vos and L. Aarden, J. Immunol. Methods, 1991, 138, 47 CrossRef CAS PubMed.
- J.-M. Fernandez-Real, M. Vayreda, C. Richart, C. Gutierrez, M. Broch, J. Vendrell and W. Ricart, J. Clin. Endocrinol. Metab., 2001, 86, 1154 CrossRef CAS PubMed.
- S. Jung and H. Yi, Langmuir, 2012, 28, 17061 CrossRef CAS PubMed.
- S. Park, H. J. Lee and W.-G. Koh, Sensors, 2012, 12, 8426 CrossRef CAS PubMed.
- J. Hu, X.-W. Zhao, Y.-J. Zhao, J. Li, W.-Y. Xu, Z.-Y. Wen, M. Xu and Z.-Z. Gu, J. Mater. Chem., 2009, 19, 5730 RSC.
- M. He and A. E. Herr, Nat. Protoc., 2010, 5, 1844 CrossRef CAS PubMed.
- H. C. Graves, J. Immunol. Methods, 1988, 111, 157 CrossRef CAS PubMed.
- A. Gertler, Eur. J. Biochem., 1971, 20, 541 CrossRef CAS PubMed.
- L. Chen, H. Z. An, R. Haghgooie, A. T. Shank, J. M. Martel, M. Toner and P. S. Doyle, Small, 2016, 12, 2001 CrossRef CAS PubMed.
- D. C. Pregibon and P. S. Doyle, Anal. Chem., 2009, 81, 4873 CrossRef CAS PubMed.
- P. Wambua, J. Ivens and I. Verpoest, Compos. Sci. Technol., 2003, 63, 1259 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17 RSC.
- C. N. Salinas and K. S. Anseth, Macromolecules, 2008, 41, 6019 CrossRef CAS.
- L. H. Stanker, P. Merrill, M. C. Scotcher and L. W. Cheng, J. Immunol. Methods, 2008, 336, 1 CrossRef CAS PubMed.
- M. W. Steward and A. M. Lew, J. Immunol. Methods, 1985, 78, 173 CrossRef CAS PubMed.
- G. Dekker and B. M. Sibai, Am. J. Obstet. Gynecol., 1991, 165, 160 CrossRef CAS PubMed.
- S. Monte, J. Prenat. Med., 2011, 5, 69 Search PubMed.
- D. M. Carty, C. Delles and A. F. Dominiczak, Trends Cardiovasc. Med., 2008, 18, 186 CrossRef CAS PubMed.
- A. M. Ashour, E. S. Lieberman, L. E. W. Haug and J. T. Repke, Am. J. Obstet. Gynecol., 1997, 176, 438 CrossRef CAS PubMed.
- R. Thadhani, W. P. Mutter, M. Wolf, R. J. Levine, R. N. Taylor, V. P. Sukhatme, J. Ecker and S. A. Karumanchi, J. Clin. Endocrinol. Metab., 2004, 89, 770 CrossRef CAS PubMed.
- C.-D. Hsu, D. W. Chan, B. Iriye, T. R. Johnson, S.-F. Hong and J. T. Repke, Am. J. Obstet. Gynecol., 1994, 170, 1135 CrossRef CAS PubMed.
- F. Muller, L. Savey, B. Le Fiblec, L. Bussières, G. Ndayizamba, J. C. Colau and P. Giraudet, Am. J. Obstet. Gynecol., 1996, 175, 37 CrossRef CAS PubMed.
- R. L. Srinivas, S. C. Chapin and P. S. Doyle, Anal. Chem., 2011, 83, 9138 CrossRef CAS PubMed.
- D. Zubtsov, E. Savvateeva, A. Y. Rubina, S. Pan'kov, E. Konovalova, O. Moiseeva, V. Chechetkin and A. Zasedatelev, Anal. Biochem., 2007, 368, 205 CrossRef CAS PubMed.
- N. Sorokin, V. Chechetkin, S. Pan'kov, O. Somova, M. Livshits, M. Donnikov, A. Turygin, V. Barsky and A. Zasedatelev, J. Biomol. Struct. Dyn., 2006, 24, 57 CrossRef CAS PubMed.
- D. S. Dandy, P. Wu and D. W. Grainger, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8223 CrossRef CAS PubMed.
- S. Eda, J. Kaufmann, W. Roos and S. Pohl, J. Clin. Lab. Anal., 1998, 12, 137 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8lc01160e |
| This journal is © The Royal Society of Chemistry 2019 |