
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceValorization of aqueous waste streams from thermochemical biorefineries†
A. Nolan
Wilson
 a,
Abhijit
Dutta
a,
Brenna A.
Black
b,
Calvin
Mukarakate
a,
Kim
Magrini
a,
Joshua A.
Schaidle
a,
William E.
Michener
b,
Gregg T.
Beckham
a and
Mark R.
Nimlos
*a
a,
Abhijit
Dutta
a,
Brenna A.
Black
b,
Calvin
Mukarakate
a,
Kim
Magrini
a,
Joshua A.
Schaidle
a,
William E.
Michener
b,
Gregg T.
Beckham
a and
Mark R.
Nimlos
*a
aNational Bioenergy Center, National Renewable Energy Laboratory, Golden, CO 80401, USA. E-mail: mark.nimlos@nrel.gov
bBiosciences Center, National Renewable Energy Laboratory, Golden, CO 80401, USA
First published on 22nd July 2019
Abstract
Thermochemical conversion of lignocellulosic biomass is a promising route to produce fuels and oxygenated chemicals and could enable circular carbon utilization. In most thermochemical conversion processes, however, some chemical co-products are lost in aqueous waste streams that are both dilute and heterogeneous. Cost-competitive isolation of these chemical co-products is challenging due to the high-purity requirements typically necessary for bulk chemical production. Here, we demonstrate the production and isolation of two biomass-derived monomers, phenol and catechol, from a comprehensively characterized aqueous waste stream generated via catalytic fast pyrolysis. Specifically, we separate phenol and catechol to 97 wt% purity using the industrially relevant processes of liquid–liquid extraction, distillation, and recrystallization. Techno-economic analysis predicts that a mixed phenolics stream can be produced from the waste stream at a minimum selling price of $1.06 kg−1. Overall, this work demonstrates an approach to high-purity oxygenated aromatic compounds that is potentially economically feasible and technically achievable which increases the atom efficiency of thermochemical conversion through waste stream valorization.
Introduction
Transitioning from the current, linear economy to the not-yet-realized circular economy will require advances and contributions from all forms of renewable energy technologies.1,2 De-carbonization of energy markets through technologies like wind and solar remains a key contributor; however, many applications are fundamentally coupled to carbon (e.g., liquid fuels and physical materials) and necessarily require alternative strategies to achieve a long-term circular economy. To realize circularity, technologies with renewably sourced feedstocks that produce material precursors and enable materials recycling must be established. Biomass conversion offers unique benefits relative to other renewable technologies in that (1) it produces fungible vehicle and aviation fuels,3–5 (2) specialty and commodity chemical co-products can be concomitantly produced with biofuels,6–8 (3) and some of the conversion operations can be applied to municipal solid waste recycling.9 Biomass-derived chemicals as a source for material feedstocks is an active and broad area of research with significant efforts that span a wide range of feed substrates (e.g., whole lignocellulosic, lignin-only, sugars, cellulose, etc.) and leverages many conversion strategies (e.g., metabolic engineering or synthetic biology, wholly catalytic, thermochemical, hybrid biological and catalytic, lignin catalysis and biocatalysis, direct or near-direct utilization of biomass polymers, etc.). Recent examples of chemical products for materials include biomass conversion into aromatic hydrocarbons,10–12 cellulose conversion into α,ω-diols,13 production of platform chemicals such as 5-hydroxymethylfurfural or gamma-valerolactone,14,15 incorporation of bio-adipic acid into nylon,16 novel chemistry for renewable acrylonitrile production,17 methacrylate functionalization of lignin monomers,18 and bio-derived polymer composites.19 Oxygenated aromatic chemicals are a target for thermochemical conversion of whole biomass, lignin deconstruction, and selective biocatalytic conversion as they are derived primarily from the molecular structure of lignin, they have a high selling price relative to fuels, and they have large markets, Fig. 1.20–23 All conversion pathways have significant technical challenges. Recently, biocatalysis has shown high yields of phenol from glucose, 18.5%, but suffers from low titers, 3.62 mM.24 Thermochemical conversion can produce oils with much higher concentration of phenol, 1.4 wt% (∼150 mM), but in a highly heterogenous mixture with components that are difficult to separate.25 The oxygenated aromatic compound with the largest market, phenol, is primarily used in polycarbonate and phenol-formaldehyde resins, which have widespread use in automotive, aerospace, building material, electronics, and consumer goods applications.26,27 Catechol, a specialty chemical, is primarily used in insecticides (e.g., Baygon®, Furadan®) at approximately 50%, perfumes and pharmaceuticals (e.g., dopamine) at 35–40%, and polymerization inhibitors at 10–15%.28 Catechol can also be used in materials such as polyether ether ketones or polydopamine.29,30 Industrially, catechol is co-produced with hydroquinone at a 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio by the hydroxylation of phenol.31
:1 molar ratio by the hydroxylation of phenol.31
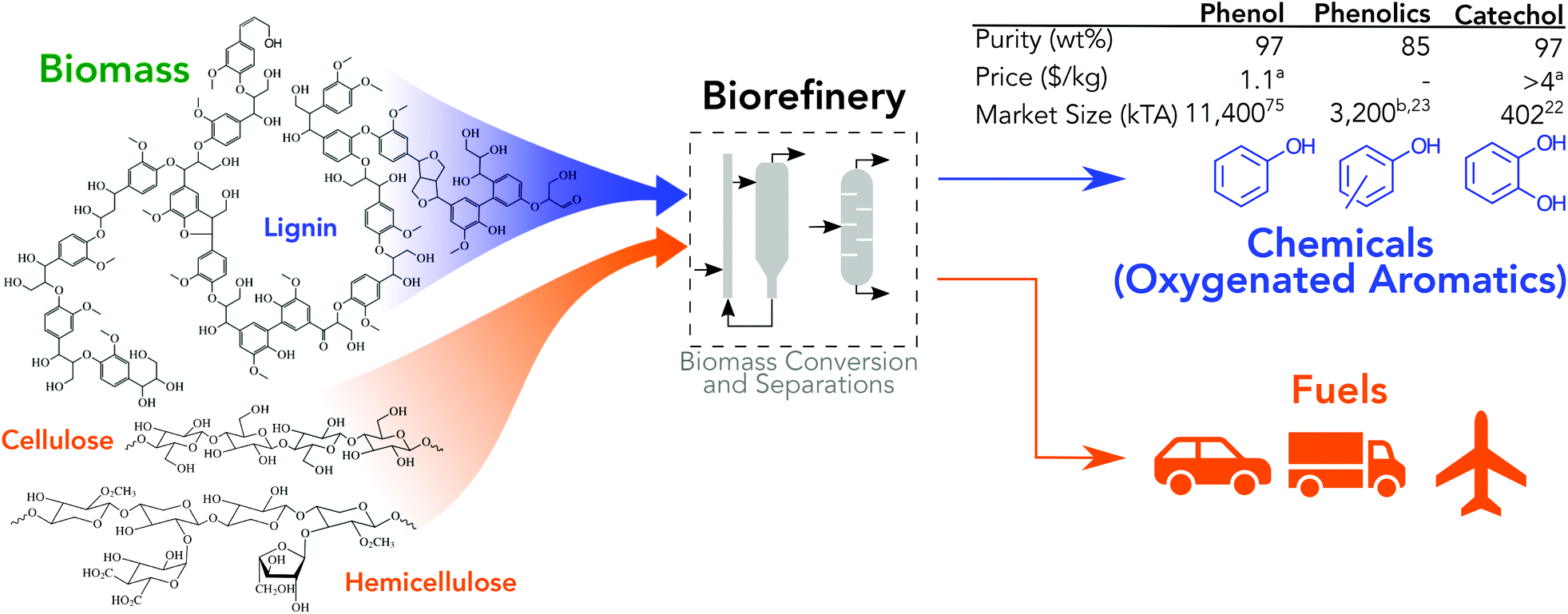 | ||
| Fig. 1 Integrated biorefinery which produces fuels and chemicals. Phenols and diols products take advantage of the structure of biomass (lignin). aInset table provides price in 2017 dollars, updated using producer price index to adjust values.19,81 Prices reflect market value of products purified to industrially accepted purities, typically >99%, and do not necessarily reflect value of co-products produced here. bEstimated as 28% of the phenol market.36 | ||
Producing oxygenated aromatics through catalytic fast pyrolysis (CFP), a thermochemical conversion strategy that deconstructs biomass in the absence of oxygen and catalytically upgrades the resulting vapors to produce an oil and aqueous product stream, is a particularly attractive strategy due to its scalability, correspondence to well understood petrochemical unit operations (fixed and fluidized bed reactors, entrained flow reactors, etc.), and high-throughput (1–2 s residence time). There has been ample work to demonstrate the positive economic impact co-products can have on the economic viability of biomass conversion processes.32–35 Iisa et al. showed biofuel production approaches economic parity with petroleum fuels when 35% or more of the carbon is converted to fuels.36 The economics of biofuel production can potentially be improved further by valorizing some of the carbon as higher valued co-products instead of fuels. While the biochemical sugar conversion platform often considers co-production of chemicals, the thermochemical platform has lagged in the area of co-products due to the production of highly heterogenous product streams, low selectivity towards a single compound, and inadequate separation strategies for processing highly oxygenated, non-ideal, and often reactive mixtures. Due to these challenges, chemicals from CFP have been limited to mixed streams of sugars, phenolics, or other pyrolysis compounds from the oil or aqueous streams.37–39 Ideally, carbon not incorporated into a fuel product can be converted into a small number of valuable co-products, which can be isolated with minimal separation unit operations. These co-products can be drop-in replacements (e.g., BTX, isoprene, succinic acid), or alternatives with improved performance (e.g., 4-n-propylguaiacol for phenol, 2,5-furandicarboxyllic acid for terephthalic acid) compared to their petroleum-derived counterparts.40–45 In typical petrochemical processes, oxygen functionality must be introduced through costly chemical syntheses and energy intensive separations.46,47 A potential benefit of biomass conversion to co-products is the intrinsic oxygen present in the feed streams, Fig. 1; however, the drawback of CFP as a conversion strategy to produce chemicals is challenging separations. Addressing this challenge to improve the atom efficiency of CFP processes can be realized through (1) process optimization and catalyst design to increase selectivity towards fuels and co-products and (2) separation of chemical co-products that are otherwise treated as costly waste or burned for heating value.
In this study, we pursue approach (2) above by examining a strategy using well-established unit operations, which eliminates the risk of novel separation technologies, for separating oxygenated aromatic molecules from a CFP aqueous stream and evaluate the impact on biorefinery costs. Biorefineries based on thermochemical conversion generate significant quantities of water with a theoretical maximum water formation (all oxygen converted to water) of over 50 wt%, but typical water streams amount to about 30 wt% of the biomass feed. Treatment is required to remove the organic molecules before discharge and options being explored have included regenerative thermal oxidation providing additional heating value to the plant, conversion to BTX and olefins, steam reforming to produce hydrogen, utilization of organisms which have engineered pathways to process biomass derived compounds, or ketonization for olefin, alcohol, and hydrogen production.48–52 The strategy being explored here is to capture phenol, catechol, and mixed phenolics from the CFP aqueous phase using a separation train. To the best of our knowledge, phenol and catechol have not been previously isolated from CFP streams, although the separation and utilization of phenolics from biomass has been widely documented.25 Separation strategies for mixed phenolics have included adsorbents for phenolic–sugar separation, fractional condensation for a phenol-formaldehyde resin precursor, and combined solvent and staged pH extraction of methoxyphenols.53–55 Reports of isolating monomers above 90 wt% purity from thermochemical streams are less prevalent, but one example leveraged steam distillation and multi-solvent extraction scheme to isolate syringol at 92.3%.56 By extracting oxygenated aromatics, known for their toxicity to wastewater treatment microorganisms,57,58 the necessity and/or severity of wastewater treatment can be minimized. Utilizing the carbon in this stream as chemicals will not affect fuel yield from the biorefinery, will directly increase atom efficiency, and will transform the costs for treating wastewater into a revenue stream. Additionally, a preliminary techno-economic analysis (TEA) is provided for a lower-value mixed phenolic stream to asses the economic viability of the initial unit operations in the separation scheme. This approach provides an industrially-relevant approach to produce phenol and catechol renewably and a valorization strategy for the “waste” aqueous stream, which would otherwise require wastewater treatment.
Results
The developed separation train, illustrated in Fig. 2, leverages well-understood separation unit operations: liquid–liquid extraction (LLE), distillation, and recrystallization. The strategy for separation was dewatering and concentrating the aqueous phase organics, distillation into crude product fractions, and finishing with recrystallization to achieve high purity product streams. The mono-component co-products are isolated at a purity of 97 wt% for both phenol and catechol. | ||
| Fig. 2 Process flow diagram for recovering organics. The separation scheme for recovering CFP derived organics comprising liquid–liquid extraction (LLE), distillation, and recrystallization. Inset images show (left) two phase system of ethyl acetate and CFP aqueous phase in separatory funnel and (right) the three resulting products from the separation process. Boxed section indicates unit operations for which TEA models were build and correspond to Fig. 6B. | ||
CFP conditions and aqueous stream
The aqueous waste stream used in this study contained 3.0 wt% organic carbon which is at the low end of the range of reported for organic carbon content, 3–14 wt%, depending on conversion conditions.49,59 The aqueous phase was generated in a Davidson Circulating Riser (DCR) reactor,59 in which the pyrolysis vapors from clean loblolly pine were upgraded using a HZSM-5 zeolite type catalyst (Johnson Matthey). Fig. 3A shows a GC chromatogram of the aqueous phase, which contains primarily phenols, catechols, acetic acid, and hydroxyacetone. Elemental analysis, used for mass balance closure, of the aqueous phase is provided in Table S1.† An overall carbon balance across the range of reported carbon content of aqueous streams, 3–14 wt%, is provided in Table S2 and Fig. S2.†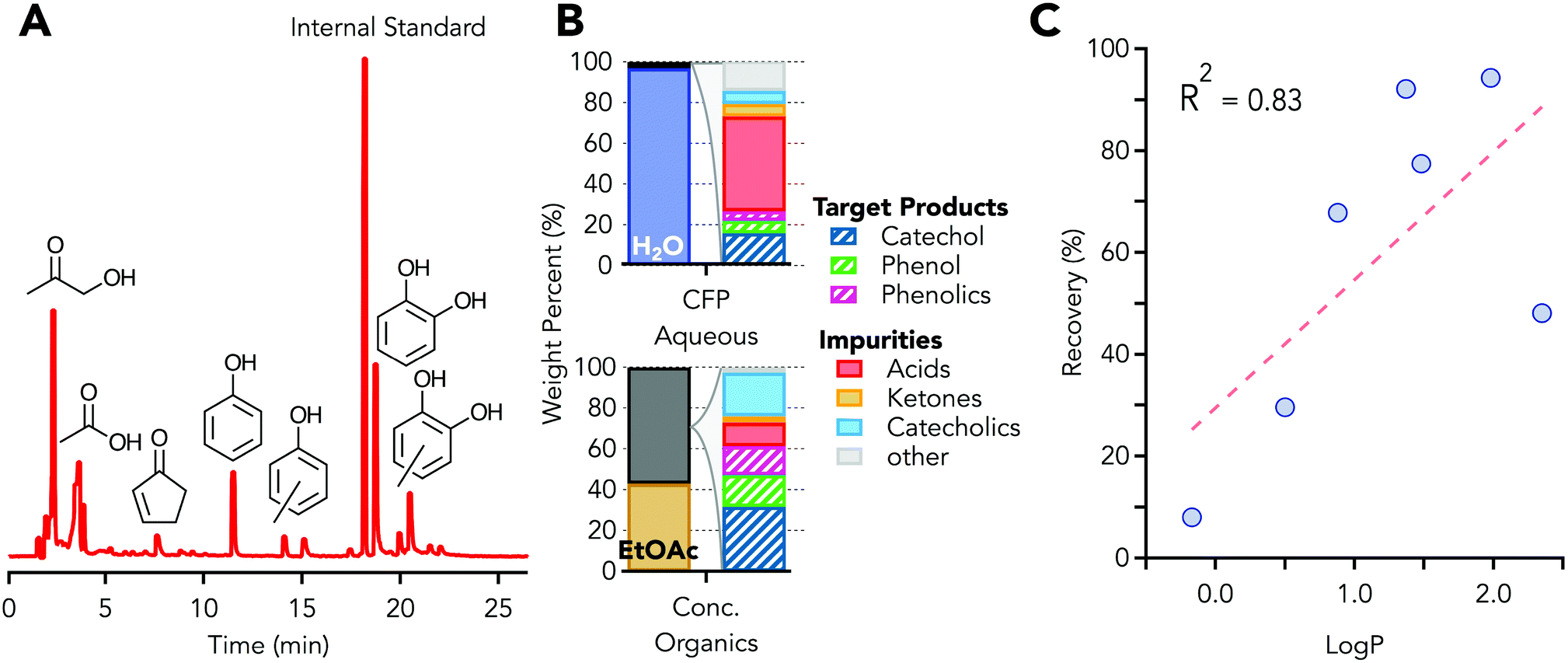 | ||
| Fig. 3 LLE of CFP aqueous stream. (A) Gas chromatogram of CFP derived aqueous phase. (B) CFP phase composition (left columns) and breakdown of organics composition (right bars). Values for stream composition are provided in Table S4.† (C) LLE recovery as a function of logP, defined as the log of the partioning coefficient between an aqueous and organic phase, showing correlation between recovery and partitioning between water and organic phases. | ||
Dewatering via liquid–liquid extraction
LLE with ethyl acetate followed by roto-vaporization was used to remove water and produce a concentrated organic stream from the CFP aqueous phase. The initial CFP aqueous phase and the concentrated organic phase (post LLE) compositions are shown in Fig. 3B. Initially, a 2:1 (v/v) ratio of aqueous phase:ethyl acetate (EtOAc) was used, and after extraction the final volumetric ratio was 3.6:1 aqueous phase:ethyl acetate. The change in the volumetric ratio was due to partitioning of ethyl acetate into the aqueous phase, and the partitioning was observed experimentally and through modeling. The ethyl acetate within the aqueous layer did not significantly retard the ability to extract oxygenated organics from the aqueous phase, but recovery of the ethyl acetate from the aqueous phase was necessary in process models for economic viability. During extraction an emulsion was formed but did not persist and a stable two phase system was observed in less than 1 hour. After extraction, the ethyl acetate phase which contains the extracted organics was 32 ± 3% smaller by volume than the original aqueous stream. The calculated energy required to remove the solvent from the organics is reduced by 95.0 ± 0.5% (2.15 ± 0.01 kJ g-CFP−1 aqueous phase), as estimated by the energy of vaporization of the pure solvents, due to the volume reduction and lower heat of vaporization of ethyl acetate relative to water. The subsequent solvent recovery unit operation, roto-vaporization here and distillation at scale, results in a stream mass reduction of 97.8 ± 0.2% relative to the original stream mass. The aqueous stream was 97 wt% solvent, water, while the final concentrated organics stream was 43 wt% solvent, ethyl acetate, as shown in Fig. 3B (left columns). The GC-MS identifiable and GC-FID/polyarc quantifiable material was 24 ± 1 g L−1 for the aqueous phase and 352 ± 4 g L−1 for the concentrated organic stream. The mass of all species can then be determined using the response factor from a single internal standard since all carbon is converted to methane in the polyarc. The majority of the species in the aqueous raffinate were acetic acid and ethyl acetate, the extraction solvent, while phenol, phenolics, and catechol were recovered at 79%, 99%, and 67%, respectively, in the concentrated organics stream. Fig. 3B (right columns), shows the weight percent distribution of the initial and final streams of the process. Table S3† shows a more extensive characterization of the organic compounds present in the predominantly aqueous starting material, using multiple analytical methods with a 95–100% organic carbon mass closure using total organic carbon (TOC) as the basis for mass balance closure. The extensive characterization was used for all techno-economic analysis; the high-throughput GC-FID with the polyarc system was used for analysis of LLE and distillation streams. In Fig. 3B, the phenolics group includes di- and tri-alkyl (methyl or ethyl) substituted phenol, the catecholics grouping includes mono-, di-, and tri-alkyl (methyl or ethyl) substituted catechol, and the “other” group includes low concentration species identified via GC-MS but have been grouped together for visual clarity. The ethyl acetate in the raffinate stream was 127 ± 26 g L−1, which was slightly higher than the solubility of ethyl acetate in pure water, 100 g L−1, and was likely due to contributions from mixed organic compounds.
The LLE unit operation exhibits a high recovery of the oxygenated aromatic compounds and some separation of acids and ketones, which were undesired in the distillation due to their reactivity and corrosivity. The acid separation was an additional benefit as stream volume and heat duty reduction were the primary purpose of the unit operation. While stream composition, solvent, temperature, and pH60,61 can affect the distribution between phases, it is empirically observed that logP, defined as the log of the partitioning coefficient, provides a reasonable initial estimate of species recovery into the organic phase. LogP accounts for 83% of the variability, with a simple linear model, as shown in Fig. 3C, for the complex but dilute aqueous phase. The general trend for species partitioning into the organic phase is:
| Oxygenated aromatics ≫ acids > ketones. |
Distillation of concentrated organics
Fractional distillation was used to obtain concentrated streams of the desired aromatic oxygenates from the organic crude from the LLE step. The concentrated organics stream from LLE was separated into the 6 fractions shown in Fig. 4A & B using distillation under batch mode operation. As will be discussed, fraction 3 is used in additional steps for phenol purification and fraction 5 is used to obtain purified catechol.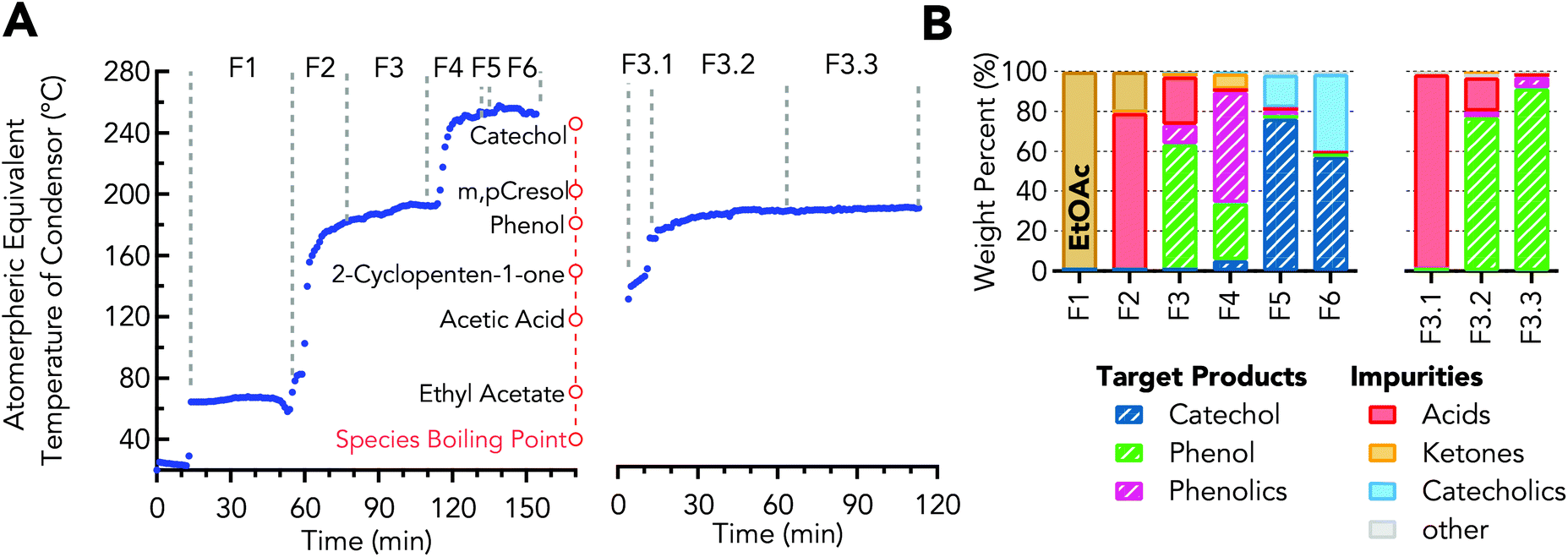 | ||
| Fig. 4 Distillation for recovering organics after LLE. (A) Vapor temperature profile during distillation of concentrated organics (left) and distillation of crude phenol (right) showing temperature ranges for each fraction. (B) Composition in weight percent for each fraction. Values for stream compositions are provided in Table S5.† | ||
Operating conditions and resulting weight distribution are provided in Table 1 – Distillation of concentrated organics. The resulting fraction F1, 27 wt% of the charged material by mass, was >99 wt% ethyl acetate. Fraction F2 was primarily acetic acid, 79 wt%, with the remainder being ethyl acetate. Phenol was 16 wt% in the initial concentrated organics and was increased to 64 wt% in fraction F3, which represents a yield of 65 wt% of the phenol originally charged to the boiler. The other major components of fraction F3 were acetic acid, propionic acid, and o-cresol. Fraction F4 was a mixture of phenol and phenolics, which comprised 84 wt% of the phase with the major impurities being 2-cyclopenten-1-one and catechol. Catechol was 32 wt% in the concentrated organics and was increased to 76 wt% in fraction F5, which represents a yield of 27 wt% of the catechol originally charged to the boiler. The other major impurities in fraction F5 were catecholics. Fraction F6 was the last fraction, which contained 57 wt% and 40 wt% catechol and catecholics, respectively. After distillation, 25 wt% of the material originally charged to the boiler remained in the bottom as residual carbon (resid). The resid results from high boiling point compounds which are generally too heavy to analyze by GC, but it is likely to be comprised of heavy biomass conversion products (e.g., pyrolytic lignin, polycyclic aromatic) and reaction products (e.g. phenolic dimers or oligomers) formed during the distillation via condensation reactions.
| Fraction | Description | AET (°CAET) | Pressure (torr) | Reflux | Wt%a |
|---|---|---|---|---|---|
| a Weight percent is the weight percent of the fraction relative to the amount charged to the boiler. Where AET is atmospheric equivalent temperature. | |||||
| Distillation of concentrated organics | |||||
| F1 | Ethyl acetate | 62–72 | atm | 30 | 27.3 ± 1.1 |
| F2 | Crude acetic acid | 72–160 | atm | 30 | 8.1 ± 0.5 |
| F3 | Crude phenol | 160–189 | 50 | 60 | 6.5 ± 0.7 |
| F4 | Phenolics | 189–229 | 50 | 60 | 7.4 ± 1.0 |
| F5 | Crude catechol | 229–247 | 5 | 60 | 4.1 ± 1.0 |
| F6 | Catecholics | 247–257 | 5 | 60 | 13.2 ± 0.8 |
| — | Bottoms | >257 | NA | NA | 25.1 ± 2.3 |
| — | Losses | NA | NA | NA | 8.3 ± 1.2 |
| Distillation of crude phenol | |||||
| F3.1 | Acids | 129–169 | 50 | 60 | 12.2 |
| F3.2 | Mix (phenol & acids) | 169–187 | 50 | 60 | 23.0 |
| F3.3 | Phenol | 187–189 | 50 | 60 | 27.9 |
| — | Bottoms | >189 | NA | NA | 10.1 |
| — | Losses | NA | NA | NA | 26.7 |
Phenol purification
Phenol was further purified by a second distillation of fraction F3 into three fractions, F3.1, F3.2, and F3.3, for which the distillation conditions are shown in Table 1 – Distillation of crude phenol. During distillation of fraction F3, a pseudo-steady state vapor temperature was observed between 140 and 149 °CAET (61–69 °C at 50 torr, AET – Atmospheric Equivalent Temperature), which was collected as fraction F3.1, as shown in Fig. 4A. Fraction F3.1 was primarily acids with 64 wt% propionic and 34 wt% acetic acid, which have boiling points of 141 and 118 °C, respectively. Fraction F3.2 was a mixture of both phenol and acids. The collection temperature of fraction F3.3 comprised of 92 wt% phenol was 187–189 °CAET (102–104 °C at 50 torr), which was in-line with the boiling point of phenol 189 °CAET (104 °C at 50 torr), as empirically determined on the distillation apparatus, shown in Fig. S1,† and calculated via Antoine's equation.62 The impurities within this fraction were propionic acid and o-cresol as determined by GC-MS. The distillation results show phenol purity was increased and acid presence was decreased prior to recrystallization. The second distillation improved phenol purity from 64 to >92 wt%, Fig. 4B.The resulting distillate, fraction F3.3, was then recrystallized. The final purified bio-derived phenol had a purity of 97 wt% and melting temperature of 41 °C, as determined by DSC and shown in Fig. 5A. Petroleum-derived phenol (Sigma-Aldrich) was determined to have a melt temperature of 42 °C and purity of 99.7%. o-Cresol at 1.4 wt% was the major impurity in the bio-phenol as determined by GC-MS with roughly 1.6 wt% of the impurities unidentified. Detection of low concentration impurities was not explored due to material limitations. The likely impurities were compounds detected in fraction F3 (e.g., propionic acid, 1,1-dimethoxycyclopentane, and 2-methylbenzofuran). The NMR spectrum of the product showed shifts between 6.7–7.2 ppm associated with aromatic protons and at 9.30 ppm associated with the hydroxyl proton. A third shift appeared at 2.11 ppm, which was associated with the alkyl protons from o-cresol. With the exception of the o-cresol impurity, the NMR spectra were comparable to spectra of petroleum-derived phenol, as shown in Fig. 5B. Recrystallization of phenol and cresols has been explored as an alternative strategy to distillation.63 Here, the phenol–hexanes system exhibits a solid–liquid or liquid–liquid phase that was dependent on the temperature of the system, 5 or 25 °C, respectively. The liquid–liquid system was not explored within this work, but there is potential to use the liquid–liquid phase equilibrium to extract impurities as it benefits from not requiring solids handling. In comparison to petrochemical derived phenol, the bio-derived phenol exhibited significantly different impurity profiles due to the upstream CFP process and separation unit operations.
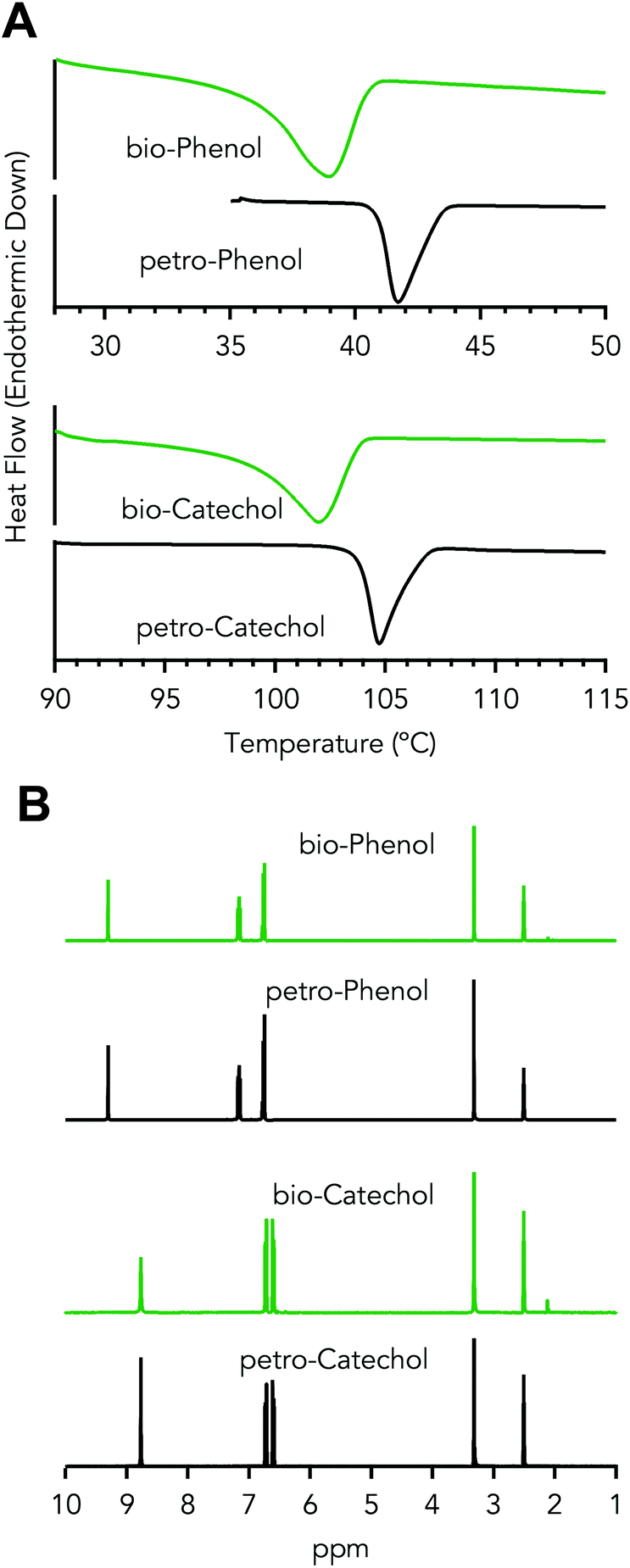 | ||
| Fig. 5 Characterization of purified oxygenated organics. (A) DSC thermograms and (B) NMR spectra of purified bioderived and petroleum derived phenol and catechol. | ||
Catechol purification
Recrystallization of the distilled crude catechol fraction, F5, was used to further purify catechol from 76 wt% to 97 wt%, with a melting temperature of 104 °C, as determined by DSC and shown in Fig. 4A. Petroleum-derived catechol (Sigma-Aldrich) exhibited a melt temperature of 105 °C and purity of 99.7%. 3-Methylcatechol and 4-methylcatechol were the major impurities in the purified bio-catechol as determined by GC-MS. The NMR spectrum of the recrystallized catechol showed shifts between 6.4–6.8 ppm, associated with aromatic protons, and at 8.77 ppm associated with the hydroxyl protons. A third shift appeared at 2.12 ppm, which was associated with the alkyl protons in the methyl groups from the two major impurities, Fig. 5B. The NMR spectra were comparable to spectra of petroleum-derived catechol with the exception of the alkyl protons.Fraction F6 contained 57% of the catechol charged to the boiler in the initial distillation. In an attempt to recover catechol from this fraction, the same recrystallization process was employed as for fraction F5; however, under the same conditions, no solid formation was observed. This was likely due to increased catecholics composition of the phase, 40 wt% vs. 17 wt% for fractions F6 and F5, respectively. The increased alkyl functionality could inhibit crystal formation by disrupting the regularity of the crystalline structure. Alternate temperatures, concentrations and/or solvent systems can be explored to further recover catechol. At full-scale operation, recrystallization may or may not be utilized depending on process economics, process complexity, and EH&S concerns.
Techno-economic analysis (TEA)
To explore the viability of separating at least one product from the aqueous stream and understand the cost implications of the initial LLE unit, the separation of a mixed oxygenated aromatic stream was considered with its potential use as a material precursor for phenol-formaldehyde resins. To simplify the TEA analysis, a single aqueous stream was considered, which was fully characterized using an array of analytical methods, as shown in Table S2.† The product stream, which would be the mixture of oxygenated aromatics in F3–F6, was chosen for analysis as it represents the lowest value co-product, it will have the largest volume, and it has minimal unit operations required for purification. This TEA model provides a base case for understanding the economics of an LLE-first approach.TEA process model
A simplified flow diagram of the CFP process is shown in Fig. 6A, and a conceptual separation process for separating and concentrating the organics in the aqueous stream is shown in Fig. 6B. The TEA model was informed by the previously discussed experimental work, and the detailed compositional analysis was used for modeling the separation; some of the experimental data was used to inform/validate the modeling. In the model, the aqueous stream was extracted with ethyl acetate with an optimal volumetric ratio of 5:1, aqueous phase to ethyl acetate. The two phases were sent to a decanter and then to two separate distillation columns, columns 1 & 2 in Fig. 6B, which were used to recover and recycle the ethyl acetate. The Aspen Plus model predicted 98.7% recovery which is in line with other acetate recovery simulations.64 The bottom stream from column 1 was the mixed phenolics product. The bottoms from column 2 was mostly water and can be sent to the wastewater treatment section of the CFP plant.48 Light organics, which may be recycled to the main CFP process, were removed from the ethyl acetate phase.
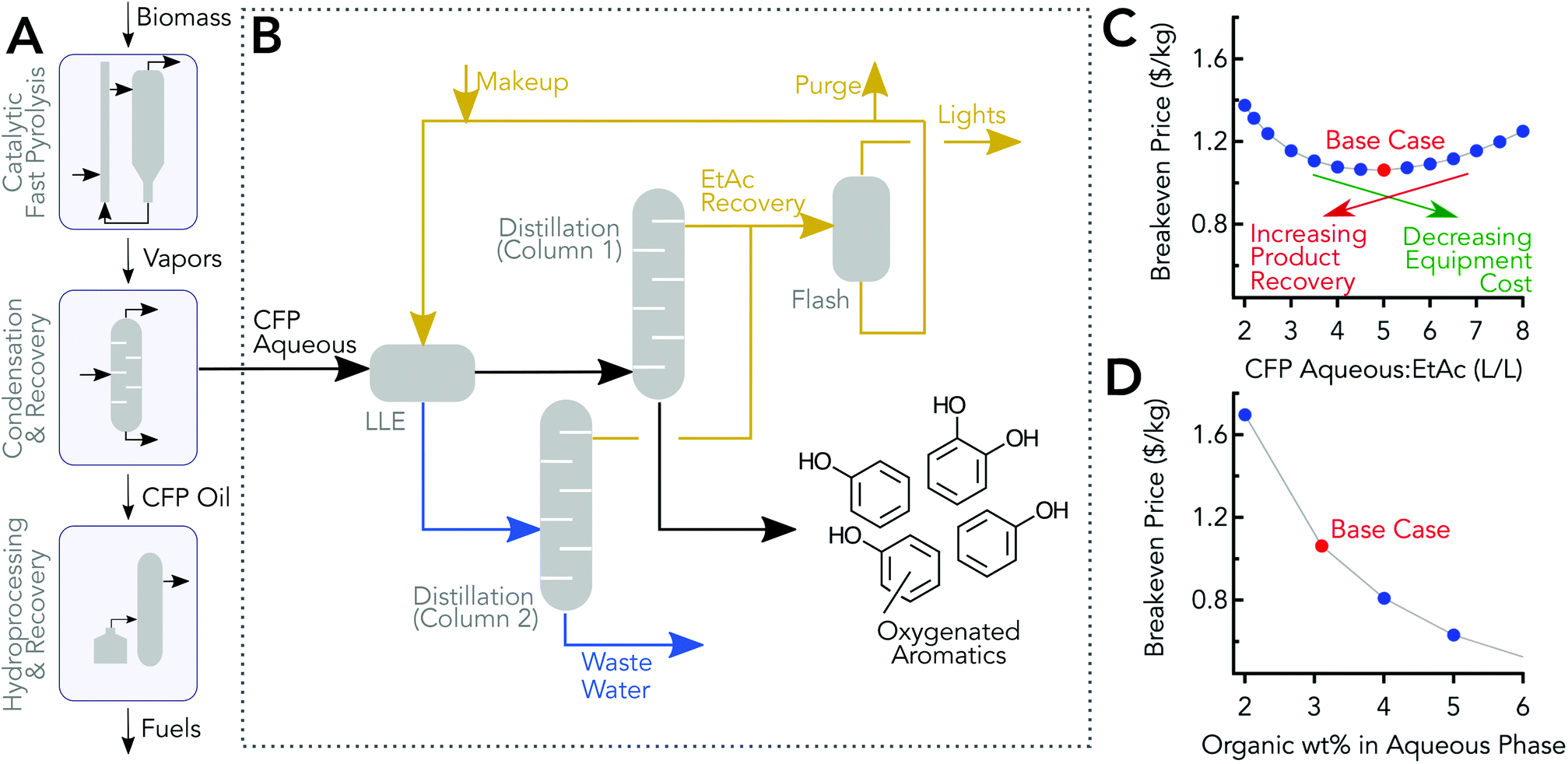 | ||
| Fig. 6 Modelled process and analysis for separating organics from the CFP aqueous stream. (A) Simplified block flow diagram of a CFP process for fuels production. (B) Simplified process flow diagram for the conceptual separation process for TEA. (C) Effect of CFP aqueous phase feed to ethyl acetate ratio on the breakeven price. (D) Effect of the concentration of organics in the CFP aqueous feed on the breakeven price. | ||
Minimum product selling price
The breakeven price, which is the minimum price to sell the product that covers capital and operating expenditures to produce the co-product, was calculated using discounted cash flow rate of return analysis, and 2014 dollars were used as the basis for the economic calculations. Assumptions for the modelled process shown in Fig. 6B are consistent with approaches taken elsewhere.48,65 The breakeven price for the base case is $1.06 kg−1. Note that this mixed product has 92% phenolics (i.e., compounds with at least one phenolic group), and 65% of the aqueous phenolics are recovered in the base case process model.Economic impact of process operating parameters
The economic impact of the ratio of volumetric flow rates of the CFP aqueous phase feed to the ethyl acetate flow rate are shown in Fig. 6C. The design specifications mentioned previously were maintained for all the cases. The analysis showed that there was a clear optimum at a ratio of 5:1 with the current set of assumptions. The recovery of oxygenated aromatics increased when more ethyl acetate was used but comes at the expense of larger flow rates which increases capital equipment costs in addition to higher heat duties and greater ethyl acetate replenishment. With higher than 5:1 ratios, the drop in the recovery has a greater impact compared to savings from capital and operating costs associated with lower flow rates. Table S7† shows operating cost contributions to the breakeven price for the base case and cost sensitivities are provided in the ESI.† Heat duties for the distillation are provided in Table S8.† The economic impact of organics composition in the aqueous phase is shown in Fig. 6D. The results are based on the assumption that the same proportion of organic species are present in the aqueous stream, but at higher concentrations. The trend shows that increasing the carbon content of the aqueous phase reduces the breakeven price. Other experimental systems with different configurations have reported significantly higher organics in the aqueous phase, which could benefit significantly from this valorization strategy.66 The overall process and economic impact of increasing organics content of the aqueous phase is not considered here, but increasing the carbon in this stream could reduce the CFP oil phase produced.
Discussion
CFP and thermochemical conversion processes are not generally considered as co-product generating technologies, but carbon present in waste streams can be recovered. Here we demonstrate a conceptual process which increases the atom-efficiency of a CFP process by separating and purifying valuable co-products from a waste stream. The process dewaters and concentrates organics in the wastewater using LLE, distills the concentrated organics into crude fractions, and further purifies phenol and catechol. Other thermochemically derived aqueous streams (e.g. hydrothermal liquefaction) are qualitatively similar in that they are high in water content and are generally comprised of ketones, acids, and oxygenated aromatics. Thus, the applicability of the approach taken, dewatering and subsequent refining to achieve high purity co-products, is proposed as a general strategy for aqueous stream valorization; however, the specific unit operations utilized may vary for streams of different carbon content, composition, or when targeting different co-products. The economics of valorizing a waste stream are explored here to provide preliminary insight into the technoeconomic viability of the unit operations used in this work.Liquid–liquid extraction
The first unit operation utilized in the aqueous separation train is key for stream valorization as it is initially dilute and has a high enthalpy of vaporization due to high water content. Here an LLE-first approach is developed that substantially reduces stream volume and heat duty for subsequent distillation, which reduces downstream equipment and energy costs. This approach has significant benefits relative to other separation approaches. Adsorptive resins or distillation have been reported53 as an approach to simultaneously achieve de-watering and separation; however, solid–liquid separation schemes require more complex unit operations (e.g., simulated moving bed, semi-batch modes) at scale and more uncertainty around the economics of the solid phase. Distillation, as a first unit operation, is associated with a high heat duty making the economics untenable. LLE is a more viable approach due to its simplicity and low energy requirement.The drawback to LLE is the addition of a solvent to the process, but this solvent can be recycled to minimize environmental and economic impact. Solvent selection for LLE affects plant economics, unit operations, safety, and environmental impact of the separation scheme. The ideal solvent will have low enthalpy of vaporization for low heat duties, low water solubility to minimize solvent partitioning to the water phase, low boiling point to separate the solvent from the higher-boiling-point oxygenated aromatic compounds, minimal environmental, health, and safety (EH&S) and life-cycle impact, and low cost to minimize capital expenses for initially charging the system and for solvent makeup. Table 2 contains values describing key parameters for solvent selection in LLE of the CFP aqueous phase. In this work, ethyl acetate was used because of its low enthalpy of vaporization, low SHE score67 which is a combined metric for EH&S and life cycle assessment (LCA) impact, and moderate water solubility. Butyl acetate has lower water solubility but higher enthalpy of vaporization while methyl acetate has an equivalent enthalpy of vaporization to ethyl acetate but is much more soluble in water. Halogenated solvents have desirable chemical properties, but these solvents have higher SHE scores and were thus not utilized. Petroleum ether and hexane were considered, but initial screening exhibited a low partitioning of the oxygenated aromatic compounds, and they also have high SHE scores making them less desirable.
| Solvent | ΔHvap80 (kJ mol−1) | T BP 81 (°C) | [C]S,H2O81 (wt%@20 °C) | $ kg−1 (82 and 83) | SHE score67 |
|---|---|---|---|---|---|
| Methyl acetate | 32 | 57 | 25% | ND | 2.44 |
| Ethyl acetate | 33 | 77 | 10% | 1.65 | 2.89 |
| Butyl acetate | 41 | 126 | 1% | 1.83 | 2.90 |
| Chloroform | 31 | 62 | 0.5% | 0.96 | 3.94 |
| Methylene Chloride | 30 | 74 | 2% | 1.13 | 3.60 |
| Petroleum ether | ND | 95–177 | Insoluble | ND | 3.91 |
| n-Hexane | 26 | 69 | 0.002% | 0.56 | 4.08 |
Identifying challenges in distilling CFP product streams
Distillation of concentrated organics from the CFP aqueous stream poses significant challenges due to (1) the diversity of compounds within the mixture, (2) incomplete vapor–liquid equilibrium (VLE) data to describe complex interactions, and (3) reactivity of compounds (e.g., aldehydes, ketones, and acids) with the mixture. Separation schemes can address highly heterogenous streams by careful selection of unit operations and order. Here, distillation of the concentrated organics produced crude fractions of the target products which allowed the subsequent unit operations to be tailored to separate the final products. With the high polarity of oxygenated compounds giving rise to strong molecular interactions, deviations from ideal behavior is expected. A prime example of such a deviation is the interaction between 2-cyclopenten-1-one and phenol, which were present in the CFP aqueous phase here. The interaction of the two compounds results in an azeotrope that will required more extensive characterization to understand the impact in steady state unit operations. A description of the binary system is provided in the ESI.† Undesired reactions, another challenge of distilling CFP product streams, is a function of composition, temperature, and time. Reactions during distillation result in species appearing in fractions collected above their boiling points. This phenomena was directly observed in this work as acetic acid appeared in fraction F3 which is well above its boiling point. A detailed description of the acid generation is given in the ESI.† Minimizing the extent of reaction can be achieved by vacuum distillation as this lowers the boiler temperature. The disadvantage of decreasing pressure is a reduction in the boiling point differences. This reduction results in requiring more trays to achieve the same separation and/or increasing the reflux ratio, which increases the residence time of the compounds. The separation of acids from phenol in fraction F3 by a second distillation, demonstrates how the generation of species due to the reactive oil can be addressed through appropriate unit operations. The second distillation allowed the acids to be collected in fractions F3.1 and F3.2 while enriched phenol was collected in F3.3. Without removing the acids from the phenol, recrystallization was not possible from fraction F3.Finished products and stream utilization
The finished product quality and market for each co-product fraction, F3–F5, must be considered for further development of the process. Fraction F3 is the first stream from which a bio-product, phenol, is separated and further purified to provide a renewable drop-in phenol. In petrochemical operations, phenol is purified from the cumene production process and typically uses 4 distillation columns68 or 3 columns and an ion exchange resin.46 The classic cumene process is used to produce 95% of the world's supply of phenol46 at purities above 99.9% (ASTM D2439). Typical impurities from this process include 2-methylbenzofuran, cresols, and carbonyls, and Table S10† provides common purity specifications. The effect of the impurities found in bio-sourced phenol on polymer performance is unknown and left for future research. The target product for fraction F4 is a feed stream for phenol-formaldehyde resins. Previous work using oxygenated aromatics from the aqueous phase demonstrated the similarity between bio-derived and petro-derived phenol-formaldehyde resins.38 Catechol which is produced from further purification of fraction F5 can serve as a functional replacement for aromatic diols (e.g., hydroquinone) or as a sustainable drop-in replacement where catechol is the primary chemical used in the application (e.g. pesticides). The primary use for catechol is in specialty chemicals, which often do not require the same purity requirements as monomers for polymeric applications. Typical purity for standard grade catechol, 98 wt%, is similar to the purity achieved here;69 however, impurity tolerance will ultimately depend on the specific chemical impurity, its concentration, and its effect on downstream processes and product performance.Fractions not intended for co-products, F1, F2 and F6, must be treated as waste streams or recycled into the process. Fraction F1 would be recovered in the solvent recovery unit operation; here the solvent recovery operation is not optimized to recover 100% of the ethyl acetate from the LLE extract, so the remaining ethyl acetate is recovered in the first fraction of the distillation. The residual ethyl acetate in fraction F2 is due to the temperature at which fraction F1 is switched to fraction F2, the reflux ratio, the mode of operation (e.g., steady state or batch), and the theoretical number of trays in the column. Further optimization of operating parameters could yield a better split between the ethyl acetate and acetic acid. Here, acetic acid was not further purified, but further purification of this stream could result in acetic acid as an additional bio-product or a carbon source for microbes in a biorefinery. More analysis is required to determine if purification of acetic acid is economically viable. Acetic acid is priced lower, $0.7 kg−1,70 than other products targeted in this work and there are other approaches for producing it from bioderived resources.71 Fraction F6 could be recycled in the separation process to improve catechol yields and the alkylated catechols in the stream could be separated as a mixed co-product stream similar to the phenolics. Applications could include utilization as hydrophobic wood resins,72 bioinspired polymer adhesives,73,74 or upgraded biocatalytically to other material precursors.16
TEA implications
The results show that the cost of concentrating the dilute CFP aqueous streams can be reduced significantly by extracting the contents into an ethyl acetate phase. The concentrated mixed phenolics product stream can be tenable only if this material can be used for making resins of acceptable quality, which remains a challenge. Additional lessons about further processing and purification may be derived from the challenges in separating phenol from coal sources.68 The economics for the base case (breakeven price of $1.06 kg−1) leave little room for additional processing costs given the price of phenol is approximately $1.1 kg−1 and is correlated with the price of benzene.75 Streams with higher carbon concentration may be significantly more attractive for co-product valorization, as shown in Fig. 6D. The relatively large phenol market size of 11400 kta in 2016, which is projected to grow to 14200 kta in 202275 leaves ample room for the growth of biomass CFP derived phenol. To put the production rate in perspective, the modelled process, shown in Fig. 6A which uses 2 dry-kt per day of woody biomass, produces approximately 3 kta of the mixed phenolics from the aqueous waste stream flowing at 31 tons per h, Table S6.† About 30% (or 3400 kta in 2016) of the world's phenol production is used for phenolic resins.76 Thus, a single biorefinery would only produce 0.1% of the global phenol demand for resins. Similarly, the maximum phenol production would be 0.6 kta, which represents a small percentage of world production. The maximum production rate of catechol from the aqueous stream is 1.1 kta, approximately 3% of world production. Increasing the co-product production from a biorefinery will require a catalyst which funnels more carbon into these products and separation of these co-products from the organic stream. The use of impure phenolics for resin-making needs to be validated, and the identification and removal of contaminates detrimental to product performance will be important. This process will not be able to penetrate the larger pure phenol market with a stringent 99.99% purity specification77 without further improving product purity. The phenol and catechol purities achieved here may be sufficient in some applications but not others, and the lower purity relative to petrochemical sources may impact the market value of the coproduct unless the product quality is improved. The growth in the phenolic resins market78 should readily sustain the production of any acceptable mixed phenolics from biomass.
Future research and conclusions
In this work phenol, catechol, and a phenolics phase were separated from a CFP aqueous stream using the following unit operations: LLE, distillation, and recrystallization. Towards reducing process heat duty requirements, LLE using ethyl acetate reduces the required vaporization energy prior to distillation by 95%. Batch mode distillation was able to purify phenol and catechol to 91 wt% and 77 wt%, respectively, while recrystallization as a final purification step was able to achieve 97 wt% purity for both compounds. These oxygenated aromatics, which have not been previously separated at high purities from thermochemical processes, are key intermediate compounds in the production of a wide range of materials (e.g., nylons, polycarbonates, polyether ether ketones, phenol-formaldehyde resins). The TEA results show the cost of concentrating the aqueous stream can be reduced using an LLE-first strategy, and the resulting products can achieve cost parity with petroleum derived oxygenated aromatics given a sufficiently high concentration of the compounds in the aqueous stream. Extending TEA modelling in the future can be used to determine the economic viability of the experimental work to isolate pure product streams. Co-production of high valued co-products from a high throughput biomass conversion process, such as CFP, offers a potential reduction in the cost of biofuels and provides a renewable source for these ubiquitous compounds.While the process proposed here was successful in economically recycling the LLE solvent, obtaining a phenolics stream for PF resin synthesis, and purifying phenol and catechol, there remain significant opportunities for improvements that can be made. Future work to improve the catalytic upgrading and separations should investigate carbon funnelling to reduce species diversity by developing and using more selective CFP catalyst or additional upgrading before the separation train. Minimization of undesired reactions during distillation of the concentrated organics may be explored through further optimization of the pressure set points and operating temperatures or through chemical pre-treatment (e.g., titration to neutral pH) to reduce the reactivity of the species. Experimental work to explore VLE data for single compounds or binary systems, which are not currently described in the literature, should be performed to improve process models, predict thermodynamic interactions which affect separations (e.g., azeotropes), and guide experimental separation strategies. Further exploration of these avenues will improve overall carbon efficiency and product recovery. For adoption of bio-derived oxygenated aromatics into the commodity market, future work will include optimization of purification strategies, more extensive impurity profiling, polymer synthesis and characterization to understand compositional effects on performance for both mixed oxygenates and pure component products, and setting impurity limits based on required performance.
Experimental procedures
Materials
Phenol, catechol and 2-cyclopenten-1-one were purchased from Sigma-Aldrich (St Louis, MO), received with purities of 99%, 99% and 98%, respectively, and used without further purification. The carbon content of the loblolly pine biomass is 51 wt% on a dry basis.CFP
CFP of pine using a HZSM-5 type catalyst, that was provided from Johnson Matthey (London, UK), was performed with a coupled pyrolyzer/DCR system with detailed methods described elsewhere.38,59,79 To generate the aqueous phase product used in the separation train, 1–2 mm pine particles were fed to the pyrolyzer with a biomass/N2 ratio of 0.5, and the resulting pyrolysis vapors were fed to the DCR at 1.0 kg vapor per h. The DCR was operated under adiabatic conditions similar to industrial fluid catalytic cracking (FCC) units with feed rates set and maintained throughout the operation. Zeolite catalyst (1.8 kg) was charged into the regenerator and moved through the system by pressure differentials. Fast pyrolysis vapors were fed into the DCR at 400 °C, while the rate of catalyst circulation was used to maintain the desired target temperatures. The product streams were collected using a reflux-style condenser, which condenses product gases swept out from the catalyst steam stripper. After condensation, the product phase separated into a hydrocarbon phase and the aqueous phase. The carbon content in the aqueous product from the DCR was determined using the Leco total carbon analysis.LLE
LLE was performed by mixing 1100 g of the aqueous stream produced by CFP with 500 g of ethyl acetate resulting in a 2:1 v/v ratio. The two-phase system was stirred vigorously for 20 min and subsequently transferred to a separatory funnel where the aqueous and ethyl acetate partitioned into two phases. The ethyl acetate phase was collected, dried with sodium sulphate and filtered to remove the salt. The organics were concentrated via rotovaporization on a Buchiemi rotovaporizer at 120 torr with a bath temperature of 50 °C. The resulting concentrated organics were stored at 4 °C until further use.
Distillation
Distillation was performed on 800-SB-A (B/R Instruments) spinning band distillation column. All distillations were performed in batch mode with a Teflon band which simulated 30 theoretical trays. Temperature profiles were collected using M690 PC Interface (B/R Instruments). During vacuum distillation the atmospheric equivalent temperature in celsius (°CAET) was calculated using BR AET Utility 1.0 software and fractions were collected based on the calculated value of the atmospheric equivalent temperature. The reported reflux ratios in Tables 1 are time based wherein the value represents the amount of time the reflux valve remains closed relative to the time it remains open. Model compound boiling points and azeotrope values were determined by operating the column under infinite reflux and plotted in Fig. S1.†Recrystallization
Recrystallization of phenol was performed by adding the purified phenol distillate, fraction F3.3 from the secondary distillation, to hexanes at 0.05 g mL−1. The solution was mixed and heated to 50 °C, and then the solution was cooled to 5 °C. Crystallized phenol was filtered from the hexanes and subsequently analysed. Recrystallization of catechol was performed by adding the crude catechol distillate, fraction F5 from the primary distillation, to toluene at 0.15 g mL−1. The solution was mixed and heated to 60 °C, and then the solution was cooled to 20 °C. Crystallized catechol was filtered from the hexanes and subsequently analysed.NMR
1H NMR spectra of petro- and bio-derived phenol and catechol were produced using a 400 MHz Bruker NMR in deuterated chloroform, Sigma-Aldrich (St, Louis, MO). 1H spectra were generated in Python 3.6 using the nmrglue-0.6 library.GC-MS
Chemical identification and compositional determination were performed on an Agilent Technologies 7890A, 5975c GC-MS, and GC-FID with a DB-5 column. A polyarc® microreactor was installed upstream of the FID detector which enabled quantification via a single internal standard, naphthalene. The polyarc converts all carbon to methane so moles of a species in the sample is proportional to the GC peak area divided by the number of carbons in the compound. All samples were prepared in methanol. Integration was performed using Agilent's Enhanced Chemstation software using the Chemstation Integrator for area determination and NIST library v2.0A 2011 for identification. The resulting areas were analysed with Python 3.6. Analysis using libraries chemspipy-1.0.5 and pyvalence-0.0.2.DSC
Melting points of petro- and bio-derived phenol and catechol were determined using a TA Q1000 DSC. Aluminium T-zero pans were purchased from TA Instruments (New Castle, DE). A 2–4 mg sample was loaded into an aluminium hermetic Tzero pan. Each sample was first annealed by thermal cycling between −20–60 °C or 20–120 °C for phenol and catechol, respectively. Modulated DSC was then used to generate thermograms by heating from 25–55 °C or 90–120 °C at 1 °C min−1 using the heat-only mode. Melting points and purities were then determined using Universal Analysis 2000 v4.5A software.TEA
The assumed base case composition of the aqueous phase, Table S3,† is from experimental results and has 96.9% water. The flow rate of this stream in the Aspen Plus process model was set at 31751 kg h−1 (70000 lb h−1); this is the approximate expected CFP aqueous phase flow rate in a 2 kt day−1 of dry-biomass plant, as detailed in a conceptual design report.48 The entire process was operated close to atmospheric pressure. Temperatures in distillation columns and flash tanks were based on modelled separation specifications. A consistent set of design criteria were maintained for the base case and all sensitivity cases for the process depicted in Fig. 6B. These assumptions include (a) a 0.25 molar reflux ratio and 0.015% ethyl acetate loss in the bottom streams of both the distillation columns, (b) two successive flash tanks were used to remove lights, 5 mol% vapor was removed from flash tank 1 and 50 mol% vapor from flash tank 2, and (c) a nominal 0.45 kg h−1 (1 lb h−1) was purged from the recycled ethyl acetate stream. The separations predicted by the Aspen Plus simulation is dependent on the physical property information. Table S6† provides the list of compounds selected in Aspen Plus based on the experimental compositional analysis. Structures for all compounds were provided to allow estimation of properties using the built-in UNIFAC-Dortmund property method in Aspen Plus. Nearest possible approximations were used when exact functional groups were not available. Table S4† shows the recovery of various components in the concentrated organic stream, in comparison to the modelled inlet flow rates.
Equipment costs and installation factors were calculated using Aspen Capital Cost Estimator, Version 9. Stainless steel (SS316) was chosen as the material of construction for most of the equipment because of the presence of ethyl acetate, and other corrosive chemicals in the aqueous stream. The total installed equipment cost along with factors for arriving at the fixed capital investment for the aqueous phase separations project are listed in Table S9.† The process model and cost information were initially developed for a 2:1 aqueous phase to ethyl acetate volumetric ratio, based on the experimental work using that proportion. The equipment costs for the other cases were scaled using flow rates, heat duties or power consumption as appropriate, with a scaling exponent of 0.7.
Flows and utility requirements from the process model were used along with unit cost information to calculate variable operating cost, Table S7.† Heat integration with the main CFP process can lead to further energy savings; however, this analysis assumes the use of steam for heating, and air and water coolers for cooling, without process heat integration which is outside the scope of the process in Fig. 6B. The total operating cost contribution was 81 cents per kg of the total breakeven price of $1.06 kg−1. Ethyl acetate was by far the biggest contributor to the variable operating costs, at 28 cents per kg followed by steam cost of 10 cents per kg.
Assumptions for determining the breakeven price include 90% plant on-stream time, 60% debt financing at 8% annual interest rate for 10 years, 10% return on the remaining 40% equity financing, a plant life of 30 years, plant depreciation over 7 years, and a three-year construction period with 6 months for start-up. It was assumed that continuing the same regenerative thermal oxidative process for treating this wastewater will be cost-neutral when compared to the original RTO.48
Conflicts of interest
The authors certify there are no conflicts of interest to declare.Acknowledgements
This work was authored by the National Renewable Energy Laboratory, operated by Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. Funding provided by the U.S. Department of Energy (DOE, Office of Energy Efficiency (EERE), Bioenergy Technology Office (BETO), under Contract DE-AC36-08GO28308 at the National Renewable Energy Laboratory (NREL), and in partial collaboration with the Chemical Catalysis for Bioenergy Consortium (ChemCatBio), a member of the Energy Materials Network (EMN).References
- International Energy Agency, Renewables information: Overview 2017, 2017 Search PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- L. Tao, J. N. Markham, Z. Haq and M. J. Biddy, Green Chem., 2017, 19, 1082–1101 RSC.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- A. Corma Canos, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef PubMed.
- A. V. Bridgwater, Chem. Eng. J., 2003, 91, 87–102 CrossRef CAS.
- A. Effendi, H. Gerhauser and A. V. Bridgwater, Renewable Sustainable Energy Rev., 2008, 12, 2092–2116 CrossRef CAS.
- D. Chen, L. Yin, H. Wang and P. He, Waste Manage., 2015, 37, 116–136 CrossRef PubMed.
- T. R. Carlson, Y. Cheng, J. Jae and G. W. Huber, Energy Environ. Sci., 2011, 4, 145–161 RSC.
- P. Karanijkar, R. Coolman, G. W. Huber, M. Blatnik, S. Almalkie, S. Bruyn Kops, T. Mountziaris and W. Conner, AIChE J., 2014, 60, 1320 CrossRef.
- S. Du, D. P. Gamliel, J. A. Valla and G. M. Bollas, J. Anal. Appl. Pyrolysis, 2016, 122, 7–12 CrossRef CAS.
- J. He, K. Huang, K. J. Barnett, S. H. Krishna, D. M. Alonso, Z. J. Brentzel, S. P. Burt, T. Walker, W. F. Banholzer, C. T. Maravelias, I. Hermans, J. A. Dumesic and G. W. Huber, Faraday Discuss., 2017, 202, 247–267 RSC.
- J. M. R. Gallo, D. M. Alonso, M. A. Mellmer and J. A. Dumesic, Green Chem., 2013, 15, 85–90 RSC.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584 RSC.
- D. R. Vardon, M. A. Franden, C. W. Johnson, E. M. Karp, M. T. Guarnieri, J. G. Linger, M. J. Salm, T. J. Strathmann and G. T. Beckham, Energy Environ. Sci., 2015, 8, 617–628 RSC.
- R. L. F. Valle, in Handbook of Polymer Synthesis, Characterization, and Processing, 2006, p. 984 Search PubMed.
- A. L. Holmberg, N. A. Nguyen, M. G. Karavolias, K. H. Reno, R. P. Wool and T. H. Epps, Macromolecules, 2016, 49, 1286–1295 CrossRef CAS.
- N. A. Rorrer, D. R. Vardon, J. R. Dorgan, E. J. Gjersing and G. T. Beckham, Green Chem., 2017, 19, 2812–2825 RSC.
-
K. Allen and P. Mehta, INEOS Phenol to expand phenol capacity of Mobile, Alabama, plant to 850000 mt per year, INEOS Phenol to expand phenol capacity of Mobile, Alabama, plant to 850000 mt per year, (accessed 25 February 2019).
- O. and G. R. F. 2017 T. 2022 Global Phenol Market Size, Price Trends, Demand, No Title, http://www.digitaljournal.com/pr/3380608.
- Catechol Faces Supply Crunch While Prices Rise.
- J. Plotkin, What's New in Phenol Production?, https://www.acs.org/content/acs/en/pressroom/cutting-edge-chemistry/what-s-new-in-phenol-production-.html, (accessed 25 February 2019).
- B. Wynands, C. Lenzen, M. Otto, F. Koch, L. M. Blank and N. Wierckx, Metab. Eng., 2018, 47, 121–133 CrossRef CAS PubMed.
- J. S. Kim, Bioresour. Technol., 2015, 178, 90–98 CrossRef CAS PubMed.
- W. O. H. Esse, H. Ag and W. K. Albert, in Ullmann's Encyclopedia of Industrial Chemistry, 2012, pp. 584–600 Search PubMed.
- J. Carvajal-diaz and P. Blanchard, Polycarbonate Resins, 2016 Search PubMed.
- H. Fiege, H.-W. Voges, T. Hamamoto, S. Umermura, T. Iwata, H. Miki, Y. Fujita, H.-J. Buysch, D. Garbe and W. Paulus, in Ullmann's Encyclopedia of Industrial Chemistry, 2012, pp. 522–582 Search PubMed.
- A. Ben-Haida, H. M. Colquhoun, P. Hodge and D. J. Williams, Macromolecules, 2006, 39, 6467–6472 CrossRef CAS.
- J. Yang, M. A. Cohen Stuart and M. Kamperman, Chem. Soc. Rev., 2014, 43, 8271–8298 RSC.
- S. Liu, Hydroquinone, 2015 Search PubMed.
- M. J. Biddy, R. Davis, D. Humbird, L. Tao, N. Dowe, M. T. Guarnieri, J. G. Linger, E. M. Karp, D. Salvachúa, D. R. Vardon and G. T. Beckham, ACS Sustainable Chem. Eng., 2016, 4, 3196–3211 CrossRef CAS.
- K. Wang, L. Ou, T. Brown and R. C. Brown, Biofuels, Bioprod. Biorefin., 2015, 9, 190–200 CrossRef CAS.
- A. Bailey, J. G. Leong and N. Fitzgerald, Bioproducts to Enable Biofuels Workshop, 2015 Search PubMed.
- W. Hu, Q. Dang, M. Rover, R. C. Brown and M. M. Wright, Biofuels, 2016, 7, 87–103 CrossRef CAS.
- K. Iisa, D. J. Robichaud, M. J. Watson, J. ten Dam, A. Dutta, C. Mukarakate, S. Kim, M. R. Nimlos and R. M. Baldwin, Green Chem., 2018, 20, 567–582 RSC.
- D. C. Elliott, H. Wang, M. Rover, L. Whitmer, R. Smith and R. Brown, ACS Sustainable Chem. Eng., 2015, 3, 892–902 CrossRef CAS.
- A. N. Wilson, M. J. Price, C. Mukarakate, R. Katahira, M. B. Griffing, J. R. Dorgan and M. R. Nimlos, ACS Sustainable Chem. Eng., 2017, 5, 6615–6625 CrossRef CAS.
- A. Sharma, V. Pareek and D. Zhang, Renewable Sustainable Energy Rev., 2015, 50, 1081–1096 CrossRef CAS.
- M. J. Biddy, C. Scarlata and C. Kinchin, NREL Tech. Rep. NREL/TP-5100-65509, 2016, p. 131.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50(10), 3733–3749 CrossRef CAS.
- W. Schutyser, S.-F. Koelewijn, M. Dusselier, S. de Vyver, J. Thomas, F. Yu, M. J. Carbone, M. Smet, P. Van Puyvelde, W. Dehaen and B. F. Sels, Green Chem., 2014, 16, 1999–2007 RSC.
- Z. Sun, B. Fridrich, A. De Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- W. Schutyser, T. Renders, S. Van Den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- S.-F. Koelewijn, S. Van den Bosch, T. Renders, W. Schutyser, B. Lagrain, M. Smet, J. Thomas, W. Dehaen, P. Van Puyvelde, H. Witters and B. F. Sels, Green Chem., 2017, 2561–2570 RSC.
- R. J. Schmidt, Appl. Catal., A, 2005, 280, 89–103 CrossRef CAS.
- S. Van de Vyver and Y. Román-Leshkov, Catal. Sci. Technol., 2013, 3, 1465–1479 RSC.
- A. Dutta, A. Sahir, E. Tan, D. Humbird, L. J. Snowden-swan, P. Meyer, J. Ross, D. Sexton, R. Yap and J. Lukas, Process Design and Economics for the Conversion of Lignocellulosic Biomass to Hydrocarbon Fuels. NREL/TP-5100-62455 and PNNL-23823, 2015.
- A. K. Starace, B. A. Black, D. D. Lee, E. C. Palmiotti, K. A. Orton, W. E. Michener, J. ten Dam, M. J. Watson, G. T. Beckham, K. A. Magrini and C. Mukarakate, ACS Sustainable Chem. Eng., 2017, 5, 11761–11769 CrossRef CAS.
- P. N. Kechagiopoulos, S. S. Voutetakis, A. a. Lemonidou and I. a. Vasalos, Energy Fuels, 2006, 20, 2155–2163 CrossRef CAS.
- J. A. Lopez-Ruiz, A. R. Cooper, G. Li and K. O. Albrecht, ACS Catal., 2017, 7, 6400–6412 CrossRef CAS.
- L. N. Jayakody, C. W. Johnson, J. M. Whitham, R. J. Giannone, B. A. Black, N. S. Cleveland, D. M. Klingeman, W. E. Michener, J. L. Olstad, D. R. Vardon, R. C. Brown, S. D. Brown, R. L. Hettich, M. Guss and G. T. Beckham, Environmental Science enhanced microbial toxicity tolerance, Royal Society of Chemistry, 2018 Search PubMed.
- J. P. Stanford, P. H. Hall, M. R. Rover, R. G. Smith and R. C. Brown, Sep. Purif. Technol., 2017, 194, 170–180 CrossRef.
- S. Conrad, J. Westermeyer and T. Schulzke, Environ. Prog. Sustainable Energy, 2017, 36(3), 662–667 CrossRef CAS.
- O. D. Mante, S. J. Thompson, S. Mustapha and D. C. Dayton, Green Chem., 2019, 21, 2257–2265 RSC.
- J. N. Murwanashyaka, H. Pakdel and C. Roy, Sep. Purif. Technol., 2001, 24, 155–165 CrossRef CAS.
- P. R. Gogate and A. B. Pandit, Adv. Environ. Res., 2004, 8, 501–551 CrossRef CAS.
- P. R. Gogate and A. B. Pandit, Adv. Environ. Res., 2004, 8, 553–597 CrossRef CAS.
- B. A. Black, W. E. Michener, K. J. Ramirez, M. J. Biddy, B. C. Knott, M. W. Jarvis, J. Olstad, O. D. Mante, D. C. Dayton and G. T. Beckham, ACS Sustainable Chem. Eng., 2016, 4, 6815–6827 CrossRef CAS.
- D. Seader and E. J. Henley, Separation Process Principles, 2006, vol. 1, pp. 778–871 Search PubMed.
- M. J. M. Wells, Principles of Extraction and the Extraction of Semivolatile Organics from Liquids, 2003, vol. 162 Search PubMed.
- R. R. Dreisbach and R. a. Martin, Ind. Eng. Chem., 1949, 41, 2875–2878 CrossRef CAS.
- V. K. Jadhav, M. R. Chivate and N. S. Tavare, J. Chem. Eng. Data, 1992, 37, 232–235 CrossRef CAS.
- R. T. P. Pinto, L. Lintomen, L. F. L. Luz and M. R. Wolf-Maciel, Fluid Phase Equilib., 2005, 447–457 CrossRef CAS.
- A. Dutta, J. A. Schaidle, D. Humbird and F. G. Baddour, Top. Catal., 2016, 59, 2–18 CrossRef CAS.
- V. Paasikallio, C. Lindfors, E. Kuoppala, Y. Solantausta, A. Oasmaa, J. Lehto and J. Lehtonen, Green Chem., 2014, 16, 3549 RSC.
- G. Koller, U. Fischer and K. Hungerbühler, Ind. Eng. Chem. Res., 2000, 39, 960–972 CrossRef CAS.
- W. Manfred, M. Weber and M. Kleien-Boymann, in Ullman's encyclopedia of industrial chemistry, 2012, vol. 26, pp. 503–519 Search PubMed.
- L. Krumenacker, M. Costantini, P. Pontal and J. Sentenac, in Kirk-Othmer Encyclopedia of Chemical Technology, 2000, pp. 1–19 Search PubMed.
- China acetic acid may rebound on demand recovery, lower supply in Q2, https://www.icis.com/resources/news/2018/03/28/10206887/china-acetic-acid-may-rebound-on-demand-recovery-lower-supply-in-q2/, (accessed 9 April 2018).
- M. Schaechter, in Encyclopedia of Microbiology, 2009, pp. 144–149 Search PubMed.
- K. Li and X. Geng, Macromol. Rapid Commun., 2005, 26, 529–532 CrossRef CAS.
- J. Duan, W. Wu, Z. Wei, D. Zhu, H. Tu and A. Zhang, Green Chem., 2018, 20, 912–920 RSC.
- J. Sedõ, J. Saiz-Poseu, F. Busqué and D. Ruiz-Molina, Adv. Mater., 2013, 25, 653–701 CrossRef PubMed.
- K. Anderson, Global Phenol Market Size, Price Trends, Demand, Outlook and Growth Rate from 2017 to 2022, http://www.abnewswire.com/pressreleases/global-phenol-market-size-price-trends-demand-outlook-and-growth-rate-from-2017-to-<?pdb_no 2022?>2022<?pdb END?>_122000.html, (accessed 18 February 2018).
- R. Peacock, Market Outlook: Phenol/acetone markets are under ressure: ICIS Consulting, https://www.icis.com/resources/news/2016/06/09/10006764/market-outlook-phenol-acetone-markets-are-under-ressure-icis-consulting/, (accessed 1 April 2018).
- M. Ghirardini and M. Tampieri, in Handbook of Petrochemical Production Processes, 2005 Search PubMed.
- Grand View Research, Phenolic Resins Market Size Worth $16.0 Billion By 2025 | CAGR: 7.7%, https://www.grandviewresearch.com/press-release/global-phenolic-resins-market, (accessed 18 February 2018).
- M. W. Jarvis, J. Olstad, Y. Parent, S. Deutch, K. Iisa, E. D. Christensen, H. Ben, S. K. Black, M. R. Nimlos and K. A. Magrini, Energy Fuels, 2018, 32, 1733–1743 CrossRef CAS.
- W. Acree and J. S. Chickos, J. Phys. Chem. Ref. Data, 2016, 45, 043101 CrossRef.
- National Institute for Occupational Safety and Health, NIOSH pocket guide to chemical hazards, 2005 Search PubMed.
- ICIS, Indicative Chemical Prices A-Z, https://www.icis.com/chemicals/channel-info-chemicals-a-z/, (accessed 12 December 2016).
- B. of L. Statistics, Producer Price Index-Commodities, http://www.bls.gov/data/, (accessed 12 December 2016).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc00902g |
| This journal is © The Royal Society of Chemistry 2019 |