DOI:
10.1039/C9GC00448C
(Paper)
Green Chem., 2019,
21, 2035-2042
Functionalised tetrahydrofuran fragments from carbohydrates or sugar beet pulp biomass†
Received
4th February 2019
, Accepted 13th March 2019
First published on 14th March 2019
Abstract
Carbohydrate biomass represents a potentially valuable sustainable source of raw materials for chemical synthesis, but for many applications, selective deoxygenation/dehydration of the sugars present is necessary to access compounds with useful chemical and physical properties. Selective dehydration of pentose sugars to give tetrahydrofurans can be achieved by treatment of the corresponding N,N-dimethylhydrazones under acidic or basic conditions, with the two approaches showing complementary stereoselectivity. The dehydration process is readily scalable and the THF hydrazones derived from arabinose, ribose, xylose and rhamnose were converted into a range of useful fragments containing primary alcohol, ketone, carboxylic acid or amine functional groups. These compounds have potentially useful physiochemical properties making them suitable for incorporation into fragment/lead generation libraries for medicinal chemistry. It was also shown that L-arabinose hydrazone could be obtained selectively from a crude sample of hydrolysed sugar beet pulp.
Introduction
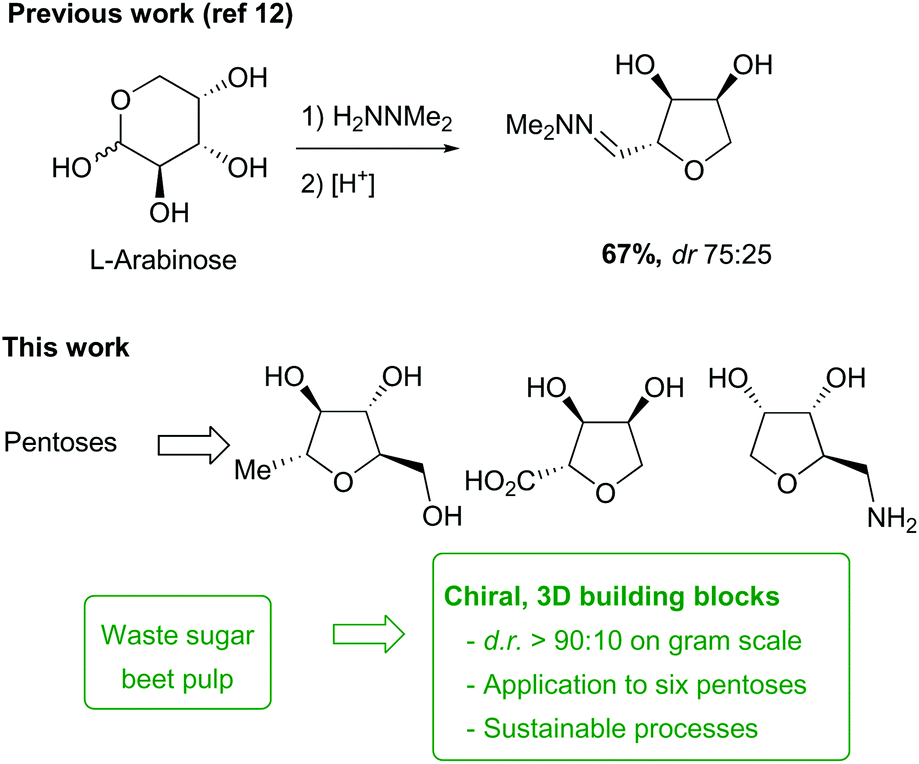
The production of chemicals for medicinal chemistry relies heavily on hydrocarbons from petrochemical sources to provide small molecule synthons. Considering the inevitable exhaustion of fossil fuels and the increased restriction of their use due to concerns relating to climate change, the key to future chemical processes will be the exploitation of renewable resources from plant biomass.1 An important sustainable feedstock is sugar beet, with a production cycle from seeds-to-seeds of only two years. Cultivated predominantly in temperate regions for the production of sucrose, the processing of the beet also generates large quantities of pulp, which is considered as a low-value waste product and is usually used as animal feed.2,3 This material is highly enriched in carbohydrates, with the pulp being composed of polymeric glucose (cellulose) and pectin (a co-polymer of hexoses and pentoses). The latter fraction contains a high proportion of L-arabinose as well as other sugars, which could potentially be exploited as valuable chemical feedstocks, but processing is required to release the monomeric sugars, and strategies for this have been reported.4,5
A variety of transformations are now available to produce small molecule building blocks from sugar monomers.4,5 However, with the exception of some partial reduction methods,6–8 carbohydrate biomass treatment often results in the loss of functional groups and chiral information, and it remains very challenging to selectively remove one or more hydroxyl groups from a polyol, without resorting to extensive protecting group manipulations.9,10 For this reason, the range of more complex targets retaining stereochemical information from biomass sugars prepared via succinct methods avoiding extensive protection–deprotection steps has remained limited.
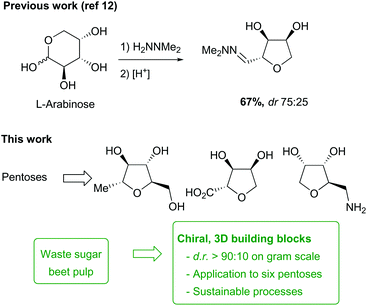
Recently, some approaches have been developed which take advantage of the intrinsic functionalities and chirality of carbohydrates to develop novel methodology to access small 3D-building-blocks.11,12 Indeed, we have recently developed a selective method for the dehydration of pentose sugars to afford hydroxy-functionalised THFs without the need for protecting groups (Scheme 1).12 In this paper, we describe the optimisation of this chemistry on a multigram scale, the development of a novel base-mediated cyclic dehydration of sugar hydrazones to access other stereoisomers, and the extension of this methodology to a range of pentoses. We also demonstrate the application of these dehydration reactions to the construction of a small library of chiral THF fragments as useful building blocks for medicinal chemistry,13,14via routes that offer selectivity, scalability, and sustainability. In addition, the direct synthesis of one of our key starting materials directly from sugar beet pulp is described, to demonstrate the feasibility of ‘up-grading’ waste from biomass into complex chiral building blocks.
 |
| | Scheme 1 Dehydration of pentose sugars for the synthesis of chiral building-blocks. | |
Results and discussion
Synthesis of chiral THFs from pentose sugars
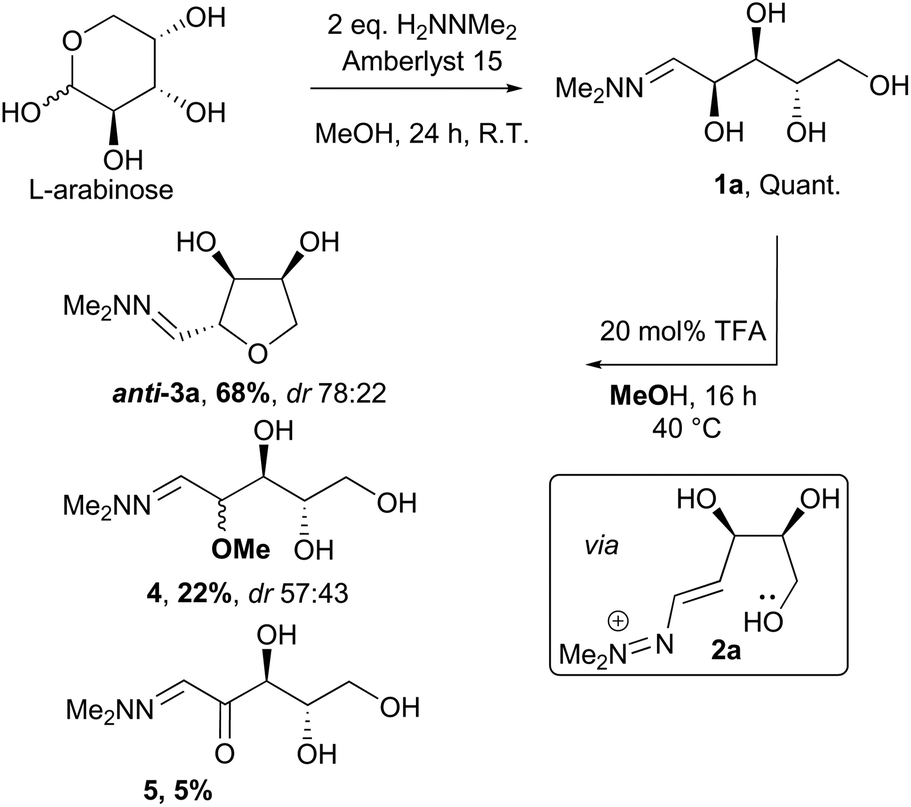
The conversion of L-arabinose into 1a–3a was conducted following the procedure described in our preliminary communication for the acid catalysed dehydration of pentoses.12 Hydrazone 1a was obtained quantitatively under acidic conditions and subsequently cyclised using catalytic TFA (Scheme 2). In our previous studies, this reaction had yielded the THF in only moderate yield (67%), but the fate of the remaining material was unclear. However, when the cyclisation was performed on a larger scale (5–20 g), formation of two side-products 4 and 5 in 22% and 5% yields respectively was observed. The formation of methyl ether 4 could be rationalised by the reaction of the vinyldiazenium intermediate 2a with methanol. The formation of ketone 5 most likely results from the aerobic oxidation of 1a at the C-2 position.
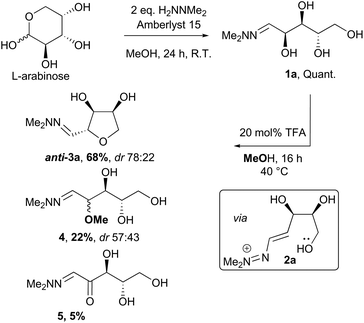 |
| | Scheme 2 Selective dehydration of L-arabinose and side-products formed. | |
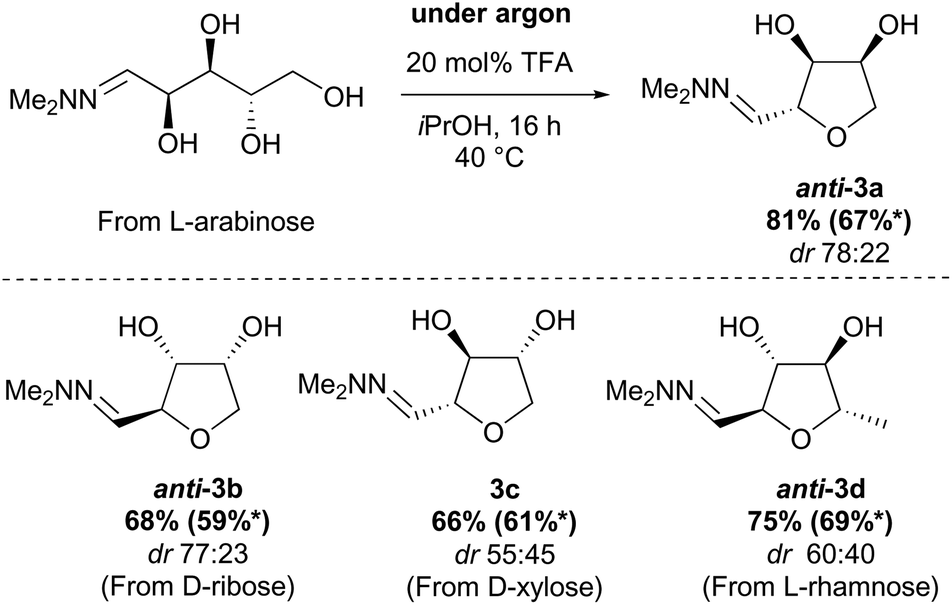
These issues were readily solved by running the cyclisation in a less nucleophilic solvent (isopropanol) and under an argon atmosphere (see ESI†). Under these conditions, 3a was obtained in 81% yield routinely on a 20 g scale. When these reaction conditions were applied to other pentose sugars, the yields were generally superior to the previously reported procedure, with no significant difference in the diastereoselectivity observed (Scheme 3). THFs anti-3a and anti-3b derived from L-arabinose and D-ribose could be recrystallised from THF/Petrol to give the pure anti-isomer in each case.
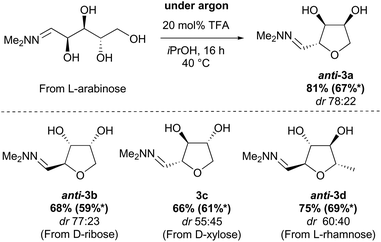 |
| | Scheme 3 Optimised synthesis of chiral THFs anti-3a–anti-3d. *Yield obtained under previous conditions.12 | |
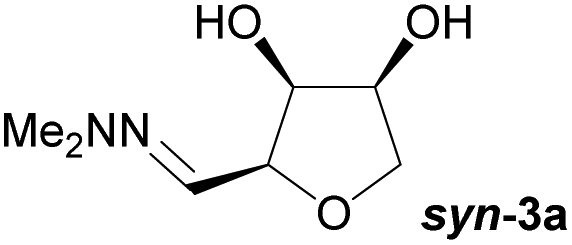
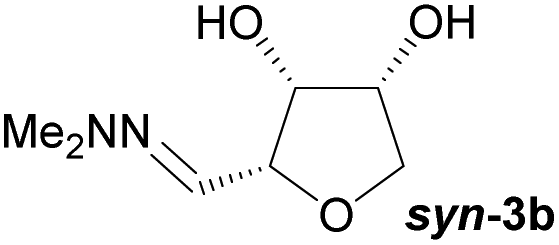
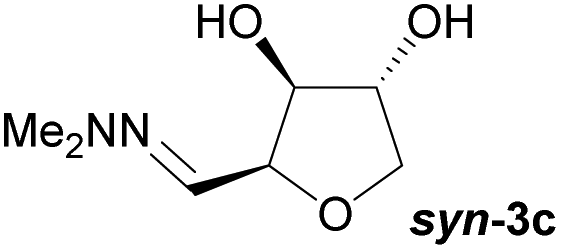
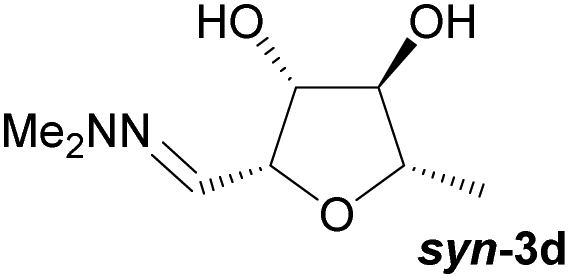
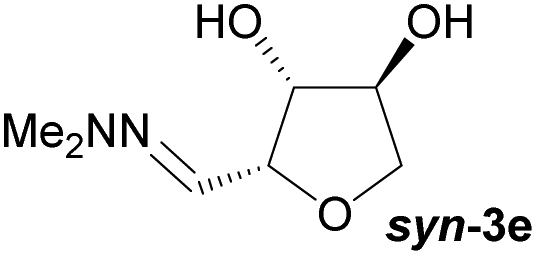
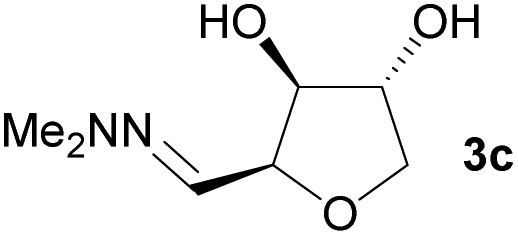
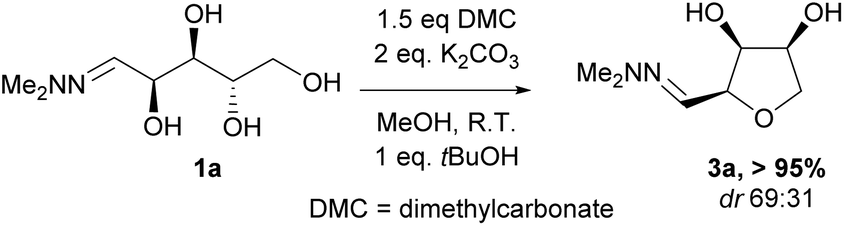
Investigations also established that the cyclisation of the sugar hydrazones could be achieved to give THFs under basic conditions, by activating the sugar-hydrazone 1a with dimethylcarbonate (DMC).15 The reaction occured at room temperature and afforded the chiral THF syn-3a from arabinose in excellent yield, with good diastereoselectivity. Importantly, this provides a complementary approach to the acid-catalysed cyclisation in terms of the THF isomer produced. These conditions were also applied successfully to other pentoses, with the major THF isomer in each case having syn-stereochemistry at the THF carbons C-1 and C-2 (Table 1).
Table 1 Synthesis of chiral THFs syn-3a to syn-3e under basic conditions
Full conversions were obtained after 4 hours and no change in the diastereomeric ratio was noted during the course of the cyclisation reaction. The presence of dimethylcarbonate and a stoichiometric amount of base were essential for the reaction to proceed in high yield. When a purified sample of the THF anti-3a was submitted to the DMC/K2CO3 reaction conditions, no epimerisation was observed after 24 hours suggesting that there was no interconversion of the two isomers and that the two products anti-3a and syn-3a were likely formed via two different reaction pathways under the basic conditions. This is in stark contrast to the acidic cyclisation conditions under which the two products can rapidly interconvert, leading to production of a thermodynamic mixture of products.
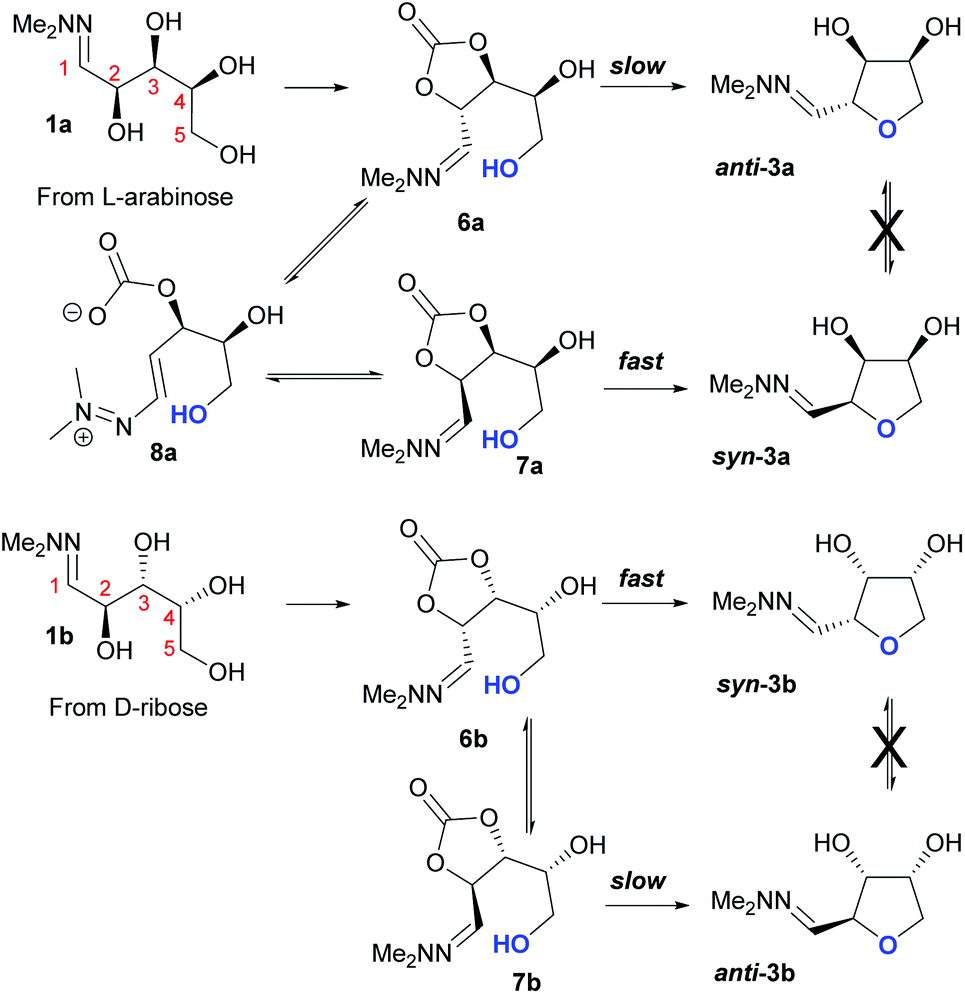
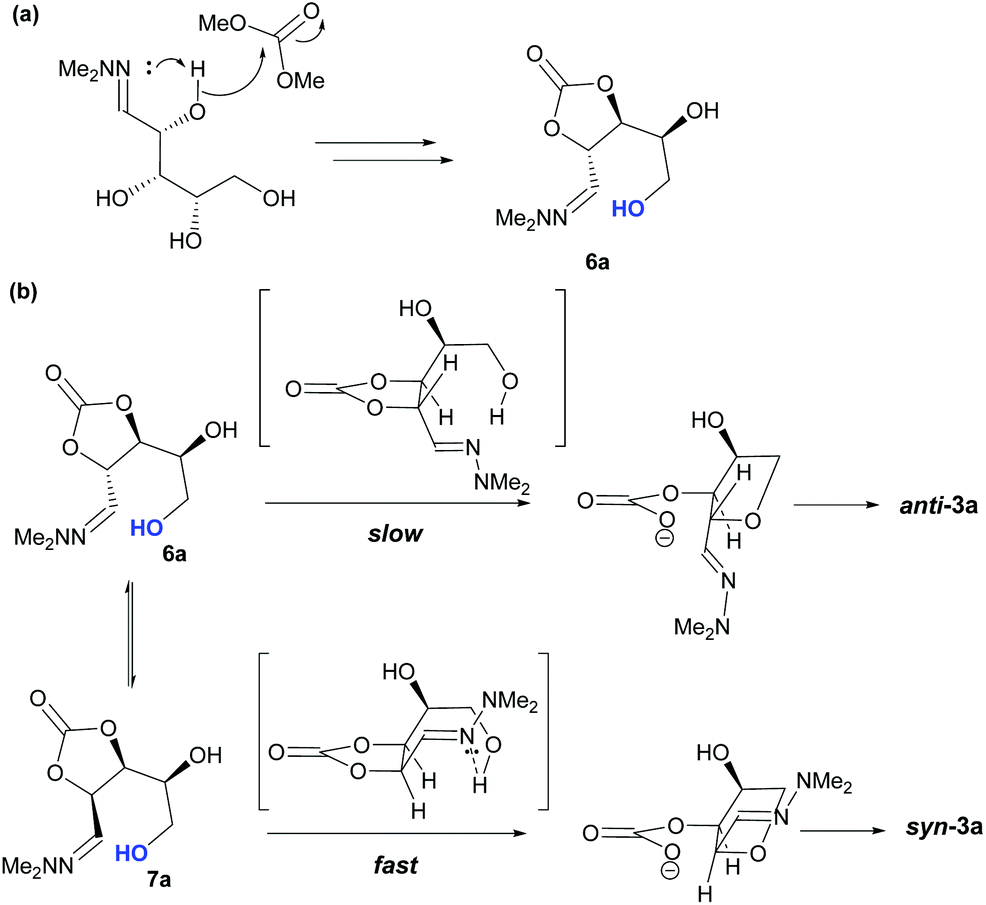
Taking this into account, along with the observed diastereomeric ratios of the products obtained from L-arabinose, D-ribose, D-xylose and D-lyxose it is evident that epimerisation of the C-2 stereocentre must take place prior to irreversible cyclisation, with the observed products resulting from the relative rates of cyclisation of the two diastereomers. Thus, arabinose hydrazone 1a must first be converted into cyclic carbonate 6a (Scheme 4), which can only cyclise relatively slowly to give anti-3a but can undergo epimerisation to 7a which cyclises more rapidly to give syn-3a. Epimerisation may take place via ring opening of the cyclic carbonate to generate zwitterion 8a. In the case of D-ribose hydrazone 1b, the initially formed carbonate 6b (the enantiomer of 7a) cyclises relatively rapidly so less epimerisation to 7b (and subsequent cyclisation to anti-3b) takes place leading to a slightly higher diastereoselectivity in this reaction in comparison to the formation of syn-3a. A similar rationale can be used to explain the observed diastereoselectivities resulting from cyclisation of D-xylose hydrazone 1c and D-lyxose hydrazone 1f. The selective formation of the cyclic carbonate at C-2/C-3 could perhaps be explained by involvement of the hydrazone group as a general base that activates the C-2 alcohol towards reaction with dimethyl carbonate (Scheme 5a). Furthermore, general base assistance could also be invoked in the cyclisation reaction (Scheme 5b), as in each case the cyclisation reaction which proceeds fastest involves reaction of an intermediate in which the hydrazone and nucleophilic primary alcohol are arranged on the same face of the 5-membered carbonate ring. Cyclisation with inversion at C-2 then leads to the THF ring in which the C-3 alcohol and the hydrazone are on the same face of the newly formed ring.
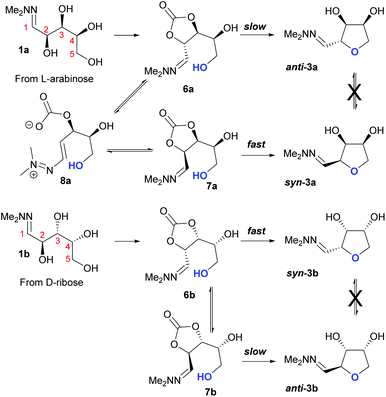 |
| | Scheme 4 Proposed mechanism for the basic cyclisation of L-arabinose hydrazone 1a and D-ribose hydrazone 1b. | |
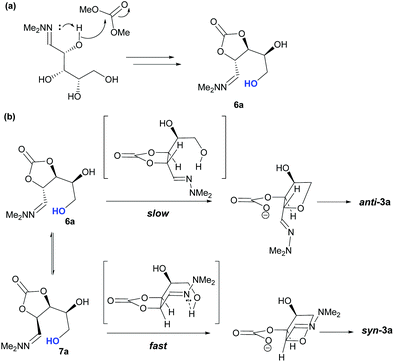 |
| | Scheme 5 Base mediated (a) cyclic carbonate 6a formation, (b) cyclisation of 7a. | |
Chiral functionalised THFs as 3D fragments for medicinal chemistry
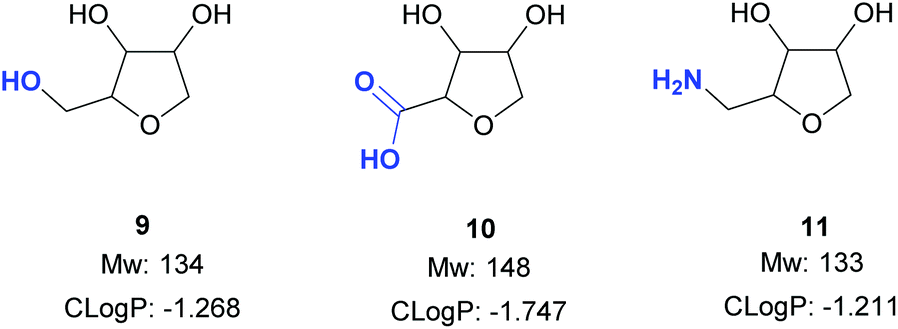
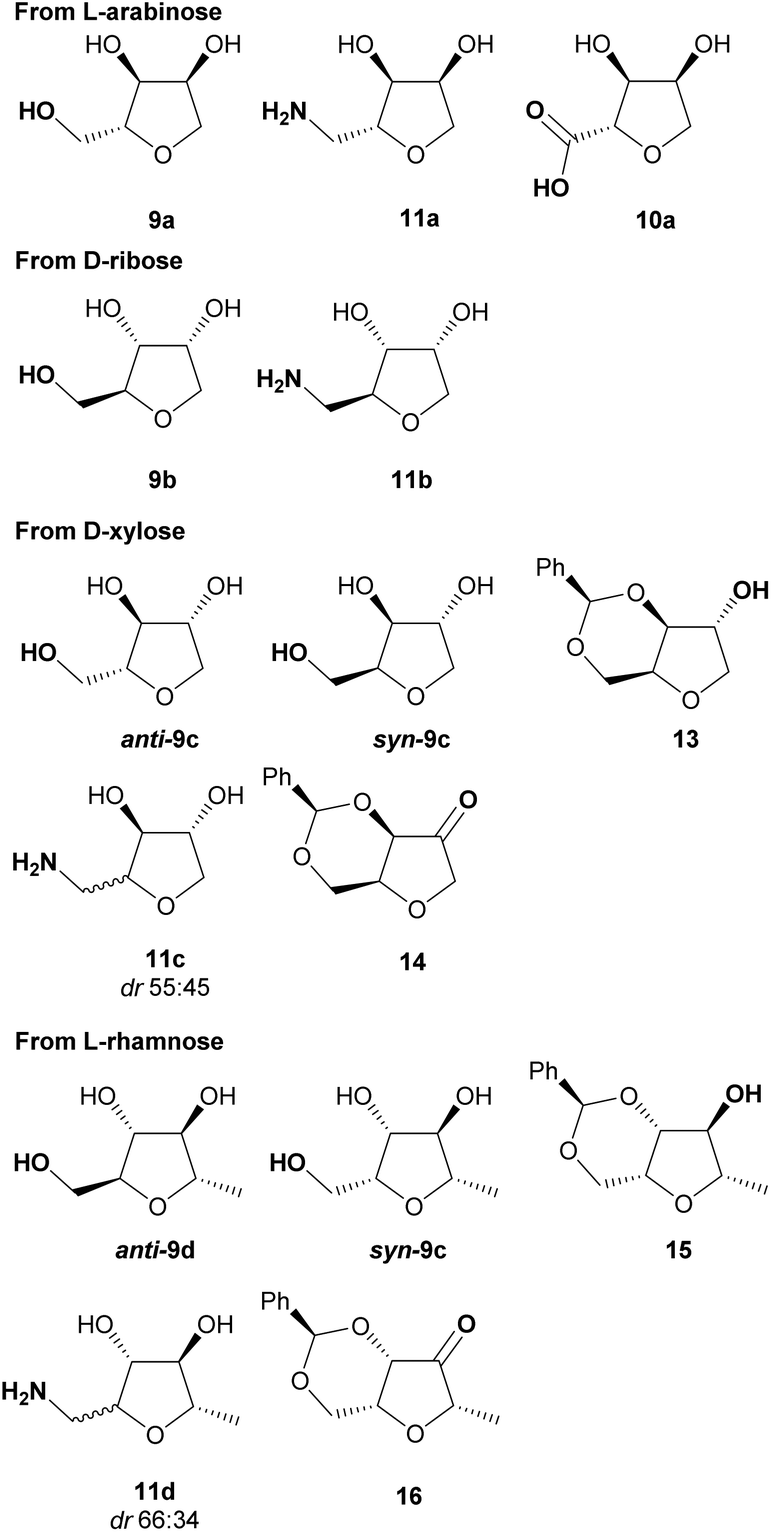
The next objective of this study was to synthesise carefully chosen targets for applications in medicinal chemistry, which contained suitable functional groups for derivatisation in order to prepare potential fragments and/or intermediates for incorporation into lead-like molecules. Three families of compounds were selected: (i) THF-triols 9, (ii) THF-carboxylic acids 10 and (iii) THF-amines 11 (Fig. 1). It was envisioned that these three compound classes would be ideal for generating libraries for fragment-based lead discovery as they are of low molecular weight (Mw < 150 g mol−1), are highly soluble in water, and contain a functional group ready for further elaboration (primary alcohol, carboxylic acid, primary amine). In addition, the targets could be used as strategic reagents16 for the final derivatisation of core molecules so as to dramatically lower log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D by incorporation of the compact polyol moiety. Finally, by using different pentose sugars, we aimed to prepare a range of different stereoisomers, each with a diastereomeric ratio >90:10, suitable for use in these applications.17
D by incorporation of the compact polyol moiety. Finally, by using different pentose sugars, we aimed to prepare a range of different stereoisomers, each with a diastereomeric ratio >90:10, suitable for use in these applications.17
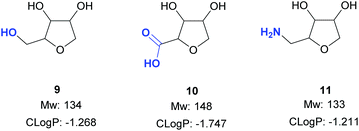 |
| | Fig. 1 Selected targets and their calculated properties. | |
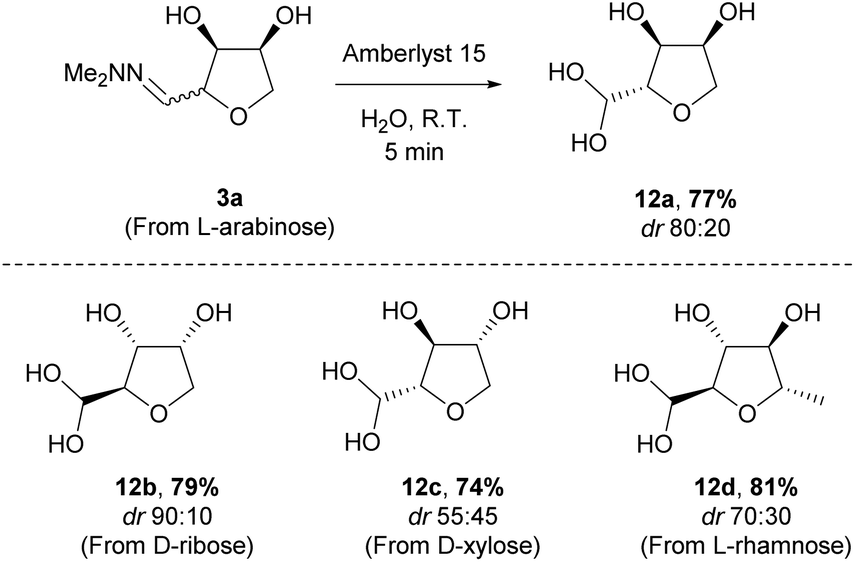
The THF-hydrazone 3 is a versatile platform for accessing several functionalities (e.g. aldehyde, alcohol, nitrile, protected-amine, alkene).12 As previously reported the hydrazone can be rapidly hydrolysed to give the aldehyde hydrate 12 using an acidic resin (Amberlyst 15). The reaction was easily scaled-up (>10 g) and extended to the other pentoses (Scheme 6). The reaction could safely be conducted on the mixture of diastereomers obtained after cyclisation, as epimerisation takes place under the acidic hydrolysis conditions. Remarkably, in the case of L-rhamnose the hydrate was obtained with a higher diastereoselectivity than the corresponding hydrazone.
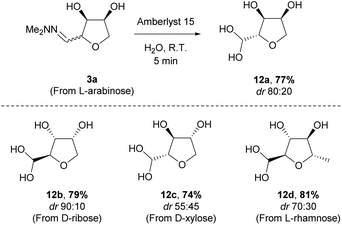 |
| | Scheme 6 Synthesis of THF-hydrates 12. | |
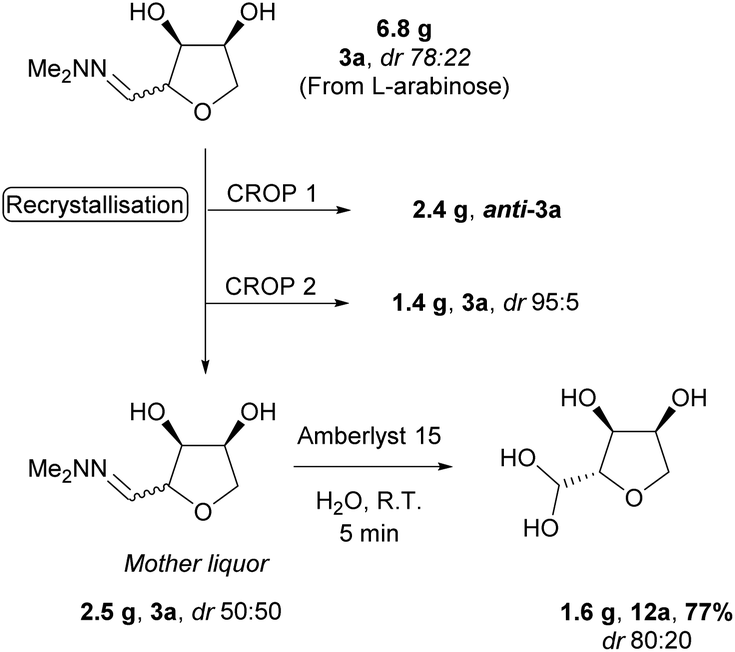
The epimerisation at C-2 during the hydrolysis of hydrazone 3a to hydrate 12a offers an opportunity to maximise the value of the material (Scheme 7). Thus, recrystallisation of 6.8 g of 3a obtained directly from the acidic cyclisation (dr 78:22) yielded 2.4 g of anti-3a as a single isomer. A second crop of high purity anti-3a was also obtained (1.4 g, dr 95:5), leaving the remaining mother liquor as an equimolar mixture of diastereomers. Submitting this material to the hydrolysis conditions led to an increase in the diastereomeric ratio (dr 80:20) in the product hydrate 12a, allowing efficient recycling of the mother liquor from the recrystallisation of anti-3a (Scheme 7).
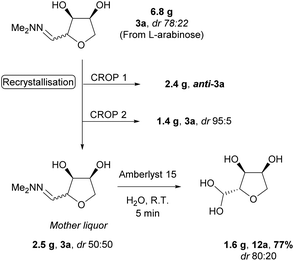 |
| | Scheme 7 Hydrolysis and increase in diastereoselectivity of 3a to 12a. | |
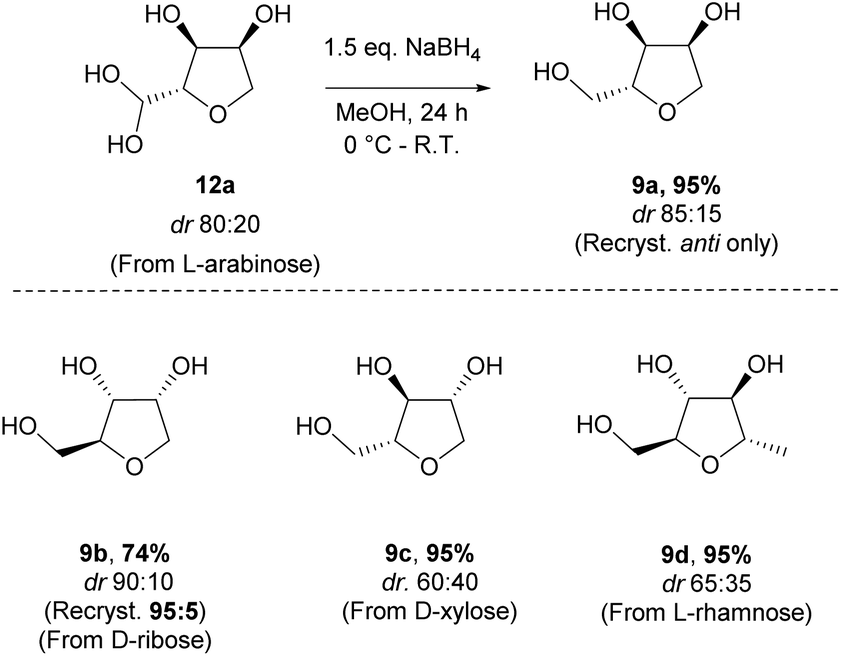
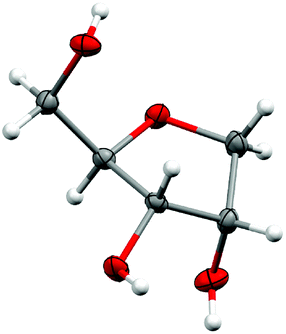
The reduction of the hydrate using sodium borohydride provided the alcohol 9a in excellent yield and without significant change in the diastereomeric ratio (Scheme 8). Alcohols 9a and 9b, respectively derived from L-arabinose and D-ribose could be recrystallised to afford the pure anti isomers in high yield. The relative stereochemistry was confirmed through an X-ray structure of ribose derivative 9b (Fig. 2).
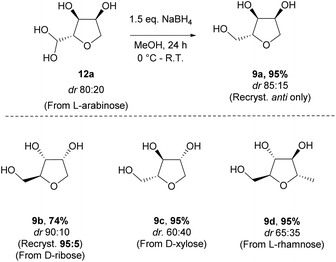 |
| | Scheme 8 Synthesis of THF-triols 9. | |
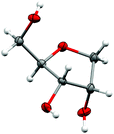 |
| | Fig. 2 X-ray crystal structure of triol 9b. The thermal ellipsoids are shown at the 50% probability level, while hydrogen atoms are drawn as fixed spheres with a radius of 0.15 Å.¶ | |
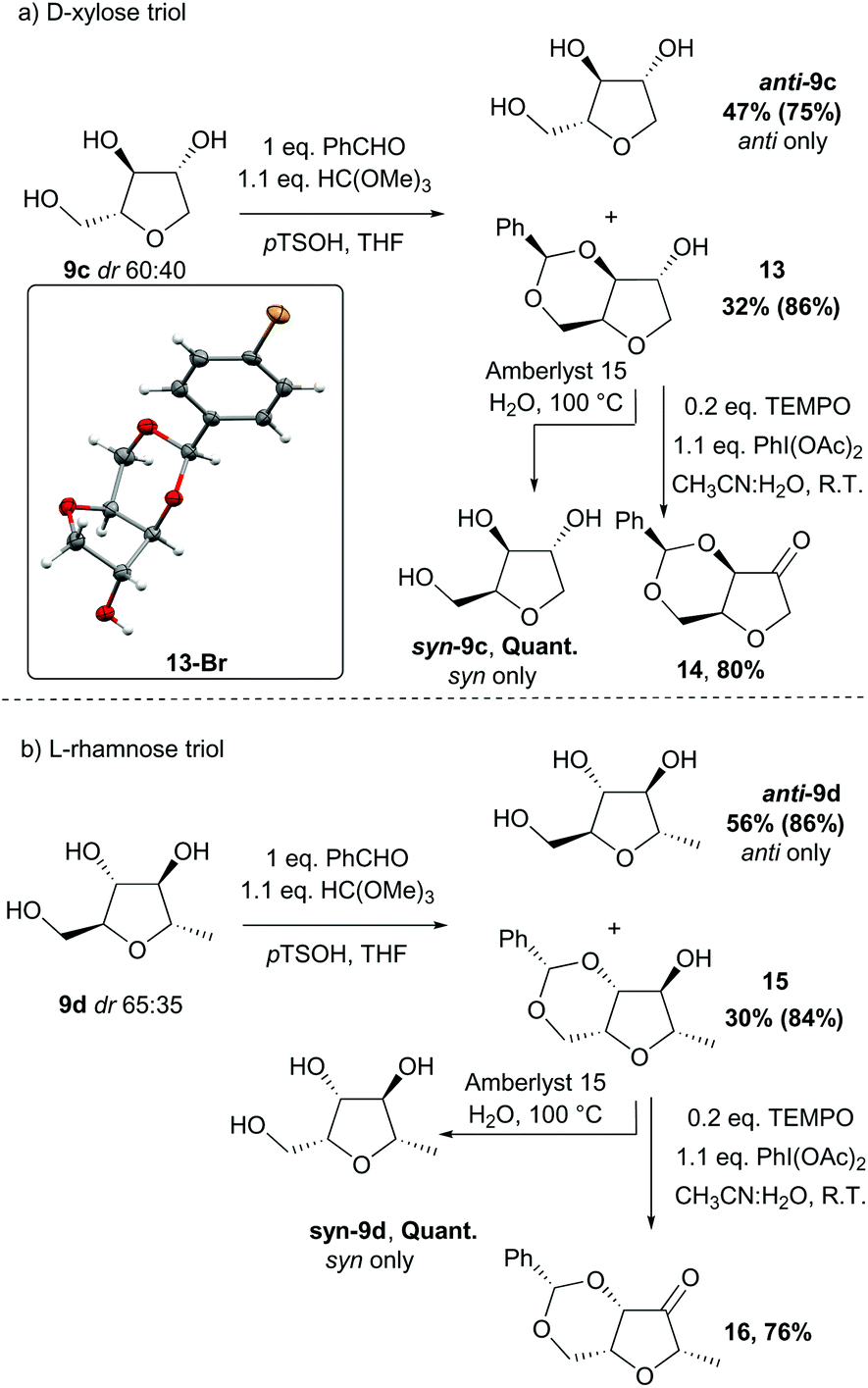
For the alcohols 9c and 9d obtained from the D-xylose and L-rhamnose hydrates 12c and 12d respectively, the diastereoselectivity was lower than required, and separation of the two isomers was necessary. In order to discriminate between the two species, formation of a benzylidene acetal on the 1,3-diol was performed as selectivity for the syn-diol should be expected.18 Indeed, the reaction of the D-xylose-triol 9c with benzaldehyde in the presence of catalytic p-toluenesulfonic acid and a dehydrating agent resulted in the selective formation of the acetal 13 along with unreacted anti-9c (Scheme 9a). The stereoselectivity of this acetal protection was confirmed by single crystal X-ray crystallography of the analogous compound 13-Br.|| The triol anti-9c and acetal 13 were easily separated by column chromatography. Finally, hydrolysis of 13 using Amberlyst 15 as a catalyst quantitatively afforded syn-9c. Acetal 13 was also oxidised with a catalytic amount of TEMPO using PhI(OAc)2 as co-oxidant to afford the ketone 14 in high yield. This reaction sequence was also applied to L-rhamnose triol 9d allowing the separation of anti-9d and syn-9dvia efficient formation of the acetal 15 (Scheme 9b). Ketone 16 was similarly obtained by oxidation of secondary alcohol 15.
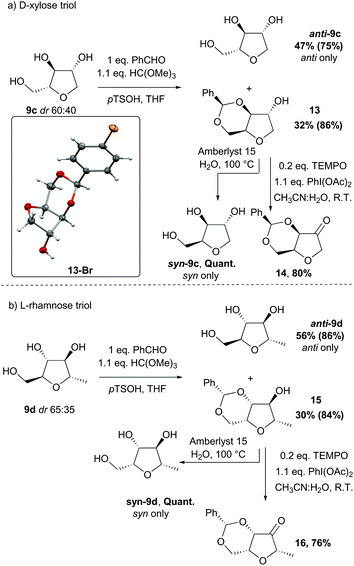 |
| | Scheme 9 Separation of the anti and syn isomers of (a) D-xylose triol 9c and (b) L-rhamnose triol 9d; yields in parentheses are based on the quantity of the relevant diastereoisomer present in the starting mixture; X-ray crystal structure of compound 13-Br with the thermal ellipsoids shown at the 50% probability level, and hydrogen atoms drawn as fixed spheres with a radius of 0.15 Å.¶ | |
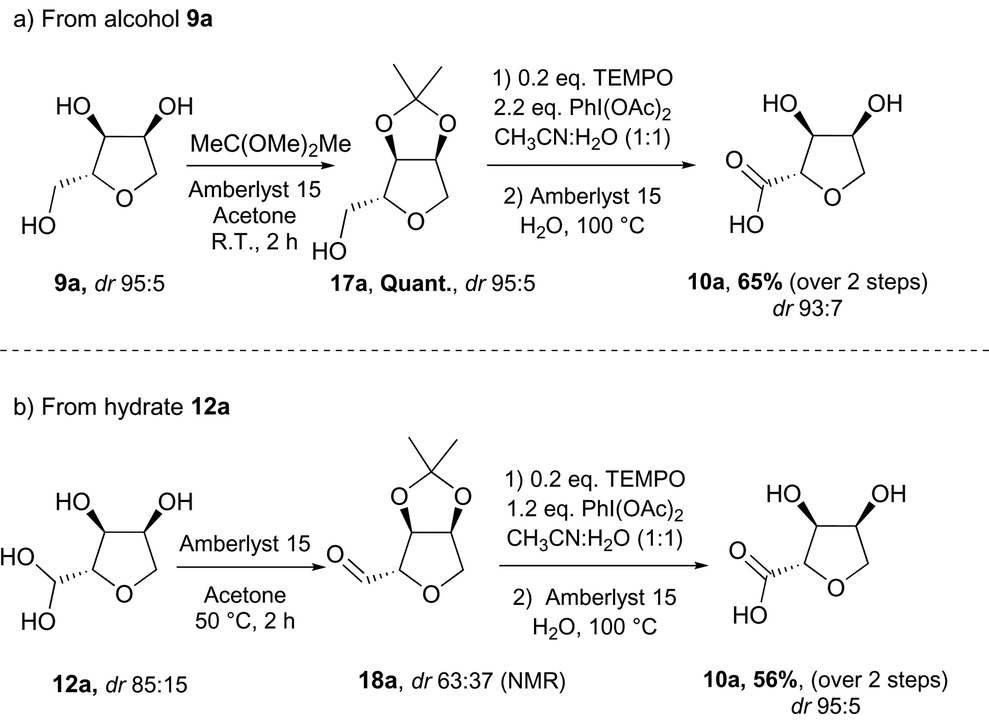
Oxidation of triol 9a into the carboxylic acid-THF 10a required a more conventional synthetic approach, using a protecting group to allow selective oxidation of the primary alcohol moiety. Protection of the syn 1,2-diol was performed with 2,2-dimethoxypropane in the presence of Amberlyst 15 (Scheme 10a). The acetal-protected triol 17a was obtained in high yield and was subsequently oxidised following a procedure previously described for the oxidation of nucleosides.19 The reaction proceeded at room temperature with a catalytic amount of TEMPO as primary oxidant, using PhI(OAc)2 as the stoichiometric oxidant. The unprotected carboxylic acid 10a was then released under acidic conditions in moderate yield over two steps and with high isomeric purity. The carboxylic acid was also accessible directly from the THF-hydrate 12a thus reducing the number of steps and the stoichiometry of oxidant required (Scheme 10b). Protection of the hydrate-THF 12b resulted in a significant loss of diastereomeric purity. However, after oxidation followed by deprotection, the carboxylic acid 10a was isolated in moderate yield with a high diastereomeric ratio in favour of the anti isomer. This rise in diastereomeric ratio during the oxidation could be explained by a higher reactivity of the anti-THF hydrate with the hindered oxidant TEMPO in comparison to the syn-THF hydrate.
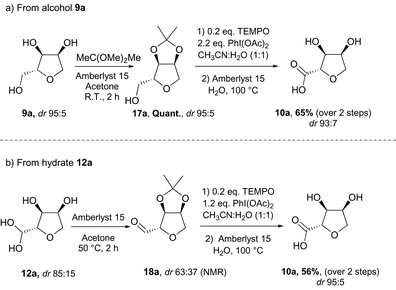 |
| | Scheme 10 Synthesis of carboxylic acid 10a from (a) triol 9a and (b) hydrate 12a. | |
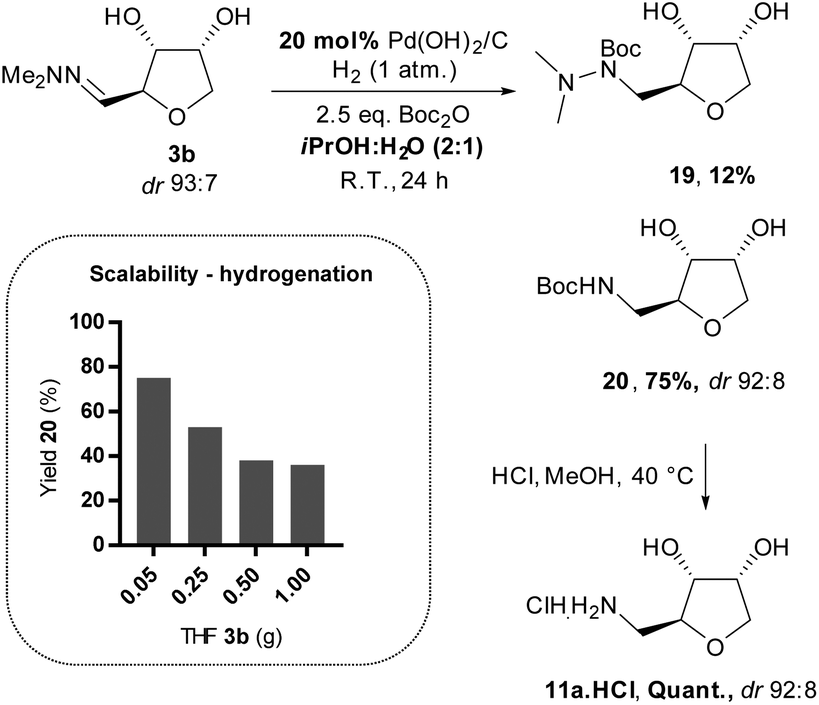
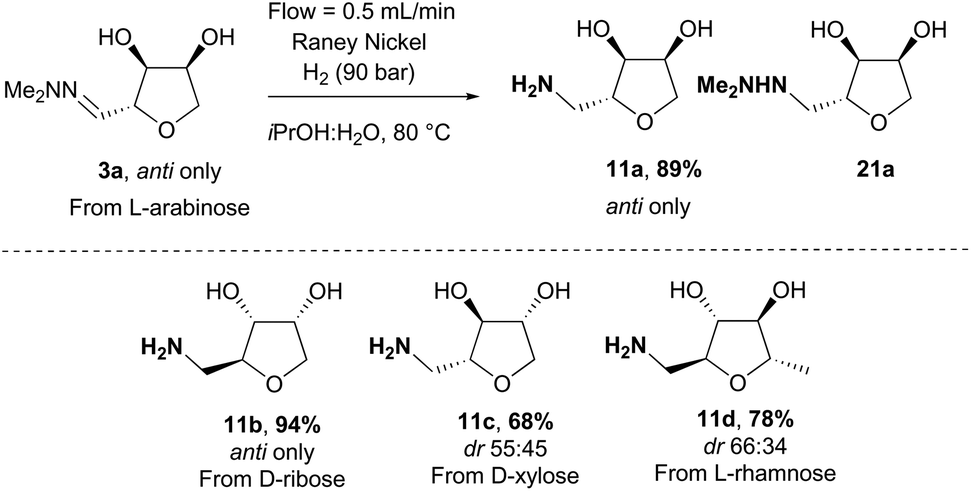
Finally, we sought an effective method for accessing the THF-amine 11 by direct hydrogenation of the THF-hydrazones 3. Although the literature is quite rich in precedent for the hydrogenation of hydrazones to hydrazines, the reduction of a hydrazone directly to an amine is rare at low hydrogen pressure.20 In our previous work, we presented a procedure to prepare the THF-amine under 1 bar of hydrogen with Boc-anhydride as an additive and Pd(OH)2/C as catalyst (Scheme 11).12 Reactions were initially conducted in CPME,21 but in subsequent studies better reproducibility was achieved in a mixture of isopropanol/water (2:1). The Boc-protected amine 20 was isolated in moderate yield on a small scale from the hydrogenation of ribose-derived hydrazone 3b, and standard deprotection using hydrochloric acid in methanol afforded the hydrochloride salt 11a·HCl (Scheme 11). The scalability of the hydrogenation proved to be problematic, however, with a dramatic loss of yield and increased reaction time as the scale was increased to gram quantities of material. The drop in the conversion can probably be explained by a poor gas–liquid–solid contact in the reaction mixture. Therefore, subsequent reductions were attempted at higher pressures of hydrogen (up to 5 bar) but did not lead to the desired amine, and instead partial reduction to the hydrazine 19 was observed (Scheme 11).
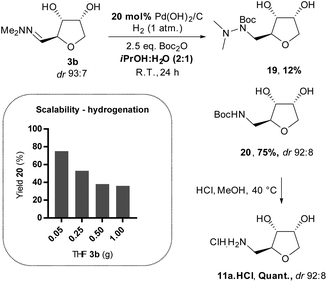 |
| | Scheme 11 Synthesis of Boc-protected amine 20 and scalability issues. | |
Hydrogenation can present serious safety issues in large scale batch reactions with high pressure, flammable solvents and pyrophoric catalysts. We therefore moved to a flow-chemistry set-up which offers several advantages in comparison with more traditional hydrogenation procedures, including the possibility to work at high pressure and temperature with an encapsulated catalyst to reduce the leaching of toxic metal into the reaction mixture (Scheme 12).22–24 As a starting point, iPrOH:H2O (2:1) was used as solvent and Pd(OH)2/C as catalyst to remain close to the initial set of reaction conditions. However even at high pressure (100 bar), the transformation did not proceed to the amine at room temperature with mostly the hydrazine 21a being formed. Moreover, when the hydrazine 21a obtained was re-submitted to a second hydrogenation-cycle, no amine product was observed. The use of Boc-anhydride as additive did not solve the issue, and as Pd(OH)2/C did not seem an efficient catalyst under these conditions we moved to RANEY® Nickel which has been reported previously as a good catalyst for the hydrogenation of hydrazones.25 At room temperature and 100 bar of hydrogen, only reduction of hydrazone 3a to the hydrazine 21a was noticeable. However, at 80 °C, 3a was fully converted to the amine 11a. Importantly no additives were required for this transformation, and the amine was isolated in high yield directly after evaporation of the solvent (Scheme 12). Moreover, no erosion of the diastereoselectivity was observed compared to the initial hydrazone 3a. This method was then applied to the synthesis of amines 11b–11d in 68–94% yield. This direct hydrogenation of the hydrazone provides an alternative route for accessing the amines 11 to our recently reported biocatalytic approach involving reaction of hydrates 12 with transaminase enzymes.26
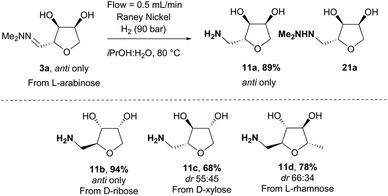 |
| | Scheme 12 Synthesis of amines 11 by reduction of the THF-hydrazones 3 using flow chemistry. | |
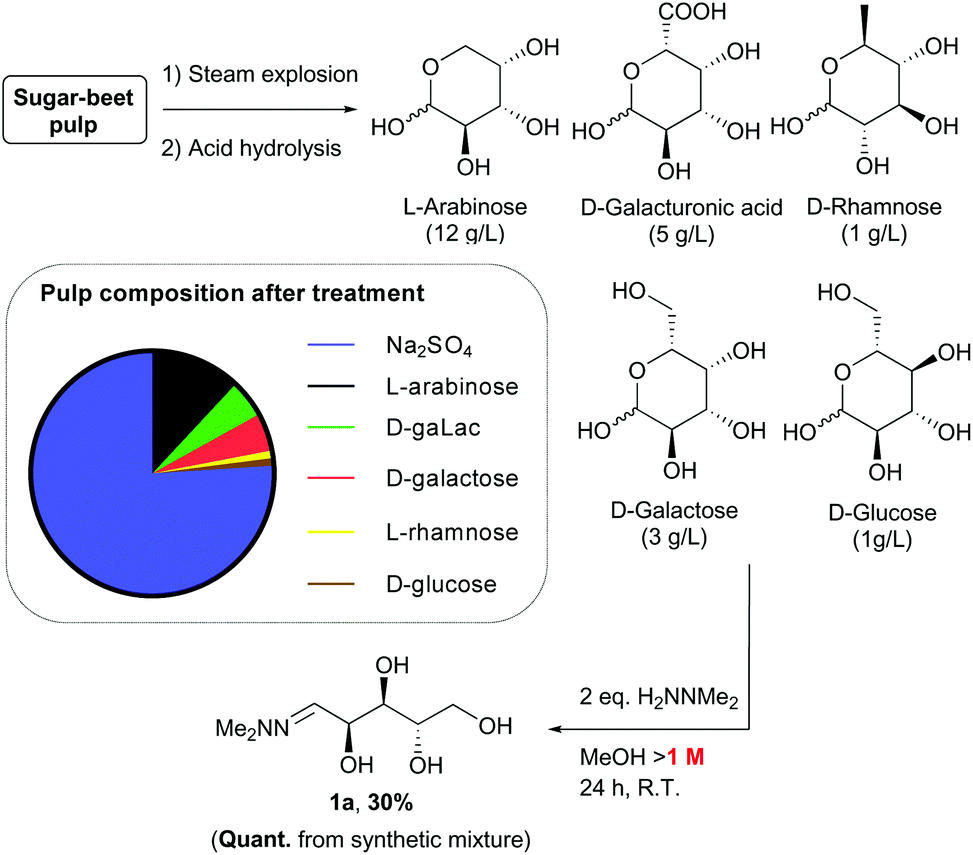
Synthesis of sugar hydrazones from sugar beet pulp
Sugar beet pulp contains significant quantities of L-arabinose, so we were interested in its potential application as a feedstock for the synthesis of these chiral THF building blocks. It was therefore important to establish that hydrazone 1a could be prepared from waste pulp material. To determine this, a solubilised sample of sugar beet pulp after steam explosion and acidic hydrolysis treatment was generated (Scheme 13).27–29 Analysis of the solution prepared in this way indicated it contained sodium sulfate (80 g L−1) and arabinose (12 g L−1), together with galacturonic acid (5 g L−1), galactose (3 g L−1), rhamnose (1 g L−1) and glucose (1 g L−1) monomers. Initially, a model synthetic mixture containing these components was treated with N,N-dimethylhydrazine (2 eq. wrt arabinose). Surprisingly, analysis of the crude product by NMR spectroscopy revealed the selective formation of the hydrazone 1a derived from L-arabinose under these conditions in quantitative yield, with only traces of the rhamnose hydrazone present. This selectivity could be explained by (i) the low solubility of galacturonic acid in methanol and (ii) the much slower reaction of hexose sugars with hydrazine compared to the pentoses. Indeed, formation of the hydrazone from galactose was previously observed to require harsher conditions for an extended period of time (3 days) to reach only moderate conversion.12 Reaction of the heterogeneous mixture derived from sugar beet pulp under similar conditions also led to selective formation of hydrazone 1a. The concentration (>1 M) was found to be a crucial parameter for achieving high conversion in this transformation, so concentration of the crude solution was necessary prior to reaction. After purification, the arabinose hydrazone 1a was isolated in high purity in 30% yield. The discrepancy in yield between the two experiments suggests that partial decomposition of the sugars is taking place during concentration of the crude starting material, and/or that impurities present in the pulp lead to loss of material during the work-up and purification of hydrazone 1a.
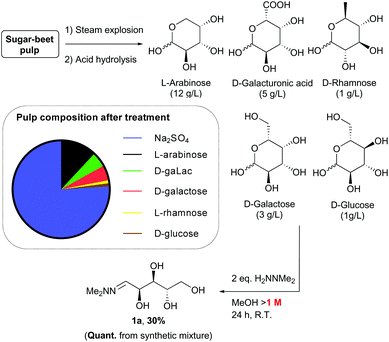 |
| | Scheme 13 Synthesis of L-arabinose hydrazone 1a from a solubilised sample of sugar beet pulp. | |
Conclusions
In conclusion, we have optimised procedures for the selective dehydration of pentose sugars on multigram scales, including: (i) improvement and scale-up (20 g) of the acid-catalysed cyclisation for L-arabinose and other pentoses; (ii) development of a novel cyclisation procedure under basic conditions which preferentially provides THFs with syn stereochemistry at C-2/C-3. These two complementary procedures offer access to a range of chiral-THFs with varied stereochemistries. The THF-hydrazones were subsequently transformed into THF-triols 9, bicyclic ketones 14/16, THF-carboxylic acids 10 and THF-amines 11 to build a library of functionalised THFs (Fig. 3) for potential application in medicinal chemistry. Finally, we were able to prepare arabinose hydrazone 1a selectively from a pre-treated sample of sugar beet pulp, thus demonstrating the feasibility of preparing many of these chiral THF building blocks from an abundant renewable feedstock.
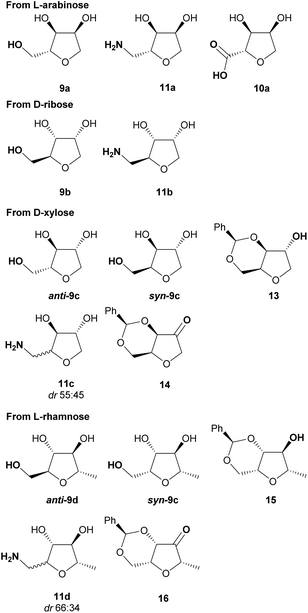 |
| | Fig. 3 Chiral THFs prepared in this work. All compounds shown above were obtained in >95:5 dr (except 11c/11d). | |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We would like to thank the Engineering and Physical Sciences Research Council (EPSRC) and the University College London Impact Acceleration Account for providing PDRA funding to L. B. (EP/K503745/1) for a collaboration with AstraZeneca and GlaxoSmithKline. We would like to thank David Andrews (AstraZeneca), Tony Dean and Rob Young (GlaxoSmithKline) for their help and support on this project. We would also like to thank GlaxoSmithKline for providing an EPSRC CASE studentship for RWF. Finally, we thank Maciej Kucharski and Lee Edwards (GlaxoSmithKline) for assistance with the flow hydrogenation experiments.
Notes and references
- Y. Xu, M. A. Hanna and L. Isom, Open Agric. J., 2008, 2, 54–61 CrossRef CAS.
- C. V. Boucque, B. G. Cottyn, J. V. Aerts and F. X. Buysse, Anim. Feed Sci. Technol., 1976, 1, 643–653 CrossRef.
- E. Evans and U. Messerschmidt, J. Anim. Sci. Biotechnol., 2017, 8, 25–35 CrossRef PubMed.
-
M. Dusselier, M. Mascal and B. F. Sels, in Top. Curr. Chem., Springer-Verlag, Berlin Heidelberg, 2014, vol. 353, pp. 1–31 Search PubMed.
- F. W. Lichtenthaler and S. Peters, C. R. Chim., 2004, 7, 65–90 CrossRef CAS.
- L. L. Adduci, T. A. Bender, J. A. Dabrowski and M. R. Gagné, Nat. Chem., 2015, 7, 576–581 CrossRef CAS PubMed.
- Y. Seo and M. R. Gagné, ACS Catal., 2018, 8, 6993–6999 CrossRef CAS.
- J. M. Lowe, Y. Seo and M. R. Gagné, ACS Catal., 2018, 8, 8192–8198 CrossRef CAS.
- T. Saloranta and R. Leino, Synlett, 2015, 26, 421–425 CrossRef CAS.
- I. S. Young and P. S. Baran, Nat. Chem., 2009, 1, 193–205 CrossRef CAS PubMed.
- T. A. Bender, J. A. Dabrowski and M. R. Gagné, Nat. Rev. Chem., 2018, 2, 35 CrossRef CAS.
- R. W. Foster, C. J. Tame, D. K. Bučar, H. C. Hailes and T. D. Sheppard, Chem. – Eur. J., 2015, 21, 15947–15950 CrossRef CAS PubMed.
- G. Romeo, U. Chiacchio, A. Corsaro and P. Merino, Chem. Rev., 2010, 110, 3337–3370 CrossRef CAS PubMed.
- L. P. Jordheim, D. Durantel, F. Zoulim and C. Dumontet, Nat. Rev. Drug Discovery, 2013, 12, 447–464 CrossRef CAS PubMed.
- K. M. Tomczyk, P. A. Gunka, P. G. Parzuchowski, J. Zachara and G. Rokicki, Green Chem., 2012, 14, 1749–1758 RSC.
- F. W. Goldberg, J. G. Kettle, T. Kogej, M. W. D. Perry and N. P. Tomkinson, Drug Discovery Today, 2015, 20, 11–17 CrossRef PubMed.
- G. M. Keserű, D. A. Erlanson, G. G. Ferenczy, M. M. Hann, C. W. Murray and S. D. Pickett, J. Med. Chem., 2016, 59, 8189–8206 CrossRef PubMed.
- H. Y. Zhang, H. W. Yu, L. T. Ma, J. M. Min and L. H. Zhang, Tetrahedron: Asymmetry, 1998, 9, 141–149 CrossRef CAS.
- J. B. Epp and T. S. Widlanski, J. Org. Chem., 1999, 64, 293–295 CrossRef CAS PubMed.
- G. Bégis, D. E. Cladingboel, L. Jerome, W. B. Motherwell and T. D. Sheppard, Eur. J. Org. Chem., 2009, 1532–1548 CrossRef.
- P. M. Murray, F. Bellany, L. Benhamou, D.-K. Bučar, A. B. Tabor and T. D. Sheppard, Org. Biomol. Chem., 2016, 14, 2373–2384 RSC.
- R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25 CrossRef CAS.
- P. J. Cossar, L. Hizartzidis, M. I. Simone, A. McCluskey and C. P. Gordon, Org. Biomol. Chem., 2015, 13, 7119–7130 RSC.
- G. Jas and A. Kirschning, Chem. – Eur. J., 2003, 9, 5708–5723 CrossRef CAS PubMed.
-
The Handbook
of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, WILEY-VCH Verlag, Weinheim, 2008 Search PubMed.
- F. Subrizi, L. Benhamou, J. Ward, T. Sheppard and H. C. Hailes, Angew. Chem., Int. Ed., 2019, 58, 3854–3858 CrossRef CAS PubMed.
- C. Hamley-Bennett, G. J. Lye and D. J. Leak, Bioresour. Technol., 2016, 209, 259–264 CrossRef CAS PubMed.
- D. P. Ward, P. Hewitson, M. Cárdenas-Fernández, C. Hamley-Bennett, A. Díaz-Rodríguez, N. Douillet, J. P. Adams, D. J. Leak, S. Ignatova and G. J. Lye, J. Chromatogr. A, 2017, 1497, 56–63 CrossRef CAS PubMed.
- M. Cárdenas-Fernández, M. Bawn, C. Hamley-Bennett, P. K. V. Bharat, F. Subrizi, N. Suhaili, D. P. Ward, S. Bourdin, P. A. Dalby, H. C. Hailes, P. Hewitson, S. Ignatova, C. Kontoravdi, D. J. Leak, N. Shah, T. D. Sheppard, J. M. Ward and G. J. Lye, Faraday Discuss., 2017, 202, 415–431 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Procedures for the preparation of all compounds, together with characterisation data and X-ray crystallographic data. CCDC 1877937 and 1877938. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9gc00448c |
| ‡ New address: AstraZeneca PLC, 2 Riverside, Milstein Building, Granta Park, Cambridge, CB21 6GP, UK. |
| § New address: Benevolent AI, 4-8 Maple St, London, W1T 5HD, UK. |
| ¶ Tables of selected crystallographic data for 9b and 13-Br can be found in the ESI.† CCDC 1877937 and 1877938 contain the supplementary crystallographic data for compounds 9b and 13-Br respectively. |
| || The selectivity of the acetal protection was confirmed by obtaining an X-ray crystal structure of 13-Br,¶ an analogue of 13 prepared from 4-bromobenzaldehyde (see ESI† for full details). |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Robert W.
Foster‡
a,
David P.
Ward
*a,
Robert W.
Foster‡
a,
David P.
Ward