
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic fungal multifunctional cellulases for enhanced biomass conversion†
Roman
Brunecky
 *a,
Venkataramanan
Subramanian
a,
John M.
Yarbrough
a,
Bryon S.
Donohoe
a,
Todd B.
Vinzant
a,
Todd A.
Vanderwall
a,
Brandon C.
Knott
a,
Yogesh B.
Chaudhari
b,
Yannick J.
Bomble
a,
Michael E.
Himmel
a and
Stephen R.
Decker
a
*a,
Venkataramanan
Subramanian
a,
John M.
Yarbrough
a,
Bryon S.
Donohoe
a,
Todd B.
Vinzant
a,
Todd A.
Vanderwall
a,
Brandon C.
Knott
a,
Yogesh B.
Chaudhari
b,
Yannick J.
Bomble
a,
Michael E.
Himmel
a and
Stephen R.
Decker
a
aBiosciences Center, National Renewable Energy Lab, Golden, CO 80401, USA. E-mail: roman.brunecky@nrel.gov
bLife Sciences Division, Institute of Advanced Study in Science and Technology (IASST), Guwahati, India
First published on 18th December 2019
Abstract
Significant effort has been expended toward the discovery and/or engineering of improved cellulases. An alternative to this approach is utilizing multifunctional enzymes; however, essentially most if not all relevant bacterial enzymes of this type do not express well in fungi. Therefore, developing a systematic understanding of how to construct multifunctional enzymes that are expressible in commercial fungal hosts is crucial to developing next generation enzymes for biomass deconstruction. Multifunctional cellulolytic enzymes, such as CelA from Caldicellulosiruptor bescii, show extremely high cellulolytic activity; however, a systematic understanding of its mechanism of action does not exist and it is not readily expressed in current industrial hosts. CelA is comprised of GH 9 and GH 48 catalytic domains connected by three type III cellulose-binding modules (CBMs). We have engineered several multifunctional enzymes designed to mimic CelA, and successfully expressed them in T. reesei. We then assessed their biophysical and kinetic performance parameters. The CBM3b-containing construct demonstrated increased initial binding rate to cellulose, enhanced digestion of biomass, and was able to increase the activity of a commercial cellulase formulation acting on pretreated biomass. The same construct containing CBM1 also demonstrated enhancement of a commercial cellulase formulation in the digestion of pretreated biomass; however, it had lower initial binding rates and did not demonstrate improved activity on its own. Interestingly, a CBM-less construct decreased the activity of the commercial cellulase formulation slightly. Examination of the mechanism of the CBM3b-containing construct revealed a novel biomass deconstruction behavior similar to, but yet distinct from that of native CelA.
Introduction
Multifunctional cellulolytic enzymes, such as CelA from Caldicellulosiruptor bescii,1–3 represent a novel intermediate class of biomass deconstructing enzymes – falling between the well-known free enzyme systems found in most fungi and bacteria and the highly complexed, bacterial cellulosome systems produced by microbes such as Clostridium thermocellum.4–6 The extremely high cellulolytic activity, unique pit-forming mechanism, and recently reported cellulose crystallinity-agnostic behavior of CelA makes it a very important enzyme to investigate further.1–3 However, fully native CelA has not been successfully expressed in fungal systems.We have designed and built a set of synthetic multifunctional enzymes based on the architecture of CelA. In order to enable expression in Trichoderma reesei, we chose enzyme components known to be expressible in fungal systems and genetically linked them to mimic the CelA domain architecture. We chose the catalytic domain of cellobiohydrolase I (Cel7A) from Penicillium funiculosum (PfCel7Acat)7,8 to serve as the N-terminal exocellulase domain. The Y245G mutant of the catalytic domain of Acidothermus cellulolyticus AcCel5A (AcCel5Acat)9 served as the C-terminal endocellulase domain. The PfCel7A enzyme has higher activity than the native Cel7A in T. reesei and the E1Y245G mutation has been shown to have increased resistance to end-product inhibition by cellobiose.7,10 Both enzymes are known to express well in T. reesei. Moreover, they possessed the correct N to C orientation to be combined into one larger construct. To parse out the functionality of critical substructures, we designed three different constructs, keeping the exo and endo catalytic domains constant while varying the linker and CBM arrangement connecting them (see Fig. 1):
• Construct 1 (C1): PfCel7Acat-PfCel7Alinker-PfCel7ACBM1-TrCel6Alinker-AcCel5Acat
• Construct 2 (C2): PfCel7Acat-PfCel7Alinker-TrCel6Alinker-AcCel5Acat
• Construct 3 (C3): PfCel7Acat-PfCel7Alinker-CbCelACBM3b-TrCel6Alinker-AcCel5Acat.
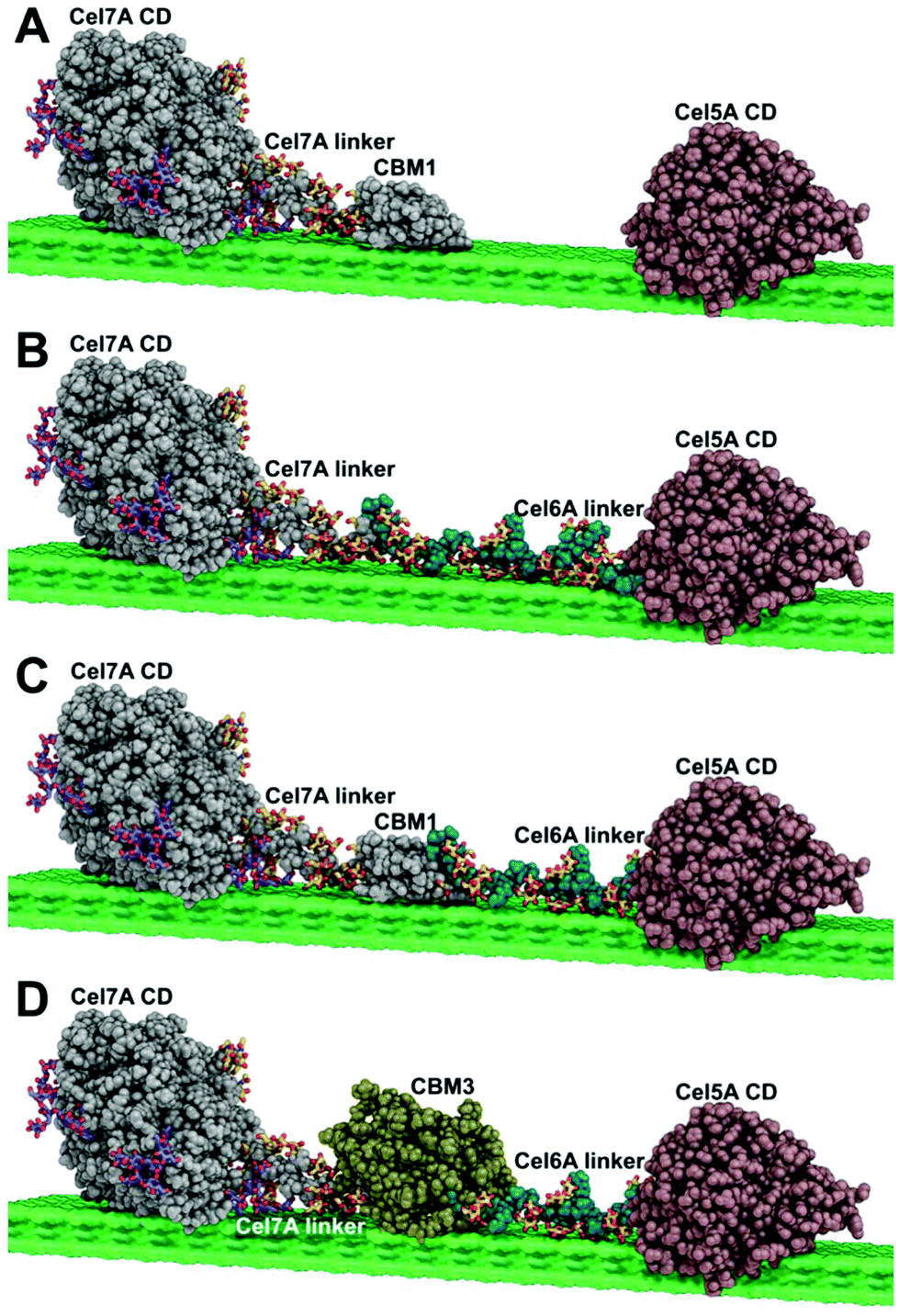 | ||
| Fig. 1 Multifunctional enzyme design (A) free enzyme system (B) Cel7A-Link-Cel5A (C) Cel7A-CBM1-Cel5A (D) Cel7A-CBM3b-Cel5A. | ||
The multifunctional constructs were expressed in T. reesei and assayed using Avicel, cotton linters modified to have variable levels of crystallinity, and pretreated corn stover. Activity was compared to mixtures of PfCel7A and AcCel5Acat in equimolar concentration and found the PfCel7A-CBM3b-AcCel5A construct to exhibit superior performance. The binding of these constructs to crystalline cellulose surfaces was examined using QCMD11 techniques, where we discovered a novel non-catalytic biomass disruption property for the PfCel7A-CBM3b-AcCel5A construct that is distinct from any other enzyme we have previously examined, including the native CelA enzyme. And finally, we observed distinct digestion patterns for each of the multifunctional enzymes using both transmission electron microscopy and scanning electron microscopy which showed some similarity, but also important differences when compared to the pit formation digestion mechanism of CelA.1
Results and discussion
We found that the activity of the multifunctional PfCel7A-CBM1-AcCel5A and PfCel7A-Link-AcCel5A enzymes were roughly similar or slightly inferior when compared to the binary component system comprised of PfCel7A and AcCel5A when both systems were assayed in the presence of β-glucosidase on crystalline cellulose. However, when the PfCel7A-CBM3b-AcCel5A construct was tested on the crystalline Avicel substrate, it significantly outperformed the binary mixture of PfCel7A (with its native CBM1) and AcCel5Acat enzymes by 40%, as well as significantly outperforming PfCel7A alone (Fig. 2A). We also generated a PfCel7A-CBM3b construct to determine if this CBM family was responsible for the improved activity of the linked system. To test the effect of CBM3b on performance, the CBM3b containing PfCel7A construct was also compared, on an equal active site basis, to PfCel7A with its native CBM1. The PfCel7A-CBM3b construct demonstrated an inferior activity to the native enzyme. This is not a surprising, but important, given that the PfCel7A catalytic domain has evolved to perform in concert with the CBM1 (Fig. 2B).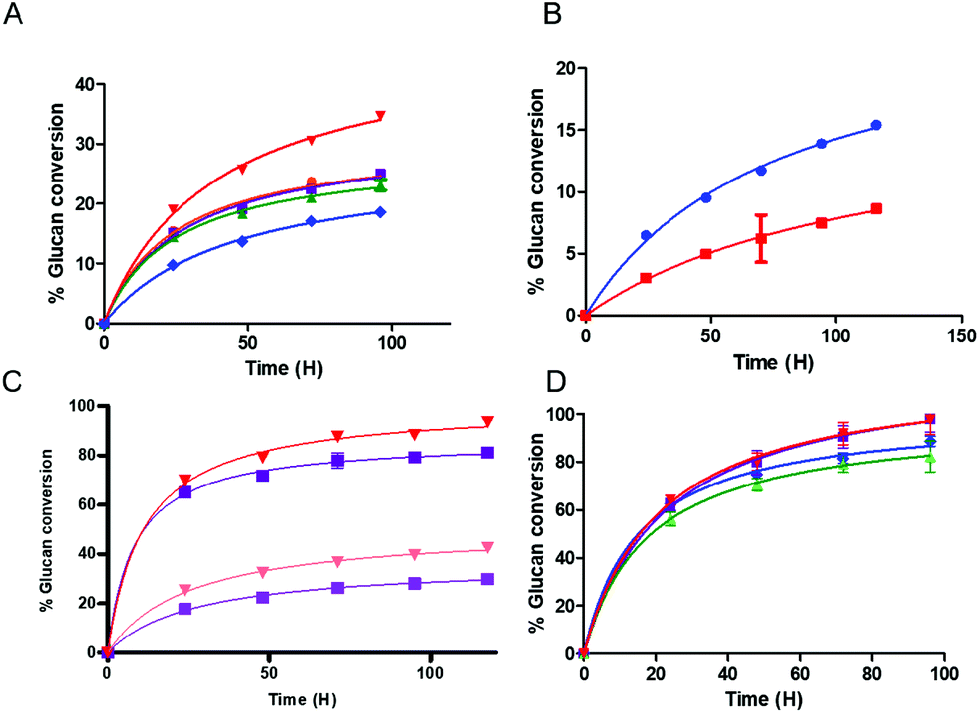 | ||
| Fig. 2 Multifunctional enzyme performance data (A) multifunctional enzyme digestions of crystalline Avicel substrate, Cel7A-CBM3b-Cel5A (red), Cel7A-CBM1-Cel5A (purple), Cel7A-Link-Cel5A (green), Cel7A + Cel5A (orange), Cel7A (blue) (B) Avicel digestion comparing a Cel7A with native CBM1 (blue) versus a Cel7A with a CBM3b attached (red). (C) High and low crystallinity cotton linter digest, Cel7A-CBM3b-Cel5A 66% crystalline material (pink), Cel7A-CBM3b-Cel5A 33% crystalline material (red), Cel7A-CBM1-Cel5A 66% crystalline material (light purple) Cel7A-CBM1-Cel5A 33% crystalline material (purple) (D) multifunctional supplemented enzyme digestions of DDR substrate, Cel7A-CBM3b-Cel5A + Ctec2 (red), Cel7A-CBM1-Cel5A + Ctec2 (purple), Cel7A-Link-Cel5A + Ctec2 (green), Cel7a + Cel5A + Ctec2 (blue). | ||
The performance of the hyperthermophilic CelA enzyme from C. bescii has recently been reported to be agnostic to the crystalline content of cellulose, converting high crystallinity cotton linters (66% CI) cellulose at the same rate as low crystallinity cotton linters cellulose (33% CI).2 To determine if the co-localization of exo- and endocellulase activity domains could replicate this behavior, we tested the PfCel7A-CBM1-AcCel5A and PfCel7A-CBM3b-AcCel5A multifunctional enzymes on both high and low crystallinity cellulose materials. The results shown in Fig. 2C indicate that whereas the PfCel7A-CBM3b-AcCel5A construct performs better than the PfCel7A-CBM1-AcCel5A construct as previously observed, there remains a distinct preference for the low crystallinity materials in both systems.
To evaluate whether a multifunctional enzyme could enhance a commercial cellulase formulation and therefore lower overall enzyme loading and formulation costs, we augmented a commercial cellulase (Cellic® CTec2, Novozymes) with either a multifunctional enzyme or the free binary enzyme system (PfCel7A and AcCel5A). The substrate tested (DDR) was a dilute NaOH treated, then mechanically refined corn stover as described by Chen et al.12 Both CBM-containing multifunctional enzymes improved the performance of the overall formulation; however, the linker-only multifunctional enzyme performed only as well as the free enzyme system (Fig. 2D). It should be noted that while the overall extent of conversion at the endpoint is only somewhat elevated (e.g., ∼10% improvement), the actual rate of conversion is significantly faster with the PfCel7A-CBM3b-AcCel5A and PfCel7A-CBM1-AcCel5A enzyme additions, which achieve in only two days what it takes the binary enzyme system four days to complete. This result represents a 50% reduction in time to our chosen target conversion (DOE BETO Base Case metric) of 85% glucose production from glucan.
Multifunctional enzyme binding and biomass interaction
Quartz Crystal Microbalance-Dissipation (QCMD) experiments were conducted to evaluate the initial rate and extent of enzyme binding and to monitor enzyme-induced changes to the physical properties of cellulose. These experiments were carried out under inhibitory concentrations of cellobiose to limit cellulose hydrolysis from impacting the measurements. The PfCel7A-CBM3b-AcCel5A enzyme had the fastest initial binding rate, whereas PfCel7A-CBM1-AcCel5A, PfCel7A-Link-AcCel5A, and Cel5A had a binding rate comparable to CelA (Fig. 3). The Cel7A initial binding rate is between these latter enzymes and the PfCel7A-CBM3b-AcCel5A construct.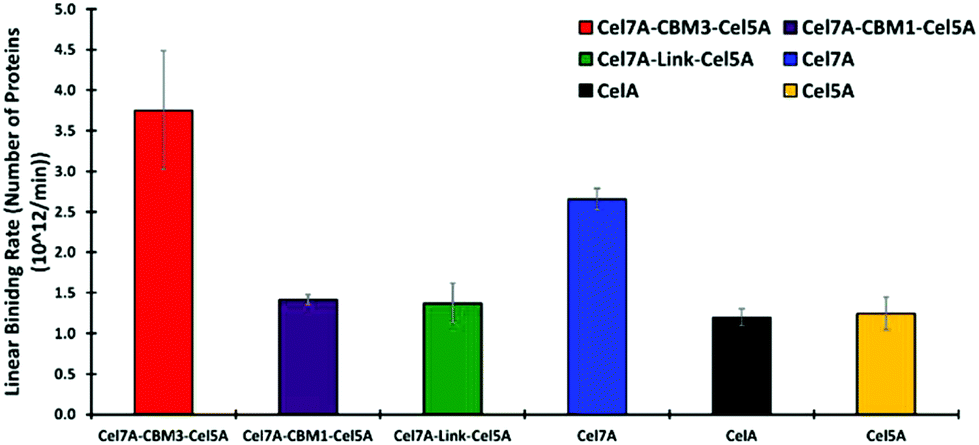 | ||
| Fig. 3 Linear binding rates of different synthetic multifunctional enzymes compared to native CelA and Cel7A and Cel5A. | ||
In Fig. 4A, we observe that the PfCel7A-CBM3b-AcCel5A enzyme rapidly binds to the cellulose surface (nearly vertical line), followed by decreasing mass (downward trend). PfCel7A-CBM1-AcCel5A and PfCel7A-Link-AcCel5A demonstrate a slower initial binding and then come to an equilibrium state (flat portion of curve). When compared to CbCelA (Fig. 4B-black line) we do not see this mass loss behavior, nor do individual enzyme components AcCel5A or PfCel7A exhibit this behavior (Fig. 4B and D). The initial binding of PfCel7A is identical to that of the PfCel7A-CBM3b-AcCel5A construct; however, the mass continues to rise for PfCel7A (Fig. 4B blue), indicating additional enzyme binding, in contrast to mass loss demonstrated by PfCel7A-CBM3b-AcCel5A.
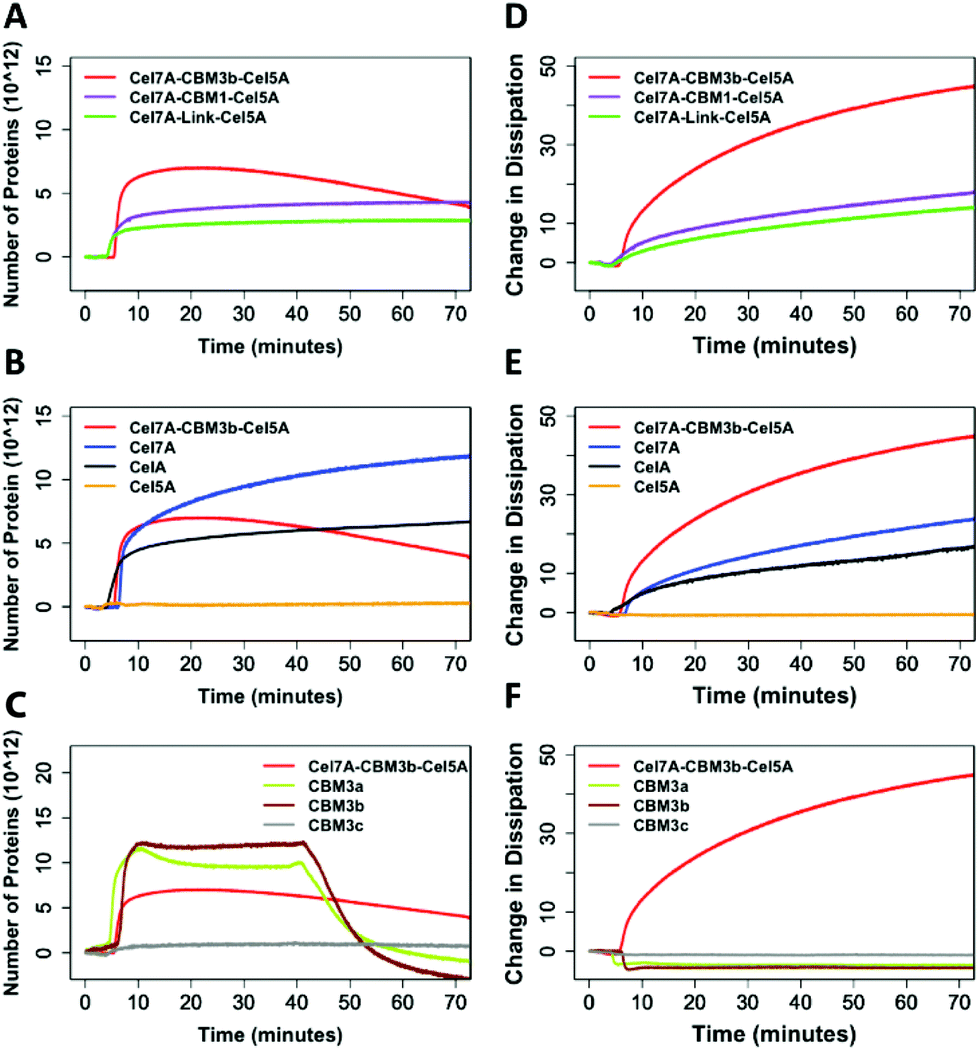 | ||
| Fig. 4 QCMD of multifunctional enzymes compared to individual enzyme components. Mass changes normalized on a number protein basis (A) Cel7A-CBM3b-Cel5A, Cel7A-CBM1-Cel5A, Cel7A-Link-Cel5A (B) Cel7A-CBM3b-Cel5A, Cel7A, CelA and Cel5A, (C) Cel7A-CBM3b-Cel5A, CBM 3a, 3b and 3c. Dissipation characteristics (D) comparison of the dissipation of Cel7A-CBM3b-Cel5A, Cel7A-CBM1-Cel5A, Cel7A-Link-Cel5A (E) comparison of the dissipation of Cel7A-CBM3b-Cel5A, Cel7A, CelA and Cel5A (F) comparison of the dissipation of Cel7A-CBM3b-Cel5A, CBM 3a, CBM 3b and CBM 3c. | ||
The PfCel7A-CBM3b-AcCel5A enzyme also “softens” the biomass as seen in Fig. 4A, where the change in dissipation is proportional to a change in the viscoelasticity of the cellulose surface. This behavior is not present in any other enzymes tested (Fig. 4D and E). Given that the major difference between PfCel7A-CBM1-AcCel5A, PfCel7A-Link-AcCel5A, and PfCel7A-CBM3b-AcCel5A is the CBM, it is logical to assume that this result may be the consequence of the CBM3b; however, when CelA (which contains three CBM3b domains) is tested, this phenomenon does not occur (Fig. 4B). To further explore this effect, we tested each component of these constructs as shown in Fig. 4B. Here, we see that PfCel7A-CBM1 binds to the surface but removes no mass, while AcCel5A binds only very minimally. Finally, we examined three different CBM3 modules (CBM3a, CBM3b, CBM3c) alone for their ability to remove cellulose. Under static conditions, none of the CBMs tested demonstrated mass loss; however, when buffer was flowed over the surface after 50 min of binding, CBM3b and CBM3a demonstrated a potential mass loss as seen by the negative characteristic to the binding curve. CBM3c, which does not bind cellulose, exhibits no such effect (Fig. 4C).
Difference in deconstruction mechanisms
Imaging of partially digested Avicel substrates was performed by both scanning transmission electron microscopy (TEM) and electron microscopy (SEM). The three different enzyme constructs appear to have different patterns of biomass deconstruction as seen in Fig. 5 and 6. PfCel7A-CBM1-AcCel5A and to some extent PfCel7A-Link-AcCel5A appear to have a very diffuse digestion pattern as viewed by TEM (Fig. 5A and B). When viewed by SEM; this sample looks like a smooth surface (Fig. 6A and B). In contrast, the PfCel7A-CBM3b-AcCel5A construct digested cellulose has a much more scalloped appearance, with particles that appear to flake off the surface of the Avicel. This is similar to, but yet distinct from the CelA treated material, which shows the unique pit-formation mechanism1 (Fig. S9†). No pit formation phenotype was detected by TEM for the PfCel7A-CBM3b-AcCel5A construct (Fig. 5C). However, the deconstruction pattern visualized by SEM does have some CelA like features such as the deconstruction front seen in both samples. The other notable feature was that the PfCel7A-CBM3b-AcCel5A SEM digestion showed a large number of cellulose nano-crystal-like structures (Fig. 6C).13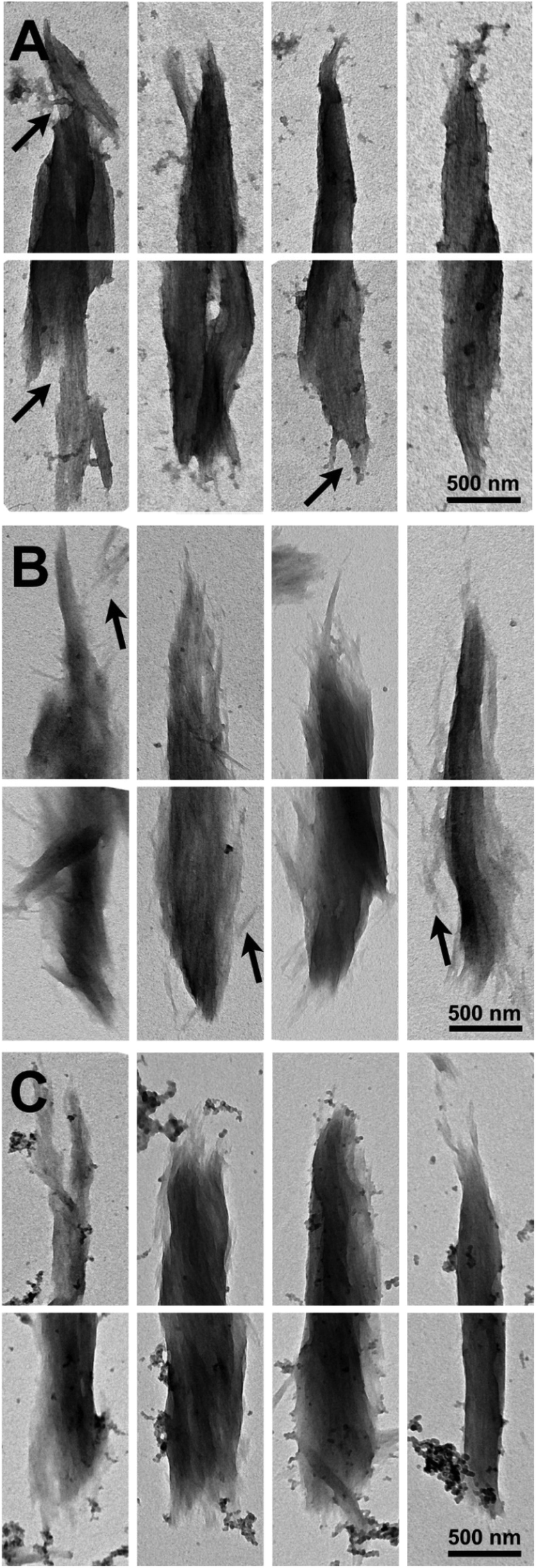 | ||
| Fig. 5 TEM micrographs of Avicel particles digested by (A) Cel7A-CBM3b-Cel5A, (B) Cel7A-CBM1-Cel5A, or (C) Cel7A-Link-Cel5A. The particle morphologies are similar overall. The Cel7a-CBM3-Cel5A digested samples show more examples of notched or scalloped surfaces (A, arrows), and the Cel7A-CBM1-Cel5A materials display and abundance of nanocrystalline particles and protuberances (B, arrow). | ||
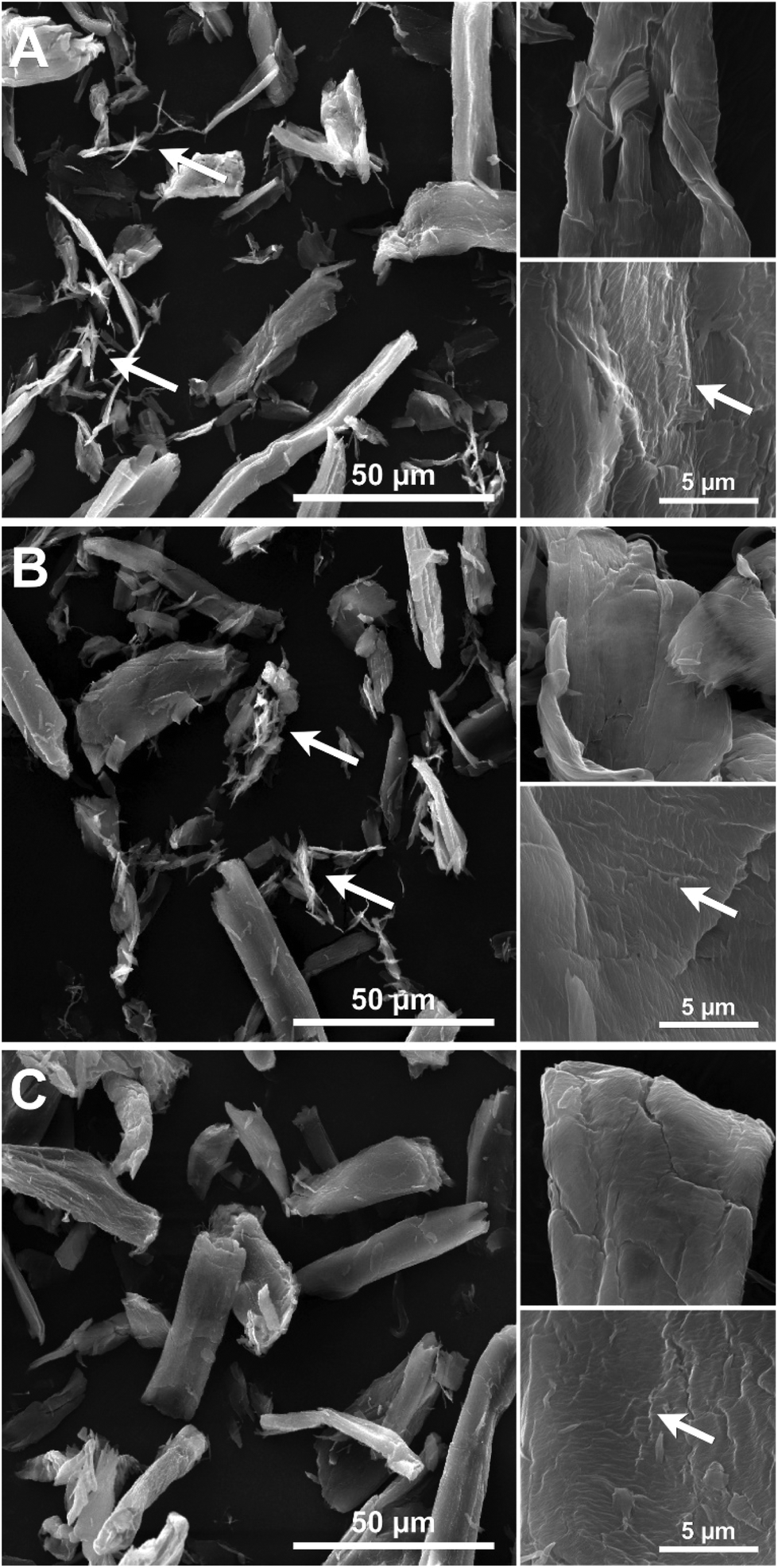 | ||
| Fig. 6 SEM micrographs of Avicel particles digested by (A) Cel7A-CBM3b-Cel5A, (B) Cel7A-CBM1-Cel5A, (C) Cel7A-Link-Cel5A. Both the Cel7A-CBM3b-Cel5A and Cel7A-CBM1-Cel5A samples had an abundance of nanocrystalline particle (A and B arrows). All of the particles displayed a notched or stepped surface morphology when examined at higher magnification (arrows). | ||
Discussion
There are several key findings from this work; the primary conclusion is that multi-modular, multifunctional enzyme systems can be successfully engineered to function in fungal expression systems and that they display activities superior to that of their component enzymes (Fig. 2A). Previous attempts to create multidomain enzymes have been predominantly focused on building much larger enzymes in bacterial systems using mini-cellulosome type systems.14 These systems utilize a scaffold containing cohesin domains and enzymes containing dockerin domains.15 Notably, none of these systems have been produced in a fungal expression system, which would likely be required to produce commercial quantities of enzymes. We have also discovered that there is a unique cellulose deconstruction mechanism that occurs with the CBM3b containing enzyme (PfCel7A-CBM3b-AcCel5A). Moreover, this novel mechanism is distinct from enzymes such as native CelA.When considering the design of multifunctional enzymes, our work suggests that emergent properties of these systems cannot easily be predicted. It also challenges some of the long-held theories about intra-molecular synergies between endo- and exoglucanases. As we see in Fig. 2A, proximity synergy, a long-discussed concept, does not appear to grant much benefit to the PfCel7A-CBM1-AcCel5A or PfCel7A-Link-AcCel5A enzymes when compared to the binary free enzyme mixture of Cel7 and Cel5. However, we do see a significant improvement when the CBM3b is added to these multifunctional enzymes. However, just adding a CBM3b to PfCel7A does not grant any improvement in the performance; rather, the opposite is seen in the case of the PfCel7A-CBM3b construct shown in Fig. 2B. Therefore, we conclude that the improved performance of the PfCel7A-CBM3b-AcCel5A enzyme is due to some novel emergent property of the entire linked enzyme system (very much like CelA).
To further understand the improved performance of these constructs, we examined their binding behavior. Indeed, the PfCel7A-CBM3b-AcCel5A enzyme does seem to bind Avicel a bit more tightly than the other multifunctional enzymes, and about as well as the native PfCel7A-CBM1 system; however, binding alone is unlikely to fully explain this enhancement in performance (see Fig. 3).
The QCM data provides us with the best evidence that something unique is occurring with the PfCel7A-CBM3b-AcCel5A enzyme. Regardless of the domain composition of the other enzymes tested, the combination of the CBM3b domain with the AcCel5A and PfCel7A domains appears to soften the cellulose surface; and may also non-catalytically remove cellulose from the surface of the sensor (Fig. 4A). The QCM data provides us with the best evidence that something unique is occurring with the PfCel7A-CBM3b-AcCel5A enzyme. Regardless of the domain composition of the other enzymes tested, the combination of the CBM3b domain with the AcCel5A and PfCel7A domains appears to soften the cellulose surface as well as non-catalytically removed cellulose from the surface of the sensor.
Multifunctional enzyme binding and biomass interaction
We found that there is a distinct difference between PfCel7A-CBM3b-AcCel5A and all other proteins and CBMs within this study. From the binding curves associated with mass, all the proteins and CBMs excluding PfCel7A-CBM3b-AcCel5A reach a state of equilibrium on the sensor surface (excluding the CBMs), wherein after 40 min, the buffer was passed over the sensor causing the CBMs to desorb from the surface of the cellulose nanocrystals (CNCs). However, the PfCel7A-CBM3b-AcCel5A system experiences mass loss around 20 min after the enzyme was introduced (Fig. 4A). After this, there is a steady decline in the mass even without a buffer desorption step. This loss of mass can be associated with either the protein desorbing from the surface of the cellulose nanocrystals or that portions of the cellulose nanocrystals are leaving the surface, or possibly both.The dissipation curve associated with PfCel7A-CBM3b-AcCel5A also suggests that a loss of cellulose occurs. Each of the cellulases within this study show a small change in dissipation as compared to PfCel7A-CBM3b-AcCel5A, which can be associated with the flexibility of the proteins sitting on the surface of the cellulose (Fig. 4D). Because these proteins are in a 10 mM cellobiose solution, their enzymatic activity on cellulose is inhibited and we can assume that there is no mass loss of cellulose. The mass loss observed with PfCel7A-CBM3b-AcCel5A construct suggest that this protein is still somehow “active” on cellulose. This conclusion is further supported with the lack of a decreasing signal in the dissipation curve associated with PfCel7A-CBM3b-AcCel5A. As the mass decreases within the PfCel7A-CBM3b-AcCel5A system, the dissipation continues increasing (Fig. 4A and D). This effect has been shown in the literature where there is an increase in dissipation as the mass of the film is decreased due to the absorption of water and the increased relaxation of the cellulose thin film.16–18 The dissipation continues increasing until enough of the cellulose thin film is gone and the bare sensor is exposed. It is at this point that the dissipation curve begins to decrease. However, it should be noted that the other two multifunctional enzymes have same catalytic domain architecture and are not “active” under identical conditions. Also, while the CBM3a/b alone also appear to be able to remove mass from the system, though only when a buffer flow is applied, this may be due to non-hydrolytic cellulose disruption. We note the reports for the cellulose binding domain of endoglucanase A (GenA) CBM2A from C. fimi and the CBM of P. janthinellum19,20 that are reported as cellulose disrupting. Moreover, the CBM of P. janthinellum was reported to release short fibers non-catalytically.19 Crystalline disruption and targeting has also been reported for CBM3 domains; however, this is not a non-hydrolytic mass loss mechanism.14,21 There is also a key difference between the CBM3 domains we used, which appears to have a negative dissipation effect, suggesting that they are making the system more rigid rather than less rigid, as is the case for the PfCel7A. Therefore, some as yet unknown emergent property of the PfCel7A-CBM3b-AcCel5A allows this construct to possibly be a non-catalytic cellulose disruptor as well as a hydrolytic enzyme.
Difference in deconstruction mechanisms
To further explore this idea of cellulose disruption and novel multifunctional enzyme cellulose deconstruction mechanisms we have employed both TEM and SEM imaging techniques to visualize the substrate as it is being digested.The images of the multi-functional digested particles all show some differences, but also some similarities to the control digested particles. Notably, tip sharpening is a phenotype associated with processive PfCel7A enzymes, such as PfCel7A (Fig. S9†). Blunt end splaying and pit formation morphologies have previously been associated with CelA type multi-functional enzymes (Fig. S9†). What is clear is that PfCel7A-CBM1-AcCel5A, PfCel7A-Link-AcCel5A and PfCel7A-CBM3b-AcCel5A to some extent share both hydrolysis morphologies. Furthermore, PfCel7A-CBM1-AcCel5A and PfCel7A-Link-AcCel5A appear to impart extensive nanofibrillation/fuzzing when compared to the control sample (Fig. 5b and c). The PfCel7A-CBM3b-AcCel5A digested particles, while at the SEM length scale appear somewhat similar to the CelA digested particles, also share some similarities at the TEM length scale1. Although we do observe tip sharpening, which is characteristic of PfCel7A (Fig. S9†). However, we also see some level of end splaying as well (Fig. 5a). Notably, no “pits” such as those previously observed for CelA were observed (Fig. 5a).
We also utilized SEM and it is clear from the lower magnification images that all three enzymatic configurations tested have an effect on reducing the physical size of the Avicel as compared to the larger particle size samples characterized in the control Avicel (PH 101) substrate, which are nominally between 50 to 100 μm (Fig. 6). In most fields of view, the average reduction in particle size appear to be in the 40 to 60 μm range suggesting effective digestion in all cases. The images of PfCel7A + AcCel5A, PfCel7A-Link-AcCel5A and PfCel7A-CBM1-AcCel5A all present a similar “smooth” digestion morphology at this magnification (Fig. 6B and C). However, the enzymatic mechanisms in which the CelA and PfCel7A-CBM3b-AcCel5A cocktails appear to digest cellulose are of a delamination and separation method which is illustrated in Fig. 6C. This observation is consistent with our prior findings utilizing TEM and tomography.1
Materials and methods
Construct design and architecture
In creating multifunctional enzymes, we elected to exploit the synergy found between exo- and endo-acting cellulases found in natural enzyme systems. Such synergy has been well-established in free fungal and bacterial cellulase systems and this architecture is clearly used in multifunctional enzymes, such as the hyperthermophilic CelA from C. bescii, which utilizes a GH9 endo-cellulase domain, a GH48 exo-cellulase domain, and three GH Family 3 cellulose binding modules (CBMs) all connected by a series of linker domains. To date; however, CelA has not been expressed in fungal systems, presumably due to its large size and/or incompatible glycosylation patterns.To explore whether we could produce a similar multifunctional enzyme system in a fungal host and to systematically study the factors that are important for multifunctional enzyme function we chose to use well-characterized, highly active enzyme domains that we knew were expressible in T. reesei, specifically the PfCel7A exo-cellulase from P. funiculosom (PfCel7A), and the Y245G mutant of the AcCel5A endoglucanase catalytic domain from A. cellulolyticus (AcCel5A) that is well known to synergize with cellobiohydrolases.22 Three different multifunctional cellulases were designed, constructed, expressed, and purified. Each had an N-terminal PfCel7A catalytic domain and linker connected to a C-terminal Trichoderma reesei Cel6A linker and AcCel5A catalytic domain through either the native PfCBM1, a CbCBM3, or the linkers alone without a CBM. In addition, we generated a PfCel7A-CBM3b mutant by swapping the native CBM1 for a CbCBM3. All enzymes were expressed in T. reesei and purified to homogeneity as described below. The overall enzyme scheme of the multifunctional enzymes tested is shown in Fig. 1.
Molecular image creation
The molecular images in Fig. 1 were generated with PyMOL version 2.2.0 (PyMOL reference). The molecular coordinates for the Cel7A catalytic domain (CD) are taken from the crystal structure for TrCel7A (PDB code 4C4C).23 The glycosylation pattern displayed on the Cel7A CD and linker are the same as those utilized in computational studies of TrCel7A,24,25 which were based on the experimental evidence of Amore et al. (CD)24 and Harrison et al. (linker).26 The Cel7A linker coordinates are taken from a computational model utilized in Amore et al.24 The molecular coordinates for the CBM1 are from the original NMR structure presented by Kraulis et al.27 Atomic coordinates for Cel5A CD are from the Acidothermus cellulolyticus endoglucanase “E1” crystal structure (PDB code 1ECE).28 The atomic coordinates for the Cel6A linker configuration originated from the computational study of Payne et al.25 The coordinates for CBM3 represent a homology model from SWISS-MODEL29 generated for the sequence of Clostridium thermocellum CBM3b. The template utilized was the structure of the CBM3a in the main scaffoldin from C. thermocellum (PDB code 4B9F), which has 54% sequence identity with C. thermocellum CBM3b. Finally, the coordinates for the cellulose surface originate from the computational study of Payne et al.25 Please note that the actual enzymes utilized in the study have, in some cases, different organisms of origin from those used to make the images in Fig. 1 (for convenience in figure making), but at the level of detail displayed in Fig. 1, this has no significant effect.Cloning and transformation
For generation of the three constructs, PfCel7A-CBM1-AcCel5A, PfCel7A-Link-AcCel5A and PfCel7A-CBM3b-AcCel5A, we used the parent vector pTreno-PF (Linger et al. 2015).30 This vector contained the P. funiculosum Cel7A under the control of the T. reesei cbh1 promoter and a cbh2 terminator. The pTreno-PF vector was amplified using the primers SV-32 (AAT CTA GAG GCT TTC GTG ACC G) and SV-30 (AAG GAT CCT CCA GTG CCA GTA GAG) such that only the P. funiculosum cel7A catalytic- and linker-domains were retained in the PCR product. The two primers also introduced a BamHI at the 3′ end of the P. funiculosum linker and an XbaI site at the 5′ end of the cbh2 terminator. The vector was then restricted with BamHI and XbaI enzymes to be used as the base vector.Three different insert sequences were generated for construction of PfCel7A-CBM1-AcCel5A, PfCel7A-Link-AcCel5A and PfCel7A-CBM3b-AcCel5A plasmids as follows: for the PfCel7A-CBM1-AcCel5A construct, the P. funiculosum CBM1 sequence along with T. reesei Cel6A linker and the A. cellulolyticus E1 sequence was synthesized using the BioXP 3200 System (SGI-DNA), such that a BamHI and XbaI restriction sites were introduced at the 5′ and 3′ ends, respectively. The E1 sequence contained the Y245G mutation that was previously shown to increase its enzymatic activity.9 Similarly, for PfCel7A-Link-AcCel5A and PfCel7A-CBM3b-AcCel5A constructs, the Cel7Alinker-E1 fusion sequence and the Caldicellulosiruptor bescii (cb) CBM3b-Cel7ALinker-E1 fusion sequences were synthesized using the same instrument with built-in 5′ BamHI and 3′ XbaI sites. The cbm3 sequence was codon optimized for expression in T. reesei prior to synthesis. The three fragments were independently cloned into a temporary vector pUCGA-1.0 followed by confirmation of their nucleotide sequences by DNA sequencing analysis. The three fragments were excised from the temporary vector using BamHI and XbaI enzymes and cloned into the base vector obtained above. The plasmid vector maps are presented in ESI Fig. S1, S2 and S3.† As a E1-negative control plasmid, we also generated a PfCel7A-CBM3b-AcCel5A construct lacking the E1 sequence. In order to generate this plasmid, the PfCel7A-CBM3b-AcCel5A construct was amplified with primers SV-75 (GTA GTA ATC TAG AGG CTT TCG TGA C) and SV-165 (ATC TAG ATT ACG CTC CGG AAG GTT CTT GG) so as to eliminate the T. reesei Cel6A linker and the E1 sequences from the PfCel7A-CBM3b-AcCel5A construct, while introducing an XbaI sequence at the 3′ end of the cbCBM3 sequence. The obtained vector PCR fragment was restricted with XbaI enzyme followed by self-ligation of this restricted PCR product to obtain a circularized product (Fig. S4†).
Each of these plasmid vectors were linearized with Sbf1 enzyme, purified using the DNA Clean and Concentrator-5 kit (Zymo Research Corp. Irvine, CA) and transformed into either the catabolite repressed T. reesei QM6A mutant strain, QM9414, or the NREL Cel7A knock-out strain of QM6A, AST1116, using an established protocol (Subramanian et al. 2017).31 Transformants were plated on PDHX (potato-dextrose agar containing 0.1% (v/v) Triton X-100 and hygromycin (100 μg mL−1)) plates and incubated in light at 30 °C for 2–3 d to allow colony development. Individual transformant mycelial fragments were transferred to 2 mL of minimal medium containing 5% glucose (MAG) containing hygromycin (100 μg mL−1) in a 24-well microtiter plate and incubated for 3 d. Fifteen μL of cell-free culture broth was mixed with 5 μL of SDS-PAGE loading buffer in a microcentrifuge tube and boiled for 10 min at 95 °C. This protein extract was separated on 4 to 12% NuPAGE gel, followed by transferring the proteins onto PVDF membrane. Membranes were probed with anti-Cel7A polyclonal antibody raised against the P. funiculosum Cel7A protein in rabbit. Cel7A-fusion proteins were detected using alkaline phosphatase-conjugated anti-rabbit secondary antibody (Thermo Fisher Scientific, Inc. Grand Island, NY, USA, ESI Fig. 5A, 6A, 7A, and 8A†). Upon confirmation of protein expression in a transformant, the best expressing transformant was subjected to clonal isolation procedure. This involved obtaining a conidial spore preparation from the protein expressing transformant, spreading the spore suspension to obtain single isolated colonies on PDHX media plates. Small mycelial fragments from five independent colonies were then subjected to the protein screening protocol as explained above to obtain a clonal isolate (ESI Fig. S5B, S6B, S7B, and S8B†), which was used for large scale protein expression in a batch fermenter.
Enzyme expression
For the multifunctional enzymes, transformed T. reesei clonal isolates were streaked onto Potato Dextrose Agar plates and grown for 2 to 3 d until a well-lawned plate of spores was achieved. A ∼0.5 cm plug was extracted from the plate and deposited into 1.0 L of Mandel's Media with 1.0% glucose and 0.5% tryptone in a 2.8 L Fernbach flask. The culture was grown at 28 °C at 150 rpm for 24 h, after which the entire 1.0 L was transferred to 6.5 L of 1.15× of the same Mandel's media (yielding 1× after inoculum addition) in a 14 L New Brunswick bioreactor controlled by the New Brunswick BioFlo310 system. Operational parameters were 300 rpm using dual down-flow marine style impellers, 1.5 vvm filtered air, 28 °C, and pH controlled to pH 4.8 using 2.0 M HCl and 2.0 M KOH. The culture was grown for ∼24 h until the glucose concentration was less than 0.5%, after which 24% (w/v) glucose was added semi-continuously to maintain between 0.1 and 0.5% glucose until 1.0 L was added, usually about 36 h. The culture was harvested, filtered through a ∼1 μm nylon mesh filter bag (McMaster-Carr) to remove all cell mass, concentrated by tangential flow ultrafiltration with a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Dalton MWCO membrane (GE Health Sciences), buffer exchanged into 20 mM Bis–Tris pH 6.5 to a final volume of ∼200 mL, filtered through a 0.45 PES membrane, and stored at −20 °C.
000 Dalton MWCO membrane (GE Health Sciences), buffer exchanged into 20 mM Bis–Tris pH 6.5 to a final volume of ∼200 mL, filtered through a 0.45 PES membrane, and stored at −20 °C.
Enzyme purification
For PfCel7a and multifunctional enzymes, frozen concentrated/diafiltered broth was thawed, adjusted to 1.5 M (NH4)2SO4, filtered through a 0.2 μM PES filter, loaded onto a 26/10 Phenyl Sepharose Fast Flow column (GE Healthcare), and eluted with a descending 1.6 to 0 M (NH4)2SO4 gradient in 20 mM Bis–Tris pH 6.5 over 20 column volumes. Active fractions were identified as para-nitrophenyl-β-D-lactopyranoside (pNPL) activity by screening 25 μL fractions in 100 μL of 50 mM acetate pH 5.0 buffer with 2 mM pNPL in a microtiter plate incubated for 30 min at 45 °C. Reactions were quenched with 25 μL of 1 M NaCO3 and the absorbance at 405 nm (A405) was measured. Standard curve concentrations ranged from 0 to 250 μM pNP.pNPL-active fractions were pooled, concentrated as needed, desalted into 20 mM Bis–Tris pH 6.5 buffer, and loaded onto a Tricorn 10/100 Source 15Q anion exchange column. Bound proteins were eluted with a 0 to 0.5 M NaCl gradient over 30 column volumes in 20 mM Bis–Tris pH 6.5. pNPL activity was followed again to identify active fractions. After concentrating, the pooled active fractions were subjected to size exclusion chromatography (SEC) using over a 26/60 Superdex 75 column in 20 mM acetate pH 5.0 buffer containing 100 mM NaCl.
SDS-PAGE and αCel7A immunoblotting were performed to assess purity. For SDS-PAGE, samples were diluted 4:1 in 4× LDS sample buffer and run on a NuPAGE 4–12% Bis–Tris gel for 50 min at 200 V constant (Invitrogen). For western blots, the gel was transferred to a PVDF membrane using an iBlot2 (Invitrogen) operated sequentially at 20 V, 23 V, and 25 V for 1, 4, and 2 min, respectively. Immuno-detection of PfCel7A was achieved using the SNAP i.d. Protein Detection System (Millipore Corp., Billerica MA). The PVDF membrane was blocked using SuperBlock PBS (Thermo Fisher Scientific Inc., Rockford, IL) for 20 min. Rabbit anti-Cel7A polyclonal (custom antibody, Robert Sargeant, Ramona CA) was used as the primary antibody (1:20000 dilution of crude serum), with alkaline phosphatase-conjugated goat anti-rabbit IgG (Thermo Fisher Scientific Inc., Rockford, IL) as secondary. The alkaline phosphatase localization was visualized using BCIP/NBT (Life Technologies Corp., Carlsbad, CA).
T. maritima β-D-glucosidase was purchased from Megazyme (Bray, Ireland) and desalted using a Hi-Trap 26/10 (GE life sciences) desalting column to remove the ammonium sulfate stabilizer. Recombinant Penicillium funiculosum Cel7A and Acidothermus cellulolyticus AcCel5A-Y245G-catalytic domain were purified as described previously.1 For experiments requiring pure CelA (QCMD experiments), CelA was tagged with 6× His tag, expressed in C. bescii, and purified from the C. bescii secretome using a 5 mL HisTrap fast flow column (GE) followed by Superdex 26/60 200 prep-grade size exclusion in 100 mM NaCl 20 mM acetate pH 5.0 buffer mobile phase as described previously.2 This was utilized in the QCM experiments.
Substrate preparation
Generation of cellulose at varying degrees of crystallinity
Amorphous cellulose and cellulose I at reduced degree of crystallinity were prepared from cotton linters (CAS Number 9004-34-6; catalogue number 22183) obtained from Sigma-Aldrich by using a method described by Hall et al., 2010.33 Briefly, 1 g of dry cellulose powder was added to 30 mL of ice-cold concentrated phosphoric acid. The slurry was to reacted at 0 °C for 40 min with occasional stirring. After 40 min, 20 mL of ice-cold acetone was added to the slurry followed by stirring and filtration on a sintered glass crucible. The filtered sample was further washed three times each with 20 ml of ice-cold acetone and DI water.Cellulose nanocrystal generation
Cellulose nanocrystals were generated from Avicel PH101 via acid hydrolysis. The techniques utilized incubating Avicel in 4 N hydrochloric acid heated to 80 °C for 4 h. The reaction is stopped with a 10-fold dilution with DI water. The cellulose suspension is washed via centrifugation at 1600g until the pH of the solution is close to pH 4. Following this, the cellulose suspension is sonicated and pelleted with the supernatant collected. This process was continued until the supernatant is relatively clear. The CNCs in the pooled supernatants were not furthered concentrated.Cellulose thin films
Thin films of cellulose I nanocrystals were used as a model substrate for the QCMD work in measuring the binding and hydrolysis of proteins to cellulose. QCMD resonators were purchased from Filtec and consisted of 5 MHz-AT cut quartz crystals sensors with a SiO2 coating on top of the gold conductive layer. These sensors were rinsed with water, followed by an ethanol rinse and finally a dry etching with argon ion plasma. Following the cleaning procedure, these sensors were incubated in the 0.02% solution of PDADDMAC for thirty minutes, followed by washing in water in an orbital manner. The PDADDMAC is used as an anchoring polymer to attach the cellulose nanocrystals to the surface of the ACM resonator. The QCMD resonators were then coated with CNC'sx using a variable thin film spin coater with a pre-cycle spin set to spin for 15 seconds at 1500 rmp followed by the main cycle spin set for 60 seconds at 3500 rpm. This procedure was repeated 3 times to obtain a film thickness of approximately 25 nm (measured by QCMD).X-ray diffraction measurements
The crystallinity indexes (CI) of cellulose samples were measured by X-ray diffraction (XRD) by using a Rigaku (Tokyo, Japan) Ultima IV diffractometer with CuKα radiation having a wavelength λ (Kα1) = 0.15406 nm generated at 40 kV and 44 mA. The diffraction intensities of dried samples placed on a quartz substrate were measured in the range of 8 to 42° 2θ using a step size of 0.02° at a rate of 2° min−1. The crystallinity indexes of the cellulose samples were measured according to the amorphous subtraction method described by Park et al., 2010.34 A diffractogram of amorphous cotton linter cellulose sample mentioned above was subtracted from the other cellulose samples to remove the influence of the amorphous component in the diffractograms. The ratio of the integrated area of each subtracted diffractogram to the area of the original was then calculated and multiplied by 100 to give the CI value of the sample. The starting cotton linter material had a CI of 66% and the reduced crystallinity material had a CI of 33%.Compositional analysis of pretreated solids
To determine the structural carbohydrates and lignin components of the pretreated solids, the compositional analysis was conducted according to the NREL method previously described.35Activity assays and characterization
Heterologously expressed PfCel7A and AcCel5Acat were used as controls after production and purification as described previously.10,36 In using these controls, we chose to compare the synthetic multifunctional enzymes to their parent components on a molar equal active site loading, i.e., a multifunctional enzyme with two active sites would be compared an equal number of active sites from its parent components. Example: 100 nmol of a multifunctional enzyme composed of PfCel7A and AcCel5A would have 200 nmol equivalents of active sites and would be compared to 100 nmol of PfCel7A and 100 nmol of AcCel5A (total 200 nmol).Several cellulosic substrates were used in evaluating the three multifunctional cellulase constructs; as well as component mixtures of purified PfCel7A and AcCel5-AY245Gcat. Avicel was used as a standard cellulose for baselining the activities of the various enzyme systems shown in Fig. 2A. Two additional cellulose substrates of reduced crystallinity (66% and 33%) were generated as described previously and used to probe the enzymes for crystallinity-dependent performance2 (see Fig. 2C). Deacetylated Disc Refined (DDR) corn stover was used as a model biomass conversion feedstock substrate12 (see Fig. 2D). The imaging experiments were carried out using Avicel as the substrate.
The initial enzyme activity comparisons were performed on Avicel at 200 nmol enzyme active site per gram cellulose. For Fig. 2A, this means that the PfCel7a-CBM1-AcCel5a, PfCel7a-CBM3b-AcCel5a and PfCel7a-Link-AcCel5a were each loaded at 100 nmol g−1 glucan, but because these enzymes have two active sites, its effectively 200 nmol g−1 glucan, for the free enzyme control experiment we loaded the PfCel7a at 100 nmol g−1 glucan and the AcCel5a at 100 nmol g−1 glucan. On a mg g−1 glucan basis the loadings were: 9.2 mg g−1 glucan, 11.34 mg g−1 glucan, 9.6 mg g−1 glucan and 5.8 + 4.2 mg g−1 glucan respectively. All experiments were run in the presence of 0.5 mg beta glucosidase per g glucan. Note: The apparently higher mg g−1 loading for PfCel7a-CBM3b-acCel5a is due its higher mass and is equal on a molar basis to all the other enzymes. In subsequent experiments, we utilized equal mass loadings, as this is more typical for the field and therefore PfCel7a-CBM3b-AcCel5a is slightly underloaded compared to the other enzymes (i.e., approximately 15%).
For the differential crystallinity material digests (Fig. 2C), loadings were 15 mg total enzymes per g glucan. 14.5 mg g−1 Pf Cel7a-CBM1-AcCel5a and PfCel7a-CBM3b-AcCel5a and 0.5 mg g−1 beta glucosidase.
For the DDR material digestion (Fig. 2D), we loaded 10 mg Ctec2 (Novozymes) per g glucan and supplemented this basic loading with 5 mg g−1 of PfCel7a-CBM1-AcCel5a, PfCel7a-CBM3b-AcCel5a, PfCel7a-Link-AcCel5a, and 2.5 mg g−1 of PFCel7a + 2.5 mg g−1 AcCel5a. The Ctec2 only control was loaded at 15 mg g−1 glucan.
Digestions were run continuously for 5 d with sampling at various time points. Enzymes were inactivated by boiling for 15 min after which samples were filtered through 0.45 μm Acrodisc syringe filters. The released sugars were analyzed by HPLC. Samples were injected at 20 μL volume and run on an Agilent 1100 HPLC system equipped with a BioRad Aminex HPX-87H 300 mm x 7.8 mm column heated to 55 °C. A constant flow of 0.6 mL min−1 was used with 0.1 M H2SO4 in water as the mobile phase to give optimal sugar separation. Glucose, xylose, cellobiose and xylobiose were determined against independent standard curves and converted to anhydrous glucan equivalent and the results are reported as anhydrous glucan converted. All experiments were performed in triplicate and the resulting extents of conversion are shown as percent glucan or xylan converted.
All digestions were conducted at a total initial solid loading of ∼1% and pH 5.0. Digestions were run continuously for 5 d with sampling at various time points. Enzymes were inactivated by boiling for 15 min after which samples were filtered through 0.45 mm Acrodisc syringe filters. The released sugars were analyzed by HPLC following the protocol described above.
Biomass substrate microscopy
Digested Avicel PH101 samples were prepared for SEM imaging by freezing in liquid nitrogen followed by lyophilization. The dry samples were placed on aluminium stubs using carbon tape and sputter coated with 10 nm Iridium metal. Imaging was performed using a FEI Quanta 400 FEG SEM (FEI, Hillsboro, OR) instrument operating under vacuum (0.45 Torr) at a beam accelerating voltage of 20 keV and capturing secondary electrons with an Everhart–Thornley detector. For TEM analysis, digested Avicel PH101 samples were drop cast directly on carbon-coated 200 mesh copper grids (SPI Supplies, West Chester, PA) and negatively stained with 2% aqueous uranyl acetate. Images were taken with a four mega-pixel Gatan UltraScan 1000 camera (Gatan, Pleasanton, CA) on a FEI Tecnai G2 20 Twin 200 kV LaB6 TEM (FEI, Hillsboro, OR).Quartz crystal microbalance with dissipation (QCMD)
Binding experiments were carried out on a Q-Sense E4 system that measures both the frequency (Hz) and the dissipation simultaneously. For all binding experiments, the quartz sensors were initially characterized bare in both air and the appropriate buffer solution to measure their intrinsic frequencies. Following this, the sensors were coated with lignin, re-characterized to measure their new frequencies, allowing the mass of the lignin films to be calculated. All odd harmonic overtones were collected but the third harmonic overtone was used to measure the adsorbed mass of both the cellulose film and the adsorbed proteins. The dissipation data was not utilized in these experiments.During the binding experiments, the cellulose-coated sensors were allowed to come to equilibrium (resonance frequency reaches steady state) by flowing the buffer solution at 0.1 mL min−1 during the equilibrium phase of the experiment. Once equilibrium was reached, the enzyme solution was passed over the sensors for set amount of time at a rate of 0.1 mL min−1 during the adsorption phase. Finally, the sensors went through the rinsing phase where they were rinsed with the buffer solution for an addition set of time.
Conclusions
PfCel7A-CBM3b-AcCel5A is the first highly active, synthetic multi-functional fungal bacterial fusion enzyme to be expressed in a fungal host. This non-natural enzyme shows significant improvement compared to the PfCel7A + AcCel5A free enzyme mixture when digesting crystalline cellulose. It is also the best performing multi-functional enzymes combination we tested. Moreover, incorporating this specific enzyme into existing cellulase formulations provides a significant improvement in the digestion of DDR treated corn stover. The improvement of nearly 50% using a time to target metric suggests that these enzymes are synergistic with the other components in the Ctec2 mixture. Moreover, the architecture of this new enzyme may represent a general framework for future enzyme improvement [i.e., if catalytic domains that are more active or more thermostable than PfCel7A are discovered then they may operate even better in this construct]. A secondary finding was that in all cases tested, the multi-functional constructs outperformed PfCel7a alone on an equal active site basis. Before this study, PfCel7A was the best performing free fungal enzyme tested by these authors.The second set of findings pertains to the novel mass loss mechanism observed, which may explain the superior performance of the PfCel7A-CBM3b-AcCel5A multifunctional enzyme when compared to the other enzymes. This effect is also confirmed by its visually distinct fibrillation deconstruction mechanism as observed by TEM and SEM. In fact, it appears that all three of the different multi-functional enzymes have novel and distinct deconstruction mechanisms as observed by TEM when compared to the canonical PfCel7A and CelA mechanisms, possibly indicating that the deconstruction properties of cellulases and other hydrolytic enzymes are tunable to some extent, resulting in novel, even non-natural deconstruction paradigms.
Our progress to date indicates that it is possible to produce multifunctional enzymes in fungal hosts that are both less complex than CelA and much more efficient than the native fungal enzymes. These are both significant findings as heterologous enzyme expression in T. reesei has historically been difficult and producing better PfCel7A enzymes has also been challenging in the past. Furthermore, the results show that we can modulate bulk properties such as extent of cellulose binding of multifunctional enzymes by varying their modular composition. Also, testing indicates that we may be able to improve the overall performance of multifunctional enzymes by altering their domain architecture, i.e., specifically tailoring enzymes to work on highly crystalline substrates. We have also demonstrated that by generating these multi-functional enzymes, we can also modify the meso-scale deconstruction mechanism of cellulose by utilizing different combinations of catalytic domains and carbohydrate binding modules.
Author contributions
RB: Wrote the paper, designed the multifunctional constructs, purified proteins, performed the digestion experiments; VS: Generated the plasmids, performed the transformations into T. reesei, screened transformants; JY: Performed QCMD experiments; BH: Performed TEM experiments; TV: Performed the SEM experiments; TAV: Grew T. reesei to produce culture broths; BCK: Generated images used in Fig. 1; YBC: Screened fungal transformats for activity; YJB: Purified some of the proteins, contributed to experimental design, and writing; SD: Contributed to experimental design and writing; MH: Contributed to experimental design and in writing.Conflicts of interest
A patent was filed on the recombinant enzyme.Acknowledgements
This work was authored by Alliance for Sustainable Energy, LLC, the Manager and Operator of the National Renewable Energy Laboratory for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. Funding provided by U.S. Department of Energy Office of Energy Efficiency and Renewable Energy Bioenergy Technologies Office. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. We thank the U.S. Department of Energy Bioenergy Technologies Office for funding under Contract DE-AC36-08GO28308 with the National Renewable Energy Laboratory. Funding also provided by the Center for Bioenergy Innovation (CBI), a U.S. Department of Energy Bioenergy Research Center supported by the Office of Biological and Environmental Research in the DOE Office of Science.YBC was supported by Indo-US Science & Technology Forum (IUSSTF) – Bioenergy-Awards for Cutting Edge Research (B-ACER) grant, Govt. of India.
Notes and references
- R. Brunecky, M. Alahuhta, Q. Xu, B. S. Donohoe, M. F. Crowley and I. A. Kataeva, Science, 2013, 342(6165), 1513–1516 CrossRef CAS PubMed.
- R. Brunecky, B. S. Donohoe, J. M. Yarbrough, A. Mittal, B. R. Scott, H. Ding, L. E. Taylor Ii, J. F. Russell, D. Chung, J. Westpheling, S. A. Teter, M. E. Himmel and Y. J. Bomble, Sci. Rep., 2017, 7, 9622 CrossRef PubMed.
- R. Brunecky, D. Chung, N. S. Sarai, N. Hengge, J. F. Russell, J. Young, A. Mittal, P. Pason, T. Vander Wall, W. Michener, T. Shollenberger, J. Westpheling, M. E. Himmel and Y. J. Bomble, Biotechnol. Biofuels, 2018, 11, 22 CrossRef PubMed.
- N. D. Gold and V. J. J. Martin, J. Bacteriol., 2007, 189, 6787–6795 CrossRef CAS PubMed.
- K. Hirano, M. Kurosaki, S. Nihei, H. Hasegawa, S. Shinoda, M. Haruki and N. Hirano, Sci. Rep., 2016, 6, 35709 CrossRef CAS PubMed.
- R. Lamed and E. A. Bayer, in Advances in Applied Microbiology, ed. A. I. Laskin, Academic Press, 1988, vol. 33, pp. 1–46 Search PubMed.
- L. E. Taylor, B. C. Knott, J. O. Baker, P. M. Alahuhta, S. E. Hobdey, J. G. Linger, V. V. Lunin, A. Amore, V. Subramanian, K. Podkaminer, Q. Xu, T. A. VanderWall, L. A. Schuster, Y. B. Chaudhari, W. S. Adney, M. F. Crowley, M. E. Himmel, S. R. Decker and G. T. Beckham, Nat. Commun., 2018, 9, 1186 CrossRef PubMed.
- F. E. Ogunmolu, N. B. K. Jagadeesha, R. Kumar, P. Kumar, D. Gupta and S. S. Yazdani, Biotechnol. Biofuels, 2017, 10, 71 CrossRef PubMed.
- J. O. Baker, J. R. McCarley, R. Lovett, C.-H. Yu, W. S. Adney, T. R. Rignall, T. B. Vinzant, S. R. Decker, J. Sakon and M. E. Himmel, Appl. Biochem. Biotechnol., 2005, 121, 129–148 CrossRef PubMed.
- J. O. Baker, J. R. McCarley, R. Lovett, C. H. Yu, W. S. Adney, T. R. Rignall, T. B. Vinzant, S. R. Decker, J. Sakon and M. E. Himmel, Appl. Biochem. Biotechnol., 2005, 121–124, 129–148 CrossRef CAS PubMed.
- M. C. Dixon, J. Biomol. Tech., 2008, 19, 151–158 Search PubMed.
- X. Chen, J. Shekiro, T. Pschorn, M. Sabourin, M. P. Tucker and L. Tao, Biotechnol. Biofuels, 2015, 8, 173 CrossRef PubMed.
- J. M. Yarbrough, R. Zhang, A. Mittal, T. Vander Wall, Y. J. Bomble, S. R. Decker, M. E. Himmel and P. N. Ciesielski, ACS Nano, 2017, 11, 3101–3109 CrossRef CAS PubMed.
- H.-P. Fierobe, E. A. Bayer, C. Tardif, M. Czjzek, A. Mechaly, A. Bélaïch, R. Lamed, Y. Shoham and J.-P. Bélaïch, J. Biol. Chem., 2002, 277, 49621–49630 CrossRef CAS PubMed.
- Y. Vazana, Y. Barak, T. Unger, Y. Peleg, M. Shamshoum, T. Ben-Yehezkel, Y. Mazor, E. Shapiro, R. Lamed and E. A. Bayer, Biotechnol. Biofuels, 2013, 6, 182 CrossRef PubMed.
- S. Ahola, X. Turon, M. Österberg, J. Laine and O. J. Rojas, Langmuir, 2008, 24, 11592–11599 CrossRef CAS.
- F. Jiang, J. D. Kittle, X. Tan, A. R. Esker and M. Roman, Langmuir, 2013, 29, 3280–3291 CrossRef CAS PubMed.
- X. Turon, O. J. Rojas and R. S. Deinhammer, Langmuir, 2008, 24, 3880–3887 CrossRef CAS PubMed.
- P. J. Gao, G. J. Chen, T. H. Wang, Y. S. Zhang and J. Liu, Acta Biochim. Biophys. Sin., 2001, 33, 13–18 CAS.
- N. Din, N. R. Gilkes, B. Tekant, R. C. Miller Jr., R. A. J. Warren and D. G. Kilburn, Bio/Technology, 1991, 9, 1096 CrossRef CAS.
- J. Sakon, D. Irwin, D. B. Wilson and P. A. Karplus, Nat. Struct. Biol., 1997, 4, 810–818 CrossRef CAS PubMed.
- J. O. Baker, C. I. Ehrman, W. S. Adney, S. R. Thomas and M. E. Himmel, Appl. Biochem. Biotechnol., 1998, 70–72, 395–403 CrossRef CAS PubMed.
- B. C. Knott, M. Haddad Momeni, M. F. Crowley, L. F. Mackenzie, A. W. Götz, M. Sandgren, S. G. Withers, J. Ståhlberg and G. T. Beckham, J. Am. Chem. Soc., 2014, 136, 321–329 CrossRef CAS PubMed.
- A. Amore, B. C. Knott, N. T. Supekar, A. Shajahan, P. Azadi, P. Zhao, L. Wells, J. G. Linger, S. E. Hobdey, T. A. Vander Wall, T. Shollenberger, J. M. Yarbrough, Z. Tan, M. F. Crowley, M. E. Himmel, S. R. Decker, G. T. Beckham and L. E. Taylor, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13667 CrossRef CAS PubMed.
- C. M. Payne, M. G. Resch, L. Chen, M. F. Crowley, M. E. Himmel, L. E. Taylor, M. Sandgren, J. Ståhlberg, I. Stals, Z. Tan and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14646 CrossRef CAS PubMed.
- M. J. Harrison, A. S. Nouwens, D. R. Jardine, N. E. Zachara, A. A. Gooley, H. Nevalainen and N. H. Packer, Eur. J. Biochem., 1998, 256, 119–127 CrossRef CAS PubMed.
- J. Kraulis, G. M. Clore, M. Nilges, T. A. Jones, G. Pettersson, J. Knowles and A. M. Gronenborn, Biochemistry, 1989, 28, 7241–7257 CrossRef CAS PubMed.
- J. Sakon, W. S. Adney, M. E. Himmel, S. R. Thomas and P. A. Karplus, Biochemistry, 1996, 35, 10648–10660 CrossRef CAS PubMed.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2018, 46, W296–W303 CrossRef CAS PubMed.
- J. G. Linger, L. E. Taylor II, J. O. Baker, T. Vander Wall, S. E. Hobdey, K. Podkaminer, M. E. Himmel and S. R. Decker, Biotechnol. Biofuels, 2015, 8, 45 CrossRef PubMed.
- V. Subramanian, L. A. Schuster, K. T. Moore, L. E. Taylor II, J. O. Baker, T. A. Vander Wall, J. G. Linger, M. E. Himmel and S. R. Decker, Biotechnol. Biofuels, 2017, 10, 34 CrossRef PubMed.
- A. Sluiter, R. Ruiz, C. Scarlata, J. Sluiter and D. Templeton, Determination of structural carbohydrates and lignin in biomass, NREL Laboratory Analytical Procedure, National Renewable Energy Laboratory, Golden, CO, 2006 Search PubMed.
- M. Hall, P. Bansal, J. H. Lee, M. J. Realff and A. S. Bommarius, FEBS J., 2010, 277, 1571–1582 CrossRef CAS PubMed.
- S. Park, J. O. Baker, M. E. Himmel, P. A. Parilla and D. K. Johnson, Biotechnol. Biofuels, 2010, 3, 1–10 CrossRef PubMed.
- A. Sluiter, R. Ruiz, C. Scarlata, J. Sluiter and D. Templeton, Determination of extractives in biomass, NREL: Laboratory Analytical Procedure (LAP) 1617, 2005 Search PubMed.
- L. E. Taylor, 2nd, B. C. Knott, J. O. Baker, P. M. Alahuhta, S. E. Hobdey, J. G. Linger, V. V. Lunin, A. Amore, V. Subramanian, K. Podkaminer, Q. Xu, T. A. VanderWall, L. A. Schuster, Y. B. Chaudhari, W. S. Adney, M. F. Crowley, M. E. Himmel, S. R. Decker and G. T. Beckham, Nat. Commun., 2018, 9, 1186 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc03062j |
| This journal is © The Royal Society of Chemistry 2020 |