
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical oxidation of benzylic primary and secondary alcohols utilizing air as the oxidant†
Nikolaos F.
Nikitas
,
Dimitrios Ioannis
Tzaras
,
Ierasia
Triandafillidi
and
Christoforos G.
Kokotos
 *
*
Laboratory of Organic Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis, Athens 15771, Greece. E-mail: ckokotos@chem.uoa.gr
First published on 11th December 2019
Abstract
A mild and green photochemical protocol for the oxidation of alcohols to aldehydes and ketones was developed. Utilizing thioxanthenone as the photocatalyst, molecular oxygen from air as the oxidant and cheap household lamps or sunlight as the light source, a variety of primary and secondary alcohols were converted into the corresponding aldehydes or ketones in low to excellent yields. The reaction mechanism was extensively studied.
Introduction
Carbonyl compounds are one of the most important functionalities in pharmaceuticals and their synthesis via alcohol oxidation is a key reaction in chemical science.1 Among the traditional and most common methods for the oxidation of alcohols are the use of stoichiometric amounts of oxidants, such as MnO2, hypochlorite, permanganate, osmium oxide, activated dimethylsulfoxide (DMSO), hypervalent iodine reagents, peroxide-based or salt-based protocols, corresponding to a number of named reactions in organic synthesis, that nowadays are common practice (Scheme 1A).1,2 Unfortunately, these processes create large volumes of waste that deteriorate the climate on Earth, while most of them can be also considered quite toxic. In order to avoid the use of large amounts of toxic and hazardous reagents, a variety of homogeneous and heterogeneous catalytic systems have been reported. In an effort to make these reactions friendlier to the chemical industry and more environmentally-friendly, new, green and sustainable protocols have been developed utilizing air or oxygen as the oxidant (Scheme 1B).3,4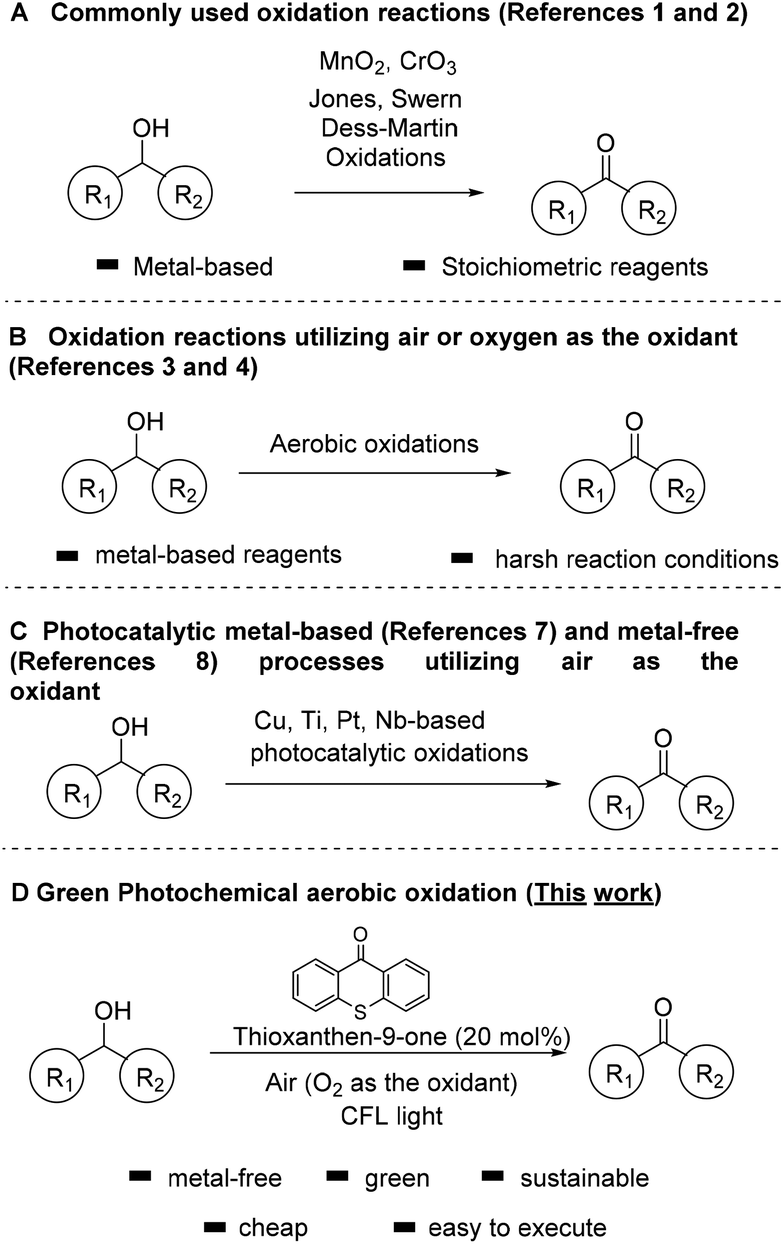 | ||
| Scheme 1 Different approaches for the oxidation of alcohols to carbonyl compounds. | ||
These catalytic systems use either metal-complexes or metal nanoparticles or Tempo-like radicals as catalysts which, unfortunately, lead to a variety of byproducts and wastes. For example, the group of Iwabuchi have elegantly demonstrated the oxidation of alcohols to carbonyl compounds, utilizing a balloon of oxygen or air as the oxidant and a synthetic catalyst, which is not commercially available (TEMPO-like reagent).4d Unfortunately, the use of acetic acid as the solvent can be considered harsh for sensitive groups and not environmentally friendly. Moreover, biocatalysis has also provided significant advances in oxidation reactions.5
Over the past two decades, photocatalysis has made a powerful impact in the activation of molecules and its applications have increased significantly.6 Inspired by the potential of photocatalysis, several metal-based photocatalysts have been reported, based on Cu, Ti, Pt, Nb, Pt-functionalized porphyrinic MOF, supported nanoparticles, etc. for the oxidation of alcohols (Scheme 1C).7 Although most of these processes are very elegant, the requirement of a metal cluster can be considered as a disadvantage. In an effort to prevent the use of toxic metallic reagents in oxidation reactions, there is a strong requirement for alternative metal-free photocatalysts.8 Photoorganocatalysis is a low-cost and environmentally friendly alternative.9 Photocatalysis has already been used for the oxidation of alcohols to carbonyl compounds, utilizing various oxidants, such as DDQ or TBHP.10 Small organic molecules have also shown their activity towards the activation of O2 under visible light, and this concept has already shown powerful applications in organic synthesis.11 A part of these applications constitutes the oxidation of alcohols to carbonyl compounds.12
We have a long interest in photocatalysis13 and in sustainable processes for oxidations.14 In an effort to develop a green catalytic protocol, herein, we report a mild photochemical system for the selective oxidation of alcohols, without the use of toxic metal clusters and complexes or the use of stoichiometric oxidation. The presented protocol herein, focuses on an eco-friendly metal-free process, which avoids the use of toxic and hazardous reagents, minimizing the need for energy consumption (since the reaction can be also performed by sunlight irradiation) and utilizes air, which is considered by green metrics the best source, as the oxidant (Scheme 1D).
Results and discussion
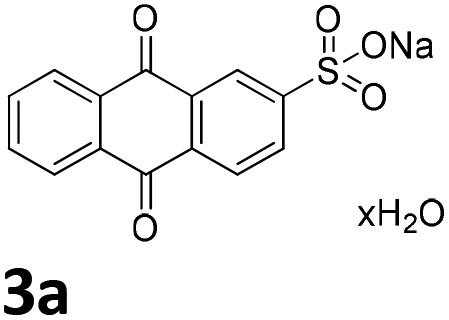
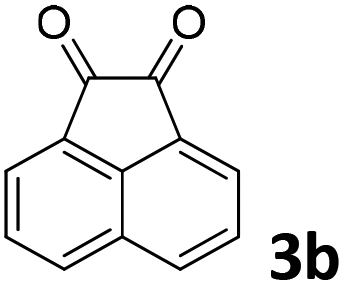
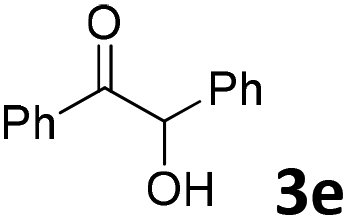
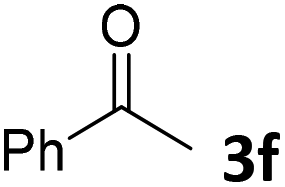
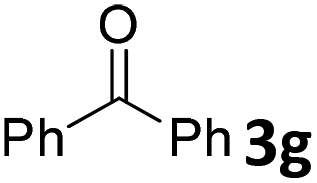
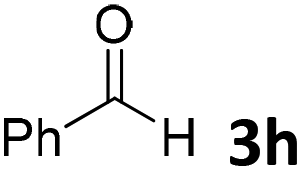
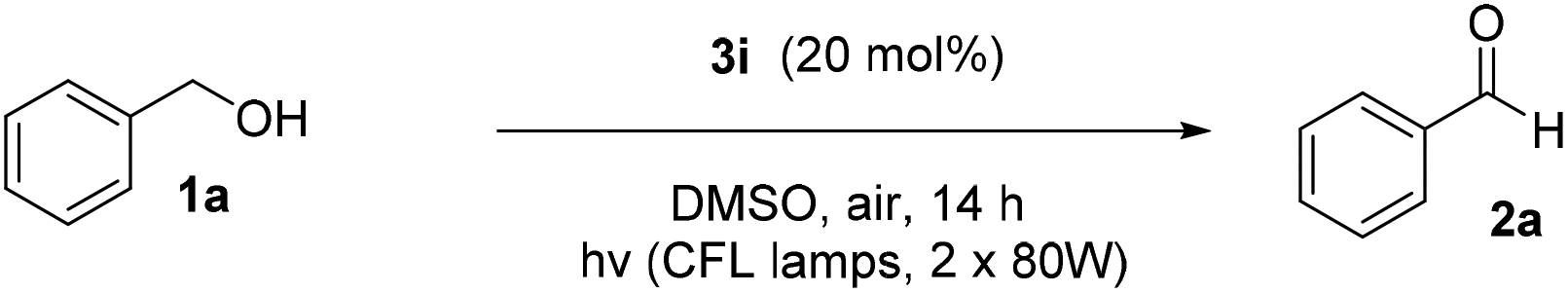
We initiated our study utilizing benzyl alcohol (1a) as the substrate and air as the oxidant. In the optimization of the reaction conditions, a variety of different catalysts and solvents were tested (Table 1). As shown in Table 1, aromatic ketones, in the role of the catalyst, such as sodium anthraquinone-2-sulfonate (3a) or acenaphthenquinone (3b) provided low yields (Table 1, entries 1 and 2). A common photocatalytic organic dye, eosin Y (3c), showed similar reactivity (Table 1, entry 3). Different type of catalysts, such as acetophenone (3f) or benzophenone (3g), which are usually used as photoinitiators, afforded the product in low yields (Table 1, entries 4–8). In contrast, the use of thioxanthenone (3i) as the photocatalyst, provided the desired aldehyde in 61% yield (Table 1, entry 9). Further increase in the reaction time dropped the yield in 55% (Table 1, entry 10). This result was very interesting, and by monitoring the reaction outcome by 1H-NMR, it was possible to detect the production of benzoic acid. This meant that overoxidation to the corresponding carboxylic acid was occuring, a common problem in the selective oxidation of primary alcohols.15 Therefore, we decided to reduce the reaction time and examine whether we could obtain a higher yield of the product. To our delight, when the reaction time dropped to 14 h, we managed to obtain the aldehyde in 85% yield (Table 1, entry 11). For more details regarding the ratio of overoxidation, see ESI.† We also examined if benzaldehyde could act as the photocatalyst (autocatalytic reaction), which gave a poor 5% yield (Table 1, entry 8). Since DMSO is still considered a practical solvent for chemical industries, but it would be highly desirable if we could substitute it with a greener solvent, our next step was to examine the role of the solvent in the reaction. Acetonitrile provided the product in 33% yield (Table 1, entry 12), while other polar solvents, such as water or DMF, afforded lower results (Table 1, entries 13 and 14). When non-polar solvents, such as toluene, were used no product was detected (Table 1, entry 15). When a mixture of MeCN/DMSO (instead of MeCN alone) was used as the solvent, the yield was increased from 33% to 58%, which indicates that DMSO plays a significant role in the reaction outcome and cannot be replaced (Table 1, entry 16 vs. 12). Trying to determine the role of the solvent, it is known that DMSO can stabilize the excited state of diaryl ketones (of similar chemical type with 3i), so it is rational to assume that the excited state of thioxanthene-9-one (3i) has a prolonged lifetime in the presence of DMSO as the solvent, making it an important part of the reaction.16|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Catalyst | Time (h) | Yielda (%) |
| a All reactions were carried out with 1a (0.20 mmol), catalyst (20 mol%) and solvent (0.6 mL) in an open-air vessel, under household bulb irradiation. b 5 mol% catalyst loading. c Isolated yield. | ||||
| 1 | DMSO |
|
18 | 34 |
| 2 | DMSO |
|
18 | 11 |
| 3b | DMSO | Eosin Y 3c | 18 | 29 |
| 4 | DMSO |
|
18 | 6 |
| 5 | DMSO |
|
18 | 12 |
| 6 | DMSO |
|
18 | 26 |
| 7 | DMSO |
|
18 | 5 |
| 8 | DMSO |
|
18 | 4 |
| 9 | DMSO |
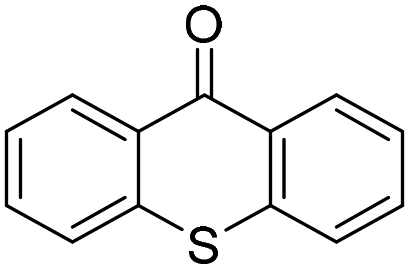
|
18 | 61 |
| 10 | DMSO | 19 | 55 | |
| 11 | DMSO | 14 | 85(82)c | |
| 12 | MeCN | 14 | 33 | |
| 13 | H2O | 14 | 0 | |
| 14 | DMF | 14 | 25 | |
| 15 | Toluene | 14 | 0 | |
| 16 | MeCN/DMSO | 14 | 58 | |
Having in hand the optimized reaction conditions, we then explored the substrate scope (Scheme 2). Various mono- or disubstituted benzyl alcohols were used and provided the desired aldehydes 2a–2c in good yields. Furthermore, ortho-substituted benzyl alcohols afforded the desired aldehydes 2d and 2e in lower yields, probably due to steric hindrance close to the center of the reaction. The electronics on the aromatic ring plays a significant role in the reaction outcome, since products 2f–i were obtained in various yields (from 14 to 70%). Unfortunately, non-benzylic primary alcohols, like 3-phenylpropanol and 1-hexanol, did not afford the oxidation. When the photochemical protocol was applied in an allylic alcohol (cinnamyl alcohol), the desired α,β-unsaturated aldehyde 2j was formed in 36% yield. In the case of benzyl secondary alcohols, we determined that we could lower the catalyst loading to 5 mol%, in order to highlight the green character of the protocol. In some cases we could purify the desired ketone just by an ice-water workup-extraction, in order to remove the DMSO from the reaction mixture. The best results were provided from the oxidation of substituted 1-phenylethan-1-ols. para-Substituted alcohols, with either electron-donating or electron-withdrawing groups, provided the desired ketones 2k–2s in moderate to excellent yields. In the case of 2l, it was possible to isolate the desired product by simple solvent evaporation, without the need of column chromatography. This also applies in all cases where the reaction reached completion (for example, compounds 2m–p). ortho-Substituted alcohols, like 1r, provided the desired product 2r in 39% yield, presumably due to steric hindrance close to the center of the reaction. Double and triple bond-bearing alcohols also afforded the desired products 2u and 2w in very good yields, highlighting the chemoselectivity of the method. Aromatic substituted alcohols, such as benzhydrol, and xanthydrol, provided ketones 2t and 2aa in excellent yields. In order to expand the substrate scope, we attempted the oxidation of cyclic secondary alcohols, which gave the ketones 2ac–2ag in very good yields. Aliphatic secondary alcohols, such as 2-pentanol, provided the carbonyl compound 2ab in 49% yield, due to volatility issues. In order to examine the chemoselectivity of the method, we chose to oxidize an 1,3-diol to the corresponding β-hydroxy ketone (2ah), as the only product in 70% yield, showing the chemoselectivity of the method to the oxidation of the benzylic position versus the aliphatic one. In order to apply the oxidation in more complicated real-life examples, cholesterol (1ai) was employed as the substrate and the desired ketone 2ai was obtained in 63% yield.
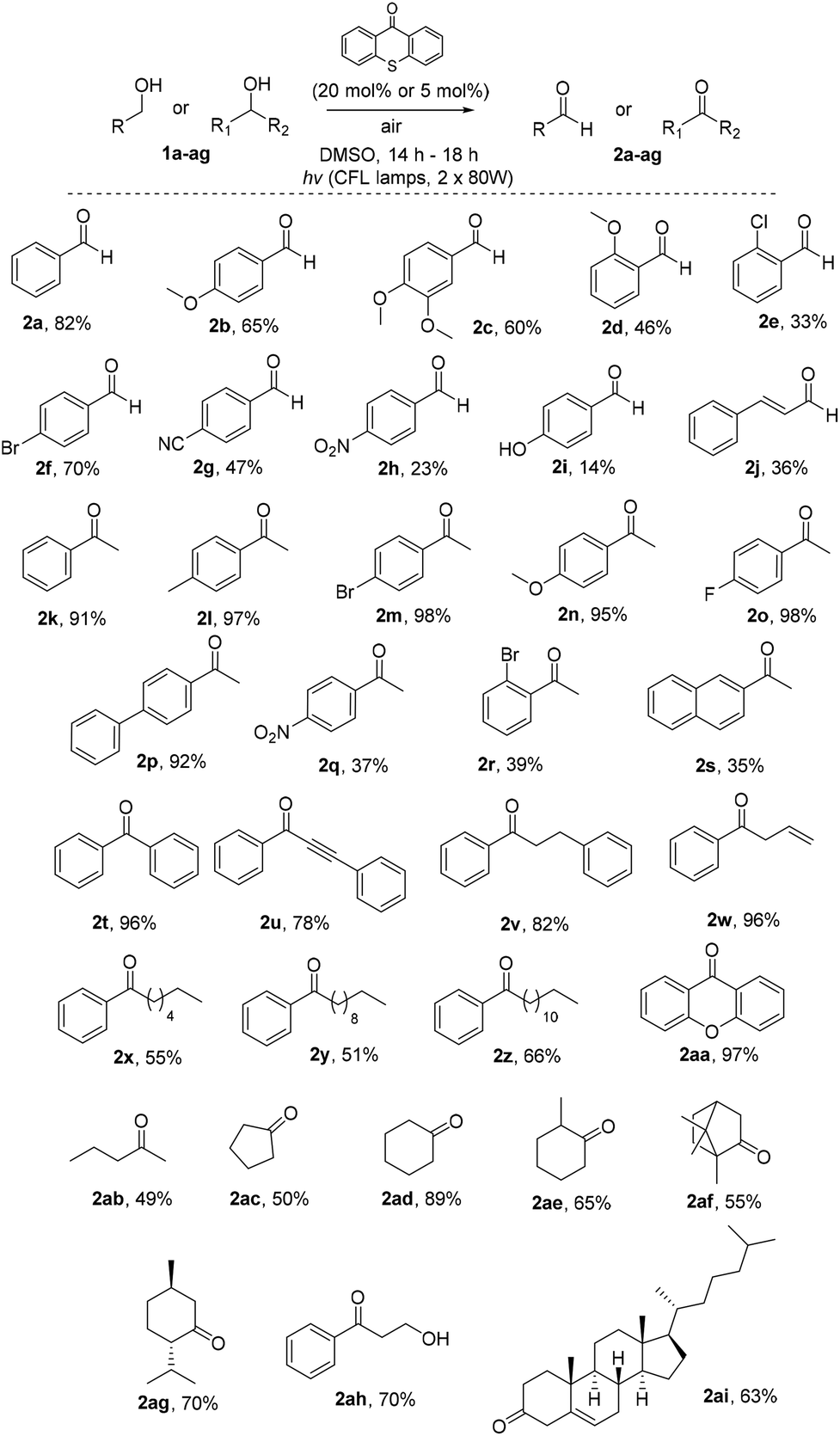 | ||
| Scheme 2 Substrate scope for the photochemical oxidation of benzylic and secondary alcohols. | ||
After substrate scope evaluation, we became interested in determining the actual role of the catalyst and the light source. Performing control experiments for the oxidation reaction of benzyl alcohol (1a) to benzaldehyde (2a), we extracted some very important results, in order to understand the different parameters of the reaction (Tables 2 and 3). These experiments showed that no product was obtained under dark (Table 2, entry 2) and thermal conditions (Table 2, entry 3), indicating a “clean” photochemical reaction, while in the absence of catalyst, no product was obtained (Table 2, entry 4). In addition, when the reaction was performed under argon atmosphere, no product was observed, which highlighted the importance of oxygen in the reaction (Table 2, entry 5). When Tempo was added in the reaction mixture as a radical trap, no oxidation product was formed (Table 3, entry 1). The intermediate compound formed between Tempo and alcohol 1k was observed by GC-MS, hinting the generation of a benzyl radical.17 Furthermore, quenching reagents were used, in order to recognize the oxygen species that take part in the reaction mechanism (Table 3). As an oxygen species quencher, NaN3 was used and partially inhibited the process, which as a fact shows that the presence of singlet oxygen species in the reaction mechanism is strong (Table 3, entry 3). An additional experiment, that strongly indicates the association of singlet oxygen, is the use of anthracene and its derivatives as singlet oxygen traps, which was introduced in literature by Fukuzumi.18 The addition of anthracene in the standard reaction mixture furnished the corresponding anthraquinone in 90% yield, which indicates the presence of singlet oxygen. The same conclusion can be drawn with the use of DABCO (Table 3, entry 4). Additionally, utilizing 2,2,6,6-tetramethylpiperidine, which is a known singlet oxygen quenching reagent,19 no product was formed (Table 3, entry 5).

Another tool that contributes to the understanding of the reaction mechanism, is the determination of the quantum yield (Φ) of the photocatalytic reaction. Based on literature,20 the quantum yield (Φ) of the photocatalytic reaction was calculated: [Φ = 12 (Φ > 1)], which hints that a radical propagation mechanism is taking place.
In order to gather further information about the reaction mechanism, fluorescence quenching studies took place. Stern–Volmer plots for the fluorescence quenching experiments revealed that the excited photocatalyst does not interact directly with benzyl alcohol or benzhydrol, since the slope of the quenching is minimal (Scheme 3).8e Further support for this notion, that a direct single electron transfer between excited thioxanthenone and benzyl alcohol is not possible, is given by the comparison of the reduction potentials of the two compounds (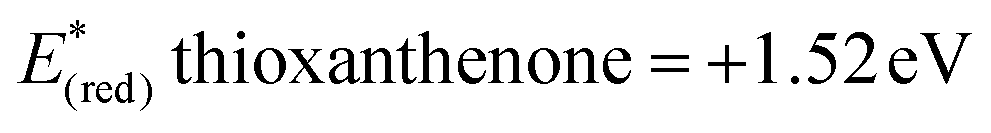 vs. SCE,6e while E(ox) benzyl alcohol = +2.16 eV vs. SCE21). Furthermore, bubbling oxygen in a cuvette with a solution of thioxanthenone led to diminished fluorescence, validating that air quenches the catalyst.17 Further control fluorescence quenching experiments with the quenchers of Table 3 were also performed.17
vs. SCE,6e while E(ox) benzyl alcohol = +2.16 eV vs. SCE21). Furthermore, bubbling oxygen in a cuvette with a solution of thioxanthenone led to diminished fluorescence, validating that air quenches the catalyst.17 Further control fluorescence quenching experiments with the quenchers of Table 3 were also performed.17
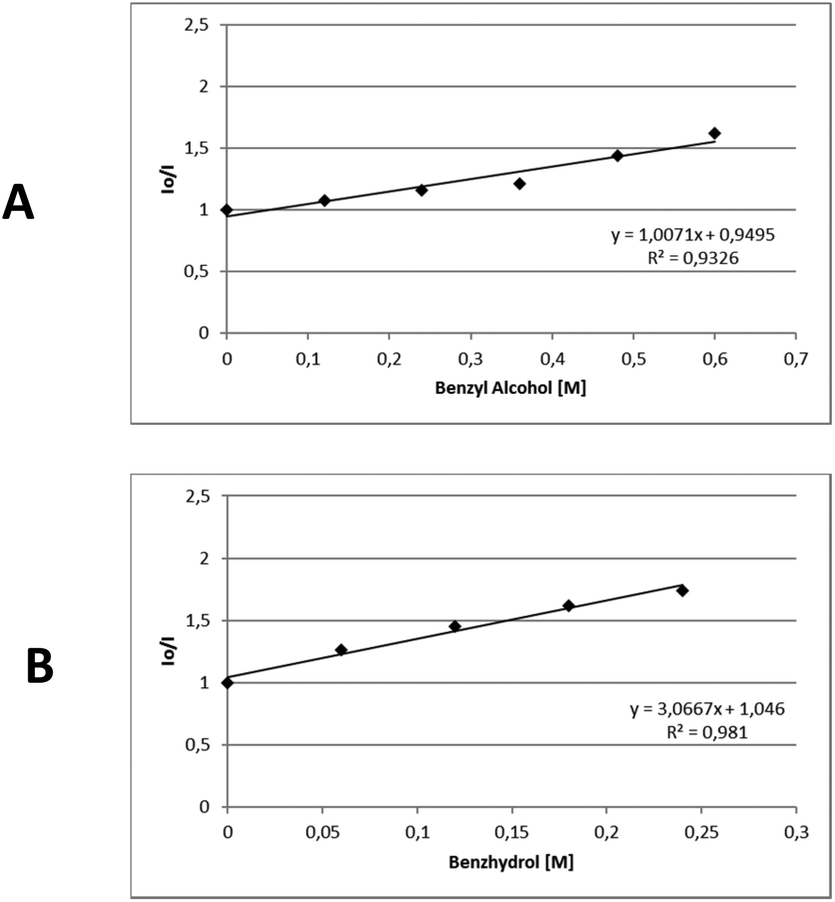 | ||
| Scheme 3 Stern–Volmer plots for the fluorescence quenching of thioxanthenone (1 mM in DMSO) with A. Benzyl alcohol, B. Benzhydrol. | ||
Based on literature, Orfanopoulos and coworkers performed a series of experiments, in order to distinguish between SET or HAT processes for the oxidation of alcohols.22 In that work, a control experiment utilizing 1-(4-methoxyphenyl)-2,2-dimethyl-1-propanol (1aj) as the substrate was demonstrated. Due to its substitution pattern, when a SET process is occurring in the Cα–H of the hydroxyl group, a Cα–Cβ bond cleavage follows, leading to the formation of the corresponding aldehyde and carboxylic acid. On the other hand, if the photooxidation of this substrate proceeds through a HAT event, then, the reaction furnishes the tert-butyl ketone (2aj), as the sole product.22 Applying this test reaction in our method, we examined the photooxidation of 1-(4-methoxyphenyl)-2,2-dimethyl-1-propanol (Scheme 4, 1aj). In our case, after 18 h of irradiation, the starting alcohol was converted completely in a mixture of 2aj![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :benzaldehyde:benzoic acid in a ratio of 68:3:29, respectively. As we expected, the major product is ketone 2aj, which indicates that our method does not involve a SET process, but mainly a HAT event (Scheme 4). Moreover, similar conclusions derived from quenching experiments with 1,3,5-trimethoxybenzene (TMB).17 TMB has a relative low oxidation potential (E(ox) TMB = +1.12 eV vs. SCE)22 and subsequently can act as an electron donor against the excited form of thioxathenone, quenching the excited state and as a result the termination of the radical procedure. We performed the oxidation of benzyl alcohol 1a to aldehyde 2a in the presence of TMB. The result of the reaction was, as we assumed, the oxidation of the alcohol to aldehyde, verifying that the reaction does not proceed through a SET pathway.22
:benzaldehyde:benzoic acid in a ratio of 68:3:29, respectively. As we expected, the major product is ketone 2aj, which indicates that our method does not involve a SET process, but mainly a HAT event (Scheme 4). Moreover, similar conclusions derived from quenching experiments with 1,3,5-trimethoxybenzene (TMB).17 TMB has a relative low oxidation potential (E(ox) TMB = +1.12 eV vs. SCE)22 and subsequently can act as an electron donor against the excited form of thioxathenone, quenching the excited state and as a result the termination of the radical procedure. We performed the oxidation of benzyl alcohol 1a to aldehyde 2a in the presence of TMB. The result of the reaction was, as we assumed, the oxidation of the alcohol to aldehyde, verifying that the reaction does not proceed through a SET pathway.22
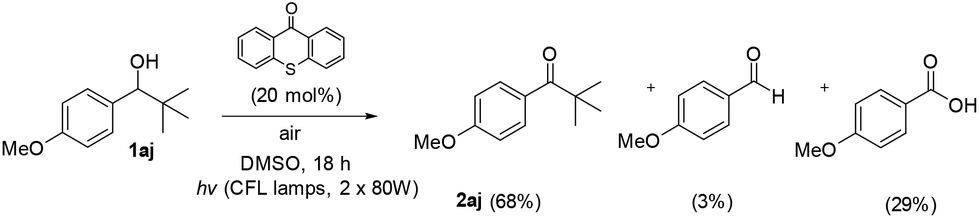 | ||
| Scheme 4 Mechanistic test reaction for SET or HAT processes. | ||
Utilizing the above mechanistic studies, we can propose a plausible mechanism for the photochemical oxidation (Scheme 5). The first step is the excitation of the photocatalyst, thioxanthene-9-one, by light. Next, based on the results of the fluorescent quenching studies, excited thioxanthene-9-one interacts with molecular oxygen, generating singlet oxygen and returning thioxanthene-9-one to its ground state. This is in agreement with literature precedent, regarding excited thioxanthenone as singlet oxygen generator.23 Then, singlet oxygen, as an activated form of oxygen, interacts with benzyl alcohol, either through a two-step mechanism, in which the first step is the abstraction of a hydrogen atom, leading to the formation of the benzylic radical I followed by a fast radical recombination,24 leading, to the formation of peroxy hemiacetal II or through an one-step insertion in the C–H bond of the benzylic position, leading again to II. This activated compound decomposes with further irradiation, leading to benzaldehyde and hydroxy radicals, which can lead to propagation or can decompose to hydrogen peroxide. Indication for the presence of hydrogen peroxide in the reaction is the formation the corresponding dimethylsulfone, as a byproduct, the product of oxidation of the solvent (Scheme 6). In order to be sure for the origin of the oxidant source, we examined the power of hydrogen peroxide as the oxidation reagent, firstly for the oxidation of dimethylsulfoxide to dimethylsulfone and secondly for the oxidation of 1-phenylethanol (1k) to acetophenone (2k) (Scheme 6). DMSO can be oxidized either by aqueous hydrogen peroxide solution itself or hydrogen peroxide-derived from singlet oxygen,23b,25 the latter can be formed by thioxanthenone through a photocatalytic process (Scheme 6A). On the other hand, aqueous hydrogen peroxide solution cannot oxidize alcohol 1k, confirming the hypothesis that singlet oxygen is the main oxidant source (Scheme 6B). Finally, the use of alternative energy source in our photochemical process was envisaged, in order to have a significant industrial applicability. Therefore, a variety of primary and secondary alcohols was tested as substrates, utilizing sunlight as the irradiation source, instead of CFL lamps, and the desired carbonyl compounds were formed in very good yields (Scheme 7). Secondary benzylic alcohol, like 1k and 1t afforded the desired ketones in similar yields as the household lamps. Unfortunately, the oxidation of 1a under sunlight led to diminished yield (48%), since overoxidation to benzoic acid could not be controlled.
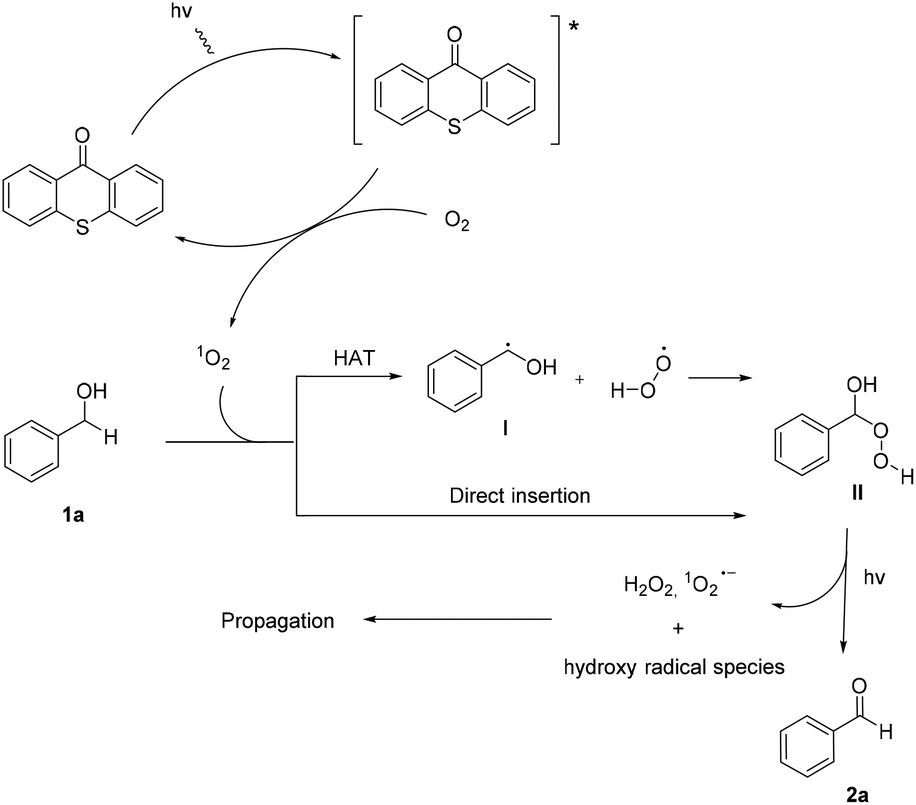 | ||
| Scheme 5 Proposed reaction mechanism. | ||
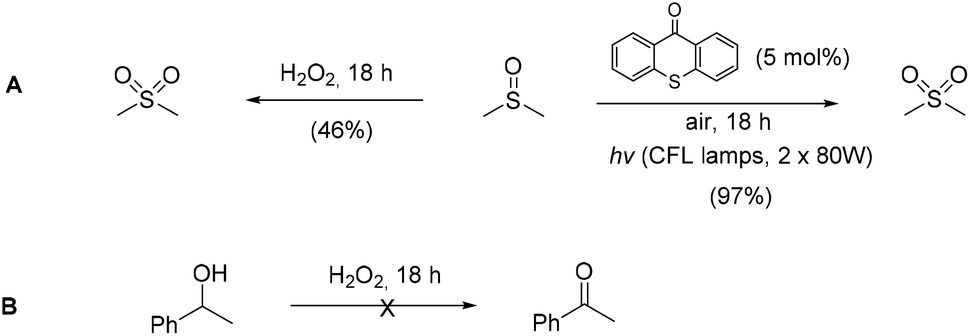 | ||
| Scheme 6 Test reactions for the oxidant source. | ||
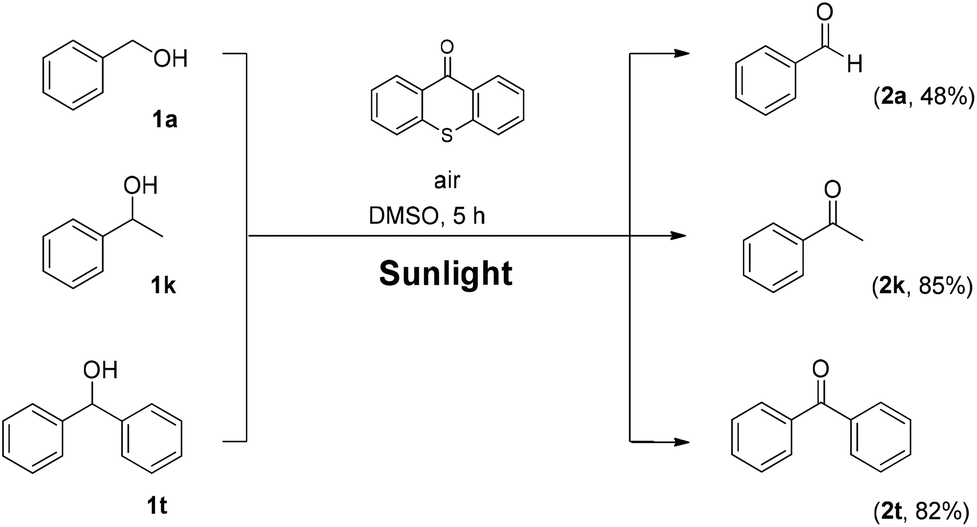 | ||
| Scheme 7 Photochemical oxidation utilizing sunlight. | ||
Conclusions
In conclusion, a green and sustainable photochemical protocol for the oxidation of alcohols was developed. Since a plethora of stoichiometric and catalytic systems have been used for this reaction, our approach was to find an environmentally friendly way to promote this transformation, by utilizing air as the oxidant and a cheap and commercially available organic molecule as the photocatalyst. After an extensive study, a variety of benzylic and secondary alcohols were transformed to the corresponding carbonyl compounds and mechanistic studies were performed in order to better understand our catalytic system.Experimental
General procedure for the oxidation of alcohols
In a glass vial containing thioxanthene-9-one (3i) (8.5 mg, 0.04 mmol) in DMSO (0.6 mL), benzyl alcohol or secondary alcohol (0.20 mmol) was added. The vial was left open in air and left stirring under household bulb irradiation (2 × 80 W household lamps) for 14–18 h. The desired product was isolated via purification of the crude mixture by flash column chromatography or just by ice-water workup-extractions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the John Latsis Foundation for the financial support through the program ‘EPISTHMONIKES MELETES 2015’ (PhotoOrganocatalysis: Development of new environmentally-friendly methods for the synthesis of compounds for the pharmaceutical and chemical industry). Also, the authors gratefully acknowledge COST Action C–H Activation in Organic Synthesis (CHAOS) CA15016 for helpful discussions. N. F. N. would like to thank the State Scholarships Foundation (IKY) for financial support through a PhD fellowship through the “Strengthening of Human Resources through Doctoral Research” (MIS-5000432) program of the Operational Program “Human Resource Development, Education and Lifelong Learning” 2014-2020, co-financed by the European Union (European Social Fund ESF) and Greek national funds. I. T. would like to thank the Hellenic Foundation for Research and Innovation (HFRI) and the General Secretariat for Research and Technology (GSRT), for a HFRI PhD Fellowship grant (GA14476).Notes and references
- For a book, see:
(a)
G. Tojo, Oxidation of Alcohols to Aldehydes and Ketones: A Guide to Current Common Practice, Springer Science, New York, 2006 Search PubMed
. For a recent review, see: (b) D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Chem. Rev., 2018, 118, 2636–2679 CrossRef CAS PubMed
- For selected reviews, see:
(a) T. T. Tidwell, Synthesis, 1990, 857–870 CrossRef CAS
- For reviews, see:
(a) C. Parmeggiani and F. Cardona, Green Chem., 2012, 14, 547–564 RSC
- For selected examples, see:
(a) R. Mu, Z. Liu, Z. Yang, Z. Liu, L. Wu and Z.-L. Liu, Adv. Synth. Catal., 2005, 347, 1333–1336 CrossRef CAS
- J. Dong, E. Fernandez-Fueyo, F. Hollmann, C. E. Paul, M. Pesic, S. Schmidt, Y. Wang, S. Younes and W. Zhang, Angew. Chem., Int. Ed., 2018, 57, 9238–9261 CrossRef CAS
- For selected examples and reviews, see:
(a) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed
- For selected examples, see:
(a) S. Yurdakal, G. Palmisano, V. Loddo, V. Augugliaro and L. Palmisano, J. Am. Chem. Soc., 2008, 130, 1568–1569 CrossRef CAS PubMed
- For metal-free photocatalytic examples, see:
(a) S. Gazi and R. Ananthakrishnan, RSC Adv., 2012, 2, 7781–7787 RSC
- For selected reviews, see:
(a) D. Ravelli, S. Protti and M. Fagnoni, Acc. Chem. Res., 2016, 49, 2232–2242 CrossRef CAS
-
(a) S. Devari, M. A. Rizvi and B. A. Shah, Tetrahedron Lett., 2016, 57, 3294–3297 CrossRef CAS
- For reviews, see:
(a) M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS
-
(a) S. Fukuzumi, S. Kuroda and T. Tanaka, J. Am. Chem. Soc., 1985, 107, 3020–3027 CrossRef CAS
- For selected photochemistry examples, see:
(a) G. N. Papadopoulos, D. Limnios and C. G. Kokotos, Chem. – Eur. J., 2014, 20, 13811–13814 CrossRef CAS
- For selected organocatalytic examples, see:
(a) I. Triandafillidi, M. G. Kokotou and C. G. Kokotos, Org. Lett., 2018, 20, 36–39 CrossRef CAS PubMed
- For highlighting this problem and providing an excellent solution, see: E. Gaster, S. Kozuch and D. Pappo, Angew. Chem., Int. Ed., 2017, 56, 5912–5915 CrossRef CAS PubMed
- I. Ghosh, A. Mukhopadhyay, A. L. Koner, S. Samanta, W. M. Nau and J. Narasimha, Phys. Chem. Chem. Phys., 2014, 16, 16436–16445 RSC
- For more information and data, see ESI.†.
- H. Kotani, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2004, 126, 15999–16445 CrossRef CAS PubMed
- D. Bellus and H. Lind, J. Chem. Soc., Chem. Commun., 1972, 1199–1200 RSC
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC
- S. Higashimoto, N. Suetsugu, M. Azuma, H. Ohue and Y. Sakata, J. Catal., 2010, 274, 76–83 CrossRef CAS
- I. N. Lykakis, C. Tanielian and M. Orfanopoulos, Org. Lett., 2003, 5, 2875–2878 CrossRef CAS PubMed
-
(a) L. Shen and H.-F. Ji, Int. J. Mol. Sci., 2009, 10, 4284–4289 CrossRef CAS
-
(a) M. Sankar, E. Nowicka, E. Carter, D. M. Murphy, D. W. Knight, D. Bethell and G. J. Hutchings, Nat. Commun., 2014, 5, 3332–3338 CrossRef PubMed
- S. L. Mali, V. Vaidya, R. L. Pitliya, V. K. Vaidya and S. C. Ameta, Asian J. Chem., 1994, 6, 796–800 CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data, compound characterization, UV-Vis absorbance spectra, fluorescence quenching and mechanistic studies. See DOI: 10.1039/c9gc03000j |
| This journal is © The Royal Society of Chemistry 2020 |