
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of N-heterocycles from diamines via H2-driven NADPH recycling in the presence of O2†
Ammar
Al-Shameri‡
 a,
Niels
Borlinghaus‡
b,
Leonie
Weinmann
b,
Philipp N.
Scheller
b,
Bettina M.
Nestl
*b and
Lars
Lauterbach
*a
a,
Niels
Borlinghaus‡
b,
Leonie
Weinmann
b,
Philipp N.
Scheller
b,
Bettina M.
Nestl
*b and
Lars
Lauterbach
*a
aTechnische Universität Berlin, Institute of Chemistry, Strasse des 17. Juni 135, 10623 Berlin, Germany. E-mail: lars.lauterbach@tu-berlin.de
bUniversitaet Stuttgart, Institute of Biochemistry and Technical Biochemistry, Department of Technical Biochemistry, Allmandring 31, 70569 Stuttgart, Germany. E-mail: bettina.nestl@itb.uni-stuttgart.de
First published on 23rd January 2019
Abstract
Herein, we report an enzymatic cascade involving an oxidase, an imine reductase and a hydrogenase for the H2-driven synthesis of N-heterocycles. Variants of putrescine oxidase from Rhodococcus erythropolis with improved activity were identified. Substituted pyrrolidines and piperidines were obtained with up to 97% product formation in a one-pot reaction directly from the corresponding diamine substrates. The formation of up to 93% ee gave insights into the specificity and selectivity of the putrescine oxidase.
Saturated N-heterocycles are widespread in biologically active molecules. In particular, pyrrolidines and piperidines are increasingly attractive scaffolds found in agrochemicals, pharmaceuticals and natural alkaloid products.1 Among the different strategies reported for the preparation of saturated nitrogen heterocycles,2 the biocatalytic synthesis via cascades has attracted particular interest. Biocatalytic cascades coupling several enzymatic transformations in a one-pot process allow the formation of complex molecular architectures under ambient conditions and without the need of protecting groups and purification of intermediates.3,4 To date, current enzymatic cascade strategies for the synthesis of pyrrolidine and piperidine heterocycles involve ω-transaminases, monoamine oxidases, carboxylic acid reductases and imine reductases including also artificial imine reductases.5–7 Although these cascades have been mainly used for the transformation of keto acids, diketones and keto aldehydes to the corresponding heterocyclic products, only one example has been described for the preparation of N-heterocycles from the corresponding diamines. The group of Turner has described the transamination of terminal aliphatic diamines and polyamines using putrescine transaminase and pyruvate as amine acceptor to provide different N-heterocycles in good yields.8,9 Highest activity was observed towards the natural substrate putrescine (1,4-diaminobutane). However, highly decreased activity using a methyl-substituted diamine has been obtained. The low activity of putrescine transaminase towards this compound might result from the rather narrow substrate access channel to the active site pocket. In the transformation of substituted diamines, there is still a requirement for an efficient oxidation. We wondered if we could access pyrrolidine and piperidine N-heterocycles in a biocatalytic cascade from diamines employing a diamine oxidase (putrescine oxidase) for the first oxidation step followed by the reduction of the in-situ formed cyclic imine using an imine reductase (IRED) (Scheme 1).
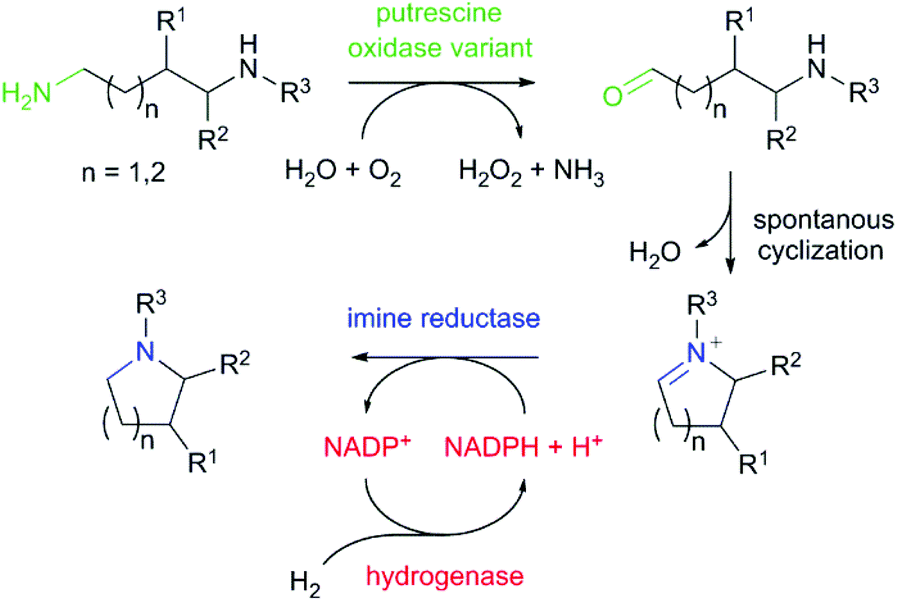 | ||
| Scheme 1 Synthesis of pyrrolidine and piperidine heterocyclic scaffolds from diamines by an enzymatic cascade. In the first step one amino group is oxidized with molecular oxygen to a carbonyl group using putrescine oxidase variant (green). Subsequently a spontaneous cyclisation forms the cyclic imine, which is reduced by the imine reductase (blue) to obtain a saturated N-heterocycle. The consumed NADPH cofactor is regenerated via H2-driven enzymatic cofactor regeneration without by-product formation, using a NADP+-reducing hydrogenase (red). Diamine substrates with methyl substituents either on site R1, R2 or R3 were selected. | ||
The asymmetric reduction of prochiral imines with NADPH-dependent IREDs has enjoyed particular success for the generation of N-heterocyclic compounds including pyrrolidines, piperidines, indolines and tetrahydroisoquinolines.10–17 Given the high cost, stoichiometric use, and instability of NADPH, regeneration of this cofactor is essential for practical application. Therefore, we have based our approach on emerging “green” chemistry. This involves no utilization of toxic compounds and solvents and 100% atom efficiency by applying the H2 hydrogenase cofactor regeneration system.18
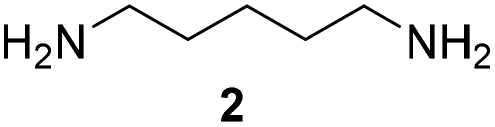
For the initial oxidation of diamines and polyamines, we selected the putrescine oxidase (PuO) from Rhodococcus erythropolis.19 PuO is a flavin-dependent amine oxidase that catalyses the oxidation of its natural substrate putrescine (1) with molecular oxygen (O2) as electron acceptor. This makes oxidases inexpensive and straightforward in usage compared to other redox enzymes. Unfortunately, PuO possesses a narrow substrate specificity accepting only small aliphatic diamines as substrates such as putrescine (1) and cadaverine (2). In an attempt to increase the substrate spectrum, we initially engineered PuO through directed evolution using error prone PCR (epPCR). We exchanged an average number of four nucleotides that corresponds to one or two amino acid substitutions. For the screening of variants with improved activity, a solid phase assay was used.20 We examined the altered oxidation activity by screening libraries against 1,5-diamino-2-methylpentane (3) and 1,5-diaminohexane (4).
Interestingly, the best variant PuOE203G for the oxidation of substituted substrate 4 possesses a single amino acid substitution of glutamic acid at position 203 to glycine. Analysis of the crystal structure revealed that this residue is located at the access channel to the active site pocket of PuO. We assume that the introduction of a smaller amino acid residue at this position expands the access channel to the active site and facilitates the passage of substituted or elongated diamine substrates (Fig. 1). In this light, we tested a panel of nine diamines (1–9) and two polyamines (21–22) with purified PuOnative and PuOE203G (Fig. 1). Both wildtype and variant were equipped with a Strep-tag II and purified by using affinity chromatography (Fig. S1†). We could demonstrate that the activity of both enzymes was decreased when one amino group harboured a methyl substituent (5) and was abolished when the diamine was highly substituted (9), indicating that the less hindered ω-amino group is oxidized by PuO.
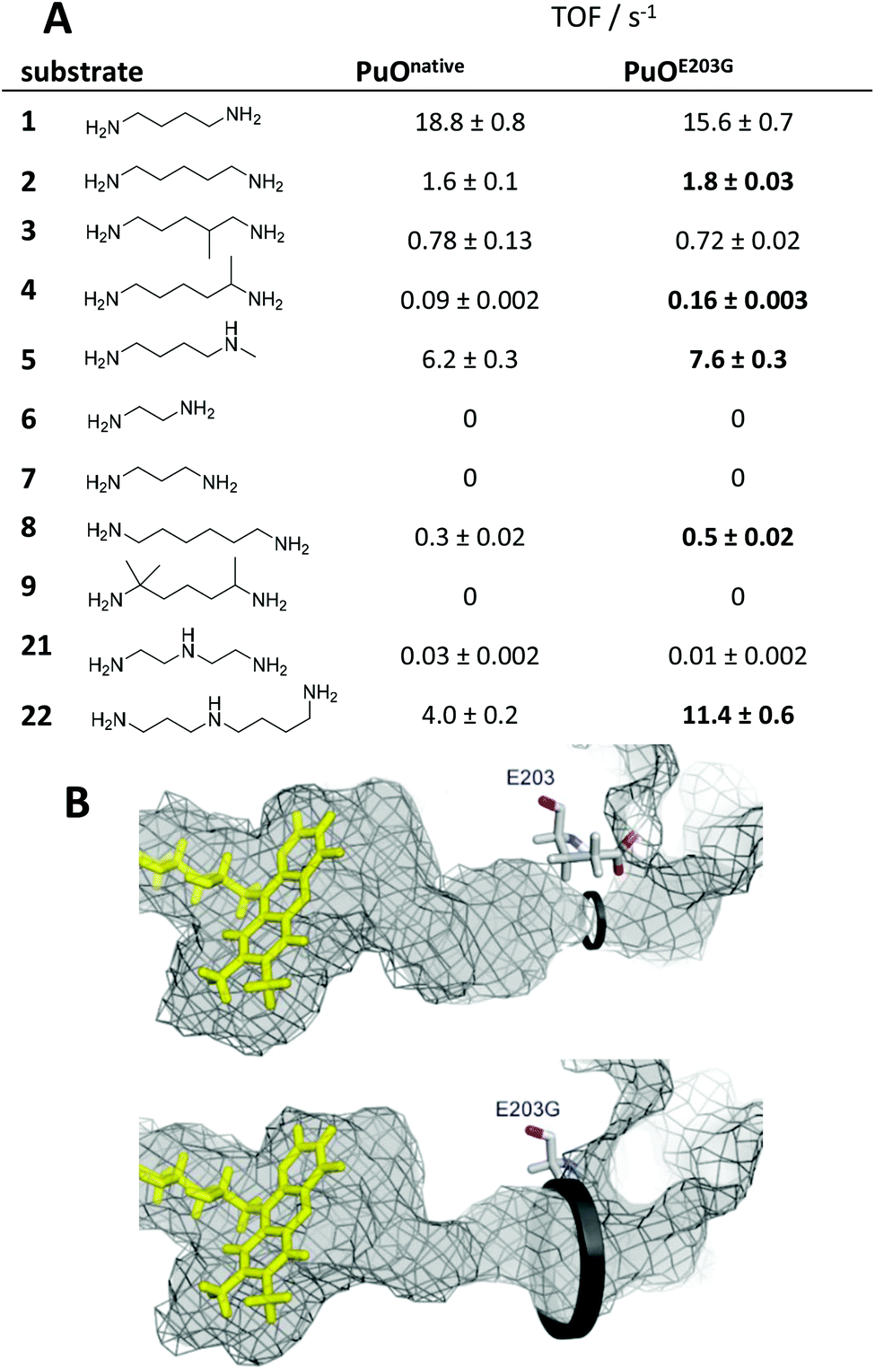 | ||
| Fig. 1 Engineering of putrescine oxidase and substrate scope. (A) Turnover frequencies of PuOnative and PuOE203G are shown for nine different diamines (1–9) and two polyamine substrates (21–22). Improved activities of the variant are highlighted. (B) Comparison of substrate channels in PuOnative (top, PDB code: 2YG4) and PuOE203G (bottom, homology model via SWISS-MODEL-server).23 Inner surfaces of channels are shown in grey visualized with PyMOL. The structures were calculated with Yasara,24 running energy minimizations in water and subsequent MD refinements (0.5 ns, force field: YAMBER3). The steric bottleneck of the channel next to glutamic acid at position E203 is opened by introducing glycine at this position (indicated with black circles). | ||
For the variant, increased activities where observed with five different non-natural substrates including 1,5-diaminopentane (2), 1,5-diaminohexane (4) and N-methyl putrescine (5). We speculate that the configuration of the active site in PuOE203G enables almost exclusively the oxidation of the less-hindered amino group. Photometric analysis of the cofactor content revealed 0.94 ± 0.2 and 1.2 ± 0.14 FAD per PuOnative and PuOE203G, respectively, which is in contrast to previous studies with only 0.5 FAD per enzyme.19,21 The previous low FAD loading is related to competing binding of the inhibitor adenosine diphosphate to the active site.22 The nearly total FAD occupancy of purified PuO derivatives can be explained by optimized PuO production conditions at low temperature (18 °C) in TB complex media. The UV/Vis spectra showed typical characteristics of flavoenzymes (Fig. S2†).
We next addressed the enzyme kinetics of PuOE203G compared to PuOnative using four model diamine substrates 1–4 (Table 1, Fig. S3A–D†) and evaluated the Michaelis constant for the co-substrate O2 (Fig. S3E†). In order to shed light on the optimal biotransformation conditions, we first measured the affinity of PuO to O2 using a peroxidase-coupled assay similar to Hellemond and co-workers.19
| Substrate | PuOnative | PuO E203G | ||||
|---|---|---|---|---|---|---|
| K M (μM) | k cat (s−1) | k cat/KM (s−1 mM−1) | K M (μM) | k cat (s−1) | K cat/KM (s−1 mM−1) | |
| 1 | 54 | 18 | 331 | 213 | 34 | 160 |
| 2 | 69 | 1.1 | 16 | 315 | 3.4 | 11 |
| 3 | 30 | 0.5 | 15 | 177 | 1.5 | 8 |
| 4 | 11 | 0.1 | 0.9 | 201 | 0.1 | 0.5 |
The O2 concentration was precisely determined by an optical sensor before measuring the kinetics with 1. The steady state kinetic for 1 as a function of O2 (Fig. S4†) indicated an apparent ping-pong reaction mechanism, where no ternary complex of enzyme–substrate–oxygen is formed similar to Desa et al.21 The Km value of 30 μM of native PuO for 1 at ambient O2 concentration was similar to the result obtained by Kopacz and co-workers.25 In contrast to ambient O2 concentration, a higher KM (51.8 μM) for 1 at 1.24 mM O2 (100% O2) was observed (Fig. S4†) in line with a ping-pong mechanism. Stopped-flow spectroscopic experiments indicated a bifurcated mechanism at ambient O2 concentrations due to comparable rate constants of product release from the reduced enzyme and reoxidation of the reduced enzyme–product complex.25 Thus, the results support the assumption that at lower or higher O2 concentrations, the contribution of the ping-pong and ordered sequential mechanism can change.25KM values for O2 using PuOnative and PuOE203G were 246 μM and 296 μM, respectively. It is worth mentioning that affinities for O2 are at levels similar to those observed for other flavoenzymes and the atmospheric O2 concentration of 260 μM. Due to the high KM for O2, all further kinetic determinations were performed under 100% O2 saturation (1.24 mM). Under these conditions the glucose-6-phosphate dehydrogenase/glucose-6-phosphate system for cofactor regeneration was used since no H2 is present for SH-mediated regeneration.
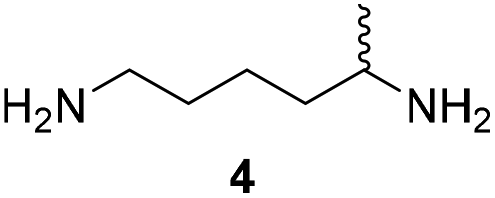
Furthermore, up to 3-fold improved kcat values were observed for variant PuOE203G compared to PuOnative (Table 1). However, due to high KM values the catalytic efficiency was reduced. Interestingly, catalytic efficiencies were 16-fold reduced for 1,5-diaminohexane (4) compared to 1,5-diamino-2-methylpentane (3), which indicates that the position of the methyl group in the molecule influences the affinity and the acceptance of the substrate.
To assess the potential for the application of variant PuOE203G in the preparation of different pyrrolidine and piperidine hetereocycles, we combined PuOE203G with an R-selective imine reductase from Streptosporangium roseum (R-IRED_Sr).16 The IRED-catalysed reduction of imines requires a sufficient supply of NADPH as a reducing equivalent. For the regeneration of NADPH, the enzyme glucose-6-phosphate dehydrogenase is frequently used converting glucose-6-phosphate by NADP+ reduction to 6-phosphoglucono-δ-lactone. A highly attractive alternative for NADPH regeneration constitutes the O2-tolerant NAD+-reducing hydrogenase (SH) from Ralstonia eutropha H16, which utilises only molecular hydrogen as the reductant. In contrast to commonly used cofactor regeneration systems, the H2-based NADH recycling is 100% atom-efficient and relies on a cheap, carbon-free reducing agent.18 Unlike other hydrogenases, the R. eutropha enzyme is O2-tolerant with its activity remaining unchanged even at ambient O2 concentrations.26 The new SH-based NADH regenerating enzyme was able to be successfully applied in many multi-cascade reactions in vitro as well as in whole cell systems.27–30 Recently, the cofactor specificity of SH was altered from NADH to NADPH by rational design (manuscript submitted). The engineered SHE341A/S342R variant retained its O2 tolerance and permits the use of SH in numerous of reactions where NADPH is required as cofactor. For the regeneration of NADPH both the glucose-6-phosphate dehydrogenase (1 U mL−1)/glucose-6-phosphate system (20 mM) and the NADP+-dependent variant SHE341A/S342R (1 U mL−1) with H2 as reducing agent were examined. In this context it is worth noting that primary amines are described to react with aldehydes including also the open-chain form of glucose-6-phosphate to generate imine derivatives.31 Indeed, we found that glucose-6-phosphate reacted with 1 and 2 in solution overnight (Fig. S09–S13, Schemes S1 and S2†). Similar observations are reported for different sugars that might react with primary and secondary amines. Even though, those reactions are reversible in aqueous solution and no drastic negative effects were observed in the overall enzyme activity (Table S4†), these side reactions will hamper the isolation and purification of desired products. The glucose/glucose dehydrogenase cofactor regeneration system was reported to show promiscuous enzyme activity towards the reduction of imine compounds,32 which would complicate IRED activity analysis and was therefore not applied. The presented alternative H2-driven cofactor regeneration circumvents these problems, since the only needed substrate is H2. In this setup, biotransformations were performed combining PuOE203G, R-IRED-Sr and SHE341A/S342R in a one-pot process (Table 2). Furthermore, we employed catalase for the decomposition of H2O2, which is generated by PuO. Due to the low solubility of H2 and O2 in aqueous solutions, biotransformations were performed in small explosive secured and enclosed vessels with 1 mL reaction mixture and excess of headspace (7 mL) containing a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of H2 and O2 gas mixtures. This ensured sufficient supply as well as O2 and H2 concentrations for maximum amine oxidase and hydrogenase activity. With this setup, the transformations of five model diamine substrates to the corresponding N-heterocyclic products were employed with up to 97% of product formed (Table 2). Thereby, reaction conditions emerged as crucial for the overall performance of the cascade as well as for the enantiomeric excess (ee) of generated products. For substrates which are just poorly accepted by PuOE203G, for example in the transformation of diamine 4, the product formation can be improved either by higher O2-concentrations (Table S2†) or by increasing the ratio of catalysts PuOE203G/R-IRED_Sr.
:1 ratio of H2 and O2 gas mixtures. This ensured sufficient supply as well as O2 and H2 concentrations for maximum amine oxidase and hydrogenase activity. With this setup, the transformations of five model diamine substrates to the corresponding N-heterocyclic products were employed with up to 97% of product formed (Table 2). Thereby, reaction conditions emerged as crucial for the overall performance of the cascade as well as for the enantiomeric excess (ee) of generated products. For substrates which are just poorly accepted by PuOE203G, for example in the transformation of diamine 4, the product formation can be improved either by higher O2-concentrations (Table S2†) or by increasing the ratio of catalysts PuOE203G/R-IRED_Sr.
| Substrate | Product | Product formationa (%) | Optimized conditions for increased product formationb (%) | Optimized conditions for increased selectivityc (%) |
|---|---|---|---|---|
| a Enzyme cascades were performed in Tris-HCl buffer (50 mM, pH 7.5) with 10 mM diamine and 2 mM NADP+ in 8 mL glass-vials (horizontal shaking, 180 rpm) for 4 hours at 25 °C. Concentrations of PuOE203G and R-IRED_Sr were adapted to substrate related activities (PuO: 0.05–1.5 mg mL−1; IRED: 1–2 mg mL−1) and combined with SHE341A,S342R and catalase. Samples were aerated with O2 and H2 in the ratio 1:1. b Increased product formations were obtained under modified reaction conditions with 5 mM diamine after 2 hours (Table S2, Fig. S5 and S6). c Increased enantiomeric excesses were obtained when reactions were stopped after 30 minutes under modified reaction conditions with 5 mM diamine (Table S2). n.d. not determined. | ||||
|
|
71 ± 7 | n.d. | — |

|
|
30 ± 4 | 34 ± 2 | — |
|
|
97 ± 2 | n.d. | — |

|
|
56 ± 6 (ee = 25% R) | 99 ± 1 (racemic) | 22 ± 3 (ee = 93% R) |
|
|
24 ± 12 (ee = 40% S) | 86 ± 3 (ee = 40% S) | 16 ± 4 (ee = 57% S) |
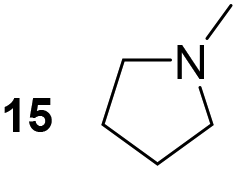
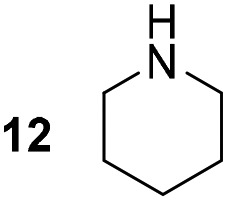
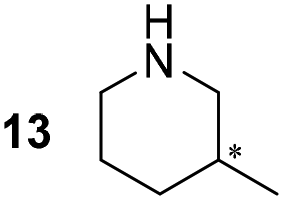
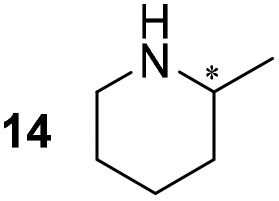
Pyrrolidine (11), piperidine (12) and methylpiperidines (13, 14) were formed in high to excellent product formations (up to 97%) starting from the corresponding diamines (1–4). In addition, chiral methylpiperidines 13 and 14 were obtained after 30 min reaction time with 93% ee (R) and 57% ee (S), respectively. It is worth mentioning that selectivities are not induced by R-IRED_Sr, because the oxidation of diamines mainly causes the formation of the non-prochiral imine intermediate species (Fig. S7 and S8†). Instead, PuOE203G is able to distinguish between both enantiomers from the racemic diamine substrate with one enantiomer being faster converted than the other. In the course of the reaction, however, the concentration of the preferred substrate enantiomer decreases and thus affects the ee due to the conversion of the slower-reacting remaining substrate enantiomer (Table S2†). The resulting kinetic resolution allowed for example the asymmetric formation of 13 with up to 93% ee. In contrast to 13, the ee was just slightly improved for 14 after 30 min reaction time (Table S2†). This reveals that PuOE203G is more selective in the kinetic resolution of 3 compared to 4 and that the enzymatic cascade can be tuned towards productivity or selectivity but not in both directions. Moreover, the secondary amine substrate N-methyl 1,4-butanediamine (5) was successfully converted (30% product formation) to the corresponding tertiary amine product N-methylputrescine (15). Such products are not accessible via other reported enzyme cascades starting from dicarbonyls.
Conclusions
In conclusion, we have developed an enzymatic cascade reaction, allowing access to pyrrolidine and piperidine heterocycles from substituted diamines. The importance of substituted nitrogen heterocycles, in particular pyrrolidine and piperidine types as subunits of bioactive molecules or pharmaceuticals stimulates the development of new synthetic methods. Directed evolution of the diamine oxidase resulted in variants with improved oxidation activity towards non-natural substrates. The presented cascade enabled the production of various N-heterocycles and gave insights into the selectivity and specificity of the putrescine oxidase. By optimized conditions for increased selectivity, the enantiomeric excess was enhanced up to 93% ee for 13 and demonstrated the kinetic resolution behaviour of the applied oxidase. Importantly, the H2-driven NADPH regeneration by soluble hydrogenase was well sufficient and surpassed the commonly used glucose-6-phosphate dehydrogenase system with no by-product formation and 100% atom efficiency. Further benefits of the described enzymatic cascade include the prevention of organic solvents and toxic compounds demonstrating the “greeness” of our approach. To the best of our knowledge, this work constitutes an important advance in the field by opening a one-pot and selective access to pyrrolidine and piperidine nitrogen heterocycles from diamines, thus definitely increasing the attractiveness of the H2-driven regeneration platform.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. B. and A. A-S. have received funding from the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG, project number 284111627). L. L. was supported through the Cluster of Excellence “Unifying Concepts in Catalysis” (UniCat, EXC314-2). We thank Reinhard Schömäcker and Gabrielle Vetter at the Technische Universität Berlin for product analysis by gas chromatography.References
- C. Lamberth and J. Dinges, in Bioactive Heterocyclic Compound Classes, John Wiley & Sons, Ltd, 2012, pp. 1–20 Search PubMed.
- C. V. T. Vo and J. W. Bode, J. Org. Chem., 2014, 79, 2809–2815 CrossRef CAS PubMed.
- J. H. Schrittwieser, J. Sattler, V. Resch, F. G. Mutti and W. Kroutil, Curr. Opin. Chem. Biol., 2011, 15, 249–256 CrossRef CAS PubMed.
- E. Ricca, B. Brucher and J. H. Schrittwieser, Adv. Synth. Catal., 2011, 353, 2239–2262 CrossRef CAS.
- S. P. France, L. J. Hepworth, N. J. Turner and S. L. Flitsch, ACS Catal., 2017, 7, 710–724 CrossRef CAS.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed.
- M. Genz, V. Köhler, M. Krauss, D. Singer, R. Hoffmann, T. R. Ward and N. Sträter, ChemCatChem, 2014, 6, 736–740 CrossRef CAS.
- I. Slabu, J. L. Galman, N. J. Weise, R. C. Lloyd and N. J. Turner, ChemCatChem, 2016, 8, 1038–1042 CrossRef CAS.
- J. L. Galman, I. Slabu, N. J. Weise, C. Iglesias, F. Parmeggiani, R. C. Lloyd and N. J. Turner, Green Chem., 2017, 19, 361–366 RSC.
- G. A. Aleku, S. P. France, H. Man, J. Mangas-Sanchez, S. L. Montgomery, M. Sharma, F. Leipold, S. Hussain, G. Grogan and N. J. Turner, Nat. Chem., 2017, 9, 961 CrossRef CAS PubMed.
- M. Gand, H. Müller, R. Wardenga and M. Höhne, J. Mol. Catal. B: Enzym., 2014, 110, 126–132 CrossRef CAS.
- S. Hussain, F. Leipold, H. Man, E. Wells, S. P. France, K. R. Mulholland, G. Grogan and N. J. Turner, ChemCatChem, 2015, 7, 579–583 CrossRef CAS PubMed.
- F. Leipold, S. Hussain, D. Ghislieri and N. J. Turner, ChemCatChem, 2013, 5, 3505–3508 CrossRef CAS.
- H. Man, E. Wells, S. Hussain, F. Leipold, S. Hart, J. P. Turkenburg, N. J. Turner and G. Grogan, ChemBioChem, 2015, 16, 1052–1059 CrossRef CAS PubMed.
- D. Wetzl, M. Berrera, N. Sandon, D. Fishlock, M. Ebeling, M. Müller, S. Hanlon, B. Wirz and H. Iding, ChemBioChem, 2015, 16, 1749–1756 CrossRef CAS PubMed.
- P. N. Scheller and B. M. Nestl, Appl. Microbiol. Biotechnol., 2016, 100, 10509–10520 CrossRef CAS PubMed.
- P. N. Scheller, S. Fademrecht, S. Hofelzer, J. Pleiss, F. Leipold, N. J. Turner, B. M. Nestl and B. Hauer, ChemBioChem, 2014, 15, 2201–2204 CrossRef CAS PubMed.
- L. Lauterbach, O. Lenz and K. A. Vincent, FEBS J., 2011, 280, 3058–3068 CrossRef PubMed.
- E. W. van Hellemond, M. van Dijk, D. P. H. M. Heuts, D. B. Janssen and M. W. Fraaije, Appl. Microbiol. Biotechnol., 2008, 78, 455–463 CrossRef CAS PubMed.
- M. Alexeeva, R. Carr and N. J. Turner, Org. Biomol. Chem., 2003, 1, 4133–4137 RSC.
- R. J. DeSa, J. Biol. Chem., 1972, 247, 5527–5534 CAS.
- E. W. van Hellemond, H. Mazon, A. J. Heck, R. H. H. van den Heuvel, D. P. H. M. Heuts, D. B. Janssen and M. W. Fraaije, J. Biol. Chem., 2008, 283, 28259–28264 CrossRef CAS PubMed.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2018, 46, W296–W303 CrossRef PubMed.
- E. Krieger, K. Joo, J. Lee, J. Lee, S. Raman, J. Thompson, M. Tyka, D. Baker and K. Karplus, Proteins: Struct., Funct., Bioinf., 2009, 77, 114–122 CrossRef CAS PubMed.
- M. M. Kopacz, D. P. H. M. Heuts and M. W. Fraaije, FEBS J., 2014, 281, 4384–4393 CrossRef CAS PubMed.
- L. Lauterbach and O. Lenz, J. Am. Chem. Soc., 2013, 135, 17897–17905 CrossRef CAS PubMed.
- A. K. Holzer, K. Hiebler, F. G. Mutti, R. C. Simon, L. Lauterbach, O. Lenz and W. Kroutil, Org. Lett., 2015, 17, 2431–2433 CrossRef CAS PubMed.
- J. Ratzka, L. Lauterbach, O. Lenz and M. B. Ansorge-Schumacher, Biocatal. Biotransform., 2011, 29, 246–252 CrossRef CAS.
- M. Andersson, H. Holmberg and P. Adlercreutz, Biotechnol. Bioeng., 2000, 57, 79–86 CrossRef.
- T. H. Lonsdale, L. Lauterbach, S. Honda Malca, B. M. Nestl, B. Hauer and O. Lenz, Chem. Commun., 2015, 51, 16173–16175 RSC.
- M. A. Sprung, Chem. Rev., 1940, 26, 297–338 CrossRef CAS.
- S. Roth, A. Präg, C. Wechsler, M. Marolt, S. Ferlaino, S. Lüdeke, N. Sandon, D. Wetzl, H. Iding, B. Wirz and M. Müller, ChemBioChem, 2017, 18, 1703–1706 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8gc03798a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |