
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Centrifugal fractionation of softwood extracts improves the biorefinery workflow and yields functional emulsifiers†
Fabio
Valoppi
 *ab,
Maarit H.
Lahtinen
a,
Mamata
Bhattarai
a,
Satu J.
Kirjoranta
a,
Venla K.
Juntti
a,
Leena J.
Peltonen
c,
Petri O.
Kilpeläinen
d and
Kirsi S.
Mikkonen
ab
*ab,
Maarit H.
Lahtinen
a,
Mamata
Bhattarai
a,
Satu J.
Kirjoranta
a,
Venla K.
Juntti
a,
Leena J.
Peltonen
c,
Petri O.
Kilpeläinen
d and
Kirsi S.
Mikkonen
ab
aDepartment of Food and Nutrition, P.O. Box 66 (Agnes Sjöbergin katu 2), FI-00014 University of Helsinki, Finland. E-mail: fabio.valoppi@helsinki.fi; Tel: +358-2941-58223
bHelsinki Institute of Sustainability Science, Faculty of Agriculture and Forestry, FI-00014 University of Helsinki, Finland
cDivision of Pharmaceutical Chemistry and Technology, Drug Research Program, P.O. Box 56 (Viikinkaari 5 E), FI-00014 University of Helsinki, Finland
dNatural Resource Institute Finland (Luke), Tietotie 2, FI-02150 Espoo, Finland
First published on 1st August 2019
Abstract
With the emerging bio-based technologies, the fractionation of complex biomass is essential to obtain value-added functional molecules for material, chemical, and energy production. Softwood extracts obtained through environmentally friendly pressurized hot water extraction are heterogeneous mixtures rich in hemicelluloses and lignin. Here we developed a simple, fast, organic solvent-free, and sustainable method to fractionate softwood extracts using centrifugal forces. The characteristics of each obtained fraction in terms of composition, macromolecular properties (particle size, molar mass, charge), interfacial activity, and stabilization capacity were highly dependent on the centrifugal force and time applied. The hemicellulose and lignin contents in the fractions were balanced by centrifugal forces to obtain functional emulsifiers that efficiently stabilized the oil/water interface. Through fractionation of softwood extracts, we also found that both the hemicelluloses and lignin particles are involved in emulsion interface formation and stabilization. We demonstrated that centrifugation at low centrifugal forces (<20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g) can effectively separate softwood extracts into hemicellulose-rich and lignin-rich fractions. Organic-solvent free centrifugation is a scalable concept that can be feasibly and easily introduced into the biorefinery system and used to optimize the composition of biomass fractions for targeted purposes, reducing at the same time biorefineries’ environmental impact.
000g) can effectively separate softwood extracts into hemicellulose-rich and lignin-rich fractions. Organic-solvent free centrifugation is a scalable concept that can be feasibly and easily introduced into the biorefinery system and used to optimize the composition of biomass fractions for targeted purposes, reducing at the same time biorefineries’ environmental impact.
1. Introduction
Cellulose, hemicelluloses, and lignin constitute the most abundant biomass source on Earth.1 Cellulose makes up roughly 40–50% of the wood dry mass, while hemicelluloses and lignin constitute 25–35% and 18–35% of the wood dry mass, respectively.2 Cellulose has been the main refining target from wood from the end of the 19th century.3 It is commonly processed using chemical pulping such as Kraft and sulfite pulping, thermomechanical pulping (TMP), or chemi-thermomechanical pulping (CTMP) to produce, for example, fibers, nanocrystals, and derivates for paper, material, pharmaceutical, and food applications.4–6 During cellulose refining a large part of lignin and hemicelluloses often ends up in waste pulping liquor. The latter is then used for combustion to regenerate pulping chemicals (NaOH and Na2S) and produce steam and electricity for the pulping operation.7–9 Lignin and hemicelluloses are low-value by-products remaining outside the circular economy concept. The need to improve the sustainability of wood bioeconomy and to shift from an oil-based to a biomass-based society is fueling the transformation of traditional pulp mills into biorefineries.10 Biorefineries are expected to find efficient utilization and conversion methods for the valorization of all lignocellulosic components.9Biorefineries are facilities in which renewable biomass is transformed into value-added components for material, chemical, and energy production.9,11–13 The workflow of biorefineries is based on a primary refining where biomass is converted into intermediary products and a secondary refining where intermediary products are upgraded into final profitable products.13 It is thus evident that to obtain sustainable and economical processes, a careful design of the biorefinery workflow is necessary.12 Moreover, the characteristics (e.g. composition, purity, and molar mass) of biorefinery products should be designed according to their target application. This means that not only the wood biorefinery workflow must be conceived to effectively recover/separate cellulose, hemicelluloses, and lignin, but the characteristics of the fractions need to be tailored. In this case, a cascade utilization of lignocellulosic biomass would increase the value of each single component.14 To further increase the value of the whole biorefinery chain, new products should mainly derive from hemicelluloses and lignin rather than from cellulose, using a cascading biorefinery model.7,14
Hemicelluloses are a group of heterogeneous polysaccharides that are formed in plants and act as a supporting material in their cell walls.2 The most abundant hemicelluloses in softwoods are galactoglucomannans (GGMs). GGMs consist of a linear backbone composed of β-(1→4)-D-glucopyranosyl and partially acetylated β-(1→4)-D-mannopyranosyl units, branched with α-(1→6)-D-galactopyranosyl side groups.15 Lignin, on the other hand, is a macromolecule formed by polymerization of p-coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol.16 Lignin function in plants is related to the integrity of the cellulose/hemicellulose/pectin matrix in the cell wall, formation of xylem vessels, and protection of the plant from wounding and pathogens.17,18
In the cascading biorefinery concept of lignocellulosic biomass,14 hemicelluloses and lignin can be recovered, for example, from the process water or spent liquor after TMP17,19 or pre-extracted from sawdust or from wood chips before pulping using pressurized hot water extraction (PHWE),20 organosolv and steam-explosion extraction,17,21 or a vacuum-enhanced aqueous extraction method called BLN (from the initials of the inventors’ names), where cellulose, hemicelluloses, and lignin are efficiently separated.22 The fractionation of wood biomass into its individual components is a fundamental step towards the cascade utilization of biomass.14 Depending on the wood species and recovery method, hemicelluloses and lignin differ in terms of their composition, molar mass, and purity (i.e. presence of lignin and extractives in hemicelluloses).23 Hemicellulose-rich extracts can be further purified by adding an ethanol precipitation step and/or ultrafiltration.15 The isolation method and resulting characteristics determine the functionality and applicability of hemicelluloses and lignin and thus also the sustainability and profitability of the hemicellulose biorefinery concept. For example, the PHWE hemicelluloses, which have an intermediate average molar mass of 5000–10000 g mol−1 and a notable fraction of phenolic co-components, have been identified as effective novel bio-based emulsifiers for oil-in-water emulsion stabilization.23–28 Emulsifiers are surface-active substances able to adsorb at the oil/water interface and stabilize emulsified droplets, protecting them from aggregation. Emulsifiers can be divided into synthetic (e.g. Tweens, Spans, and Brijs) and bio-based (e.g. proteins, polysaccharides, saponins, and phospholipids) depending on their origin.29 The common characteristic of all emulsifiers is their amphiphilic property, that is the presence of hydrophilic and hydrophobic parts. The phenolic residues are responsible for hemicelluloses’ amphiphilic characteristics and superior stabilizing capacity against emulsion droplet breakdown and creaming compared to other commonly used biopolymers such as gum Arabic or synthetic small-molecular surfactants such as Tween20,25,26 as well as for the enhanced protection of oil against oxidation in emulsified systems compared to gum Arabic.24,30 Oil-in-water emulsions stabilized by hemicelluloses have been suggested for different potential applications such as environmentally compatible paints,23 delivery systems for essential fatty acids in food products,28 and essential oil carriers in beverages.31 Lignin particles have also been proposed as emulsion stabilizers. In this case, oil-in-water or water-in-oil Pickering emulsions are formed.32,33 Besides the extraction and purification methods, the particle preparation method also greatly affects the emulsifying performance of lignin particles.32 Moreover, stable oil-in-water emulsions can be obtained using lignin-containing food grade materials such as cocoa particles34 and spent coffee grounds.35 It is evident that hemicelluloses and lignin, extracted using, for example, the PHWE method, can be used to increase the sustainability of the food chain because woods can be grown on, for example, non-arable land, introducing a wood-to-food approach.
The presence of impurities such as phenolic compounds plays an important role in hemicelluloses’ functionality. However, hemicelluloses extracted with, for instance, the PHWE method are a complex mixture comprising different chemical compounds, including hemicelluloses, free and hemicellulose-bound phenolics, and residual lignin.24 There is no information available on the role of each fraction in the functionality of hemicelluloses, although this information could be critical in optimizing the workflow of a cascading biorefinery model. Thus, to increase the value added to hemicelluloses and lignin, to increase the profitability margin of wood biorefineries, and to improve the functionality of hemicelluloses, we studied a simple and sustainable organic solvent-free method to enhance separation efficiency in the biorefinery chain. The separation method described here does not use any organic solvent to fractionate hemicelluloses (such as ethanol), therefore recycling of chemicals is not needed. Specifically, we investigated the effect of centrifugal force-based methods on PHWE GGM with the aim of separating the GGM into fractions with different chemical compositions (i.e. different lignin-rich and hemicellulose-rich fractions). We then evaluated the ability of selected fractions to form and stabilize oil-in-water emulsions, following the evolution of the droplets’ size and distribution over time. Centrifugation is an industrially scalable concept that can be introduced into the cascading biorefinery workflow without disrupting/subverting the whole chain.
2. Experimental section
2.1 Materials
GGMs were extracted from spruce sawdust using a pressurized hot water flow-through extractor pilot plant.20 In the present work, around 96.9 kg of spruce sawdust (43.5 kg on dry basis) obtained from Herralan Saha Oy (Herrala, Finland) were extracted at 170 °C for 60 min at a flow rate of 20 L min−1 using tap water. The spruce sawdust was composed of 26% lignin, 23% hemicelluloses, 42% cellulose, and 3% extractives.20 At the end of the process, 1000 L of extract were collected. On dry basis, 24% of the initial sawdust was extracted, while 76% remained as a residue. The extract was composed of 17% lignin, 75% hemicelluloses, and 7.5% extractives, while the residue sawdust was composed of 30% lignin, 7% hemicelluloses, 58% cellulose, and 2% extractives.20 Following this, the extract pH was adjusted to neutral with NaOH and concentrated by means of ultrafiltration using tubular modified polyethersulfone membranes (EM006). The pH value of the extract was kept neutral during ultrafiltration using NaOH. The concentrated extract was then spray-dried (sGGM) by using a Buchi Mini Spray Dryer B-290 (Buchi, Switzerland). Spray drying was carried out at an inlet temperature of 170 °C, an outlet temperature of 65 °C, and a drying air flow rate of 667 L h−1. The concentrated extract was also precipitated using ethanol (1:8 concentrate:ethanol v/v) and then dried (eGGM), as described by Bhattarai et al.27 Around 1/3 of the concentrate was recovered after ethanol precipitation, accounting for 8% of the initial sawdust. Both sGGM and eGGM powders were stored in pouches protected from light. sGGM and eGGM had a water content of 7.5% and 6.5% (gwater/gsample), respectively.
Citric acid monohydrate, gallic acid, sodium hydroxide, dry methanol, D-glucuronic acid sodium monohydrate, D-sorbitol, lithium bromide, sodium hydroxide, and hydrochloric acid were purchased from Sigma-Aldrich (St Louis, MO, USA). Sodium azide, sodium carbonate, pyridine, bis(trimethylsilyl)trifluoroacetamide (BSTFA), L(+) arabinose, D(+) xylose, D(+) galactose, D(+) glucose, D(+) mannose, and Folin–Ciocalteu's phenol reagent were purchased from Merck (Darmstadt, Germany). Acetyl chloride, trimethylsilyl chloride (TMSCl), L(+) rhamnose monohydrate, and D(+) galacturonic acid monohydrate were purchased from Fluka (St Louis, MO, USA). Pullulan standards were obtained from Postnova Analytics (Landsberg am Lech, Germany). HPLC-grade methanol was purchased from JT Baker (Deventer, Netherlands), HPLC-grade heptane was purchased from Rathburn (Walkerburn, UK), and HPLC-grade DMSO was purchased from Lab-Scan (Dublin, Ireland). D2O and d6-DMSO were purchased from Eurisotop (Saint-Aubin, France). Rapeseed oil (Keiju, Bunge Finland Ltd, Raisio, Finland) was purchased in a local supermarket.
2.2 Sample preparation
700g (12500 rpm) for 20 min were carried out using an RC5C Sorvall (DuPont, Newtown, CT, USA) centrifuge mounted with a Fiberlite F-21 fixed angle rotor (Piramoon Technologies, Inc., Santa Clara, CA, USA). Ultracentrifugation was carried out using an Optima L-90K Ultracentrifuge (Beckman Coulter, Inc., Brea, CA, USA) mounted with a swinging-bucket rotor type SW 28 (Beckman Coulter) operating at 82700g (25000 rpm) for 20 min, or mounted with a fixed angle rotor type 50.2 Ti (Beckman Coulter) operating at 146000g (40000 rpm) for 20 and 130 min. Pellets and supernatants were immediately collected after centrifugation and ultracentrifugation. An additional fractionation was carried out, recovering the supernatant after centrifugation at 18700g for 20 min and ultracentrifuging it at 146000g for 130 min. From this step, only the pellet was collected. All centrifugations and ultracentrifugations were carried out at 20 °C. Finally, the obtained supernatants and pellets were freeze-dried for 72 h. The dried samples were stored in light-protected containers. Dry supernatants and pellets were labeled as S and P, respectively, with subscripts indicating the average relative centrifugal field and centrifugation time, for example, S146k_130 is the supernatant obtained by centrifuging the starting sGGM solution at 146000g for 130 min. The pellet obtained by centrifugation followed by ultracentrifugation was labeled as CUP. All samples are summarized in Table 1.
| Starting material | Fractionation parameters | Fraction code | Emulsion code | |
|---|---|---|---|---|
| Relative centrifugal force (×g) | Time (min) | |||
| eGGM = ethanol precipitated GGM; sGGM = spray-dried GGM; S = supernatant; P = pellet; E- = emulsion; CUP = pellet obtained after ultracentrifugation of S18.7k_20. | ||||
| eGGM | — | — | — | E-eGGM |
| sGGM | — | — | — | E-sGGM |
| 3000 | 20 | S3k_20 | — | |
| 3000 | 20 | P3k_20 | — | |
| 18700 |
20 | S18.7k_20 | E-S18.7k_20 | |
| 18700 |
20 | P18.7k_20 | — | |
| 82700 |
20 | S82.7k_20 | — | |
| 82700 |
20 | P82.7k_20 | — | |
| 146000 |
20 | S146k_20 | — | |
| 146000 |
20 | P146k_20 | — | |
| 146000 |
130 | S146k_130 | E-S146k_130 | |
| 146000 |
130 | P146k_130 | — | |
| S18.7k_20 | 146000 |
130 | CUP | E-CUP |
000 rpm for 2 min was used to obtain coarse emulsions. Fine emulsions were obtained by homogenizing the coarse emulsions with four passes at a pressure of 800 bar using a Microfluidizer 110Y high-pressure homogenizer (Microfluidics, Westwood, MA, USA) configured with 75 μm Y-type F20Y and 200 μm Z-type H30Z chambers in series, recirculating the sample in the homogenizer for 32 s (corresponding to three passes) and withdrawing the sample with an extra pass. All emulsions contained 1% (w/w) polysaccharide and 5% (w/w) oil. Sodium azide was added soon after homogenization at a 0.02% (w/w) final concentration to avoid microbial spoilage. Emulsions were stored in the dark at 40 °C for up to 8 weeks. Emulsions were labeled adding the prefix “E-” to the GGM or GGM fraction used, for example, the emulsion obtained using S146k_130 was labelled as E-S146k_130. All samples are summarized in Table 1.
2.3 Analytical determinations
:10 with Milli-Q water, and 600 μL of 7.5% (w/v) Na2CO3 solution were mixed and kept in the dark for 30 min. The samples were then read at 765 nm by means of a UV-1800 UV-Visible Spectrophotometer (Shimadzu, Kyoto, Japan), using water as a blank. Phenolic compounds were expressed as gallic acid equivalents (GAEs) in mg per g of dry sample using a 6-point gallic acid standard calibration curve.
800, 47300, 112000, and 212000 g mol−1 were used for column calibration. They were dissolved in the same eluent at a concentration of 1–2 mg mL−1. The SEC data were processed using OmniSEC 4.5 software (Viscotek Corp., Malvern Instruments, Malvern, UK).
:99). The instrument details and method for GC analysis are described by Chong et al.40 Quantification was performed using six levels of concentration of each monosaccharide. Methyl glucuronic acid was quantified based on the D-glucuronic acid standard as described by Chong et al.40 The total polysaccharide content was calculated from the monosaccharide content of triplicate samples applying correction factors to consider the condensation reaction among pentoses (0.88), hexoses (0.9), and uronic acids (0.91).
:10–1:100 using the same sodium citrate buffer to avoid multiple scattering effects. After loading the samples in the folded capillary cells and inserting into the instrument, they were equilibrated at 25 °C for 2 min. Next, three repeated measurements obtained from 30 continuous readings on each sample were recorded. ζ-Potential was calculated applying the Smoluchowski model on the acquired electrophoretic data using Dispersion Technology Software v. 5.10 (Malvern Instruments). Each sample was read at least twice.
:2–1:100 to avoid multiple scattering effects. After loading the samples in the folded capillary cells and inserting into the instrument, they were equilibrated at 25 °C for 2 min. Three repeated backscattering measurements at 173° obtained from 10 continuous readings on each sample were then recorded using Dispersion Technology Software v. 5.10 (Malvern Instruments). Each sample was read at least twice.
2.4 Data analysis
All determinations were expressed as the mean ± standard error (SE) of at least two measurements from two experimental replicates (n ≥ 2 × 2), if not otherwise specified. Statistical analysis was performed using R v. 3.5.1 (The R Foundation for Statistical Computing). Median-based Levene's test was used to check the homogeneity of variance, one-way ANOVA was carried out, and Tukey's-test was used as a post-hoc test to determine significant differences among means (p < 0.05). Linear regression analysis by least squares minimization was performed using Microsoft Excel 365 ProPlus (Microsoft Corporation, Redmond, WA, USA). The goodness of fit was evaluated based on statistical parameters of fitting (R2adj, p-value, standard error) and the residual analysis. Data were plotted using GraphPad Prism v. 5.03 (GraphPad Software, San Diego, CA, USA).3. Results and discussion
Galactoglucomannans (GGMs) extracted using the PHWE method are a heterogeneous mixture containing hemicelluloses, phenolic residues, and lignin.41 To improve the cascading biorefinery workflow of GGM to enable hemicelluloses to be used as an emulsifier, further fractionation can be applied after PHWE. We developed a simple, fast, organic solvent-free, and sustainable method to fractionate GGM into hemicellulose-rich and lignin-rich fractions using centrifugal forces. Composition, structure, and emulsifying ability of each fraction were tested, resulting in the development of a scalable method that can improve the cascading biorefinery system to obtain functional compounds for targeted applications.3.1 GGM fractionation
In the first part of this research, we fractionated spray-dried PHWE GGM (sGGM) by centrifugation and ultracentrifugation. We used the starting sGGM as a reference sample, while ethanol-precipitated GGM (eGGM) was used as an example of a current method for purification/fractionation. Centrifugation separated sGGM into a pellet rich in lignin and a supernatant rich in polysaccharides, as will be outlined in the following sections.Fig. 1 shows (A) the phenolic content of sGGM, eGGM, and supernatants after centrifugation and ultracentrifugation and (B) the recovery yield after centrifugation and ultracentrifugation.
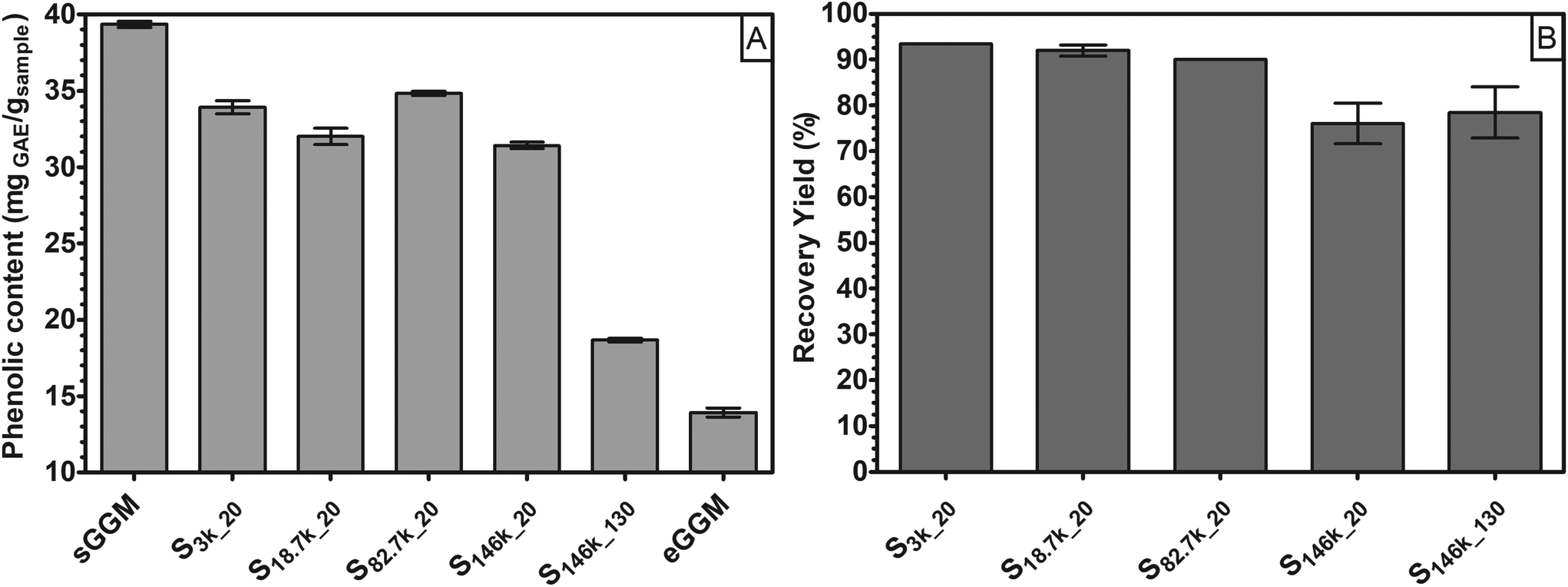 | ||
| Fig. 1 (A) Phenolic content (mgGAE gsample−1) for sGGM, supernatants obtained after centrifugation (S3k_20 and S18.7k_20) and ultracentrifugation (S82.7k_20, S146k_20, and S146k_130), and eGGM, and (B) recovery yield (%) for supernatants after centrifugation and ultracentrifugation. | ||
The sGGM showed a phenolic content of around 40 mgGAE gsample−1, in line with the value reported by Ebringerova et al.42 and Mikkonen et al.23 The application of centrifugal forces decreased the phenolic content of the supernatant to some extent. After the most severe centrifugal treatment (S146k_130), the phenolic content in the supernatant diminished markedly, although not as much as after ethanol precipitation (eGGM). The precipitation of GGM by ethanol removes the ethanol-soluble, low molar mass GGM oligo/polysaccharides and free phenolic residues.24
The samples treated with the highest centrifugal force (S146k_20 and S146k_130) showed a recovery yield between 75% and 80%, regardless of the centrifugation time. All other samples showed a yield greater than 90%, meaning that most of the GGMs was soluble and present in the supernatant. Differences between ultracentrifuged samples can be explained by the possible sedimentation of GGM aggregates. Indeed, hemicelluloses above a critical concentration tend to aggregate in water.43,44 These aggregates could have sedimented only at the highest centrifugal force applied. Interestingly, only when applying the harshest condition was a simultaneous reduction of both recovery yield and phenolic content in supernatant observed. The pellets accounted for 3.9 ± 0.4%, 6.0 ± 0.8%, and 15.9 ± 1.7% for 3000g for 20 min, 18700g for 20 min, and 146000g for 130 min, respectively. The mass balance of these selected samples led to a total recovery of around 97.5% of the initial GGM for both centrifuged samples, while ultracentrifugation led to a total recovery of around 93.8% of the initial GGM.
This means that by using centrifugal forces it is possible to fractionate sGGM into polysaccharide-rich and phenolic-rich fractions.
A first insight into the nature of the fractions obtained was achieved using FT-IR (Fig. 2).
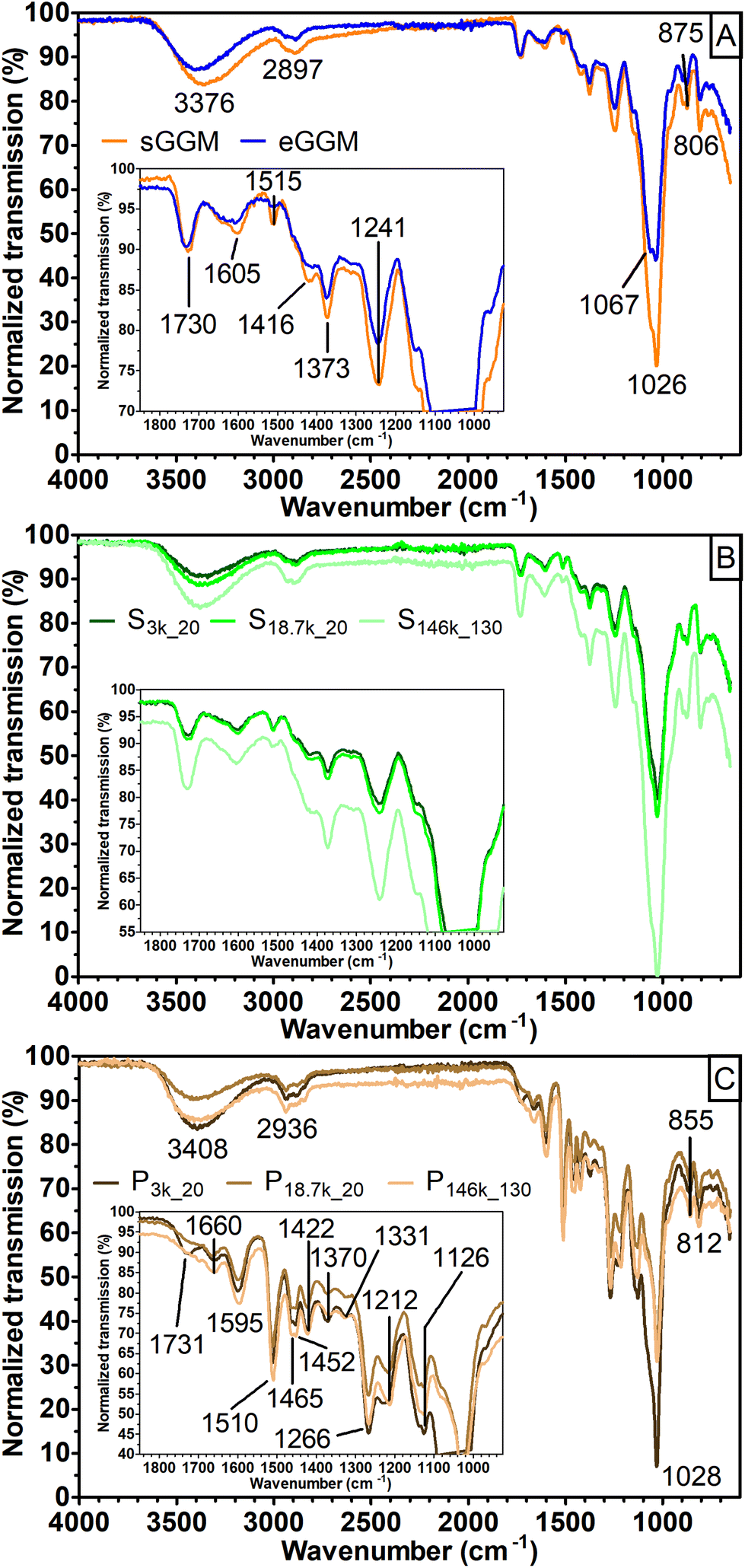 | ||
| Fig. 2 FT-IR transmission spectra for (A) sGGM (orange) and eGGM (blue), (B) selected supernatants (green) obtained after centrifugation (S3k_20 and S18.7k_20) and ultracentrifugation (S146k_130), and (C) selected pellets (brown) obtained after centrifugation (P3k_20 and P18.7k_20) and ultracentrifugation (P146k_130). In panels B and C, color darkening indicates a reduction of centrifugation force and time applied. Magnification of spectra between 1850 and 925 cm−1 is shown as an inset in each panel. | ||
The spectra obtained for both sGGM and eGGM (Fig. 2A) showed an intense absorption band at 1026 cm−1 and less intense absorption bands in the range of 1800–1200 cm−1. These bands are caused by the abundant fraction of hemicelluloses and the minor fraction of phenolic compounds.45–48 A characteristic peak at 1515 cm−1 related to the aromatic skeleton vibrations (polymerized phenolic compounds49) was present in both samples, meaning that phenolic compounds are present even after ethanol precipitation, in accordance with the phenolic content data (Fig. 1). sGGM, eGGM, and all supernatants shared similar IR absorption spectra (Fig. 2A and B, and Fig. S1†). On the other hand, pellets (Fig. 2C) showed totally different absorption bands, which are characteristic of lignin.49 It is reasonable that lignin is present in the pellet because the PHWE method is able to extract both hemicelluloses and lignin from wood sawdust,20 and lignin is insoluble at neutral pH50 and can form particles51 that can be sedimented using centrifugal forces. Finally, P3k_20 showed an additional peak at around 1735 cm−1. This peak was also present in supernatants, sGGM, and eGGM, but absent in all other pellets, indicating that P3k_20 had an intermediate composition between that of other pellets and supernatants. Thus, all centrifugation conditions tested in this work, except centrifugation at 3000g, are capable of effectively fractionating sGGM into hemicellulose-rich (supernatant) and lignin-rich (pellet) fractions.
Based on the great difference in phenolic content and recovery yield obtained, and FT-IR data, we selected 18700g for 20 min as the centrifugation method and 146000g for 130 min as the ultracentrifugation method.
To further characterize the fractions obtained with these centrifugation conditions, we carried out molar mass, carbohydrate composition and particle size analyses. Then, with the goal of using the hemicellulose-rich fractions as emulsifiers, we also determined their surface tension and ζ-potential in sodium citrate buffer.
Selected samples were then analyzed using size exclusion chromatography to gain information on their molar mass (Fig. 3A and B). The eGGM was characterized by a nearly unimodal RI distribution, with a peak maximum at 20 mL, corresponding to an estimated Mp of 6500 g mol−1 (Fig. 3A) and two low UV-absorbing fractions (Fig. 3B). The resulting weight-average molar mass Mw for this sample was estimated to be 8100 g mol−1, in accordance with the data reported by Bhattarai et al.27 All other samples showed bi- and polymodal distributions in the RI mode. In particular, pellets (P18.7k_20 and P146k_130) were characterized by an abundant fraction of small-sized molecules with an estimated Mp of 1500 g mol−1 able to highly absorb UV light. Their estimated Mw was approximately 2650 g mol−1. Pellets mainly contained lignin as observed using FT-IR, which is known to highly absorb in the UV region, with a local absorption maximum at 280 nm.52 On the other hand, sGGM and supernatants (S18.7k_20 and S146k_130) showed convoluted peaks in the RI mode, with the peaks being the sum of the two main peaks described for pellets and eGGM. The intensities of the supernatants’ peaks differed depending on the centrifugal force applied, giving rise to the peaks shown in Fig. 3A. Their estimated Mw was around 7200 g mol−1. Interestingly, S146k_130 had a smaller fraction of small-sized molecules and the RI peak resembled the eGGM peak, while S18.7k_20 resembled the sGGM RI peak. As expected, supernatants were also characterized by less UV-absorbing compounds than sGGM (Fig. 3B) due to the removal of the lignin-rich fraction, in accordance with the phenolic content data. The intensity of the UV signal decreased with increasing centrifugation force. Centrifugal forces and ethanol precipitation reduced the intensity of the UV signal, meaning that less phenolic compounds were present in the material after purification. Moreover, using ultracentrifugation it is possible to sediment a lower molar mass fraction that absorbs UV light, absent in the centrifuged pellet. It should be noted that the negative peak towards the end of retention volume in Fig. 3A is a typical system peak.53
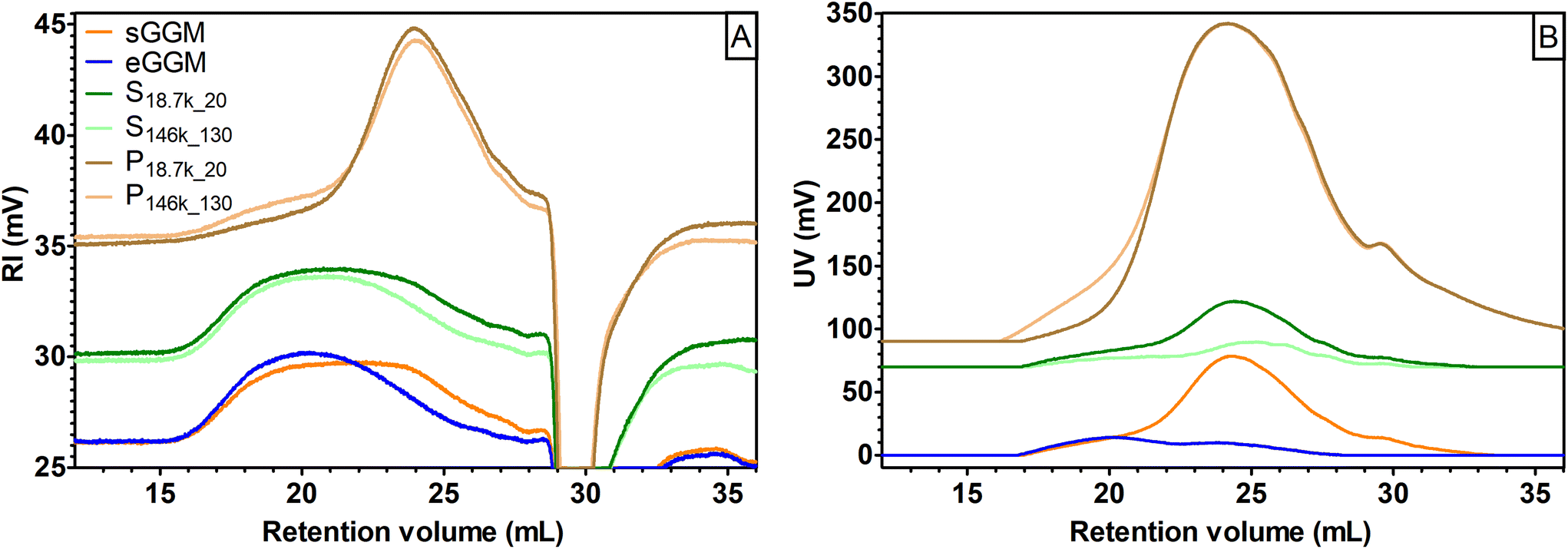 | ||
| Fig. 3 Size exclusion chromatograms in (A) RI and (B) UV absolute signal intensity for sGGM (orange) and eGGM (blue) – bottom curves, S18.7k_20 and S146k_130 (green) – middle curves, and P18.7k_20 and P146k_130 (brown) – top curves. For pellets and supernatants, color darkening indicates a reduction of centrifugation force and time applied. Curves were shifted vertically for clarity. | ||
Lignin can form insoluble particles and the velocity of sedimentation in gravitational and centrifugal fields is also governed by particle diameter as described by Stokes’ law.54Fig. 4 shows the intensity-based particle size distribution from dynamic light scattering analysis. A main peak between 500 and 600 nm is present for sGGM, eGGM, and the pellets. Interestingly, also supernatants contained particles, which were smaller than in the other samples and their size decreased with increasing centrifugal force (the main peak for S18.7k_20 and S146k_130 was 120 and 40 nm, respectively). Since both sGGM and pellets contain lignin, which can form insoluble particles of μm size during spray drying,55 we also analyzed the samples by using static light scattering. All samples showed signals above 2 μm (Fig. S2†), confirming the presence of bigger particles. In this case, the results are expressed as volume-based distributions, and caution should be paid when comparing these distributions with intensity-based distributions (Fig. 4). It is not surprising that even though pellets seemed to have the lowest molar masses, they sedimented first due to their bigger particle size.
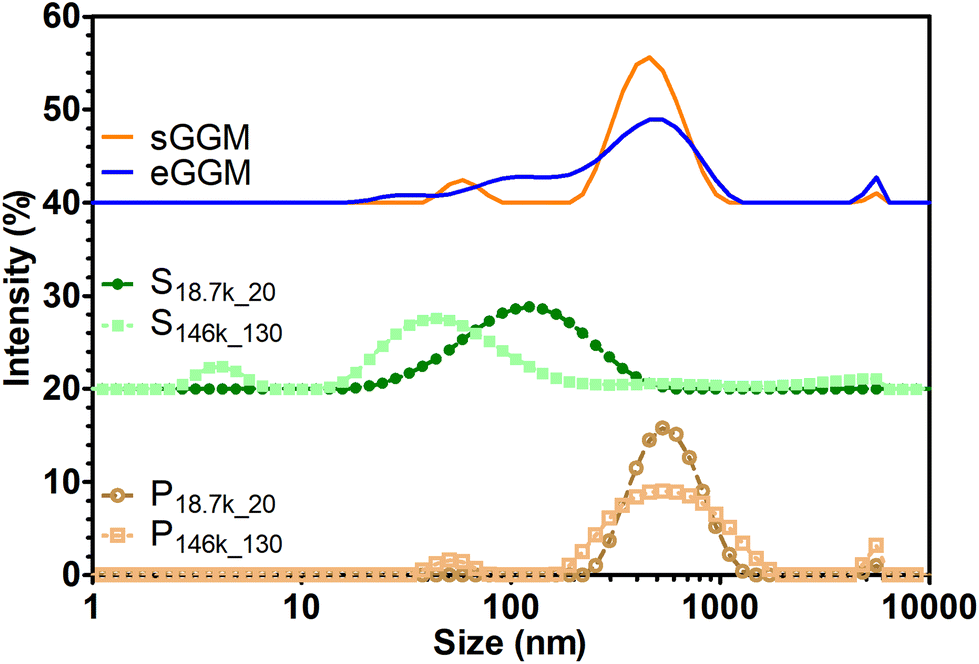 | ||
| Fig. 4 Intensity-based particle size distribution for sGGM (orange) and eGGM (blue) – top curves, S18.7k_20 (green, circle) and S146k_130 (green, square) – middle curves, and P18.7k_20 (brown, circle) and P146k_130 (brown, square) – top curves. In pellets and supernatants, color darkening indicates a reduction of centrifugation force and time applied. Curves were shifted vertically for clarity. | ||
Because sGGM is heterogeneous, fractionation or precipitation into completely pure fractions was not expected. Table 2 shows that sGGM is composed of around 72% carbohydrates, in agreement with Mikkonen et al.23 Ethanol precipitation and centrifugation increased the share of carbohydrates because free phenolic compounds and lignin particles were removed. On the other hand, ultracentrifugation did not affect the total carbohydrate content. This is because after ultracentrifugation, another fraction containing both carbohydrates and lignin was sedimented. The total carbohydrate composition calculated on the initial wood was lower for eGGM and ultracentrifugation compared to sGGM and centrifuged samples. The extract obtained from PHWE accounts for 24% of the initial wood material.20 Further fractionation using ethanol or ultracentrifugation reduced the yield to 8% and 18.8%, respectively, further reducing the share of carbohydrates. Interestingly, even if pellets were composed of lignin, as highlighted by FT-IR data (Fig. 2C), carbohydrates were also present at a concentration between 21% and 27%.
| Sample | Arabinose | Xylose | Rhamnose | Galactose | Glucose | Mannose | Galacturonic acid | Methyl glucuronic acid | Total sample-based | Total wood-based |
|---|---|---|---|---|---|---|---|---|---|---|
| n.d. = not detected; glucuronic acid was not detected in any of the samples. a,b,c,d = means with different letters in the same column are significantly different (p < 0.05). | ||||||||||
| sGGM | 1.28 ± 0.00b | 10.14 ± 0.07bc | 0.97 ± 0.01b | 7.35 ± 0.01b | 14.01 ± 0.01c | 60.05 ± 0.11a | 2.78 ± 0.04a | 3.43 ± 0.15ab | 734.7 ± 12.5bc | 176.3 ± 3.0a |
| eGGM | n.d. | 16.34 ± 0.11a | n.d. | 11.57 ± 0.07a | 16.73 ± 0.03bc | 60.72 ± 0.05a | 3.36 ± 0.00a | 2.69 ± 0.02bc | 857.4 ± 18.5a | 68.6 ± 1.4c |
| S18.7k_20 | 1.17 ± 0.03b | 10.04 ± 0.30bc | 0.86 ± 0.02b | 7.60 ± 0.11b | 13.93 ± 0.08c | 60.34 ± 0.13a | 2.80 ± 0.01a | 3.26 ± 0.04ab | 844.6 ± 15.0ab | 186.5 ± 3.3a |
| P18.7k_20 | 2.60 ± 0.43a | 8.66 ± 0.31c | 2.22 ± 0.34a | 6.99 ± 0.26b | 18.96 ± 1.52ab | 54.30 ± 1.04b | 2.02 ± 0.56b | 4.25 ± 0.55a | 251.3 ± 53.6d | 2.6 ± 0.6d |
| S146k_130 | 1.34 ± 0.02b | 11.21 ± 0.28b | 0.98 ± 0.03b | 7.50 ± 0.15b | 14.07 ± 0.16c | 59.16 ± 0.14a | 2.93 ± 0.06a | 2.81 ± 0.08bc | 730.0 ± 16.7c | 137.4 ± 3.1b |
| P146k_130 | 2.91 ± 0.06a | 8.73 ± 0.84c | 2.04 ± 0.02a | 8.10 ± 0.64b | 21.83 ± 1.12a | 52.89 ± 0.65b | 1.78 ± 0.26b | 1.71 ± 0.06c | 215.5 ± 4.8d | 8.2 ± 0.2d |
Not only the carbohydrate content was affected by centrifugal forces, but also the carbohydrate composition varied depending on the fractionation method used (Table 2). In particular, eGGM was richer in xylose, galactose, and galacturonic acid than the other samples. Compared with sGGM, pellets were richer in arabinose, rhamnose, and glucose, but depleted in xylose, mannose, and galacturonic acid. In particular, P18.7k_20 was rich in methyl glucuronic acid. Supernatants had a carbohydrate composition similar to that of sGGM.
Hemicelluloses extracted from spruce have been recently identified and studied as novel, bio-based, natural, value-added emulsion stabilizers by our group.23–27,30 To elucidate the emulsifying performances of sGGM, eGGM, S18.7k_20, and S146k_130 in oil-in-water emulsions, surface tension and ζ-potential were analyzed (Table 3).
| Sample | Surface tension ± SE (mN m−1) | ζ-Potential ± SE (mV) |
|---|---|---|
| a,b,c,d,e = means with different letters in the same column are significantly different (p < 0.05). n.d. = not determined. | ||
| Sodium citrate buffer | 66.82 ± 0.15a | n.d. |
| sGGM | 53.14 ± 0.40c | −2.45 ± 0.16b |
| eGGM | 59.64 ± 0.37b | −5.66 ± 0.30a |
| S18.7k_20 | 50.41 ± 0.51e | −4.13 ± 0.38a |
| S146k_130 | 55.74 ± 0.05d | −5.07 ± 0.18a |
All samples had reduced surface tension with respect to sodium citrate buffer. The magnitude of the reduction depended on the fractionation technique used and followed the order S18.7k_20 > sGGM > S146k_130 > eGGM. This means that ethanol precipitation and ultracentrifugation removed surface active molecules, while centrifugation removed the fraction that had lower or no surface activity. Low surface tension (i.e. high surface activity) is beneficial for emulsion stabilization because during homogenization less energy is required to overcome the Laplace pressure (i.e. the differential pressure between the inside and outside of a curved surface)29 and to obtain fine dispersed oil droplets. Alternatively, at a constant energy input and similar diffusion rate of surface active molecules between the aqueous phase and the oil/water interface, smaller oil droplets are expected soon after preparation in samples containing molecules with higher surface activity.29 Regarding ζ-potential, all samples exhibited negative values. Centrifugal forces and ethanol precipitation increased the number of negatively charged molecules. ζ-potential is used to understand the possible emulsion stabilization via electrostatic repulsions.29 In this case, however, the magnitude of the ζ-potential is not sufficient to guarantee an electrostatic stabilization of oil droplets because the absolute value of the ζ-potential is lower than 30 mV.25
3.2 Emulsions
To characterize the fractionated samples as emulsifiers, we prepared 5% rapeseed oil in water emulsions stabilized with 1% sGGM, eGGM, S18.7k_20, and S146k_130. Emulsions were then stored at 40 °C and analyzed over time for droplet size distribution and droplet mean diameters (Fig. 5).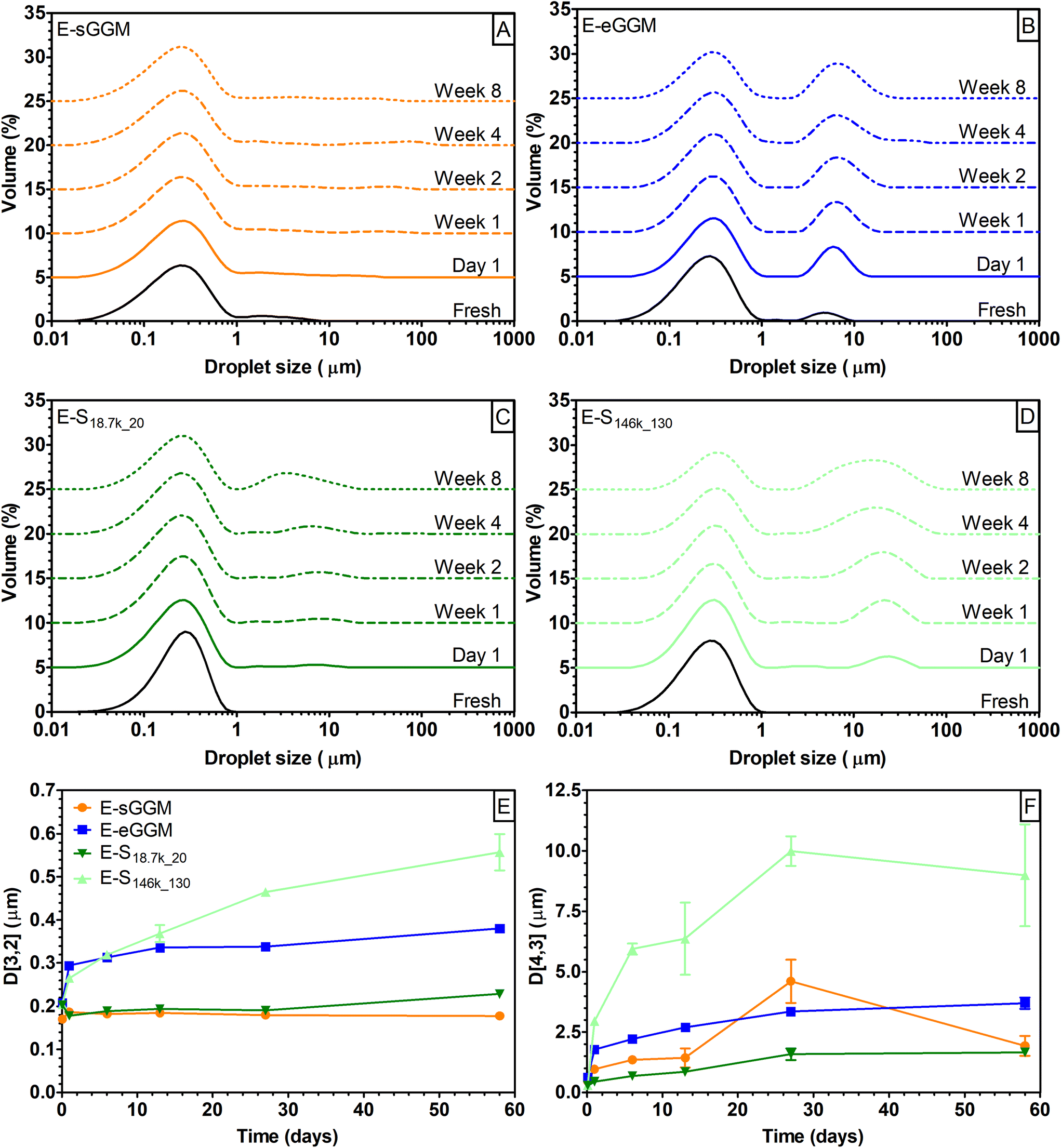 | ||
| Fig. 5 Evolution over time of droplet size distribution for emulsions (E-) obtained using (A) sGGM, (B) eGGM, (C) S18.7k_20, and (D) S146k_130 stored at 40 °C in the dark. Curves were shifted vertically for clarity. Evolution over time of (E) surface- and (F) volume-weighted mean diameter for E-sGGM (orange, circle), E-eGGM (blue, square), E-S18.7k_20 (dark green, reversed triangle), and E-S146k_130 (light green, triangle). | ||
Freshly prepared E-sGGM showed a bimodal droplet distribution with a main peak at around 0.3 μm and a shoulder at higher droplet size. During storage the main peak was stable while minor changes were recorded in the shoulder peak (Fig. 5A), in agreement with our previous results.24–26,28 On the other hand, E-eGGM showed two defined peaks at around 0.3 and 5 μm. During storage the main peak remained stable, while the peak at the higher droplet diameter became more intense and shifted to slightly higher values (Fig. 5B), in accord with the results of Bhattarai et al.27 The two hemicellulose-rich fractions obtained after centrifugation (S18.7k_20) and ultracentrifugation (S146k_130) produced emulsions with different characteristics (Fig. 5C and D). Regardless of the centrifugal force applied during fractionation, both supernatants produced monodisperse emulsions with droplet size distribution centered at 0.3 μm. Differences were noted during storage: a second peak appeared after 1 day of storage at dimensions <10 μm for E-S18.7k_20 and >10 μm for E-S146k_130. The second peak grew over time, indicating coalescence of the oil droplets during storage. However, the magnitude of the change was much more evident for E-S146k_130 (Fig. 5E and F). A rapid increase of the D[4,3] for E-S146k_130 was noted within the first week of storage, which indicates the formation of bigger oil droplets due to coalescence, confirming that E-S146k_130 was the most unstable emulsion. E-sGGM and E-S18.7k_20 were the most stable ones, while E-eGGM was situated between these extremes. Based on the evolution of droplet size distribution and mean droplet diameter over time, the stability of the emulsions followed the order: E-sGGM ≈ E-S18.7k_20 > E-eGGM ≫ E-S146k_130. This order agrees with the surface tension and phenolic content data, indicating that intense purification (ethanol precipitation or ultracentrifugation) of hemicelluloses gives rise to unstable emulsions. However, even though eGGM had a lower phenolic content than S146k_130, the obtained emulsions were more stable. This is because ethanol-precipitated hemicelluloses have covalently bound phenolic compounds in their polysaccharide backbone structure.18,56–58 GGM containing bound phenolic compounds can anchor at the oil/water interface, stabilizing the emulsion.24 In S146k_130, the separation of particles was based on their size, thus the supernatant is not only formed by GGM molecules bound to phenolic residues but also by free phenolic compounds and simple sugars that might decrease the overall interfacial activity of S146k_130. In S146k_130 and S18.7k_20, the absence of lignin-rich microparticles allowed us to obtain an emulsion with unimodally distributed oil droplets. This means that the lignin-rich microparticles are responsible for the shoulder observed in E-sGGM. The shoulder can represent either the microparticles or a fraction of oil droplets with bigger diameters. However, lignin-rich microparticles are also responsible for the stability of the small oil droplets and the aggregation/flocculation/coalescence of the large oil droplets in E-sGGM. The results suggest that in E-sGGM the stabilization mechanism is not only due to the GGM containing bound phenolic compounds, as we previously hypothesized,24,25,30 but also lignin-rich microparticles and possibly nanoparticles have a role in emulsion formation and stabilization. Finally, comparing the stability of E-S18.7k_20 and E-S146k_130, it is evident that the smaller particles present in the supernatants (Fig. 4) were not efficient in stabilizing the emulsions.
3.3 The surface-active fraction – the CUP sample
To better understand the differences between centrifuged and ultracentrifuged supernatants and their performances as emulsifiers, we further fractionated the sGGM. First, the sGGM was centrifuged at 18700g for 20 min to remove the lignin-rich fraction and obtain S18.7k_20. Next, S18.7k_20 was ultracentrifuged at 146000g for 130 min, and the pellet was collected (CUP). This sequential centrifugation/ultracentrifugation allowed separation of the fraction responsible for the differences observed between S18.7k_20 and S146k_130.
The CUP fraction represented around 10% (w/w) of sGGM and had a total phenolic content of 162.7 ± 2.0 mgGAE gsample−1. The phenolic content was more than four times that of sGGM (Fig. 1). Fig. 6 shows (A) the FT-IR spectrum, (B) the refractive index and UV signals during size exclusion chromatography, and (C) intensity-based particle size distribution for the CUP sample.
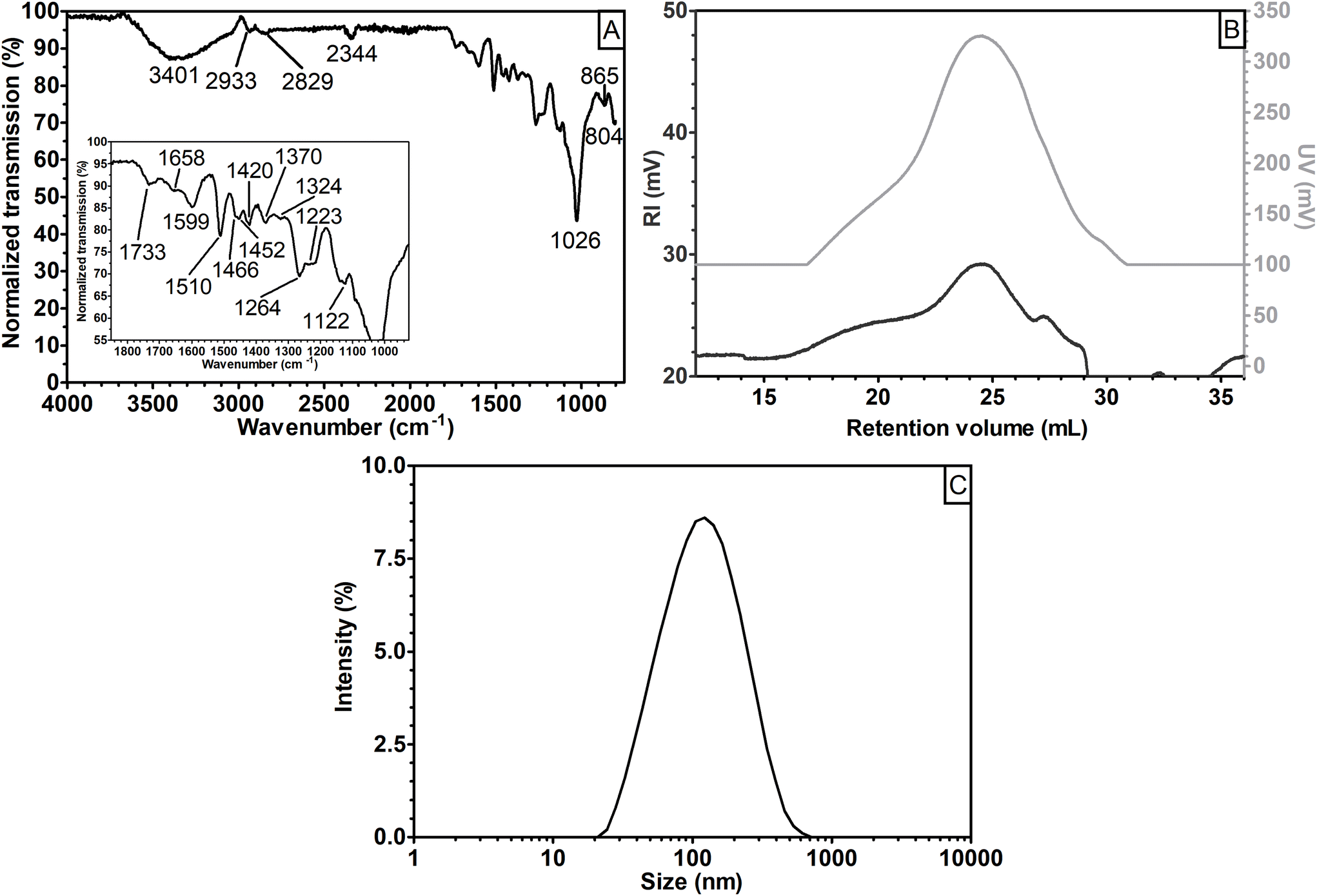 | ||
| Fig. 6 (A) FT-IR transmission spectrum, (B) the refractive index (dark gray) and UV (light gray) signals during size exclusion chromatography, and (C) intensity-based particle size distribution for the CUP sample. In panel A, the inset shows the magnification of the spectrum between 1850 and 925 cm−1. In panel B, curves were shifted vertically for clarity. | ||
Interestingly, the CUP sample, which was a fraction of the S18.7k_20 sample, showed an FT-IR spectrum similar to that obtained for the other pellets (cf.Fig. 6A with Fig. 2C), revealing that the CUP sample was composed of lignin. The RI signal in Fig. 6B shows a polymodal distribution with three convoluted peaks at an estimated Mp = 6500 g mol−1 (retention volume of 20 mL), Mp = 1000 g mol−1 (24 mL), and Mp < 342 g mol−1 (27.5 mL). The RI chromatogram had intermediate characteristics between those of eGGM and the pellets (Fig. 3A), while the UV signal (Fig. 6B) was similar to that of the pellets (Fig. 3B). The estimated Mw of CUP was calculated to be 3500 g mol−1, which was higher than the Mw of pellets. Finally, Fig. 6C shows the CUP particle size distribution. A unimodal distribution with a peak maximum at 120 nm was obtained. The CUP particle size distribution perfectly overlaps with that of the S18.7k_20 sample (Fig. 4). Based on these data, it is evident that the CUP sample (and partially S18.7k_20) was composed of lignin-rich nanoparticles. Probably, also S146k_130 contained a lignin-rich nanoparticle fraction with a smaller dimension. Even if S18.7k_20 contained lignin-rich nanoparticles, it can be inferred that the FT-IR signal related to the nanoparticles is suppressed by the signal generated by the abundant carbohydrates. Indeed, S18.7k_20 was composed of around 85% carbohydrates, while CUP was composed of 23% carbohydrates, similarly to pellets (cf.Table 4 with Table 2). Even if the total amount of carbohydrates in CUP was similar to that of the other pellets, their composition was different regarding xylose and mannose. The concentration of these two units was higher than that of pellet samples, but lower than that of sGGM and supernatants, indicating that CUP had an intermediate composition.
| Arabinose | Xylose | Rhamnose | Galactose | Glucose | Mannose | Galacturonic acid | Methyl glucuronic acid | Total sample-based | Total wood-based |
|---|---|---|---|---|---|---|---|---|---|
| 3.28 ± 0.22 | 9.77 ± 0.28 | 2.27 ± 0.16 | 7.90 ± 0.67 | 15.85 ± 0.31 | 57.87 ± 0.99 | 1.82 ± 0.28 | 1.24 ± 0.25 | 231.1 ± 9.9 | 5.4 ± 0.2 |
The surface tension of CUP was 49.29 ± 0.22 mN m−1. CUP showed low surface tension (i.e. high surface activity), which was not statistically different from that of S18.7k_20 (p > 0.05). On the other hand, the ζ-potential of CUP was −4.07 ± 0.01 mV, similar to that of all fractionated/purified samples, and thus, no electrostatic repulsions are expected among oil droplets after emulsion formation. To gain a better understanding of the identity of the CUP sample, the chemical structure was characterized using 2D NMR techniques, and the resulting HSQC spectrum in d6-DMSO is shown in Fig. 7. The signals were tentatively identified based on existing data.59–61
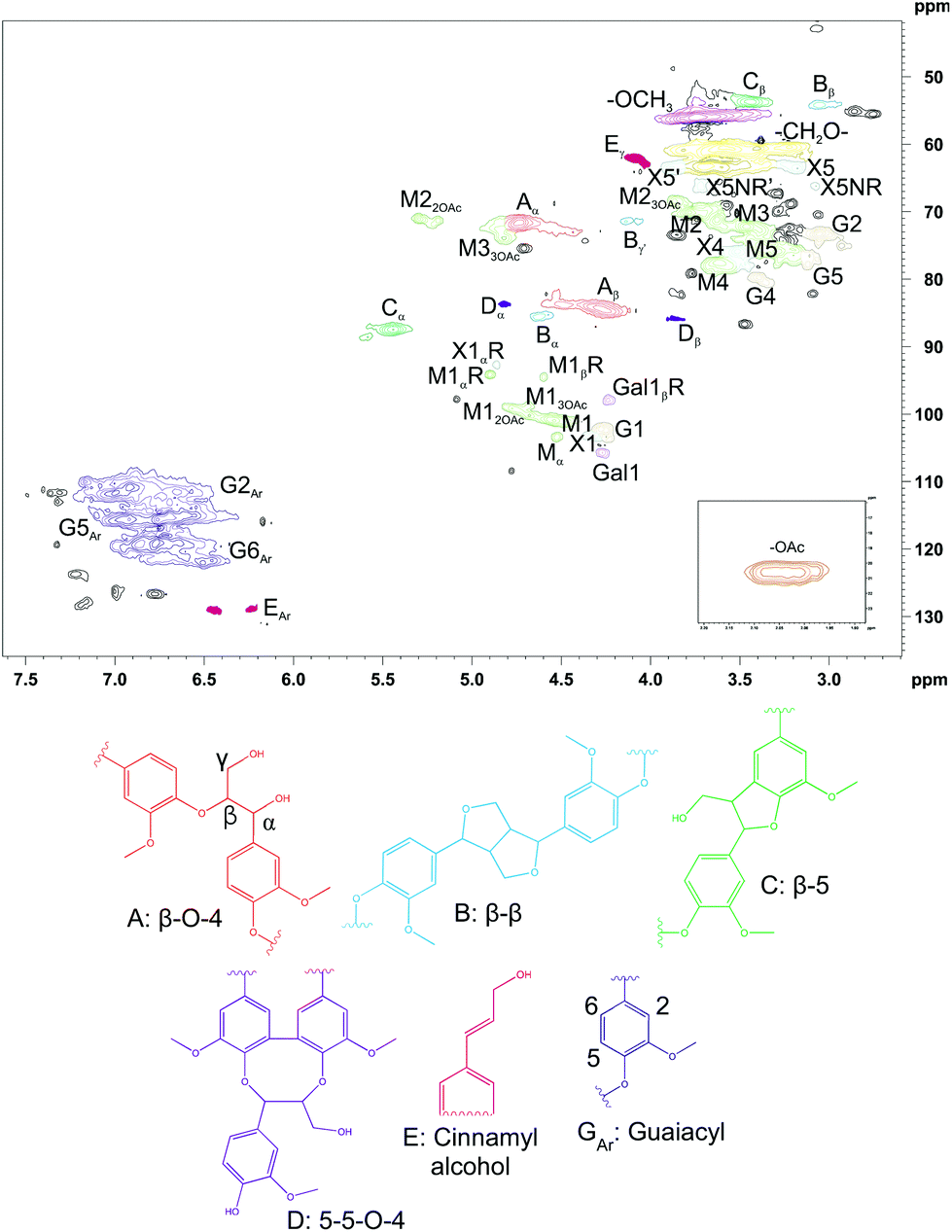 | ||
| Fig. 7 2D HSQC NMR spectrum of CUP in d6-DMSO. Structures related to lignin are presented as drawings in A–E and GAr. M = mannose, G = glucose, Gal = galactose, X = xylose. | ||
In general, the structure of lignin in CUP is consistent with the previously published HSQC spectrum of milled wood lignin.61 The signals for the main carbohydrates in GGM were also found, and the result of NMR analysis was comparable to that of the quantitative analysis of monosaccharides. In addition, there were some signals that could not be identified. For example, the signals at 2.75/54.82 ppm and 2.85/54.43 ppm might belong to some type of β–β structure,62,63 but the structure could not be confirmed because of overlapping signals.
In conclusion, the CUP sample was composed of slightly negative highly surface-active lignin-rich nanoparticles with the composition and molar mass lying between that of the pellets and the supernatants.
The CUP sample was then used as an emulsifier, similarly to the other samples (section 3.2). CUP produced homogeneous emulsions (E-CUP), and it is reasonable to assume that they were of Pickering type since CUP is composed of insoluble lignin-rich nanoparticles.
Freshly prepared E-CUP showed a clear bimodal distribution, with peaks at around 0.2 and 4 μm (Fig. 8A). The peak at the lower dimension shifted to 0.35 μm within the first day of storage and remained constant throughout the storage period. On the other hand, the peak at the higher dimension progressively decreased over time to reach a value of 2 μm after 14 days of storage. The overall effect of peak shifts can also be observed in the changes of Sauter and De Brouckere diameters (Fig. 8B). D[3,2] and D[4,3] rapidly increase within the first day of storage to then decrease and reach a constant value after 14 days.
 | ||
| Fig. 8 (A) Evolution over time of droplet size distribution for emulsion obtained using CUP stored at 40 °C in the dark. Curves were shifted vertically for clarity. (B) Evolution over time of surface- (cross, black) and volume-weighted (star, gray) mean diameter. | ||
Oil-in-water macro- and nanoemulsions are thermodynamically unstable systems, which tend to separate over time.29 During storage coalescence can take place and oil droplets merge together, increasing their sizes. In the present case, however, the progressive shift towards lower oil droplet size of the peak at 4 μm can be due to a rearrangement of lignin-rich nanoparticles at the interface, resulting in a reduction of the hydrodynamic diameter of the droplet. Particles at the oil/water interface can move between droplets and the interface can be subjected to structural rearrangements.64
In comparing E-CUP with the other emulsions analyzed in this work, it can be noted that the droplet size distribution was similar to that of E-eGGM (cf.Fig. 8A and Fig. 5B). However, even if E-CUP showed a clear bimodal distribution, it was as stable as E-sGGM and E-S18.7k_20 or even more stable during storage (cf. variations of D[3,2] and D[4,3] over time in Fig. 8B and Fig. 6E and F). It can be concluded that even if the particle size of the lignin-rich nanoparticles in CUP was the same as that of S18.7k_20, its smaller molar mass and the lower amount of carbohydrates did not permit the formation of unimodally distributed oil droplets in emulsion.
These results highlight that emulsion formation and stabilization via GGM is a complex phenomenon, comprising the anchoring of GGM bound with phenolic compounds and lignin-rich micro- and nanoparticles at the droplet interface, followed by a spatial reorganization of the molecules and particles at the interface over time.
These results are important in understanding GGM emulsifying mechanisms and also from the perspective of cascading biorefinery workflow improvement. To obtain a product that has good emulsifying and stabilizing ability, hemicellulose extracts containing lignin residues are desired. Centrifugation can be used to fractionate the extracts and to optimize the composition of fractions for targeted purposes. Centrifugation leads to the separation of lignin-rich microparticles, which can be further used to produce high value-added products such as vanillin, DMSO, carbon fibers and nanofibers, colloidal lignin particles, and controlled size lignin nanoparticles.17,21,65,66 The supernatant obtained after centrifugation can then be used as a high value-added effective, bio-based, green, and natural emulsifier for food, pharmaceutical, cosmetic, and chemical applications.
In this work we used PHWE as an example of the extraction method for hemicelluloses. However, some challenges related to this emerging technology must be addressed before PHWE becomes part of a cascading wood biorefinery model. Indeed, during extraction, the precipitation of highly reactive lignin can clog the system sticking onto, for example, pipes, heat exchanges, and receiving tanks.20 To overcome this problem, possible solutions can include cleaning of the system on a daily basis using, for example, an integrated cleaning in place system (CIP), a continuous fractionation inside the reaction vessel (by integrating the centrifuge into the reaction vessel) to separate lignin and hemicelluloses soon after extraction, or modulating the process parameters such as the ratio between wood sawdust and water, sawdust particle size, flow rate, and pH profile during extraction, to control the amount of extracted reactive lignin. Even though in this work we used the PHWE method as an example, the centrifugation concept can be extended to other existing extraction/recovery methods currently used in cascading biorefinery systems.
Centrifugation is a scalable concept that can be feasibly introduced into the cascading biorefinery system, leading to an increase of the value of products obtained in biorefineries.
4. Conclusions
In this study, we developed a centrifugal-based fractionation method to separate pressurized hot water softwood extracts into hemicellulose-rich and lignin-rich fractions. The successful approach described here demonstrates that (i) centrifugation can be used to improve the cascading biorefinery process using low centrifugal forces (<20000g), and a commercially available (e.g. conical plate) centrifuge can be used, (ii) centrifugation can modulate the composition and characteristics of softwood extract fractions, and consequently, the functionality and performances of the fraction can be tailored according to the intended end use, and (iii) centrifugal force-based fractionation sheds light on the complex mechanism underlying interfacial stabilization of softwood extracts.
The application of organic solvent-free centrifugation to the cascading biorefinery workflow model can improve the sustainability of biorefineries and reduce their environmental impact.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Maija Tenkanen for fruitful discussion on the manuscript, Dr Ndegwa Maina for fruitful discussion on size exclusion chromatography data, and Ms Julia J. Varis for drawing the graphical abstract. We also thank the NMR core facility supported by the University of Helsinki and Biocenter Finland. We gratefully acknowledge the Academy of Finland (project number 305517) for providing the funding for this project.References
- L. Klass, Biomass for Renewable Energy, Fuels, and Chemicals, Academic Press, New York, 1998 Search PubMed.
- R. C. Pettersen, The Chemical Composition of Wood, in The Chemistry of Solid Wood, ed. R. Rowell, Advances in Chemistry 207, American Chemical Society, Washington DC, 1984, vol. 207, pp. 57–126 Search PubMed.
- M. Brennan, Chem. Eng. News, 2000, 78, 42 CrossRef.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3359–3387 CrossRef PubMed.
- H. Shaghaleh, X. Xu and S. F. Wang, RSC Adv., 2018, 8, 825–842 RSC.
- S. M. F. Kabir, P. P. Sikdar, B. Haque, M. A. R. Bhuiyan, A. Ali and M. N. Islam, Prog. Biomater., 2018, 7, 153–174 CrossRef CAS PubMed.
- A. van Heiningen, Pulp Pap. Can., 2006, 107, T141–T146 Search PubMed.
- H. Tran and E. K. Vakkilainen, Tappi, 2008. Available online: https://www.tappi.org/content/events/08kros/manuscripts/1-1.pdf Search PubMed.
- G. Gellerstedt, E. Sjöholm and I. Brodin, Open Agric. J., 2010, 4, 119–124 CrossRef CAS.
- Ministry of Economic Affairs and Employment. http://www.bioeconomy.fi/publication-wood-based-bioeconomy-solving-global-challenges/ (accessed 18th September 2018).
- H. Q. Lê, Y. Ma, M. Borrega and H. Sixta, Green Chem., 2016, 18, 5466–5476 RSC.
- G. Mongkhonsiri, R. Gani, P. Malakul and S. Assabumrungrat, Comput. Chem. Eng., 2018, 119, 70–84 CrossRef CAS.
- T. Schröder, L. P. Lauven, T. Sowlati and J. Geldermann, J. Cleaner Prod., 2019, 211, 1502–1516 CrossRef.
- Y. Liu, Y. Nie, X. Lu, X. Zhang, H. He, F. Pan, L. Zhou, X. Liu, X. Ji and S. Zhang, Green Chem., 2019, 21, 3499–3535 RSC.
- S. Willför, K. Sundberg, M. Tenkanen and B. Holmbom, Carbohydr. Polym., 2008, 72, 197–210 CrossRef.
- G. Henriksson, Lignin, in Wood Chemistry and Wood Biotechnology, ed. G. Gellerstedt and G. Henriksson, De Gruyter, Berlin, Germany, 2009, pp. 121–146 Search PubMed.
- F. G. Calvo-Flores and J. A. Dobado, ChemSusChem, 2010, 3, 1227–1235 CrossRef CAS PubMed.
- H. Nishimura, A. Kamiya, T. Nagata, M. Katahira and T. Watanabe, Sci. Rep., 2018, 8, 6538 CrossRef PubMed.
- S. Willför, P. Rehn, A. Sundberg, K. Sundberg and B. Holmbom, Tappi J., 2003, 2, 27–32 Search PubMed.
- P. O. Kilpeläinen, S. S. Hautala, O. O. Byman, L. J. Tanner, R. I. Korpinen, M. K. J. Lillandt, A. V. Pranovich, V. H. Kitunen, S. M. Willför and H. S. Ilvesniemi, Green Chem., 2014, 16, 3186–3194 RSC.
- W. Fang, S. Yang, X. L. Wang, T. Q. Yuan and R. C. Sun, Green Chem., 2017, 19, 1794–1827 RSC.
- S. Von Schoultz, Method for Extracting Biomass, US Pat., 14413409, U.S. Patent and Trademark Office Patent, Washington DC, 2015 Search PubMed.
- K. S. Mikkonen, S. Kirjoranta, C. Xu, J. Hemming, A. Pranovich, M. Bhattarai, L. Peltonen, P. Kilpeläinen, N. Maina, M. Tenkanen, M. Lehtonen and S. Willför, Ind. Crops Prod., 2019, 133, 212–220 CrossRef CAS.
- M. Lehtonen, M. Merinen, P. O. Kilpeläinen, C. Xu, S. M. Willför and K. S. Mikkonen, J. Colloid Interface Sci., 2018, 512, 536–547 CrossRef CAS PubMed.
- K. S. Mikkonen, C. Xu, C. Berton-Carabin and K. Schroën, Food Hydrocolloids, 2016, 52, 615–624 CrossRef CAS.
- K. S. Mikkonen, D. Merger, P. Kilpeläinen, L. Murtomaki, U. S. Schmidt and M. Wilhelm, Soft Matter, 2016, 12, 8690–8700 RSC.
- M. Bhattarai, L. Pitkänen, V. Kitunen, R. Korpinen, H. Ilvesniemi, P. O. Kilpeläinen, M. Lehtonen and K. S. Mikkonen, Food Hydrocolloids, 2019, 86, 154–161 CrossRef CAS.
- F. Valoppi, N. Maina, M. Allén, R. Miglioli, P. O. Kilpeläinen and K. S. Mikkonen, Eur. Food Res. Technol., 2019, 245, 1387–1398 CrossRef CAS.
- D. J. McClements, Food Emulsions: Principles, Practices, and Techniques, CRC Press, Boca Raton, 3rd edn, 2016 Search PubMed.
- M. Lehtonen, S. Teräslahti, C. Xu, M. P. Yadav, A.-M. Lampi and K. S. Mikkonen, Food Hydrocolloids, 2016, 58, 255–266 CrossRef CAS.
- K. S. Mikkonen, M. Tenkanen, P. Cooke, C. L. Xu, H. Rita, S. Willför, B. Holmbom, K. B. Hicks and M. P. Yadav, LWT–Food Sci. Technol., 2009, 42, 849–855 CrossRef CAS.
- L. Bai, L. G. Greca, W. Xiang, J. Lehtonen, S. Huan, R. W. N. Nugroho, B. L. Tardy and O. J. Rojas, Langmuir, 2019, 35, 571–588 CrossRef CAS PubMed.
- L. Dai, Y. Li, F. Kong, K. Liu, C. Si and Y. Ni, ACS Sustainable Chem. Eng., 2019, 7, 13497–13504 CrossRef CAS.
- J. Gould, J. Vieira and B. Wolf, Food Funct., 2013, 4, 1369–1375 RSC.
- J. Gould, G. Garcia-Garcia and B. Wolf, Materials, 2016, 9, 791 CrossRef PubMed.
- M. T. Satue, S. W. Huang and E. N. Frankel, J. Am. Oil Chem. Soc., 1995, 72, 1131–1137 CrossRef CAS.
- L. Pitkänen, P. Tuomainen, K. S. Mikkonen and M. Tenkanen, Carbohydr. Polym., 2011, 86, 1230–1235 CrossRef.
- A. Sundberg, K. Sundberg, C. Lillandt and B. Holmbom, Nord. Pulp Pap. Res. J., 1996, 11, 216–219 CAS.
- C. Laine, T. Tamminen, A. Vikkula and T. Vuorinen, Holzforschung, 2002, 56, 607 CAS.
- S. L. Chong, S. Koutaniemi, L. Virkki, H. Pynnönen, P. Tuomainen and M. Tenkanen, Carbohydr. Polym., 2013, 91, 626–630 CrossRef CAS PubMed.
- L. Pitkänen, M. Heinonen and K. S. Mikkonen, Food Funct., 2018, 9, 1931–1943 RSC.
- A. Ebringerova, Z. Hromadkova, V. Hribalova, C. Xu, B. Holmbom, A. Sundberg and S. Willfor, Int. J. Biol. Macromol., 2008, 42, 1–5 CrossRef CAS PubMed.
- S. Kishani, F. Vilaplana, W. Xu, C. Xu and L. Wågberg, Biomacromolecules, 2018, 19, 1245–1255 CrossRef CAS PubMed.
- S. Kishani, A. Escalante, G. Toriz, F. Vilaplana, P. Gatenholm, P. Hansson and L. Wagberg, Biomacromolecules, 2019, 20, 1263–1270 CrossRef CAS PubMed.
- A. M. Raspolli Galletti, A. D'Alessio, D. Licursi, C. Antonetti, G. Valentini, A. Galia and N. Nassi o Di Nasso, J. Spectrosc., 2015, 2015, 1–12 CrossRef.
- F. Peng, J. L. Ren, F. Xu, J. Bian, P. Peng and R. C. Sun, J. Agric. Food Chem., 2010, 58, 1768–1776 CrossRef CAS PubMed.
- M. Schwanninger, J. C. Rodrigues, H. Pereira and B. Hinterstoisser, Vib. Spectrosc., 2004, 36, 23–40 CrossRef CAS.
- S. Viljamaa, E. Dikareva, J. Tolonen, J. Edesi, K. Nickolov, T. Laitinen, T. Laakso, R. Korpinen, P. Saranpaa, S. Jokipii-Lukkari, A. Karkonen and H. Haggman, Plant Cell, Tissue Organ Cult., 2018, 133, 225–235 CrossRef CAS.
- O. Faix, Holzforschung, 1991, 45, 21 CrossRef CAS.
- E. I. Evstigneev, Russ. J. Appl. Chem., 2010, 83, 509–513 CrossRef CAS.
- T. Sewring, J. Durruty, L. Schneider, H. Schneider, T. Mattsson and H. Theliander, J. Wood Chem. Technol., 2019, 39, 1–13 CrossRef CAS.
- A. J. Stamm, J. Semb and E. E. Harris, J. Phys. Chem., 1931, 36, 1574–1584 CrossRef.
- S. Levin and E. Grushka, Anal. Chem., 1986, 58, 1602–1607 CrossRef CAS.
- K. T. Whitby, J. Air Pollut. Control Assoc., 1955, 5, 120–132 CrossRef CAS.
- C. W. Miao and W. Y. Hamad, J. Appl. Polym. Sci., 2017, 134 DOI:10.1002/APP.44669.
- N. Giummarella and M. Lawoko, ACS Sustainable Chem. Eng., 2017, 5, 5156–5165 CrossRef CAS.
- N. Giummarella, Y. Pu, A. J. Ragauskas and M. Lawoko, Green Chem., 2019, 21, 1573–1595 RSC.
- A. Martinez-Abad, N. Giummarella, M. Lawoko and F. Vilaplana, Green Chem., 2018, 20, 2534–2546 RSC.
- J. Berglund, S. Azhar, M. Lawoko, M. Lindström, F. Vilaplana, J. Wohlert and G. Henriksson, Cellulose, 2019, 26, 2155–2175 CrossRef CAS.
- H. Kim and J. Ralph, RSC Adv., 2014, 4, 7549–7560 RSC.
- T. M. Liitiä, S. L. Maunu, B. Hortling, M. Toikka and I. Kilpeläinen, J. Agric. Food Chem., 2003, 51, 2136–2143 CrossRef PubMed.
- L. Zhang, G. Henriksson and G. Gellerstedt, Org. Biomol. Chem., 2003, 1, 3621–3624 RSC.
- F. Lu and J. Ralph, Novel β-β structures in lignins incorporating acylated monolignols, in Proceedings of the 13th International Symposium on Wood, Fiber, and Pulping Chemistry, May 2005, pp. 16–19 Search PubMed.
- D. J. French, A. T. Brown, A. B. Schofield, J. Fowler, P. Taylor and P. S. Clegg, Sci. Rep., 2016, 6, 31401 CrossRef CAS PubMed.
- M. H. Sipponen, M. Smyth, T. Leskinen, L. S. Johansson and M. Osterberg, Green Chem., 2017, 19, 5831–5840 RSC.
- W. W. Zhao, B. Simmons, S. Singh, A. Ragauskas and G. Cheng, Green Chem., 2016, 18, 5693–5700 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc02007a |
| This journal is © The Royal Society of Chemistry 2019 |