
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free quinolylation of the primary amino groups of amino acid derivatives and peptides with dihydrooxazolo[3,2-a]quinoliniums†
Peng
Liu
ab,
Bo
Li
 *ab,
Mengyu
Xi
ab,
Zhaoqiang
Chen
ab,
Haiguo
Sun
ab,
Xiajuan
Huan
a,
Xuejun
Xu
a,
Yong
Zhang
a,
Kun
Zou
a,
Xiangrui
Jiang
ab,
Zehong
Miao
ab,
Jinggen
Liu
ab,
Jingshan
Shen
ab,
Kaixian
Chen
abc and
Weiliang
Zhu
*abc
*ab,
Mengyu
Xi
ab,
Zhaoqiang
Chen
ab,
Haiguo
Sun
ab,
Xiajuan
Huan
a,
Xuejun
Xu
a,
Yong
Zhang
a,
Kun
Zou
a,
Xiangrui
Jiang
ab,
Zehong
Miao
ab,
Jinggen
Liu
ab,
Jingshan
Shen
ab,
Kaixian
Chen
abc and
Weiliang
Zhu
*abc
aKey Laboratory of Receptor Research, Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China. E-mail: boli@simm.ac.cn; wlzhu@simm.ac.cn
bSchool of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing 100049, China
cOpen Studio for Druggability Research of Marine Natural Products, Pilot National Laboratory for Marine Science and Technology (Qingdao), 1 Wenhai Road, Aoshanwei, Jimo, Qingdao 266237, China
First published on 12th July 2019
Abstract
The chemical modification of the primary amino groups of amino acid derivatives and peptides is an important process in the pharmaceutical industry and the field of chemical biology. However, suitable reactions that can be carried out under mild and environmentally friendly conditions are limited. We present a versatile method to selectively modify primary amino groups using novel dihydrooxazolo[3,2-a]quinoliniums in 1-butanol as solvent under mild and metal-free conditions. The application of this method to peptides with primary amino, secondary amino, amide, alcoholic hydroxyl, phenolic hydroxyl, disulfide bond, ester and cyano groups revealed that only the primary amino groups were selectively modified, suggesting that this method is compatible with other reactive moieties. We also demonstrated that the quinolylation of existing peptides affected peptide bioactivity and stability, indicating that the novel dihydrooxazolo[3,2-a]quinoliniums can be widely applied, especially in medicinal chemistry and chemical biology.
Introduction
Peptide drugs interact with target proteins with highly specific binding capacity, high versatility, low immunogenicity and low toxicity, thereby attracting considerable interest in the pharmaceutical industry.1–5 The number of approved peptide drugs has been increasing in recent decades, and the drugs cover a broad range of therapeutic areas.6–8 Reported peptide drugs include atosiban (obstetrics),9 degarelix (oncology),10 liraglutide (metabolic disease),11 tesamorelin (antiinfective),12 peginesatide (hematology),13 linaclotide (gastroenterology),14 afamelanotide (dermatology),15 taltirelin (CNS)16 and teriparatide (osteoporosis).17 Despite their advantages, peptides have a few drawbacks such as low cell membrane permeability, metabolic instability, poorly oral bioavailability and relatively short circulating half-life.18,19 Therefore, chemical modification strategies are essential to enhance the druggability of peptides. For instance, the angiotensin-converting enzyme (ACE) inhibitor captopril is obtained by optimizing a venom oligopeptide derived from the Brazilian viper.20Several strategies have been developed for the chemical modification of primary amino groups in amino acid derivatives and peptides, including acetylation,21–23 alkylation,24 oximation,25–27 arylation28 and quinonylation.29 The quinoline skeleton is a prevalent structure in small-molecule drugs, including quinine sulfate (antimalarial),30 montelukast sodium (asthma),31 and saquinavir (anti-HIV).32 Furthermore, the quinolylation of primary amino groups can also be used to generate peptide–drug conjugates (PDCs) and is thus important in the development of both peptide and PDC drugs. To the best of our knowledge, Buchwald–Hartwig amination is currently the only available quinolinylation method and requires metal catalysis, expensive ligands and high temperature (Fig. 1a).28 Furthermore, no suitable method is available for the direct quinolylation of amino acid derivatives and peptides. Accordingly, new metal-free strategies with mild reaction conditions and good group compatibility are needed to selectively modify the primary amino groups of amino acid derivatives and peptides with the goal of promoting the development of peptide drugs with green chemistry characteristics.
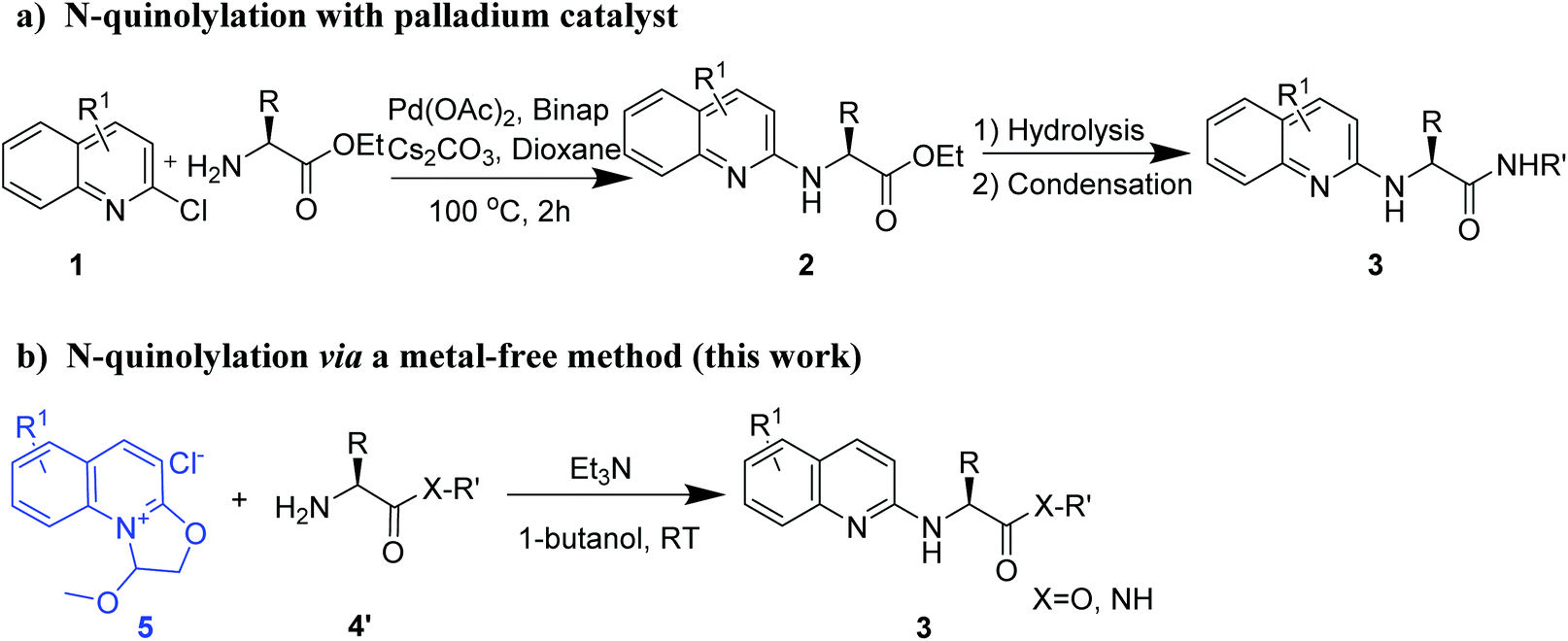 | ||
| Fig. 1 N-Quinolylation of amino acid residues: (a) palladium-catalyzed Buchwald–Hartwig reaction; and (b) metal-free click reaction (this work). | ||
Previously, we disclosed a novel oxazoline[3,2-a]pyridinium that was treated with different nucleophiles for performing regioselective and metal-free C–O and C–N bond-cleaving to afford heterocyclic N-substituted pyridones and 2-substituted pyridines.33 These results inspired us to investigate whether the oxazoline[3,2-a]pyridinium is suitable for coupling with amino acid residues. In this study, we found that the quinoline quaternary ammonium salt can be used to modify the primary amino group (Fig. 1b and S1†) under mild reaction conditions without the need for a heavy-metal catalyst. Therefore, we further optimized this reaction.
Results and discussion
Table 1 summarizes the optimization results of the reaction conditions. 5a was prepared according to the reported methods,33,34 and the synthetic details are summarized in the ESI.† After preparation, 5a was dissolved in different solvents, and 2-amino-N-methylacetamide was added to the reaction mixture to furnish the N-methyl-2-(quinolin-2-ylamino)acetamide 6. To determined the ideal reaction conditions, different bases (Table 1, entries 1–7) were screened as additives, and Et3N (Table 1, entry 7) was found to be the best. After exploration of different solvents, 1-butanol (Table 1, entries 7–18) was found to be ideal. Higher equivalents of 2-amino-N-methylacetamide led to an increased yield of 6 (Table 1, entries 7 and 19–21). Accordingly, two equivalents of 2-amino-N-methylacetamide were chosen for the reaction.|
|
||||
|---|---|---|---|---|
| Entry | Base | Eq. of amino amide | Solvent | Yieldb (%) |
| a All reactions were performed at ambient temperature. 5a was dissolved in different solvents and treated with 2-amino-N-methylacetamide and base. b The yields were determined by HPLC. | ||||
| 1 | DABCO | 1.0 | 1-Butanol | 47 |
| 2 | DMAP | 1.0 | 1-Butanol | 57 |
| 3 | DBU | 1.0 | 1-Butanol | 47 |
| 4 | CO(NH2)2 | 1.0 | 1-Butanol | 11 |
| 5 | DIPEA | 1.0 | 1-Butanol | 59 |
| 6 | Arginine | 1.0 | 1-Butanol | 22 |
| 7 | Et3N | 1.0 | 1-Butanol | 62 |
| 8 | Et3N | 1.0 | DMSO | 6 |
| 9 | Et3N | 1.0 | THF | 6 |
| 10 | Et3N | 1.0 | 1,4-Dioxane | 9 |
| 11 | Et3N | 1.0 | Acetone | Trace |
| 12 | Et3N | 1.0 | MeCN | 10 |
| 13 | Et3N | 1.0 | PhMe | 20 |
| 14 | Et3N | 1.0 | EtOH | 20 |
| 15 | Et3N | 1.0 | DMF | 17 |
| 16 | Et3N | 1.0 | CCl4 | 18 |
| 17 | Et3N | 1.0 | ClCH2CH2Cl | 18 |
| 18 | Et3N | 1.0 | CH2Cl2 | 26 |
| 19 | Et3N | 1.5 | 1-Butanol | 71 |
| 20 | Et 3 N | 2.0 | 1-Butanol | 95 |
| 21 | Et3N | 3.0 | 1-Butanol | 95 |
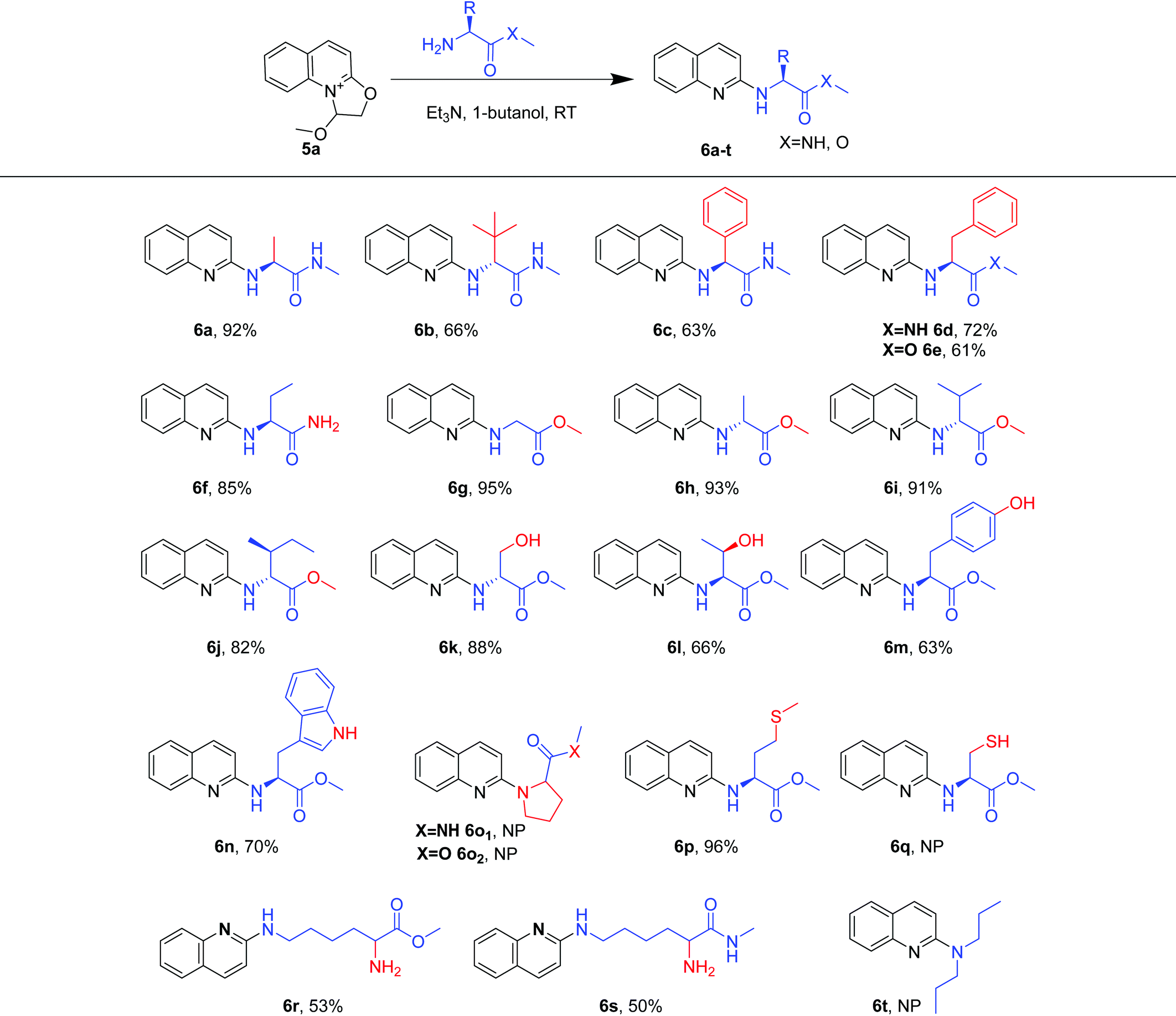
Using the optimized reaction conditions (Table 1, entry 20), we explored a variety of amino substrates bearing different substituents (Table 2). We found that α-amino amides with different substitutions at the α position gave the quinolylated products in 63%–92% yields over two steps (6a–6d, 6f). Among these, (S)-2-amino-N-methylpropanamide afforded the (S)-N-methyl-2-(quinolin-2-ylamino)propanamide 6a in 92% yield. By switching to 2-amino-N-3,3-trimethylbutanamide, which bears a hindered tertiary butyl at the α position of the amino group, the yield of 6b decreased to 66%, suggesting that steric hindrance had an obvious effect on the reaction. Similarly, the N-quinolylation of (S)-2-amino-N-methyl-2-phenylacetamide and (S)-2-amino-N-methyl-3-phenylpropanamide proceeded smoothly to provide products 6c and 6d in 63% and 72% yields, respectively. Specially, the amino group of 2-aminobutanamide was preferentially N-quinolylated rather than the acylamino to afford 6f in 85% yield. In addition, products 6g and 6e were obtained from glycine methyl ester and phenylalanine methyl ester in 95% and 61% yields, respectively. Accordingly, more amino acid esters were explored. Alanine methyl ester, valine methyl ester and isoleucine methyl ester gave the quinolylated products 6h, 6i and 6j in high yields of 82%–93%. Serine methyl ester, threonine methyl ester and tyrosine methyl ester gave 6k, 6l and 6m in moderate yields (63%–88%) without the interference of hydroxyl groups. Moreover, tryptophan methyl ester was converted into the quinolylated product 6n in 70% yield, while the indole amino group was not N-quinolylated. Neither proline methyl ester nor formamide formed quinolylated products (6o1 and 6o2), and the di-n-propylamine did not form the quinolylated product (6t). These results indicate that the quinoline quaternary ammonium salt selectively reacted with primary amino groups rather than secondary amino groups. Interestingly, methionine methyl ester furnished product 6p in 96% yield, while quinolylated product 6q was not observed with cysteine methyl ester, indicating that the thiol group needs to be protected before the quinolylation reaction. Particularly, N-quinolylation mainly occurred at the ε amino group rather than the α amino group of lysine methyl ester and amide (6r and 6s).
|
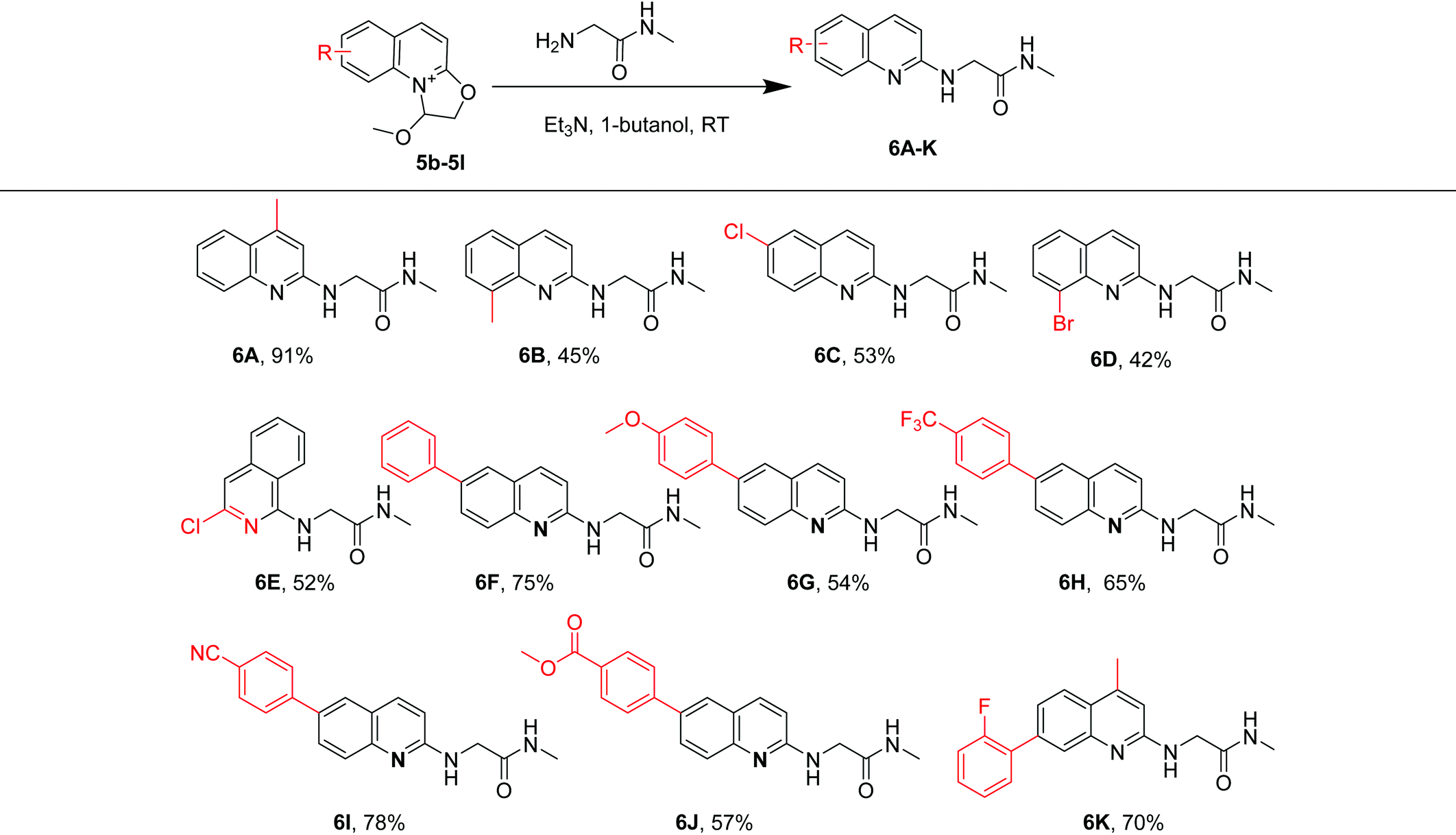
To demonstrate the utility of this developed protocol, we investigated the N-quinolylation of 2-amino-N-methylacetamide with a variety of substituted 2-(2,2-dimethoxyethoxy) quinolines and isoquinolines (Table 3). Both electron-donating (Me–, phenyl) and withdrawing (Cl–, Br–) groups at different positions of the quinoline or isoquinoline moiety gave the quinolylated products 6A–6K in 42%–91% yields. Among these, methyl group-substituted quinoline derivatives smoothly afforded 6A and 6B in 91% and 45% yields, respectively. The quinoline substrates substituted with chloro and bromo groups were also compatible under our experimental protocol, giving 6C and 6D in 53% and 42% yields, respectively. The halogens can be applied in further coupling reactions under Suzuki conditions.35,36 Moreover, when a 4-methoxyphenyl group was installed at the 6-position of the quinoline moiety, the substrate provided 6G in 54% yield. Substrates in which phenyl groups were substituted by electron-withdrawing groups (CF3–, CN–, CO2Me–) also afforded the desired products 6H, 6I and 6J in 65%, 78% and 57% yields, respectively. When the quinoline moiety was simultaneously substituted with 2-fluorophenyl and methyl, it provided 6K in 70% yield.
|
The proposed mechanism of the reaction is shown in Scheme 1. First, dihydrooxazolo[3,2-a]quinolinium 5a is generated from 4 after treatment with hydrogen chloride in diethyl ether. The nucleophilic attack of 2-amino-N-methylacetamide then affords the corresponding intermediate 9. Quantum chemistry calculation at the M06-2X/6-311+G(d) level shows that the activation energy of the transition state (TS) is 14.5 kcal mol−1, indicating that the reaction occurs easily. The subsequent removal of hydrogen chloride facilitated by triethylamine provides the intermediate 10, which ultimately undergoes aromatization to give product 6 with 2-butoxy-2-methoxyethan-1-ol as a possible byproduct.
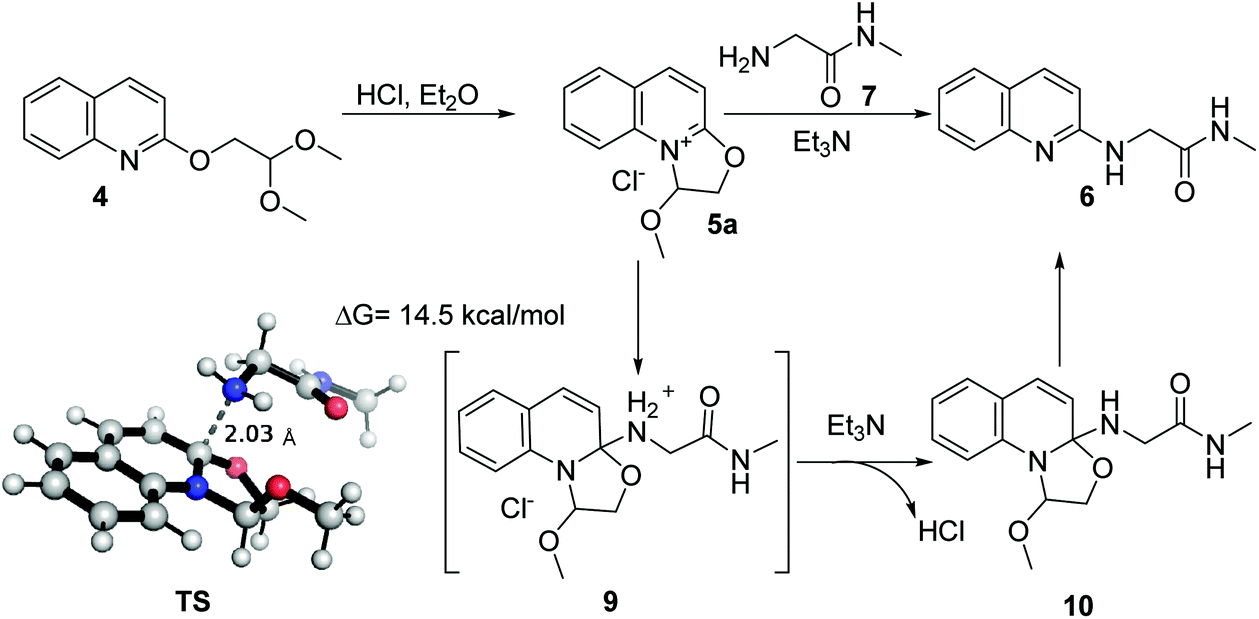 | ||
| Scheme 1 Proposed reaction mechanism. | ||
As mentioned above, 2-(2,2-dimethoxyethoxy) quinoline was treated with hydrogen chloride in diethyl ether after distilling the solvent and without any complicated purification step to give the 1-methoxy-1,2-dihydrooxazolo[3,2-a]quinolinium 5a (Table 4). 5a was reacted with 2-amino-N-methylacetamide to afford 6 in 95% yield. However, the N-quinolylation of other reported quaternary ammonium salts, viz., 1-methylquinolin-1-ium (11), quinoline 1-oxide (12) and 1-acetylquinolin-1-ium (13), with 2-amino-N-methylacetamide did not occur, indicating that the novel dihydrooxazolo[3,2-a]quinolinium has unique reaction characteristics.
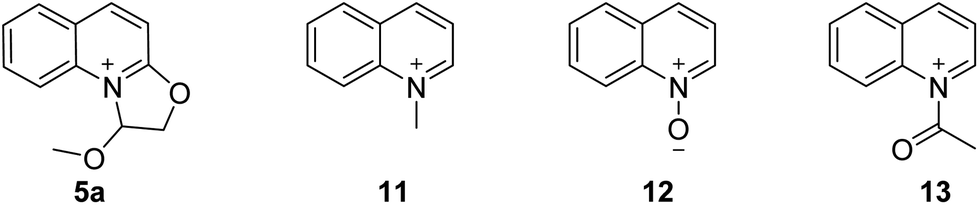
|
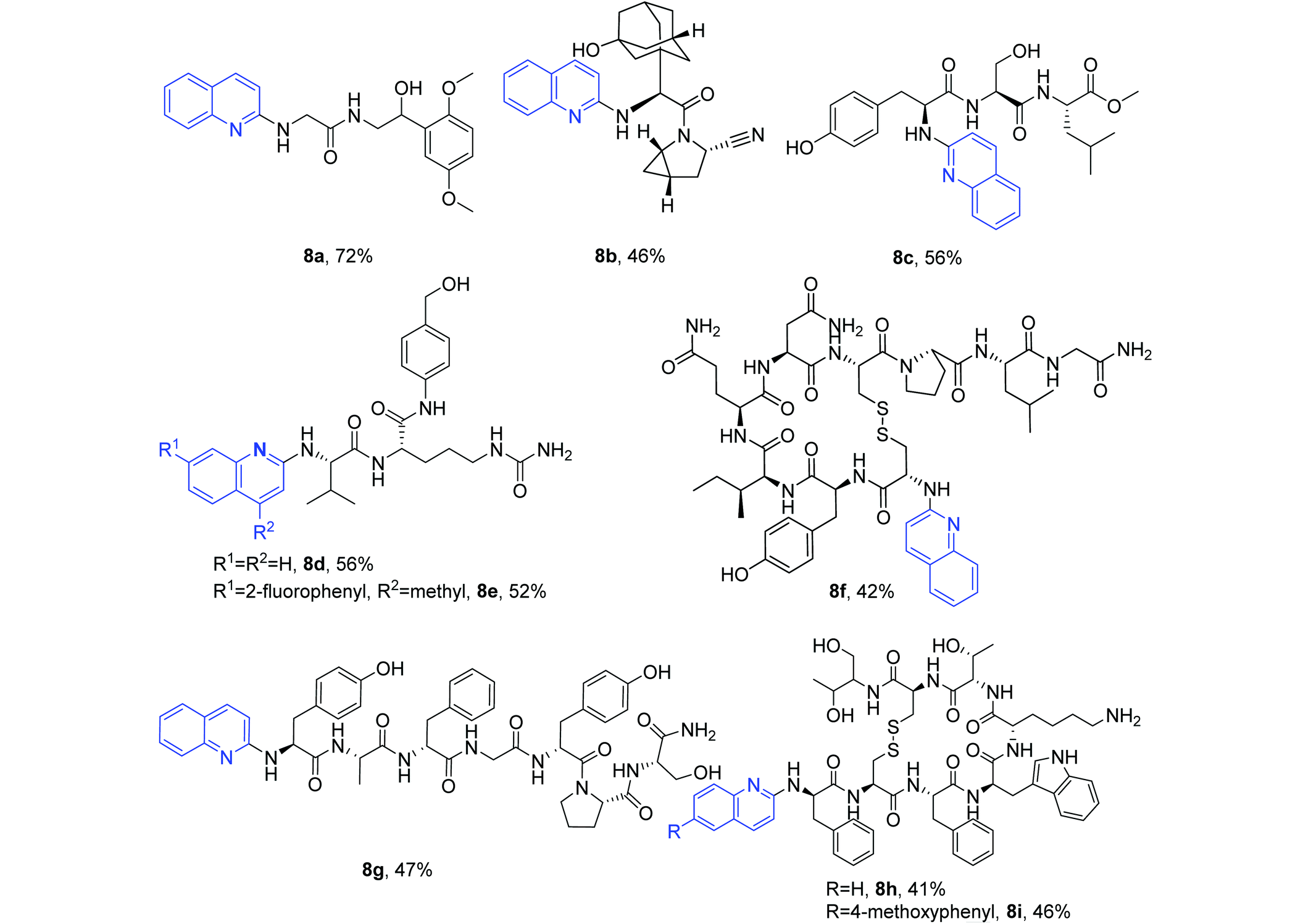
The established method was further used for the N-quinolylation of the primary amino groups of seven molecules (Table 5). Two drug molecules used to treat hypotension and type 2 diabetes, midodrine hydrochloride37 and saxagliptin,38 were successfully coupled with quinoline quaternary ammonium salt to provide the quinolylated products 8a and 8b in 72% and 46% yields, respectively. Five peptide molecules, methyl tyroserleutide (liver cancer),39,40 Val-Cit-PAB-OH (a linker for antibody–drug-conjugation),41–43 oxytocin (improvement of uterine contractions),44 dermorphin (a μ-opioid receptor agonist),45 and octreotide (functional gastrointestinal pancreatic endocrine tumors),46 were reacted to give the desired site-selectively N-quinolylated peptides 8c–8i under the same protocol.
| a Used 2.0 equiv. quinoline quaternary ammonium salt and 1.0 equiv. peptide. |
|---|
|
These structural modifications of peptide drugs indicate that reactive or sensitive groups such as secondary amino groups (6n), amides (6f, 8d, 8e, 8f, 8g, 8h and 8i), alcoholic hydroxyls (6k, 6l, 8a–8e, 8g, 8h and 8i), phenolic hydroxyls (6m, 8c, 8f and 8g), disulfide bonds (8f, 8h and 8i), ester groups (6e, 6g–6n, 6p, 6r, 8c) and cyano groups (8b) are well tolerated, and the site-selective N-quinolylation occurs at the primary amino group position under metal-free reaction conditions.
Among these peptides, tyroserleutide (YSL) has been studied in phase III clinical trials for the treatment of liver cancer. Therefore, YSL, YSL-M (tyroserleutide methyl ester) and 8c were evaluated for anticancer activity against hepatocarcinoma BEL-7402 and SMMC-7721 cells. Bioassay results revealed that 8c exhibited the highest cytotoxicity against both BEL-7402 and SMMC-7721 cells (ESI Table S1†), indicating that the introduction of a quinolyl group into the peptide changed its bioactivity. Dermorphin is a clinically used μ-opioid receptor agonist. Its quinolylated compound 8g exhibited a slightly decreased activity compared to dermorphin (ESI Table S2†), again suggesting that the modification of active peptides is of significance. 7-(2-Fluorophenyl)-4-methylquinolin-2(1H)-one was found to be a tankyrase inhibitor with an IC50 value of 0.052 μM.47 This compound can be coupled with Val-Cit-PAB-OH to afford 8evia our method. A preliminary in vitro liver stability assessment (ESI Fig. S2†) of 8e indicated good metabolic stability, suggesting the potential to develop PDC drugs based on the target peptide using our new method. N-Quinolylation mainly occurred at the α amino group of octreotide with both lysine ε amino and α amino groups (8h and 8i). Small amounts of the quinolylation products of both the α and ε amino groups were detected for this reaction, but no single ε amino group coupling product was observed.
Conclusions
In conclusion, we have developed a green method for the specific N-quinolylated modification of primary amino groups in complex molecules. This protocol was performed under room temperature and metal-free conditions in 1-butanol solvent. It has very good reactive moiety compatibility with secondary amino, amide, alcoholic hydroxyl, phenolic hydroxyl, disulfide bond, ester and cyano groups. Therefore, the developed method can be widely used, particularly in medicinal chemistry and chemical biology.Data availability
Complete experimental procedures and compound characterization data are available within the article and its ESI,† or from the corresponding author upon request.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This research was supported by grants from the National Key R&D Plan (2016YFA0502301), National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program”, China (Number: 2018ZX09711002), and National Natural Science Foundation of China (No. 81573350, 81273546, 81603270).References
- K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20, 122–128 CrossRef CAS PubMed.
- D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136–147 CrossRef CAS PubMed.
- C. Katz, L. Levy-Beladev, S. Rotem-Bamberger, T. Rito, S. G. Rudiger and A. Friedler, Chem. Soc. Rev., 2011, 40, 2131–2145 RSC.
- I. Avan, C. D. Hall and A. R. Katritzky, Chem. Soc. Rev., 2014, 43, 3575–3594 RSC.
- B. J. Bruno, G. D. Miller and C. S. Lim, Ther. Delivery, 2013, 4, 1443–1467 CrossRef CAS PubMed.
- S. S. Usmani, G. Bedi, J. S. Samuel, S. Singh, S. Kalra, P. Kumar, A. A. Ahuja, M. Sharma, A. Gautam and G. P. S. Raghava, PLoS One, 2017, 12, e0181748 CrossRef PubMed.
- L. Di, AAPS J., 2015, 17, 134–143 CrossRef CAS PubMed.
- A. Henninot, J. C. Collins and J. M. Nuss, J. Med. Chem., 2018, 61, 1382–1414 CrossRef CAS PubMed.
- S. H. Kim, O. Pohl, A. Chollet, J. P. Gotteland, A. D. Fairhurst, P. R. Bennett and V. Terzidou, Mol. Pharmacol., 2017, 91, 403–415 CrossRef PubMed.
- M. Sakai, M. Elhilali and V. Papadopoulos, Horm. Metab. Res., 2015, 47, 925–931 CrossRef CAS PubMed.
- N. Matikainen, S. Soderlund, E. Bjornson, K. Pietilainen, A. Hakkarainen, N. Lundbom, M. R. Taskinen and J. Boren, Diabetes, Obes. Metab., 2019, 21, 84–94 CrossRef CAS PubMed.
- S. Dhillon, Drugs, 2011, 71, 1071–1091 CrossRef CAS PubMed.
- S. Doss and B. Schiller, Nephrol. Nurs. J., 2010, 37, 617–626 Search PubMed.
- Y. Yang, J. Fang, X. Guo, N. Dai, X. Shen, Y. Yang, J. Sun, B. R. Bhandari, D. S. Reasner, J. A. Cronin, M. G. Currie, J. M. Johnston, P. Zeng, N. Montreewasuwat, G. Z. Chen and S. Lim, J. Gastroenterol. Hepatol., 2018, 33, 980–989 CrossRef CAS PubMed.
- T. Passeron, JAMA Dermatol., 2015, 151, 349–350 CrossRef PubMed.
- K. Eto, S. K. Kim, J. Nabekura and H. Ishibashi, Brain Res., 2011, 1414, 50–57 CrossRef CAS PubMed.
- M. N. Michalski, A. L. Seydel, E. M. Siismets, L. E. Zweifler, A. J. Koh, B. P. Sinder, J. I. Aguirre, K. Atabai, H. Roca and L. K. McCauley, FASEB J., 2018, 32, 3730–3741 CrossRef CAS PubMed.
- N. Krall, F. P. da Cruz, O. Boutureira and G. J. Bernardes, Nat. Chem., 2016, 8, 103–113 CrossRef CAS PubMed.
- L. Otvos Jr. and J. D. Wade, Front. Chem., 2014, 2, 62 Search PubMed.
- C. Y. Koh and M. Kini, Toxicon, 2012, 59, 497–506 CrossRef CAS.
- S. Tsunasawa, J. W. Stewart and F. Sherman, J. Biol. Chem., 1985, 260, 5382–5391 CAS.
- A. M. Wagner, M. W. Fegley, J. B. Warner, C. L. Grindley, N. P. Marotta and E. J. Petersson, J. Am. Chem. Soc., 2011, 133, 15139–15147 CrossRef CAS PubMed.
- W. K. Chan, C. M. Ho, M. K. Wong and C. M. Che, J. Am. Chem. Soc., 2006, 128, 14796–14797 CrossRef CAS PubMed.
- T. Chen, T. L. Muratore, C. E. Schaner-Tooley, J. Shabanowitz, D. F. Hunt and I. G. Macara, Nat. Cell Biol., 2007, 9, 596–603 CrossRef CAS PubMed.
- J. M. Gilmore, R. A. Scheck, A. P. Esser-Kahn, N. S. Joshi and M. B. Francis, Angew. Chem., Int. Ed., 2006, 45, 5307–5311 CrossRef CAS.
- L. S. Witus, C. Netirojjanakul, K. S. Palla, E. M. Muehl, C. H. Weng, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2013, 135, 17223–17229 CrossRef CAS PubMed.
- R. A. Scheck, M. T. Dedeo, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2008, 130, 11762–11770 CrossRef CAS PubMed.
- H. Hammoud, M. Schmitt, E. Blaise, F. Bihel and J. J. Bourguignon, J. Org. Chem., 2013, 78, 7930–7937 CrossRef CAS PubMed.
- A. C. Obermeyer, J. B. Jarman and M. B. Francis, J. Am. Chem. Soc., 2014, 136, 9572–9579 CrossRef CAS PubMed.
- D. Camp, Drugs Future, 2013, 38, 245–256 CrossRef.
- J. S. Barbosa, F. A. Almeida Paz and S. S. Braga, Drug Delivery, 2016, 23, 3257–3265 CrossRef CAS PubMed.
- C. J. la Porte, Expert Opin. Drug Metab. Toxicol., 2009, 5, 1313–1322 CrossRef CAS PubMed.
- B. Li, S. Xue, Y. Yang, J. Feng, P. Liu, Y. Zhang, J. Zhu, Z. Xu, A. Hall, B. Zhao, J. Shi and W. Zhu, Sci. Rep., 2017, 7, 41287 CrossRef PubMed.
- B. Li, G. Wang, Z. Xu, Y. Zhang, X. Huang, B. Zeng, K. Chen, J. Shi, H. Wang and W. Zhu, Eur. J. Med. Chem., 2014, 77, 204–210 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, J. Chem. Soc., Chem. Commun., 1979, 866–867 RSC.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- W. Smith, H. Wan, D. Much, A. G. Robinson and P. Martin, Clin. Auton. Res., 2016, 26, 269–277 CrossRef PubMed.
- K. P. Garnock-Jones, Drugs, 2017, 77, 319–330 CrossRef CAS PubMed.
- Z. Fu, L. Ren, H. Wei, J. Lv, X. Che, Z. Zhu, J. Jia, L. Wang, G. Lin, R. Lu and Z. Yao, J. Drug Targeting, 2014, 22, 146–155 CrossRef CAS PubMed.
- C. Wang, S. Wang, R. Lu, L. Zhao, Z. F. Zhu, Q. Xu, J. Q. Lv, L. L. Wang, Z. Fu, G. Lin and Z. Yao, Anticancer Drugs, 2009, 20, 534–542 CrossRef CAS PubMed.
- J. Grunewald, Y. Jin, J. Vance, J. Read, X. Wang, Y. Wan, H. Zhou, W. Ou, H. E. Klock, E. C. Peters, T. Uno, A. Brock and B. H. Geierstanger, Bioconjugate Chem., 2017, 28, 1906–1915 CrossRef CAS.
- Q. Zhou and J. Kim, Anticancer Agents Med. Chem., 2015, 15, 828–836 CrossRef CAS.
- L. J. Scott, Drugs, 2017, 77, 435–445 CrossRef CAS PubMed.
- E. N. Erickson, C. S. Lee and C. L. Emeis, J. Midwifery Womens Health, 2017, 62, 418–424 CrossRef PubMed.
- H. Mizoguchi, G. Bagetta, T. Sakurada and S. Sakurada, Peptides, 2011, 32, 421–427 CrossRef CAS PubMed.
- R. M. Borna, J. S. Jahr, S. Kmiecik, K. F. Mancuso and A. D. Kaye, Anesthesiol. Clin., 2017, 35, 327–339 CrossRef PubMed.
- E. A. Larsson, A. Jansson, F. M. Ng, S. W. Then, R. Panicker, B. Liu, K. Sangthongpitag, V. Pendharkar, S. J. Tai, J. Hill, C. Dan, S. Y. Ho, W. W. Cheong, A. Poulsen, S. Blanchard, G. R. Lin, J. Alam, T. H. Keller and P. Nordlund, J. Med. Chem., 2013, 56, 4497–4508 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc01442j |
| This journal is © The Royal Society of Chemistry 2019 |