
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemo-enzymatic pathways toward pinene-based renewable materials†
A.
Stamm
 ab,
M.
Tengdelius
a,
B.
Schmidt
ab,
J.
Engström
ac,
P. O.
Syrén
ab,
L.
Fogelström
ac and
E.
Malmström
*ac
ab,
M.
Tengdelius
a,
B.
Schmidt
ab,
J.
Engström
ac,
P. O.
Syrén
ab,
L.
Fogelström
ac and
E.
Malmström
*ac
aKTH Royal Institute of Technology, School of Engineering Sciences in Chemistry, Biotechnology and Health, Department of Fibre and Polymer Technology, Division of Coating Technology, Teknikringen 56-58, SE-100 44 Stockholm, Sweden. E-mail: mavem@kth.se
bKTH Royal Institute of Technology, School of Engineering Sciences in Chemistry, Biotechnology and Health, Science for Life Laboratory, Division of Protein Technology, Tomtebodavägen 23, Box 1031, SE-171 21 Solna, Sweden
cKTH Royal Institute of Technology, School of Engineering Sciences in Chemistry, Biotechnology and Health, Wallenberg Wood Science Center, Teknikringen 56-58, SE-100 44 Stockholm, Sweden
First published on 10th April 2019
Abstract
Sobrerol methacrylate (SobMA) was synthesized and subsequently polymerized using different chemical and enzymatic routes. Sobrerol was enzymatically converted from α-pinene in a small model scale by a Cytochrome P450 mutant from Bacillus megaterium. Conversion of sobrerol into SobMA was performed using both classical ester synthesis, i.e., acid chloride-reactions in organic solvents, and a more green approach, the benign lipase catalysis. Sobrerol was successfully esterified, leaving the tertiary alcohol and “ene” to be used for further chemistry. SobMA was polymerized into PSobMA using different radical polymerization techniques, including free radical (FR), controlled procedures (Reversible Addition Fragmentation chain-Transfer polymerization, (RAFT) and Atom Transfer Radical Polymerization (ATRP)) as well as by enzyme catalysis (horseradish peroxidase-mediated free radical polymerization). The resulting polymers showed high glass-transition temperatures (Tg) around 150 °C, and a thermal degradation onset above 200 °C. It was demonstrated that the Tg could be tailored by copolymerizing SobMa with appropriate methacrylate monomers and that the Flory–Fox equation could be used to predict the Tg. The versatility of PSobMA was further demonstrated by forming crosslinked thin films, either using the ‘ene’-functionality for photochemically initiated ‘thiol-ene’-chemistry, or reacting the tertiary hydroxyl-group with hexamethoxymethylmelamine, as readily used for thermally curing coatings systems.
Introduction
Reducing the need for petroleum-based materials is one of the main challenges in the contemporary society. Proposed practical solutions to this are either to reduce the use of fossil-based consumer products, or to develop alternative materials originating from renewable sources. Most likely, a combination of both strategies is required to accelerate the transition towards a more sustainable society. Lately, the members of the European Parliament voted for the ban of single-use plastic by 2021, increasing the demand for new materials.1Still, the vast majority of all plastics produced are based on fossil-sources; only around one percent of the about 300 million tons of plastic produced annually is bio-based.2 There are different methods for obtaining renewable polymers. One common strategy is to utilize abundant biopolymers available in biomass, mainly cellulose, and by degradation/refining produce renewable equivalents in order to replace monomers traditionally obtained from fossil-sources, e.g., ethylene and propylene. One major drawback with this is that the biopolymer has to be degraded into small molecules and then again polymerized to large molecules. Furthermore, the production of the polymers utilizes the same harsh industrial processes regardless of the origin of the monomers.3 Another strategy is to use biopolymers originating from renewable sources, such as cellulose, lignin, starch and chitosan, in their inherent state to produce sustainable materials.3,4 This approach is hampered by the fact that biopolymers are associated with large property variations as an effect of varying growth conditions (growth season, locus etc.), significantly challenging their industrial utilization. A third strategy is to use novel renewable monomers from biomass, not readily available from fossil sources, to produce renewable polymers with properties comparable to fossil-based counterparts.
Trees and plants have proven particularly useful as sources for sustainable monomers.3,5,6 Besides containing significant amounts of biopolymers, which can be used either directly for producing materials or to produce monomers4,6 by depolymerization followed by fermentation and/or upgrading,5 these sources are also rich in small molecules useful as monomers, such as furans, fatty acids, and terpenes.3,5 With the rapid emergence of modern biorefineries, focusing not only on energy recovery, an array of waste-stream products has become more available and, therefore, more interesting from a commercial point of view.7 Terpenes belong to one such group of compounds that are rendering increasing interest in this context. Terpenes constitute one of the most diversified classes of natural products with essential functions in a broad range of organisms.8,9 Terpenes/terpenoids have, for instance, found important applications, or potential applications, in areas such as fragrances and flavors,10 biofuels,11 fine chemical synthons,12 and as anticancer and antiviral agents.8 Terpenes are assembled from the universal C5-building blocks, dimethylallyl diphosphate and isopentenyl diphosphate, to yield linear polyisoprene building blocks.3 The extensive structural variations found in terpenes is an effect of enzyme-catalyzed cyclizations to result in chiral and multicyclic natural products, which presents further opportunities for producing polymers with a wide range of properties12 beyond natural rubber (i.e. polyisoprene). The main source of terpenes is the pulp industry-derived pine-tree resin turpentine, industrially available on ca. 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ton scale annually.3 The primary terpene in turpentine is α-pinene.6 It has a bicyclic unsaturated structure (Scheme 1), and has been reported to result in polymers with alternating linear and cyclic units via cationic polymerization.13–15 Still, achieving controlled polymerization of pinene monomers remains a challenge, which has spurred an increased interest towards unlocking their inherent potential by activation through oxidation by chemical and biocatalytic methods.16–18 Biocatalysis displays a great potential to replace harsh chemical synthesis with green options for the sustainable generation of medicines, biochemicals and materials.19 In particular, by capitalizing on synthetic-biology approaches, mild oxidations required for monomer activation – which would be challenging to access by traditional organic synthesis20 – can be achieved in water with oxygen as co-substrate.21 Over the last years, many studies have been devoted to the use of enzymes as mediators for different polymerization techniques.22–26 Polymers from terpenes, or chemically modified terpenes, exhibit a wide range of molecular weights and thermal properties depending on the terpene structure/composition and polymerization technique.12,27 In the pursuit of novel polymeric materials, exploring the toolbox of terpene-based polymers holds great potential.28
000 ton scale annually.3 The primary terpene in turpentine is α-pinene.6 It has a bicyclic unsaturated structure (Scheme 1), and has been reported to result in polymers with alternating linear and cyclic units via cationic polymerization.13–15 Still, achieving controlled polymerization of pinene monomers remains a challenge, which has spurred an increased interest towards unlocking their inherent potential by activation through oxidation by chemical and biocatalytic methods.16–18 Biocatalysis displays a great potential to replace harsh chemical synthesis with green options for the sustainable generation of medicines, biochemicals and materials.19 In particular, by capitalizing on synthetic-biology approaches, mild oxidations required for monomer activation – which would be challenging to access by traditional organic synthesis20 – can be achieved in water with oxygen as co-substrate.21 Over the last years, many studies have been devoted to the use of enzymes as mediators for different polymerization techniques.22–26 Polymers from terpenes, or chemically modified terpenes, exhibit a wide range of molecular weights and thermal properties depending on the terpene structure/composition and polymerization technique.12,27 In the pursuit of novel polymeric materials, exploring the toolbox of terpene-based polymers holds great potential.28
 | ||
| Scheme 1 Chemoenzymatic pathways from (−)α-pinene to PSobMA. I–II: Conversion of α-pinene into SobMA catalyzed by Cytochrome P450 from Bacillus megaterium (CYP102A1) A74G/L188Q mutant (P450-BM3/2M), cofactors have been omitted for clarity. II–IV: Enzymatic conversion or chemical synthesis of SobMA followed by radical polymerization through different polymerization techniques. | ||
One promising underutilized terpene is sobrerol, an unsaturated diol, Scheme 1, which is formed directly via biotransformation of α-pinene in the fungi Armillaria mella21 or through aqueous oxidation.29 Sobrerol has shown antimycotic properties,30 to prevent pulmonary hypertension31 and the development of breast cancer.32
The multifunctional structure of sobrerol presents several opportunities for producing polymers. Its cyclic structure furthermore holds great promise for producing higher Tg-materials, in contrast to the majority of available green monomers that predominantly have non-cyclic structures. However, despite its functional groups, offering versatility in both polymer production and post-polymerization modifications, sobrerol is yet to be further explored as a novel monomer for polymeric materials.33
Structurally similar terpenes and terpenoids have been converted to polycarbonates,34–36 polyethers,37 polyesters,38 polyamides,17 and poly(meth)acrylates27 upon modification. The diol present in sobrerol presents opportunities for producing polymers of all the aforementioned classes. (Meth)acrylate polymers are widely used in the automotive, paint, home electronics, and medical-device industries, with a steadily increasing global market.39,40 Due to these widespread industrial applications, methods for large-scale production of (meth)acrylates are becoming increasingly more sustainable.41 The industrial usage and well-developed polymerization techniques of methacrylates,42 make sobrerol-based methacrylate polymers an interesting starting point for studying the potential of sobrerol as a renewable component in materials. Fonseca et al. recently published the use of SobMA as a substituent for polystyrene in the formation of polyester resins.33 Sobrerol was obtained from α-pinene oxide in the presence of CO2 and, after (meth)acrylation, incorporated into unsaturated polyester-resin formulations as a reactive diluent. The sobrerol-based formulations exhibited similar properties to those of the styrene-based polyester resins while also improving the environmental compatibility due to its biobased origin, low volatility and thus, a reduced exposure of VOC's to humans and the environment.
In this work, we have developed a chemoenzymatic “green” route for the synthesis of SobMA. The polymerization of SobMA was studied along with the properties of the resulting polymers. We present different routes to synthesize sobrerol-based polymethacrylates (PSobMAs). The resulting polymethacrylates showed a Tg around 150 °C. It was possible to tailor the Tg by copolymerization of various methacrylate monomers. The great versatility of PSobMA allows different material applications, e.g., coatings, and was demonstrated by subsequent crosslinking of the functional side-groups, using either thiol–ene or condensation coating chemistry.
Results & discussion
Biotransformation of (−)-α-pinene to prepare trans-sobrerol
The ability of Cytochrome P450 monooxygenases to, under mild conditions, insert oxygen at activated positions (e.g. allylic) makes them a promising class of biocatalysts for oxidation of α-pinene and other monoterpenes into possible monomers.43–46 To afford trans-sobrerol from (−)-α-pinene in one-pot via the corresponding epoxide, the biotransformation was performed in vitro using purified P450-BM3/2M adopted to procedures described for glucose-6-phosphate (G6P), and G6P dehydrogenase (G6PDH) was used for the co-factor regeneration (i.e. adenine dinucleotide phosphate NADPH) (Fig. S1†).46,47 After 22 h reaction time under aqueous conditions, 42% conversion (according to GC/MS) of (−)-α-pinene into trans-sobrerol was observed without intermediate work-up, which is in good agreement with recently reported data (Fig. S2 and S3†).46 At this point, this reaction was demonstrated on a small scale, in support of previous reports46 why all further experiments were conducted on commercially available trans-sobrerol.Chemical transformation of trans-sobrerol to SobMA
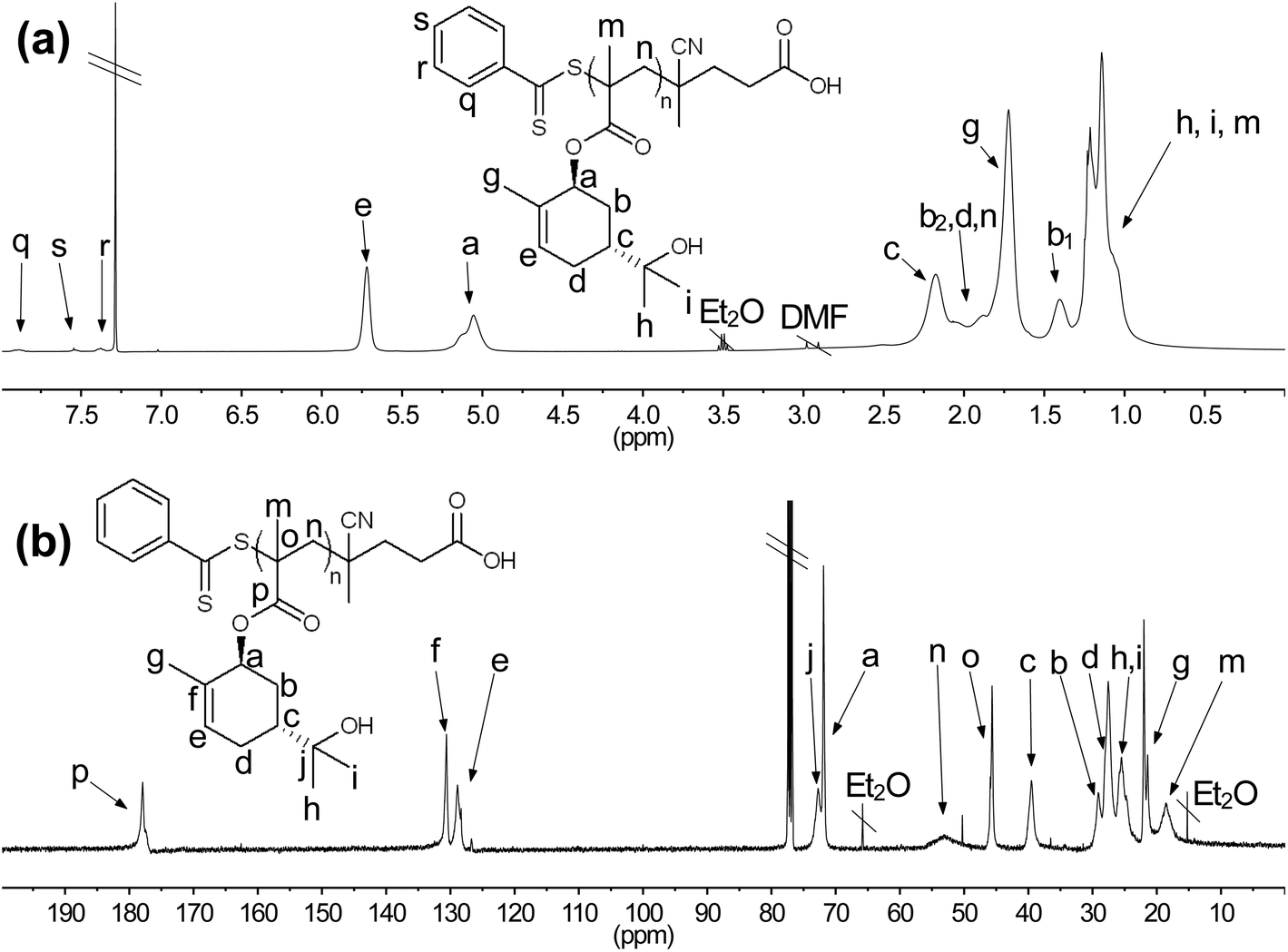
The SobMA was synthesized via a conventional chemical approach starting from commercially available sobrerol, using methacryloyl chloride and triethyl amine in DMF. DMF was not a preferred solvent but despite previous reports,48,49 sobrerol proved difficult to dissolve. Purification via medium pressure liquid chromatography (MPLC) yielded the liquid SobMA monomer in 66% yield. The monomer was sensitive to self-polymerization and was therefore stored in the mobile phase after MPLC. 1H NMR spectrum of SobMA showed a downfield shift of the signal corresponding to the methine proton neighboring the secondary alcohol (signal a in Fig. 1a and b) compared with sobrerol, corroborating the successful ester formation. This signal and the signals corresponding to the ene-protons in the ring and the methacrylate alkenes (peak e, n1, and n2 in Fig. 1b) were found to have a 1:1:1:1 integral ratio. This confirmed that the monomer was selectively mono-methacrylated. The signal of the methyl group on the ring (signal g in Fig. 1a and b) showed an upfield shift compared with sobrerol, confirming a change in electron density in the six-membered ring. 13C NMR also displayed a noticeable downfield shift for the signal corresponding to the carbon bound to the secondary alcohol (signal a in Fig. 1c, d and e), which further confirmed esterification of the secondary alcohol. GC-MS and GC-FID (Fig. S4 and 5†) confirmed methacrylation at the secondary alcohol. A previous study showed that the tertiary alcohol in sobrerol, upon ionization in the mass detector, dehydrates selectively to form a mass fragment 18 a.u. below its molecular ion.47 The mass fragment at m/z = 220 in the monomer mass spectrum thus confirmed that the methacrylation occurred selectively at the secondary alcohol, as this fragment is exactly 18 below the molecular ion of the desired monomer. These combined results show the predominant formation of a mono-methacrylated product with clear selectivity for the secondary alcohol over the tertiary, during the methacrylation. This could prove useful in industrial applications as it eliminates the need for protection/deprotection schemes.
 | ||
| Fig. 1 NMR spectra in CDCl3. (a) 1H NMR spectrum of sobrerol. (b) 1H NMR spectrum of SobMA. (c) 13C NMR spectrum of sobrerol. (d) 13C NMR spectrum of SobMA. | ||
One obvious drawback with the described synthesis is the use of DMF, which is a non-renewable solvent and associated with reproduction toxicity. A renewable and less toxic solvent is hence desirable, particularly upon scale-up. 2-Methyltetrahydrofuran (2-MeTHF) is derived from biomass50 and exhibit a low toxicity.51 Using identical conditions as described above, but 2-MeTHF instead of DMF afforded SobMA in 57% yield.
Enzymatic transformation of trans-sobrerol to SobMA
In our strive to design a ‘green chemistry-approach’ with no use of CMR-classified chemicals, we demonstrated that it is possible to accomplish SobMA from trans-Sob and vinyl methacrylate (VMA) using the enzyme Amano lipase from Pseudomonas fluorescens (P. fluorescens) at a high conversion (96%) and without the need for purification. The conditions were adopted from Chen et al., who reported high conversions for the lipase-catalyzed acetylation of sobrerol.52 The highest conversion was obtained at 80 °C and a molar ratio of Sob:VMA equal to 1:56. However, it was found that high conversions (∼90%) were possible to achieve at a lower excess of VMA (1:20) (Tables S1 and S2†). Nevertheless, it should be mentioned that in scales above 4 g of sobrerol purification by MPLC had to be conducted to isolate the pure product.
RAFT polymerization of SobMA
SobMA was polymerized by reversible addition–fragmentation chain-transfer (RAFT) polymerization to assess its applicability as a methacrylate monomer. SobMA was stored in a mixture of dichloromethane and ethyl acetate of which none is suitable for thermally initiated free radical polymerizations. The solution volume was therefore carefully reduced to prevent premature polymerization, whereafter the monomer was diluted by DMF and polymerized using 4,4′-azobis(4-cyanovaleric acid) (ACVA) as the initiator and 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPAD) as the RAFT agent. The monomer conversion was followed by 1H NMR (Fig. S6†) and the polymerization was confirmed by the disappearance of the methacryloyl alkene signals at δ = 6.07 and 5.51 ppm, as well as the upfield shift of the signal corresponding to the proton neighboring the methacrylate group from 5.29 ppm to 5.00 ppm. The ratio between the latter signals was used to determine the monomer conversion. The monomer consumption over time followed the first-order kinetics (Fig. S7†). This, in combination with the linear evolution of molecular weight with monomer conversion (Fig. S8†), corroborated a reasonably controlled RAFT polymerization. After 8 hours at 70 °C and conversions reaching above 80%, the polymer was precipitated in diethyl ether and isolated, yielding poly(sobreryl methacrylate) (PSobMAR) as a pinkish powder.Three different RAFT polymerizations were performed, using three different [SobMA]:[CPAD] ratios; 50:1, 100:1, and 200:1. The resulting polymers PSobMAR50, PSobMAR100, and PSobMAR200 were analyzed by NMR and SEC. Typical 1H NMR and 13C NMR spectra for RAFT-derived PSobMAs are displayed in Fig. 2. The molecular weights (Mn) determined by SEC (Fig. 3a) corresponded fairly well with the theoretical Mn (Table 1). The dispersities Đ were all reasonably narrow. By varying the ratio of monomer to RAFT agent it was possible to control the molecular weight.
 | ||
| Fig. 2 1H (a) and 13C (b) NMR spectra in CDCl3 of PSobMA via RAFT polymerization. | ||
 | ||
| Fig. 3 Molecular weight distributions for poly(SobMA) synthesized using (a) RAFT and (b) enzymatic polymerization. | ||
| Polymera | Method | Ratio (method) | Time (h) | Conv.b (%) | M n, theo. (g mol−1) | M n, SEC (g mol−1) | Đ | T g (°C) | T d (°C) |
|---|---|---|---|---|---|---|---|---|---|
|
a The the numbers refer to the target DP; the letter in the annotation refers to the polymerization method where R = RAFT (ratio: [M]:[CPAD]:[ACVA]), FR = free-radical (ratio: [M]:[ACVA]), A = ATRP (ratio: [M]:[HMTETA]:[EBrP]:[CuCl]) and E = enzymatic (ratio: [M]:[2,4-pentanedione]:[H2O2] (Oxidase) 80 mg mL−1 (2.9 mg enzyme per mmol monomer)).
b Determined by 1H-NMR.
c Calculated according to the RAFT and ATRP mechanisms.
d Determined by SEC using DMF as eluent and PMMA standards.
e Determined by DSC using second heating to assess the glass transition temperature.
f Determined by TGA at the temperature corresponding to 5% weight loss.
g Left figure refers to SobMA, right figure refers to MMA or BMA.
|
|||||||||
| PSobMAFR | FR | 250:1 (FR) |
5 | 91 | — | 48200 |
2.4 | 154 | 238 |
| PSobMAR50 | RAFT | 50:1:0.2 (R) |
8 | 90 | 11000 |
7200 | 1.2 | 116 | 191 |
| PSobMAR100 | RAFT | 100:1:0.2 (R) |
8 | 83 | 20100 |
20900 |
1.3 | 145 | 224 |
| PSobMAR200 | RAFT | 200:1:0.2 (R) |
8 | 81 | 38900 |
31600 |
1.2 | 155 | 238 |
| P(SobMAR100-b-MMAR100) | RAFT | (100:100)g:1:0.2 (R) |
24 | 95/66g | 29500 |
28000 |
1.2 | 141 | 227 |
| P(SobMAR100-b-BMAR100) | RAFT | (100:100)g:1:0.2 (R) |
24 | 88/70g | 31200 |
27000 |
1.1 | n.d. | 200 |
| P(SobMAR100-st-MMAR100) | RAFT | (100:100)g:1:0.2 (R) |
8 | 73/74g | 25100 |
27800 |
1.2 | 133 | 224 |
| P(SobMAR100-st-BMAR100) | RAFT | (100:100)g:1:0.2 (R) |
8 | 67/81g | 27800 |
25800 |
1.1 | 74 | 213 |
| P(SobMAR25-st-BMAR75) | RAFT | (25:75)g:1:0.2 (R) |
8 | 39/48 | 13000 |
11000 |
1.2 | 41 | 195 |
| PSobMAA50 | ATRP | 50:2:1:1 (A) |
7 | 75 | 9100 | 10500 |
1.2 | 132 | 215 |
| PSobMAA100 | ATRP | 100:2:1:1 (A) |
9 | 62 | 15000 |
16900 |
1.2 | 138 | 220 |
| PSobMAA200 | ATRP | 200:2:1:1 (A) |
9 | 71 | 34500 |
20900 |
1.2 | 141 | 224 |
| PSobMAE | Enz. | 42:1:1.48 (E) |
1 | 89 | n.d. | 39800 |
1.9 | 149 | 235 |
| P(SobMAE25-st-BMAE75) | Enz. | (10.5:31.5):1:148 (E) |
3 | 59/79 | n.d. | 51100 |
2.3 | 37 | 196 |
FR and ATRP polymerization of SobMA
To further elaborate the potential as methacrylate monomer, SobMA was polymerized using free radical (FR)- and atom transfer radical polymerization (ATRP) techniques. As for the RAFT polymerizations, the ATRP reactions were performed using three different [SobMA]:[HMTETA]:[EBrP]:[CuCl] ratios; 50:2:1:1, 100:2:1:1, and 200:2:1:1. In the FR polymerizations, similar conditions were used as in the RAFT polymerization but without the RAFT agent and with a reduced polymerization time (5 hours). All polymers were analyzed by NMR and SEC to determine molecular weights and dispersity (Fig. S7–10†). The results (Table 1) were reasonable for free-radical and ATRP polymerizations and corroborated that the polymer chain length can be tailored using ATRP, as observed for the RAFT polymerizations.
Copolymerization of SobMA with MMA and BMA
To confirm the controlled character of the RAFT polymerizations, block copolymers were synthesized using SobMA and methyl methacrylate (MMA) or butyl methacrylate (BMA). SobMA was polymerized for 6 hours, under the same conditions as the other RAFT polymers, reaching ca. 80% conversion of SobMA as assessed by 1H NMR. Thereafter, one molar equivalent of the co-monomer was added and the polymerization was left for another 18 h. 1H NMR samples acquired after a total of 24 h of polymerization showed high monomer conversions, Table 1. The MMA conversion was determined through the ratio between the signals corresponding to the methyl side groups, which show an upfield move in shift from δ = 3.69 ppm to δ = 3.54 ppm upon polymerization (Fig. S11†). Similarly, the methylene protons neighboring the alcohol part of the methacrylate ester in BMA showed an upfield shift from δ = 4.14 ppm to δ = 3.94 upon polymerization. The ratio between these signals was consequently used to determine the BMA conversion (Fig. S12†). SEC analysis shows increasing Mn after the copolymerizations and retained relatively narrow polydispersity (Fig. S13 and 14†). This confirmed the chain extension of the polymer chains, and thus the successful block formation. Analysis of the resulting P(SobMAR100-b-MMAR100) and P(SobMAR100-b-BMAR100), by SEC (Fig. S13 and 14†), again showed molecular weights comparable to the theoretical Mn (Table 1). Comonomer molar fractions of the isolated polymers were [SobMA]:[MMA] = 1:0.61 and [SobMA]:[BMA] = 1:1.37 for P(SobMAR100-b-MMAR100) and P(SobMAR100-b-BMAR100), respectively, as determined by 1H NMR (Fig. S15 and 16†).
The possibility of tailoring the properties of the polymer materials by statistical co-polymerization of SobMA with MMA or BMA was explored. Both monomers were added just prior to the polymerization, which was left to proceed for 24 h under conditions similar as for the homopolymers by RAFT. After 8 hours, monomer conversions of 73 and 74%, for SobMA and MMA respectively, and 67 and 81%, for SobMA and BMA respectively, were detected by 1H NMR. SEC analysis for the resulting P(SobMAR100-st-MMAR100) and P(SobMAR100-st-BMAR100) again showed molecular weights in good agreement with the theoretical molecular weights (Table 1). The copolymer compositions were [SobMA]:[MMA] = 1:0.99 and [SobMA]:[BMA] = 1:1.32 for P(SobMAR100-st-MMAR100) and P(SobMAR100-st-BMAR100), respectively, as determined by 1H NMR (Fig. S17 and 18†). The FTIR spectra of the four copolymers (block and statistical) showed absorbance patterns similar to those of the homo RAFT polymers (Fig. S19†).
Enzymatic polymerization of SobMA
In addition to traditional radical polymerization techniques, SobMA was also polymerized using horseradish peroxidase (HRP)53,54 in the presence of low concentrations of hydrogen peroxide and acetylacetone as radical initiator, following the procedure described by Gross et al.55 SobMA was homo- and copolymerized with BMA at room temperature in a 1:3 water:THF mixture. The reason why THF was used instead of 2-MeTHF was the low miscibility of 2-MeTHF in water. The reaction time was 1 h for the homopolymerizations and 3 h for the copolymerizations, respectively. The monomer conversion was determined by 1H NMR and found to be above 80% for both homo- and copolymerization. After precipitation, the resulting polymers were characterized by NMR and SEC (Fig. 3b and S20–22†). The obtained polymers were of high molecular weights, >45 kg mol−1, and Đ ∼ 2.2, in good agreement as reported by Gross et al. for PMMA.
Thermal characterization of polySobMA
To assess the potential material applications of PSobMA, all polymers were analyzed by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). TGA analysis showed that all polymers undergo a three-step degradation (Fig. 4 and S23†). This multistep-degradation profile is known for polymethacrylates and many studies were conducted to investigate and simulate the thermal degradation profile of polymers like PMMA.56–58 The weight ratios between the observed degradation steps correlate to the ratio of different chain-termination mechanisms (coupling or disproportionation) and therefore slightly varies from one polymerization technique to another. The degradation occurring at the lowest temperature (165 °C) is attributed to degradation through chain-scissioning of the weak head-to-head linkages.58 The second step (around 270 °C) is referred to as β-scissioning of the unsaturated chain ends formed during disproportionation and finally, the last step is random scissioning within the remaining polymer chains. A study by Yamago et al. on the effect of reaction temperature on the ratio of combination and disproportionation, showed an increased amount of combination at higher temperature. However, the ratio became constant for polymers with Mn above 104.59 This dependency is in good agreement with our results and explains the larger weight drop for PSobMAR50 at 165 °C (reaction temperature 70 °C → increased amount of coupling), as well as the higher magnitude of the second degradation step for PSobMAE (reaction temperature r.t. → increased amount of disproportionation). The degradation profiles of the prepared statistical and block copolymers generally show a decreased magnitude of the second degradation step which is in good agreement to the degradation profiles of PMMA analyzed by Ferriol et al.58 The degradation temperatures (Td, defined at 5% weight loss) of all polymers are found in Table 1. | ||
| Fig. 4 Thermograms (TGA) for enzymatic PSoBMA and RAFT PSobMAs of different chain lengths at a heating rate of 10 °C min−1. | ||
DSC analyses (Fig. S24†) showed that all homopolymers exhibited glass transition temperatures (Tg) between 110–155 °C (see Table 1), with the maximum Tg for the polymer with Mn = 30000 g mol−1. The statistical copolymer containing MMA showed a Tg comparable to those of the homopolymers, whereas the statistical incorporation of BMA into the polymer structure resulted in a markedly decreased Tg, close to 37 °C (P(SobMAE25-st-BMAE75)). As expected, no melting transitions could be detected for any of the polymers.
Coating preparation
In PSobMA, each repeating unit contains both a tertiary alcohol and a substituted alkene. These groups render possibilities for versatile post-polymerization modifications, e.g., crosslinking to form gels or coatings (Fig. 5). Crosslinking via alkenes can be conducted using thiol–ene chemistry, employing a multi-functional thiol, even though similar cyclic alkenes are reported to react relatively slowly.60 To assess the possibility of utilizing the double bond (the “ene”) in the side-chain for crosslinking purposes, PSobMAs were mixed with trimethylolpropane tris(3-mercaptopropionate) (TMTP, a trifunctional thiol), and a radical initiator (Irgacure 651). The mixture was applied on a Teflon substrate and cured under UV-irradiation. The resulting crosslinked films were analyzed by FT Raman spectroscopy which showed reductions in the “ene”-band, compared with PSobMAR50 (CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C: 1678 cm−1, normalized to the carbonyl peak at 1727 cm−1; Fig. S25 and 26†). The successful crosslinking of PSobMAs-films was further confirmed as films were insoluble in both THF and dichloromethane, two solvents which readily dissolve PSobMAs (Fig. S27†). A control experiment was conducted to further verify the observations, the film was then irradiation in the presence of the photoinitiator but in the absence of TMTP and the resulting film was readily dissolved in both THF and dichloromethane. Further attempts to crosslink PSobMA were made via transesterifications, utilizing the tertiary alcohol rather than the alkene. PSobMAR50 and P(SobMAE25-st-BMAE75) were mixed with hexamethoxymethylmelamine (HMMM), a commonly used crosslinker in thermally curable industrial coating applications and subsequently p-toluenesulfonic acid (PTSA) dissolved in a small amount of ethyl acetate was added. The coating was applied onto a Teflon substrate and cured at 140 °C to a solid film. A slight reduction in the band corresponding to the alcohol was observed by FTIR (Fig. S28†), indicating that crosslinking had occurred. The successful crosslinking were further verified by the fact that the cured coating resins were insoluble (Fig. S29†).
C: 1678 cm−1, normalized to the carbonyl peak at 1727 cm−1; Fig. S25 and 26†). The successful crosslinking of PSobMAs-films was further confirmed as films were insoluble in both THF and dichloromethane, two solvents which readily dissolve PSobMAs (Fig. S27†). A control experiment was conducted to further verify the observations, the film was then irradiation in the presence of the photoinitiator but in the absence of TMTP and the resulting film was readily dissolved in both THF and dichloromethane. Further attempts to crosslink PSobMA were made via transesterifications, utilizing the tertiary alcohol rather than the alkene. PSobMAR50 and P(SobMAE25-st-BMAE75) were mixed with hexamethoxymethylmelamine (HMMM), a commonly used crosslinker in thermally curable industrial coating applications and subsequently p-toluenesulfonic acid (PTSA) dissolved in a small amount of ethyl acetate was added. The coating was applied onto a Teflon substrate and cured at 140 °C to a solid film. A slight reduction in the band corresponding to the alcohol was observed by FTIR (Fig. S28†), indicating that crosslinking had occurred. The successful crosslinking were further verified by the fact that the cured coating resins were insoluble (Fig. S29†).
 | ||
| Fig. 5 Crosslinking of functional side-chain groups on poly(sobreryl methacrylate) via thiol–ene chemistry (left) and transesterification (right). | ||
Conclusions
Sobrerol can be readily obtained by upgrading of α-pinene. Sobrerol can be transformed into SobMA either by classical esterification, or by enzyme catalysis, and both methods show very good selectivity for the secondary alcohol over the tertiary. SobMA can readily be polymerized by RAFT, FR, and ATRP in DMF, 2-MeTHF, and THF, respectively. In contrast to the chemical catalysis, enzyme-mediated polymerization was performed in aqueous systems at room temperature showing superior activity. Copolymers could be obtained with properties as expected from stoichiometry. The synthesized polymethacrylates showed Tgs around 150 °C and narrow dispersities in the case of RAFT and ATRP. Tailoring of Tg was demonstrated by copolymerization of methacrylate monomers. Further functionalization possibilities were demonstrated by utilizing the remaining functional groups in PSobMA; thin films were photochemically cured in the presence of multifunctional thiols or thermally cured using HMMM. Our work demonstrates the paramount potential of upcycling underutilized biobased building blocks into sustainable materials through chemo-enzymatic catalysis.Materials & methods
Materials
All chemicals were obtained from Sigma-Aldrich unless otherwise noted. Methyl methacrylate (MMA) was passed through a column with Al2O3 (activated, neutral, Brockmann Activity I) before used to remove the MEHQ inhibitor.Instrumentation
GC-MS (Shimadzu, GCMS-QP2010 Ultra) was performed on an Rxi®-5 ms (30 m, 0.25 mm [inner diameter], 0.25 μm [film thickness], RESTEK). The temperature program was set at 70 °C, before increasing to 300 °C with a rate of 20 °C min−1 and finally increased to 350 °C with a rate of 5 °C min−1 before holding at 350 °C for 10 min. For MPLC, a Biotage Isolera Four system equipped with a UV detector and Biotage KP-Sil SNAP Cartridge columns was used. 1H (400 MHz) and 13C (100 MHz) NMR spectra were recorded with a Bruker Avance AM 400 instrument. The signal of the deuterated solvent CDCl3 (δ = 7.26 ppm (ref. 61)) was used as reference. For SEC, a TOSOH EcoSEC HLC-8320GPC system was used equipped with an EcoSEC RI detector and three PSS PFG 5 μm columns (microguard, 100 Å, and 300 Å). Poly(methyl methacrylate) (PMMA) standards were used for calibration and toluene was used as internal standard. DSC was performed using a Mettler Toledo DSC 820 module. Samples (5–10 mg) were prepared in 100 μL aluminum crucibles. The samples were subjected to heating from 30 to 170 °C (or 160 °C), then cooled to −60 °C, and then heated again to 170 °C (or 160 °C) at a heating/cooling rate of 10 °C min−1 under nitrogen flow (50 mL min−1). The data obtained from the second heating was used for analyses. For TGA, a Mettler Toledo TGA/DSC1 instrument was used. Samples (5–7 μg) were prepared in 70 μL alumina crucibles and heated from 40 to 700 °C at a heating rate of 10 °C min−1 under a nitrogen flow (50 mL min−1). FTIR was performed on a PerkinElmer Spectrum 100 FT-IR instrument equipped with a single reflection ATR system and a MIR-TGS detector using a MKII Golden Gate (Graseby Specac Ltd, Kent, England). Spectra were recorded over the range 4000–600 cm−1 and based on 16 scans at an average 4.0 cm−1 resolution.Sequences and strains for producing P450-BM3/2M
The vector with P450-BM3 gene inserted into pET28a(+) plasmid was generously provided by Dr Bettina Nestl, University of Stuttgart, Germany. Site directed mutagenesis to afford the P450-BM3/A74G/L188Q (P450-BM3/2M) was carried out following the Agilent Pfu Ultra II protocol with primers listed in Table S3† with the temperature program according to Table S4.† Competent E. coli BL21(DE3) [genotype: F- ompT hsdSB(rB-, mB-) gal dcm (DE3)] were transformed with P450-BM3/2M_pET28a(+) vector by heat-shock and the cells were plated on LB-agar plate [1.6% (w/v) tryptone, 0.5% (w/v) yeast extract, 1% (w/v) NaCl, 1% (w/v) agar] supplemented with 50 mg L−1 kanamycin.Protein expression and purification
Enzyme was produced by inoculating 25 ml 2xYT media [1.6% (w/v) tryptone, 1% (w/v) yeast extract, 0.5% (w/v) NaCl] supplemented with 50 mg L−1 kanamycin (2xYT-Kan media) with a single colony from the transformed BL21(DE3) cells and incubating overnight at 37 °C, 200 rpm Overnight culture was transferred to 600 ml of 2xYT-Kan media to an estimated OD600 of 0.07. The culture was incubated at 37 °C, 170 rpm until reaching an OD600 of 0.5 at which protein expression was induced by 0.35 mM IPTG and incubating the culture at 30 °C, 170 rpm for additionally 6 h. Cells were harvested by centrifugation (4500 rpm, 4 °C) and P450-BM3/2 M was purified by utilizing the N-terminal hexa-histidine-tag as previously described.62 Cells pellets were resuspended in loading buffer (50 mM K2HPO4/KH2PO4, 800 mM NaCl, pH 7.5) supplemented with Roche cOmplete, Mini Protease Inhibitor (3 ml g−1 pellet). Cells were lysed by sonication (QSonica sonificator, Q125, 5 × [50 s, amplitude: 50%, pulse: 1 s on, 2 s off]) and cell debris was discarded by centrifugation (18.000 rpm, 4 °C for 35 min). Purification of P450-BM3/2M was performed by immobilized metal affinity chromatography (IMAC, Ni-NTA agarose beads, Qiagen). After loading the crude lysate, the column was washed with loading buffer followed by washing buffer A (50 mM K2HPO4/KH2PO4, 800 mM NaCl, pH 6.2) and washing buffer B (50 mM K2HPO4/KH2PO4, 800 mM NaCl, 250 mM glycine, pH 7.5). The enzyme was subsequently eluted by elution buffer (50 mM K2HPO4/KH2PO4, 80 mM L-histidine, pH 7.5) and the purity of fractions were analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE, 10% Mini PROTEAN TGX-gels, BioRad) (Fig. S30†). Elution fractions were pooled before concentrating and exchanging buffer to reaction buffer (50 mM K2HPO4/KH2PO4, pH 7.5) using an Amicon® Ultra 15 mL Centrifugal Filters 30 kDa cut-off. Concentration was measured by Bradford63 (Expedeon BradfordUltra kit).Biotransformation to convert α-pinene into trans-sobrerol
0.5 mL scale reactions in reaction buffer were carried out in 2 mL eppendorf tubes with (−)-α-pinene (2 mM), NADPH (0.2 mM), G6P (glucose-6-phosphate, 100 mM), G6PDH (glucose-6-phosphate dehydrogenase, 222 U mL−1), DMSO (2% v/v) and purified P450-BM3/2M (4.16 μM), catalase (578 U mL−1). Reactions were incubated at 30 °C, 500 rpm for 22 h before extracting in 2 × 0.5 mL ethyl acetate and separating organic-from aqueous phase by centrifugation (10000g, 10 min, r.t.). Organic phases were analyzed by GC-MS (Shimadzu, GCMS-QP2010 Ultra) performed on an Rxi®-5 ms column (30 m, 0.25 mm [inner diameter], 0.25 μm [film thickness], RESTEK). The temperature program was set at 70 °C, before increasing to 300 °C with a rate of 20 °C min−1 and finally increased to 350 °C with a rate of 5 °C min−1 before holding at 350 °C for 10 min.
Biotransformation to convert trans-sobrerol into sobreryl methacrylate (SobMA)
trans-Sobrerol (0.1 g, 0.58 mmol, 1 eq.) and P. fluorescens (0.13 g) were mixed and heated to 80 °C under exclusion of light. The reaction was initiated by addition of vinyl methacrylate (10–40 eq.) and proceeded for 72 h. For purification, the lipase and unreacted sobrerol were removed by filtration through a thin layer of silica. Afterwards, the filtrate was stirred at 60 °C with 100 mg of P. fluorescens and 2 ml of water for 2 h. The reaction mixture was then filtered through Celite, diluted with EtOAc, washed with sodium bicarbonate and reduced to afford SobMA (0.105 g, 0.441 mmol, 76% yield). 1H-NMR and GC-FID samples were taken, using deuterated chloroform (CDCl3) as solvent for the NMR, and ethyl acetate as solvent for the GC-FID.Products were detected by GC-MS analysis as previously described (biotransformation to convert of α-pinene into prepare trans-sobrerol). 1H NMR (400 MHz, CDCl3): δ 6.09 (d, 1H, J = 1.7 Hz), 5.72 (d, 1H, J = 4.7 Hz), 5.53 (d, 1H, J = 1.6 Hz), 5.30 (t, 1H, J = 2.1 Hz), 2.23–2.15 (m, 1H), 2.05–2.00 (m, 1H), 1.94 (s, 3H), 1.88–1.69 (m, 2H), 1.68 (s, 3H), 1.47 (dt, 1H, J = 3.9, 13.0 Hz), 1.44 (s, 1H), 1.17 (2 s, 6H); 13C NMR (100 MHz, CDCl3): δ 167.2, 136.7, 131.0, 127.8, 125.1, 71.8, 71.0, 39.6, 30.1, 27.2, 26.8 (two signals), 20.6, 18.3.
Chemical synthesis of sobreryl methacrylate (SobMA)
Methacryloyl chloride (3.05 mL, 31.5 mmol) was dissolved in DMF (30 mL) on an ice-bath and added dropwise under stirring to an ice-cold solution of trans-sobrerol (4.83 g, 28.4 mmol), triethyl amine (5.90 mL, 42.6 mmol), and DMAP (0.693 g, 5.67 mmol) in DMF (50 mL). The solution was stirred for 23 hours during which it was allowed to reach room temperature. The solution wad diluted with 200 mL ethyl acetate and washed with 2 × 200 mL HCl (aq., 1 M), 2 × 200 mL NaHCO3 (aq., 10% w/w), and 200 mL de-ionized water. The organic phase was dried with MgSO4 and filtered. The solid was rinsed with 200 mL ethyl acetate and the combined organic phases were reduced. Purification by MPLC (100% dichloromethane → dichloromethane/ethyl acetate 4:1) afforded the SobMA (4.41 g, 18.7 mmol, 66% yield). Spectroscopical data were in agreement with those for the previously synthesized SobMA.
As an alternative synthesis of SobMA, methacryloyl chloride (0.63 mL, 6.51 mmol) was dissolved in 2-MeTHF (5 mL) on an ice-bath and added dropwise under stirring to an ice-cold solution of trans-sobrerol (1.00 g, 5.89 mmol), triethyl amine (1.25 mL, 9.02 mmol), and DMAP (0.144 g, 1.18 mmol) in 2-MeTHF (30 mL). The solution was stirred for 19 hours during which it was allowed to reach room temperature. Upon work-up and purification as described above, SobMA (0.80 g, 3.36 mmol, 57% yield) was afforded. Spectroscopical data were again in agreement with those for the previously synthesized SobMA.
Free-radical polymerization of SobMA
A solution of SobMA (0.824 g, 3.46 mmol) stored in dichloromethane/ethyl acetate was reduced under vacuum to roughly 80% w/w SobMA. DMF (2.90 mL, 1.20 M) was added and the solution was reduced under vacuum overnight. The solution was transferred to a round flask equipped with a magnetic stirrer containing ACVA (0.0042 mg, 0.015 mmol). The flask was sealed with a rubber septum. The solution was bubbled with argon for 60 minutes and subjected to 4 vacuum/argon cycles. The flask was put in an oil bath pre-heated to 70 °C and stirred for five hours. The polymerization was quenched by subjecting the solution to air and the solution was then diluted with 2 mL DMF. The polymer was precipitated by dropwise addition of the solution to 400 mL diethyl ether under stirring and the suspension was then stirred overnight. The polymer was recovered by filtration, rinsed with 200 mL diethyl ether and dried under vacuum overnight.RAFT polymerizations of SobMA
Specific ratios between the reagents are described in Table 1 for each RAFT-derived polymer. A solution containing SobMA (typically 0.8–1.0 g) in dichloromethane/diethyl ether was reduced under vacuum to around 80% w/w SobMA. The solution was diluted with DMF (1.20 M with regards to SobMA) and reduced under vacuum overnight. CPAD was added and the solution transferred to a round flask with ACVA and a magnetic stirrer. The flask was sealed with a rubber septum and the solution bubbled with argon for 60 minutes. After being subjected to 4 vacuum/argon cycles, the flask was put on an oil bath pre-heated to 70 °C. After stirring for 8 hours, the polymerization was quenched exposure to air and then diluted with 2 mL DMF. The polymers were precipitated by dropwise addition of the solution to 400 mL diethyl ether under stirring. The solution was stirred overnight, filtered, and the recovered polymers rinsed with 200 mL diethyl ether. The polymers were then dried under vacuum overnight.Co-polymers were synthesized in a similar fashion. For the statistical co-polymers, the co-monomers were bubbled with argon for 30 minutes in a separate round flask and added via a syringe under argon atmosphere to the SobMA-containing solution just before the vacuum/argon cycles. Statistical co-polymers containing MMA were precipitated twice in diethyl ether. Statistical co-polymers containing BMA were precipitated twice in heptane/diethyl ether 3:1. The block co-polymers were prepared by bubbling the co-monomers with argon for 30 minutes. The co-monomers were transferred via a syringe under argon atmosphere to the polymerization solution containing SobMA after 6 hours of polymerization and the polymerization then proceeded for another 18 hours. The block co-polymer containing MMA was obtained after precipitation in heptane and then re-precipitation in diethyl ether from dichloromethane. The block co-polymer containing BMA was precipitated twice in heptane/diethyl ether 3:1.
ATRP polymerizations of SobMA
Specific ratios between the reagents are described in Table 1 for each ATRP-derived polymer. Acetone was dried for 6 h on molecular sieves (3 Å) and distilled. A solution containing SobMA (typically 0.8–1.0 g) in dichloromethane/diethyl ether was carefully reduced under vacuum and transferred in a 5 mL round bottom flask. The flask, equipped with a magnetic stirrer, was sealed with a septum and degassed. Dry acetone was added (70 wt% with regards to acetone) and the solution was cooled to 0 °C. HMTETA and EBrP were added and the solution and degassed by applying three vacuum–argon cycles. The flask was then quickly opened to add Cu(I)Cl, whereafter it was sealed and subjected to three additional vacuum–argon cycles. The flask was immersed in an oil bath preheated to 50 °C to start the polymerization. After the reaction, the polymerization mixture was quenched exposure to air and then diluted with 2 mL acetone. The polymers were obtained after two precipitations in water and one precipitation in diethyl ether (250 mL each).Enzymatic polymerization
The specific ratios for homo- and copolymers are adopted from A. Gross55 and listed in Table 1. For the synthesis of PSobMA, the mixture of SobMA and ethyl acetate was dilutet in THF and reduced. This was repeated until the EtOAc was completely removed. The mixture SobMA (2.94 mmol) and THF was then added under Argon to small roundbottom flask and diluted with THF and deionized water to reach a final concentration of 3 M (in 25/75 THF/H2O). Hydrogen peroxide (0.045 mmol), and 2,4-pentanedione (0.068 mmol) were added and the reaction was initiated by the addition of 0.11 ml HRP (80 mg mL−1, 8.4 mg of enzyme). The reaction was stirred at r.t. until the reaction mixture was to viscous to be stirred. Then, the reaction mixture was slightly diluted with THF and precipitated in a large excess of diethyl ether (or chilled MeOH for BMA copolymers). The precipitate was filtered and dryd under vacuum.Preparation of PSobMA-based coatings
Coatings cross-linked via thiol–ene chemistry were prepared by dissolving PSobMAR50 (0.100 g) and TMTP (0.046 mL, 0.140 mmol, thiol/ene = 1:1) in THF (0.200 mL, containing 5 mg mL−1 Irgacure 651). The solution was applied to a glass substrate using a 150 μm applicator. Upon drying in a fume hood for 10 minutes, the coating was cured via UV-irradiation (10 × 5 s., total dose = 28.8 J cm−2).
Coatings cross-linked via transesterification were prepared by dissolving PSobMAR50 (0.100 g), p-toluenesulfonic acid (PTSA, 1 μl) and HMMM (0.022 g) in THF (0.200 mL). The solution was applied to a glass substrate using a 150 μm applicator and dried in a fume hood for 10 minutes. The coating was cured in an oven at 140 °C for 20 minutes.
Conflicts of interest
There are no conflicts to declare.References
- Chatain Babtiste, Plastic Oceans: MEPs back EU ban on throwaway plastics by 2021 | News | European Parliament. 24-10-2018, 2018. Available at: http://www.europarl.europa.eu/news/en/press-room/20181018IPR16524/plastic-oceans-meps-back-eu-ban-on-throwaway-plastics-by-2021, (Accessed: 5th February 2019).
- Market – European Bioplastics e.V., 2018. Available at: https://www.european-bioplastics.org/market/, (Accessed: 18th February 2019).
- A. Gandini and T. M. Lacerda, From monomers to polymers from renewable resources: Recent advances, Prog. Polym. Sci., 2015, 48, 1–39 CrossRef CAS.
- M. Brodin, M. Vallejos, M. T. Opedal, M. C. Area and G. Chinga-Carrasco, Lignocellulosics as sustainable resources for production of bioplastics – A review, J. Cleaner Prod., 2017, 162, 646–664 CrossRef CAS.
- S. L. Kristufek, K. T. Wacker, Y.-Y. T. Tsao, L. Su, K. L. Wooley, K. L. Wooley, K. L. Wooley, C. Boyer, T. P. Davis, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Monomer design strategies to create natural product-based polymer materials, Nat. Prod. Rep., 2017, 34, 433–459 RSC.
- Y. Zhu, C. Romain and C. K. Williams, Sustainable polymers from renewable resources, Nature, 2016, 540, 354–362 Search PubMed.
- S. Fernando, S. Adhikari, C. Chandrapal and N. Murali, Biorefineries: Current status, challenges, and future direction, Energy Fuels, 2006, 20, 1727–1737 CrossRef CAS.
- E. Oldfield and F.-Y. Lin, Terpene Biosynthesis: Modularity Rules, Angew. Chem., Int. Ed., 2012, 51, 1124–1137 CrossRef CAS PubMed.
- X. Chen, T. G. Köllner, Q. Jia, A. Norris, B. Santhanam, P. Rabe, J. S. Dickschat, G. Shaulsky, J. Gershenzon and F. Chen, Terpene synthase genes in eukaryotes beyond plants and fungi: Occurrence in social amoebae, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12132–12137 CrossRef CAS PubMed.
- M. Schalk, L. Pastore, M. A. Mirata, S. Khim, M. Schouwey, F. Deguerry, V. Pineda, L. Rocci and L. Daviet, Toward a biosynthetic route to sclareol and amber odorants, J. Am. Chem. Soc., 2012, 134, 18900–18903 CrossRef CAS PubMed.
- P. P. Peralta-Yahya, F. Zhang, S. B. del Cardayre and J. D. Keasling, Microbial engineering for the production of advanced biofuels, Nature, 2012, 488, 320–328 Search PubMed.
- P. A. Wilbon, F. Chu and C. Tang, Progress in renewable polymers from natural terpenes, terpenoids, and rosin, Macromol. Rapid Commun., 2013, 34, 8–37 Search PubMed.
- Z. Liu, T. Zhang, W. Zeng, H. Zhu and X. An, Cationic polymerization of alpha-pinene using Keggin silicotungstic acid as a homogeneous catalyst, React. Kinet., Mech. Catal., 2011, 104, 125–137 Search PubMed.
- T. Higashimura and Y. Deng, Cationic polymerization of α-pinene with the binary catalyst AIC13/SbC13, Makromol. Chem., 1992, 2321, 2311–2321 Search PubMed.
- S. W. Liu, C. X. Xie, S. T. Yu and F. S. Liu, Polymerization of alpha-pinene using Lewis acidic ionic liquid as catalyst, Catal. Commun., 2009, 10, 986–988 Search PubMed.
- H. Miyaji, K. Satoh and M. Kamigaito, Bio-Based Polyketones by Selective Ring-Opening Radical Polymerization of α-Pinene-Derived Pinocarvone, Angew. Chem., Int. Ed., 2016, 55, 1372–1376 Search PubMed.
- M. Winnacker and J. Sag, Sustainable terpene-based polyamides via anionic polymerization of a pinene-derived lactam, Chem. Commun., 2018, 54, 841 RSC.
- A. Stamm, A. Biundo, B. Schmidt, J. Brücher, S. Lundmark, P. Olsén, L. Fogelström, E. Malmström, U. T. Bornscheuer and P.-O. Syrén, A retrobiosynthesis-based route to generate pinene-derived polyesters, ChemBioChem, 2019 DOI:10.1002/cbic.201900046.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Engineering the third wave of biocatalysis, Nature, 2012, 485, 185–194 Search PubMed.
- J. C. Lewis, P. S. Coelho and F. H. Arnold, Enzymatic functionalization of carbon-hydrogen bonds, Chem. Soc. Rev., 2011, 40, 2003–2021 RSC.
- H. N. Bhatti, S. S. Khan, A. Khan, M. Rani, V. U. Ahmad and M. I. Choudhary, Biotransformation of monoterpenoids and their antimicrobial activities, Phytomedicine, 2014, 21, 1597–1626 CrossRef CAS PubMed.
- A. E. Enciso, L. Fu, S. Lathwal, M. Olszewski, Z. Wang, S. R. Das, A. J. Russell and K. Matyjaszewski, Biocatalytic “Oxygen-Fueled” Atom Transfer Radical Polymerization, Angew. Chem., Int. Ed., 2018, 57, 16157–16161 CrossRef CAS PubMed.
- M. Daoud Attieh, Y. Zhao, A. Elkak, A. Falcimaigne-Cordin and K. Haupt, Enzyme-Initiated Free-Radical Polymerization of Molecularly Imprinted Polymer Nanogels on a Solid Phase with an Immobilized Radical Source, Angew. Chem., Int. Ed., 2017, 3339–3343, DOI:10.1002/anie.201612667.
- S. Roth, I. Funk, M. Hofer and V. Sieber, Chemoenzymatic Synthesis of a Novel Borneol-Based Polyester, ChemSusChem, 2017, 10, 3574–3580 CrossRef CAS PubMed.
- Z. Liu, Y. Lv and Z. An, Enzymatic Cascade Catalysis for the Synthesis of Multiblock and Ultrahigh-Molecular-Weight Polymers with Oxygen Tolerance, Angew. Chem., Int. Ed., 2017, 56, 13852–13856 CrossRef CAS PubMed.
- A. Pellis, E. Herrero Acero, V. Ferrario, D. Ribitsch, G. M. Guebitz and L. Gardossi, The Closure of the Cycle: Enzymatic Synthesis and Functionalization of Bio-Based Polyesters, Trends Biotechnol., 2016, 34, 316–328 CrossRef CAS PubMed.
- M. F. Sainz, J. A. Souto, D. Regentova, M. K. G. Johansson, S. T. Timhagen, D. J. Irvine, P. Buijsen, C. E. Koning, R. A. Stockman and S. M. Howdle, A facile and green route to terpene derived acrylate and methacrylate monomers and simple free radical polymerisation to yield new renewable polymers and coatings, Polym. Chem., 2016, 7, 2882–2887 Search PubMed.
- Y. Zhu, C. Romain and C. K. Williams, Sustainable polymers from renewable resources, Nature, 2016, 540, 354–362 Search PubMed.
- G. G. Henderson and W. J. S. Eastburn, The conversion of pinene into sobrerol, J. Chem. Soc. Trans., 1909, 95, 1465–1466 RSC.
- A. Yuta and J. N. Baraniuk, Therapeutic approaches to mucus hypersecretion, Curr. Allergy Asthma Rep., 2005, 5, 243–251 CrossRef CAS PubMed.
- M. R. V. Santos, F. V. Moreira, B. P. Fraga, D. P. Souza, L. R. de Bonjardim and L. J. Quintans-Junior, Cardiovascular effects of monoterpenes: a review, Rev. Bras. Farmacogn., 2011, 21, 764–771 CrossRef CAS.
- P. L. Crowell, Monoterpenes in breast cancer chemoprevention, Breast Cancer Res. Treat., 1997, 46, 191–197 CrossRef CAS.
- M. S. Lima, C. S. M. F. Costa, J. F. J. Coelho, A. C. Fonseca and A. C. Serra, A simple strategy toward the substitution of styrene by sobrerol-based monomers in unsaturated polyester resins, Green Chem., 2018, 20, 4880–4890 RSC.
- O. Hauenstein, S. Agarwal and A. Greiner, Bio-based polycarbonate as synthetic toolbox, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- T. Stößer, C. Li, J. Unruangsri, P. K. Saini, R. J. Sablong, M. A. R. Meier, C. K. Williams and C. Koning, Bio-derived polymers for coating applications: comparing poly(limonene carbonate) and poly(cyclohexadiene carbonate), Polym. Chem., 2017, 8, 6099–6105 RSC.
- F. Auriemma, C. De Rosa, M. R. Di Caprio, R. Di Girolamo, W. C. Ellis and G. W. Coates, Stereocomplexed poly(limonene carbonate): A unique example of the cocrystallization of amorphous enantiomeric polymers, Angew. Chem., Int. Ed., 2015, 54, 1215–1218 CrossRef CAS.
- H. J. Park, C. Y. Ryu and J. V. Crivello, Photoinitiated cationic polymerization of limonene 1,2-oxide and alfa-pinene oxide, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 109–117 CrossRef CAS.
- M. Firdaus, L. Montero De Espinosa and M. A. R. Meier, Terpene-based renewable monomers and polymers via thiol-ene additions, Macromolecules, 2011, 44, 7253–7262 CrossRef CAS.
- J. Ackermann, M. Juda and D. Hirsch, Polymethyl Methacrylate (PMMA), Kunstst. Int., 2013, 95–100 Search PubMed.
- Grand View Research, Polymethyl Methacrylate Market Analysis And Segment Forecast To 2025, Report ID: 978-1-68038-056-9 (3AD). Available at: https://www.grandviewresearch.com/industry-analysis/polymethyl-methacrylate-pmma-industry, (Accessed: 17th April 2019).
- J. Andraos, Complete Green Metrics Evaluation of Various Routes to Methyl Methacrylate According to Material and Energy Consumptions and Environmental and Safety Impacts: Test Case from the Chemical Industry, ACS Sustainable Chem. Eng., 2016, 4, 312–323 CrossRef CAS.
- U. Ali, K. J. B. Abd Karim and N. A. Buang, A Review of the Properties and Applications of Poly (Methyl Methacrylate) (PMMA), Polym. Rev., 2015, 55, 678–705 CrossRef CAS.
- S. Janocha, D. Schmitz and R. Bernhardt, Terpene Hydroxylation with Microbial Cytochrome P450 Monooxygenases, in Biotechnology of Isoprenoids, ed. J. Schrader and J. Bohlmann, Springer International Publishing, 2015, pp. 215–250 Search PubMed.
- V. B. Urlacher and S. Eiben, Cytochrome P450 monooxygenases: perspectives for synthetic application, Trends Biotechnol., 2006, 24, 324–330 CrossRef CAS.
- V. B. Urlacher, S. Lutz-Wahl and R. D. Schmid, Microbial P450 enzymes in biotechnology, Appl. Microbiol. Biotechnol., 2004, 64, 317–325 CrossRef CAS.
- A. Hernandez-Ortega, M. Vinaixa, Z. Zebec, E. Takano and N. S. Scrutton, A Toolbox for Diverse Oxyfunctionalisation of Monoterpenes, Sci. Rep., 2018, 8, 14396 CrossRef PubMed.
- Z. B. Xu and J. Qu, Hot water-promoted SN1 solvolysis reactions of allylic and benzylic alcohols, Chem. – Eur. J., 2013, 19, 314–323 CrossRef CAS.
- Y. Zhao, Y. Wu, P. D. Clercq, M. Vandewalle and P. n. Maillos, Tetrahedron, 2000, 11, 3887–3900 CrossRef CAS.
- T. Nishii, T. Ouchi, A. Matsuda, Y. Matsubara, Y. Haraguchi, T. Kawano, H. Kaku, M. Horikawa and T. Tsunoda, Modified Markó's aerobic oxidation of alcohols under atmospheric pressure with air or molecular oxygen at room temperature, Tetrahedron Lett., 2012, 53, 5880–5882 CrossRef CAS.
- V. Pace, P. Hoyos, L. Castoldi, P. Domínguez De María and A. R. Alcántara, 2-Methyltetrahydrofuran (2-MeTHF): A biomass-derived solvent with broad application in organic chemistry, ChemSusChem, 2012, 5, 1369–1379 CrossRef CAS.
- V. Antonucci, J. Coleman, J. B. Ferry, N. Johnson, M. Mathe, J. P. Scott and J. Xu, Toxicological assessment of 2-methyltetrahydrofuran and cyclopentyl methyl ether in support of their use in pharmaceutical chemical process development, Org. Process Res. Dev., 2011, 15, 939–941 CrossRef CAS.
- Q. Wang, Y. Li and Q. Chen, A convenient, large scale synthesis of trans-(+)-sobrerol, Synth. Commun., 2003, 33, 2125–2134 CrossRef CAS.
- A. Singh, D. Ma and D. L. Kaplan, Enzyme-mediated free radical polymerization of styrene, Biomacromolecules, 2000, 1, 592–596 CrossRef CAS PubMed.
- S. Kobayashi, Enzymatic polymerization: A new method of polymer synthesis, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 3041–3056 CrossRef CAS.
- B. Kalra and R. A. Gross, Horseradish peroxidase mediated free radical polymerization of methyl methacrylate, Biomacromolecules, 2000, 1, 501–505 CrossRef CAS.
- T. Kashiwagi, J. E. Brown, A. Inaba, K. Hatada, T. Kitayama and E. Masuda, Effects of Weak Linkages on the Thermal and Oxidative Degradation of Poly(methyl methacrylates), Macromolecules, 1986, 19, 2160–2168 CrossRef CAS.
- L. E. Manring, Thermal degradation of poly (methyl methacrylate). 2. Vinyl-terminated polymer, Macromolecules, 1989, 22, 2673–2677 CrossRef CAS.
- M. Ferriol, A. Gentilhomme, M. Cochez, N. Oget and J. L. Mieloszynski, Thermal degradation of poly(methyl methacrylate) (PMMA): Modelling of DTG and TG curves, Polym. Degrad. Stab., 2003, 79, 271–281 CrossRef CAS.
- Y. Nakamura and S. Yamago, Termination Mechanism in the Radical Polymerization of Methyl Methacrylate and Styrene Determined by the Reaction of Structurally Well-Defined Polymer End Radicals, Macromolecules, 2015, 48, 6450–6456 CrossRef CAS.
- M. Claudino, M. Jonsson and M. Johansson, Thiol–ene coupling kinetics of d-limonene: a versatile ‘non-click’ free-radical reaction involving a natural terpene, RSC Adv., 2013, 3, 11021 RSC.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, NMR chemical shifts of trace impurities: Common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- S. C. Maurer, H. Schulze, R. D. Schmid and V. Urlacher, Immobilisation of P450BM-3 and an NADP(+) cofactor recycling system: Towards a technical application of heme-containing monooxygenases in fine chemical synthesis, Adv. Synth. Catal., 2003, 345, 802–810 CrossRef CAS.
- M. M. Bradford, A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc00718k |
| This journal is © The Royal Society of Chemistry 2019 |