
Acerola polysaccharides ameliorate high-fat diet-induced non-alcoholic fatty liver disease through reduction of lipogenesis and improvement of mitochondrial functions in mice
Yuanyuan
Hu
 a,
Fawen
Yin
ab,
Zhongyuan
Liu
a,
Hongkai
Xie
c,
Yunsheng
Xu
d,
Dayong
Zhou
*ab and
Beiwei
Zhu
abc
a,
Fawen
Yin
ab,
Zhongyuan
Liu
a,
Hongkai
Xie
c,
Yunsheng
Xu
d,
Dayong
Zhou
*ab and
Beiwei
Zhu
abc
aSchool of Food Science and Technology, Dalian Polytechnic University, Dalian, 116034, PR China. E-mail: zdyzf1@163.com; Fax: +86-0411-86323262; Tel: +86-0411-86323262
bNational Engineering Research Center of Seafood, Dalian, 116034, PR China
cBeijing Advanced Innovation Centre of Food Nutrition and Human Health, China Agricultural University, Beijing, 100083, PR China
dSchool of Food Science and Engineering, Hainan Tropical Ocean University, Sanya, 572022, PR China
First published on 14th November 2019
Abstract
Acerola polysaccharides (ACPs) were purified from acerola (Malpighia emarginata DC.), a tropical fruit with strong antioxidant and anti-inflammatory activities. However, the biological activities of ACPs have barely been investigated. The present study was designed to investigate the efficacy of ACPs in the treatment of high-fat diet (HFD)-induced nonalcoholic fatty liver disease (NAFLD) in C57BL/6 mice. Male C57BL/6 mice were fed with a high-fat diet and treated with different doses of ACPs for 9 continuous weeks. NAFLD was examined in terms of body weight, lipid profiles, liver function markers, and histology. Gene expression was determined by using both qRT-PCR and western blot. Our results showed that administration of ACPs significantly reduced HFD-induced hyperlipidemia and hepatic lipid deposition by inhibiting the SREBP1c pathway in mice. ACP treatment normalized oxidative stress by activating nuclear factor (erythroid-derived-2)-like 2 (Nrf2) and reduced the expressions of pro-inflammatory cytokines in HFD fed mice. Furthermore, ACPs reduced uncoupling protein 2 (UCP2) expression, restored mitochondrial ATP content, increased mitochondrial complex I, IV, and V activity, and increased mitochondrial beta-oxidation by stimulating peroxisomal proliferator-activated receptor-gamma coactivator-1α (PGC-1α) in the liver of HFD-fed mice. Our study indicated that ACPs may be an effective dietary supplement for preventing HFD-induced NAFLD by regulating lipogenesis, reducing inflammation and oxidative stress, and promoting the mitochondrial function.
1. Introduction
The initial accumulation of fat followed by subsequent inflammation is central to the development of liver damage.1,2 Nonalcoholic fatty liver disease (NAFLD), which represents a series of liver pathologies ranging from simple steatosis to steatohepatitis through to fibrosis and cirrhosis, is the most common chronic liver disease.3 The current model of NAFLD is evolved via “two-hit hypothesis”. The first is the insulin resistance (IR) that enhances dietary influx and increases hepatic lipogenesis, resulting in the accumulation of triglycerides (TG) and free fatty acids in the liver, and the second is the combination of lipid peroxidation, mitochondrial dysfunction and inflammation, leading to hepatocyte damage and development of liver fibrosis.4 It has been widely recognized that NAFLD is the hepatic manifestation of obesity. Excessive exposure to a high-fat diet (HFD) has been found as a key attribute in a growing number of individuals with NAFLD,5 which is indicated by the fact that the prevalence of NAFLD is reported to be up to 90% in obese patients.1 With the globalization of obesity and its related metabolic syndrome, the prevalence of NAFLD will likely continue to rise needing urgent attention around the world.6,7Currently, lifestyle intervention with diet and exercise is still the mainstay in the management of patients with NAFLD; the degree of liver histological improvement is directly proportional to the amount of weight lost. However, it can be difficult to implement.8 Therapeutic drug modalities for NAFLD targeted toward some induction factors such as insulin resistance, dyslipidemia and oxidative stress might also mitigate the progression of this pathology.1,8 Recently, some natural compounds, including vitamin E and omega-3 polyunsaturated fatty acids, have been demonstrated to exhibit effective effects on NAFLD.1 Thus, the regular supplementation of natural bioactive substances may become a promising strategy for NAFLD control since it exhibited effective outcomes with controlled side effects.
Polysaccharides are the most abundant group of biopolymers with a remarkable biological activity,9 such as hypolipidemic/hypoglycemic effects or antioxidative properties. Interestingly, these polysaccharides have shown appreciable effects on the amelioration of NAFLD from different sources, such as Angelica sinensis, Lycium barbarum, and Ganoderma lucidum.10 Acerola cherries, a common traditional medicinal ingredient, showed various pharmacological activities, including antioxidant, anti-inflammatory, antitumor, hypolipidemic, antihyperglycemic, hepatoprotective, and intestinal health and skin protecting activities.11 Notably, acerola contains considerable dietary fibers, a kind of polysaccharide, with approximately 10% of the dry fruit.12 Recently, pectic polysaccharides from acerola have been reported to have high antioxidant and anti-fatigue activities.13 However, their effects on NAFLD, a condition that is closely associated with oxidative stress and inflammation, have not been investigated. Thus, in this study, the beneficial effects of acerola polysaccharides on HFD-induced NAFLD were investigated through a well-established mouse model for the first time, and the mechanism behind them was also explored. This will help us to better understand the health benefits of acerola cherries, as well as to provide a theoretical basis for utilization of acerola polysaccharides as novel sources of functional foods.
2. Materials and methods
2.1. Extraction of polysaccharides
Acerola cherries were obtained from Dahailan planting base (Hainan, China), and the acerola polysaccharides were extracted by the method of hot-water extraction and ethanol precipitation as previously described in ref. 12. Briefly, after removing seeds, the acerola fruit was homogenized, freeze-dried and defatted. The polysaccharides were extracted with hot water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20, w/v) under reflux at 80 °C for 3 h. After three cycles, the aqueous extracts were combined and concentrated to 10% of the original volume under reduced pressure, and then it was precipitated with three times the volume of ethanol at 4 °C overnight. The refined pellets were dissolved in distilled water and deproteinized using the Sevag reagent (chloroform:n-butyl alcohol = 4:1, v/v), and then dialyzed for 48 hours against distilled water (cut-off Mw 14 kDa). Finally, the remaining portion was lyophilized to obtain refined acerola polysaccharides (ACPs) with a light brown fluffy shape.
:20, w/v) under reflux at 80 °C for 3 h. After three cycles, the aqueous extracts were combined and concentrated to 10% of the original volume under reduced pressure, and then it was precipitated with three times the volume of ethanol at 4 °C overnight. The refined pellets were dissolved in distilled water and deproteinized using the Sevag reagent (chloroform:n-butyl alcohol = 4:1, v/v), and then dialyzed for 48 hours against distilled water (cut-off Mw 14 kDa). Finally, the remaining portion was lyophilized to obtain refined acerola polysaccharides (ACPs) with a light brown fluffy shape.
2.2. Chemical analysis of ACPs
The total carbohydrate in ACPs was determined using the phenol–sulfuric acid colorimetric method with glucose as a standard.14 The uronic acid content was determined using the modified m-hydroxybiphenyl method with galacturonic acid as a standard.15 The determination of proteins was carried out using the Bradford method employing bovine serum albumin as a standard.16 The content of total polyphenols was determined according to the Folin–Ciocalteu method,17 adapted to microplates. Monosaccharide composition analysis of ACPs was carried out by using high performance liquid chromatography (HPLC) with 1-phenyl-3-methyl-5-pyrazolone (PMP) derivatization.18 Briefly, the ACPs (5.0 mg) were hydrolyzed with 2.0 mM trifluoroacetic acid at 120 °C for 3 h, and then labeled with PMP, and subsequently the analysis of PMP-labeled monosaccharides was performed in a HPLC system of Water Alliance coupled with a photo-diode array detector (Waters 2998, USA). The analytical column used was a Silgreen ODS C18 column (250 × 4.6 mm, 5 μm) at 30 °C. Isocratic elution of 20 mM ammonium acetate:acetonitrile (78:22) was performed at a flow rate of 1 mL min−1. The homogeneity and molecular weight (Mw) distributions of ACPs were evaluated by gel permeation chromatography (GPC) on a TSK G4000PWXL column, eluting with 100 mM ammonium acetate at a flow rate of 0.4 mL min−1 at 30 °C and detecting with a refractive index (Waters 2414, USA). For Fourier transform infrared spectroscopy (FT-IR) assay, the ACPs (2 mg) were mixed with dried KBr (100 mg) and then pressed to a transparent film. The film was scanned by using a Spectrum One-B FTIR Spectrometer (PerkinElmer, 118 Waltham, MA, USA) in the frequency range of 400–4000 cm−1 three times.
2.3. Animals and experimental design
The animal use and experimental procedure were approved by the Institutional Animal Care and Use Committee of Dalian Polytechnic University (LZ0160258) according to the Guidelines of Experimental Animal Administration issued by the State Committee of Science and Technology of People's Republic of China. All efforts were made to minimize animal suffering. Male C57BL/6 mice (SPF, 6 weeks) were purchased from the Experimental Animal Center of Dalian Medical University and housed in the Animal Center of Dalian Polytechnic University. Mice were kept under conditions of 22 ± 1 °C, relative humidity of 55 ± 5% and 12 h light/dark cycle, with ad libitum access to food and water. After one week of adaptation to the laboratory environment, the mice were randomly assigned to six groups (n = 8 per group) and fed with (1) normal-chow diet (D12450B; 10 kcal%; Research Diet Inc., USA) with administration of physiological saline as the healthy control, (2) normal-chow diet plus daily administration of 800 mg kg−1 day−1 ACPs, (3) high-fat diet (HFD; D12492; 60 kcal%; Research Diet Inc., USA) with administration of physiological saline as the model control, (4) HFD plus daily administration of 200 mg kg−1 day−1 ACPs, (5) HFD plus daily administration of 400 mg kg−1 day−1 ACPs or (6) HFD plus daily administration of 800 mg kg−1 day−1 ACPs by intragastric gavage for nine consecutive weeks. All the administrations were conducted between eight and nine o'clock in the morning. Weekly monitoring centered on body indexes, such as the body weight, food intake, and water intake. Two hours after final administration, all animals were strictly fasted but allowed free access to water for 12 h. Animals were then anesthetized with sodium pentobarbital anesthesia and sacrificed by cervical dislocation. Serum was separated by centrifuging blood samples at 3500 rpm for 15 min and then freezing at −20 °C until further use.2.4. Preparation of liver homogenates and mitochondria
At the end of treatment, liver tissues were quickly excised, weighed, and homogenized by using an automatic homogenizer in a 10-fold volume (v/w) of ice-cold saline, followed by 12000 rpm centrifugation at 4 °C for 15 min. The supernatant was then collected and frozen at −20 °C until further use. Mitochondria were isolated by differential centrifugation as previously described in ref. 19. Briefly, liver tissues were homogenized with an automatic homogenizer in ice-cold isolation buffer containing 0.25 M sucrose, 3 mM HEPES, and 0.5 mM EDTA (pH 7.4). The homogenates were centrifuged at 1000 rpm for 10 min. Then the collected supernatants were washed using isolation buffer and centrifuged at 10000 rpm for 10 min. Finally, the mitochondrial pellet was washed twice with the isolation buffer and resuspended in the same medium. The isolated mitochondria were stored at −20 °C until use. The protein concentrations were quantified by using a BCA assay kit (Beyotime, China).
2.5. Measurement of biochemical parameters
Triacylglycerol (TG), total cholesterol (TC), low-density lipoprotein cholesterol (LDL), high-density lipoprotein cholesterol (HDL), alanine aminotransferase (ALT), glucose, malondialdehyde (MDA), glutathione (GSH)/glutathione (GSSG), catalase (CAT), and superoxide dismutase (SOD) levels in the serum or liver homogenates were determined using biochemical kits according to the manufacturer's instructions (Nanjing Jiancheng Bioengineering Institute, China). Insulin, non-esterified free fatty acid (NEFA), C-reactive protein (CRP), tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), interleukin-1β (IL-1β), and adiponectin in the serum or liver homogenates were determined using commercially available ELISA kits (Nanjing Jiancheng Bioengineering Institute, China). Adenosine triphosphate (ATP) was measured in fresh liver tissue with an ATP assay kit (Beyotime, China). Total protein contents in the homogenates were determined by using the BCA assay kit (Beyotime, China).2.6. Histopathological observation
Histology of livers was examined in the tissue section with hematoxylin and eosin (H&E) staining and Oil Red O staining. For H&E staining, portions of liver were fixed in 10% paraformaldehyde solution. Fixed tissues were embedded in paraffin, sectioned (5–6 μm), and stained with H&E (Solarbio, China). For Oil Red O staining, fresh liver tissues were made into frozen sections using a cryostat (CM1950, Leika, Germany), then fixed and stained. The stained area was detected under an Olympus light microscope (DdM-LB2, Leica, Germany). Finally, images were examined and evaluated for pathological changes.2.7. RNA preparation and qRT-PCR analysis
Total RNA was extracted from 30 mg of liver homogenates using Trizol reagent (TaKaRa Bio. Inc., China) following the manufacturer's instructions. The OD260/OD280 ratio was measured to determine the quality of the RNA. cDNA was made by reverse transcription of RNA with the PrimeScript RT Master Mix (TaKaRa Bio. Inc., China). The levels of gene expressions were determined by using the SYBR Green Master Mix (TaKaRa Bio. Inc., China), and analyzed on a qTOWER3 G device (Analytik Jena) according to the protocol of the manufacturer. β-Actin was chosen as a housekeeping gene, and mean expression levels of a normal control group were set as 100%. All samples were used in duplicate in a single plate and relative quantification was calculated by the double δ cycle threshold (2−ΔΔCt) method. The primer sequences of sterol regulatory element binding protein 1c (SREBP1c), fatty acid synthase (FAS), acetyl–CoA carboxylase (ACC), stearoyl–CoA desaturase (SCD-1), peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and β-actin were obtained from Applied Biosystems (Table 1).| Gene name | Forward | Reverse |
|---|---|---|
| SREBP1c | 5′-AACCTCATCCGCCACCTG-3′ | 5′-TGGTAGACAACAGCCGCATC-3′ |
| FAS | 5′-TTGATGATTCAGGGAGTGG-3′ | 5′-AGCAGATGAGTTGTTCTTGGAC-3′ |
| ACC | 5′-TTGCCTATGAACTCAACAGCG-3′ | 5′-AGACCATTCCGCCCATCC-3′ |
| SCD-1 | 5′-CCACTCGCCTACACCAACG-3′ | 5′-GGGGTCCCTCCTCATCCT-3′ |
| PGC-1α | 5′-CCTTTCTGAACTTGATGTGA-3′ | 5′-ATGCTCTTTGCTT-TATTGCT-3′ |
| β-Actin | 5′-TCTACAATGAGCTGCGTGTG-3′ | 5′-GGTCAGGATCTTCATGAGGT-3′ |
2.8. Western blot analysis
Protein extraction and western blotting were performed as previously described in ref. 20. Briefly, liver homogenates were lysed with lysis buffer (Beyotime, China) on ice, and then were centrifuged at 10000 rpm for 10 min at 4 °C. The supernatant was transferred to a tube kept on ice and stored at −80 °C. Protein concentrations were quantified by using the BCA assay kit (Beyotime, China). After being boiled in the loading buffer for 10 min at 100 °C, protein samples were separated by electrophoresis using SEMS-PAGE, and then transferred to FL-PVDF membranes using semi-dry transfer apparatus (Bio-Rad, China). The membranes were blocked with 5% milk for 1 h at room temperature and incubated overnight with the appropriate primary antibodies, respectively, followed by incubation with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies. β-Actin was used as the internal loading control. The results were visualized with ECL-plus reagent and analyzed using a Quantity One System (Bio-Rad, CA, USA). The primary antibodies used in this study included SREBP1c, nuclear factor (erythroid-derived-2)-like 2 (Nrf2), heme oxygenase-1 (HO-1), NAD(P)H: quinone oxidoreductase 1 (NQO-1), uncoupling protein 2 (UCP2), PGC-1α, β-actin (Santa Cruz Biotechnology, USA), and complex I, II, III, IV, and V subunits (BD Biosciences, USA).
2.9. Statistical analysis
Data were presented as means ± standard error of the mean (SEM), and compared by Student's t-test (between two groups) or ANOVA (among three or more groups) followed by Bonferroni's correction where appropriate. The differences were considered significant when P < 0.05. Statistical tests were performed using GraphPad Prism software version 5.0 (GraphPad Sofware, Inc., San Diego, CA).3. Results
3.1. Chemical composition of ACPs
The crude polysaccharide fraction of ACPs was about 10.05% dry weight of the defatted acerola powder. The content of the total carbohydrate, uronic acid, protein and phenolic compounds in ACPs was 74.77, 13.93, 2.55 and 0.21% (w/w), respectively. HPLC analysis showed that ACPs were composed of mannose, rhamnose, glucuronic acid, glucose, xylose, galactose, and arabinose, with their corresponding molar percentages of 2.10, 4.37, 12.48, 7.25, 13.71, 59.81, and 0.28%, respectively (Fig. 1A and B). As a result, ACPs were shown to be a typical hetero-polysaccharide with high purity. The GPC profiles (Fig. 1C) showed that the front peak is ACPs with a Mw of 37.07 kDa. Furthermore, the FT-IR spectra of ACPs showed similar IR signal bands of typical absorption peaks assigned to the saccharide moiety (Fig. 1D). Results showed that the absorptions at 3390.12 and 2930.81 cm−1 were due to stretching vibration of O–H groups and C–H bonds, respectively. Absorptions at 1102.65 and 1068.77 cm−1 corresponded to the stretching vibration of C–O–C and C–O–H, indicating a pyranose unit. Absorptions at 1746.67, 1625.28 and 1441.44 cm−1 corresponded to carboxyl groups (COO–) arising from an asymmetrical C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration and a symmetrical C–O stretching vibration, respectively.
O stretching vibration and a symmetrical C–O stretching vibration, respectively.
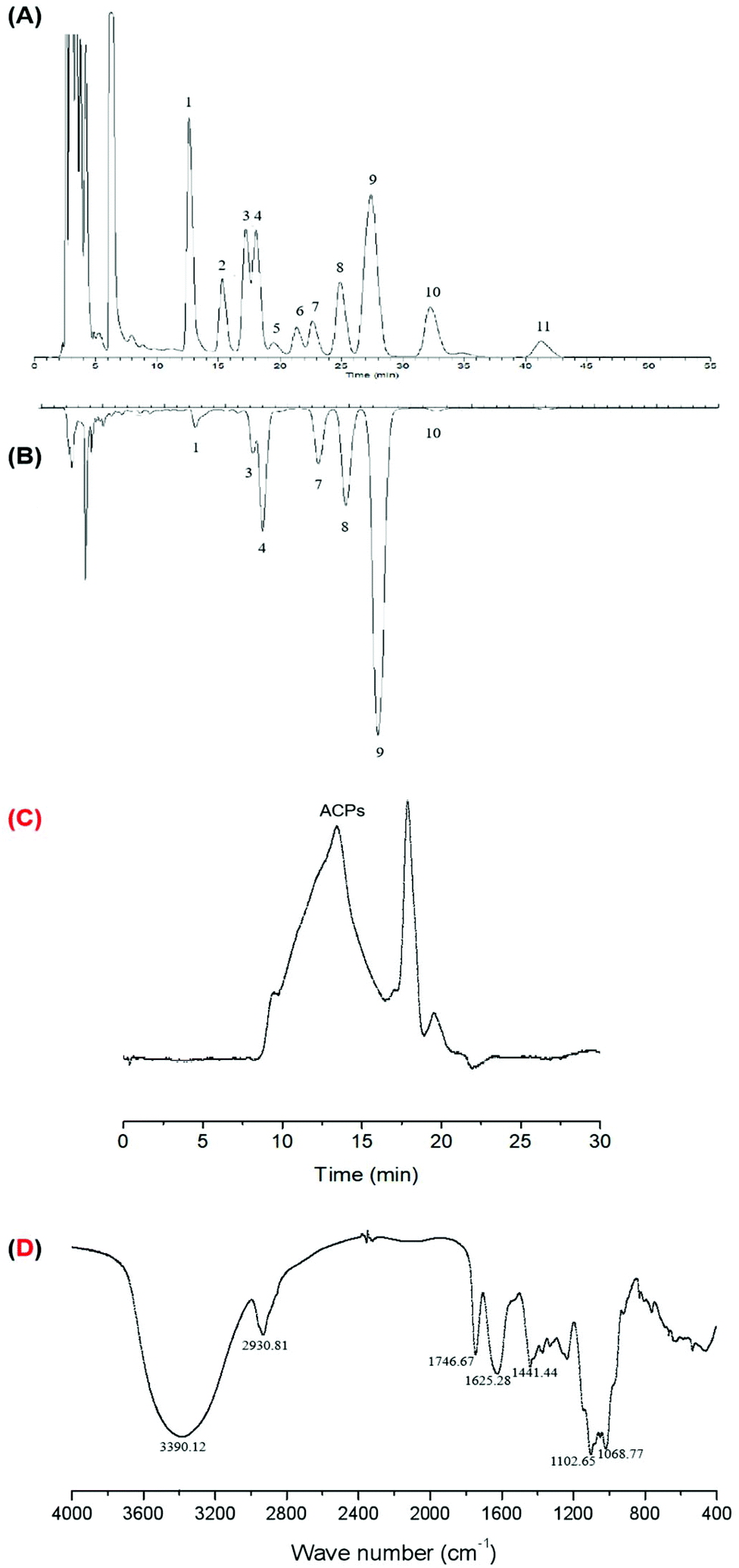 | ||
| Fig. 1 The HPLC chromatograms of PMP derivatives of (A) standard monosaccharides and (B) component monosaccharides of ACPs. Peaks: (1) mannose, (2) glucosamine, (3) rhamnose, (4) glucuronic acid, (5) galacturonic acid, (6) galactosamine, (7) glucose, (8) xylose, (9) galactose, (10) arabinose, and (11) fucose. (C) Gel permeation chromatography of ACPs. (D) FT-IR spectra of ACPs. | ||
3.2. ACPs prevented HFD-induced obesity in mice
C57BL/6J mice fed a HFD are the most commonly used animal model for the study of obesity-associated NAFLD.21 Obesity was induced by the administration of a HFD over a 9-week period. As shown in Table 2, the HFD significantly increased body and liver weights. However, oral administration of ACPs attenuated the HFD-induced increases in body and liver weights in a dose dependent manner without affecting food intake. Additionally, high concentrations of ACPs did not produce any notable effects in normal-fed mice.| Parameters | Control | Control + ACPs 800 | HFD | HFD + ACPs 200 | HFD + ACPs 400 | HFD + ACPs 800 |
|---|---|---|---|---|---|---|
| Values are expressed as means ± SEM of 8 mice in each group. Means within a line with a common letter are equal, P < 0.05. | ||||||
| Initial body wt (g) | 20.67 ± 1.03 | 20.58 ± 1.46 | 20.43 ± 0.70 | 20.34 ± 1.50 | 20.65 ± 1.70 | 20.83 ± 0.83 |
| Terminal body wt (g) | 26.72 ± 1.76a | 25.60 ± 1.78a | 37.40 ± 2.09c | 36.73 ± 3.08c | 32.48 ± 2.61b | 31.65 ± 3.24b |
| Body weight gain wt (g) | 6.05 ± 1.57a | 5.02 ± 2.28a | 16.96 ± 2.00c | 16.39 ± 3.37c | 11.83 ± 2.66b | 10.82 ± 3.49b |
| Liver wt (g) | 1.21 ± 0.09a | 1.17 ± 0.12a | 1.53 ± 0.15c | 1.42 ± 0.11b | 1.30 ± 0.11ab | 1.25 ± 0.14ab |
| Food intake (g day−1) | 4.12 ± 1.61 | 4.07 ± 1.35 | 3.16 ± 2.05 | 3.08 ± 2.33 | 3.41 ± 1.69 | 3.26 ± 1.37 |
3.3. ACPs reduced hyperglycemia and dyslipidemia in HFD-fed mice
The HFD-induced obesity model is usually accompanied by insulin resistance and lipid disorder.22 We therefore examined the effects of ACPs on serum glucose, insulin, and lipid profiles. As shown in Table 3, ACPs did not significantly affect these parameters in normal-fed mice. However, middle- and high-doses of ACPs effectively reduced the HFD-induced high glucose level, and a high-dose of ACPs inhibited the increase of the insulin level. In addition, the serum TG, TC, and LDL levels increased by the HFD, which were reversed only by treatment with the high-dose ACP supplementation. The HFD elicited no significant effect on HDL, whereas high-dose ACP treatments significantly increased its serum levels (P < 0.05; Table 3). These findings suggest that ACPs might maintain the glucose homeostasis and prevent dyslipidemia in HFD-fed mice.| Parameters | Control | Control + ACPs 800 | HFD | HFD + ACPs 200 | HFD + ACPs 400 | HFD + ACPs 800 |
|---|---|---|---|---|---|---|
| Values are expressed as means ± SEM of 8 mice in each group. Means within a line with a common letter are equal, P < 0.05. | ||||||
| Glucose (mmol L−1) | 5.46 ± 1.35a | 5.26 ± 1.35a | 8.64 ± 1.21b | 6.76 ± 2.19ab | 6.01 ± 1.42a | 5.39 ± 1.52a |
| Insulin (ng mL−1) | 2.68 ± 0.66a | 2.49 ± 0.47a | 5.93 ± 1.06b | 4.21 ± 1.41ab | 4.27 ± 0.76ab | 3.18 ± 0.81a |
| TC (mmol L−1) | 3.00 ± 0.28a | 2.86 ± 0.32a | 5.51 ± 1.17b | 4.09 ± 1.09ab | 3.66 ± 0.76ab | 3.15 ± 0.93a |
| TG (mmol L−1) | 0.89 ± 0.13a | 0.81 ± 0.19a | 1.58 ± 0.27b | 1.30 ± 0.33ab | 1.32 ± 0.35ab | 1.05 ± 0.31a |
| LDL (mmol L−1) | 0.22 ± 0.04a | 0.23 ± 0.04a | 0.39 ± 0.10b | 0.37 ± 0.08ab | 0.33 ± 0.12ab | 0.27 ± 0.06a |
| HDL (mmol L−1) | 1.46 ± 0.24ab | 1.52 ± 0.31ab | 1.03 ± 0.16a | 1.13 ± 0.24ab | 1.34 ± 0.25ab | 1.68 ± 0.36b |
3.4. ACPs reduced lipid accumulation in livers of HFD-fed mice
As shown in Fig. 2A, ALT, a major marker of liver injury,23 was significantly increased after HFD feeding, while only the high-dose of ACPs effectively prevented this injury. Together with failed effects of low- and middle-doses of ACPs in serum TG or TC, we inferred that only the high-dose of ACPs has significant protective effects on NAFLD. So the liver tissues from high-dose ACP treated mice were selected for further analysis. Obviously, high fat-diet intervention significantly elevated liver TC, TG, and NEFA levels after 9 weeks of feeding (Fig. 2B–D), whereas ACPs effectively improved the lipid profiles of livers, as evidenced by significant reductions in TC, TG, and NEFA. Furthermore, histopathological observations of H&E in livers of HFD fed mice showed apparent pathological damage, including the disorganized structure of hepatic lobules, tiny swelling in hepatocytes, heavy hepatic steatosis and cytoplasmic vacuolation. However, ACPs markedly ameliorated these hepatic lesions caused by HFD feeding, showing near normal appearance with a well-preserved cytoplasm, prominent nuclei, and legible nucleoli (Fig. 2E). Meanwhile, compared with the control group, the HFD induced massive formation of lipid droplets in the hepatocytes, while ACP intervention obviously eliminated the droplets of fat in the livers, showing near normal appearance (Fig. 2F). These results together with biochemical analysis demonstrated that administration of ACPs could prevent HFD-induced hepatic damage and lipid accumulation in mice.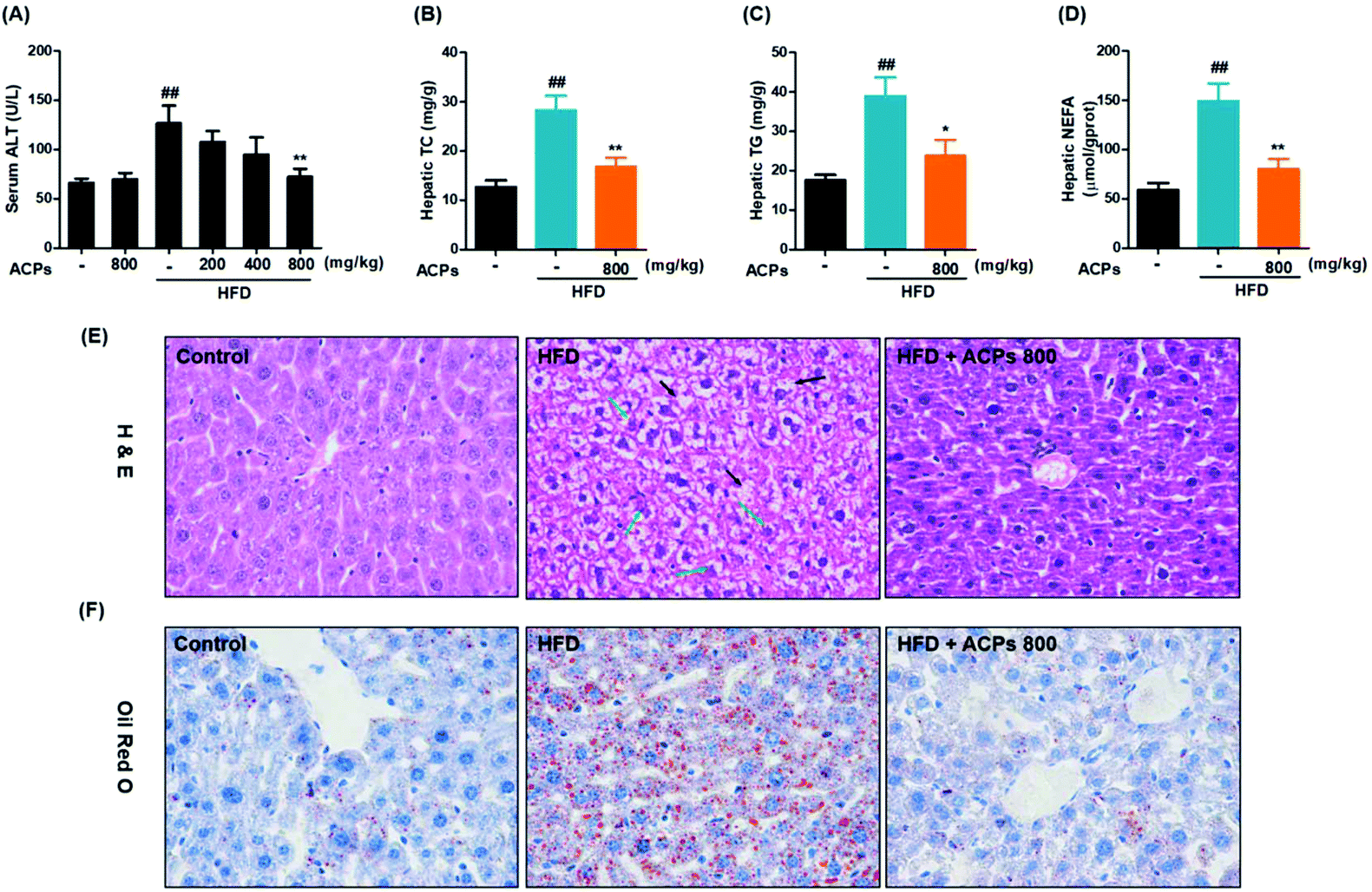 | ||
| Fig. 2 Effects of the HFD and ACPs on liver lipid accumulation in mice fed HFD for 9 continuous weeks. (A) Serum ALT, and (B) hepatic TC, (C) TG, and (D) NEFA levels. (E) Representative histological images of H&E staining in livers (original magnification of 400×). Black arrows: hepatic steatosis with vacuolation; green arrows: necrosis hepatocytes with swelling. (F) Representative histological images of Oil Red O staining in livers (original magnification of 400×). Data are expressed as means ± SEM for 8 mice in each group. #P < 0.05, ##P < 0.01, vs. the normal control group. *P < 0.05, **P < 0.01, vs. the HFD group. | ||
3.5. ACPs inhibited SREBP1c activation in HFD-fed mice
Previous studies have shown that the SREBP1c pathway activation may contribute to the progression of NAFLD.24 Hence, the effects of ACPs on the SREBP1c pathway were investigated in HFD-induced NAFLD. As shown in Fig. 3A and B, both the precursor and mature expressions of SREBP1c were significantly increased in the HFD group. However, the ACPs effectively reduced the SREBP1c levels. Then, the effects of ACPs on the mRNA levels of SREBP1c as well as its target genes FAS, ACC1, and SCD-1 were evaluated by qRT-PCR. As shown in Fig. 3C–F, the levels of hepatic SREBP1c, FAS, ACC, and SCD-1 in the HFD group increased significantly compared with the normal control group, while the ACP co-treated group exhibited marked declines in all of these lipogenic genes. These results suggested that the preventive effect of ACPs on fat accumulation appears to be mediated by inhibiting the SREBP1c pathway in HFD-fed mice.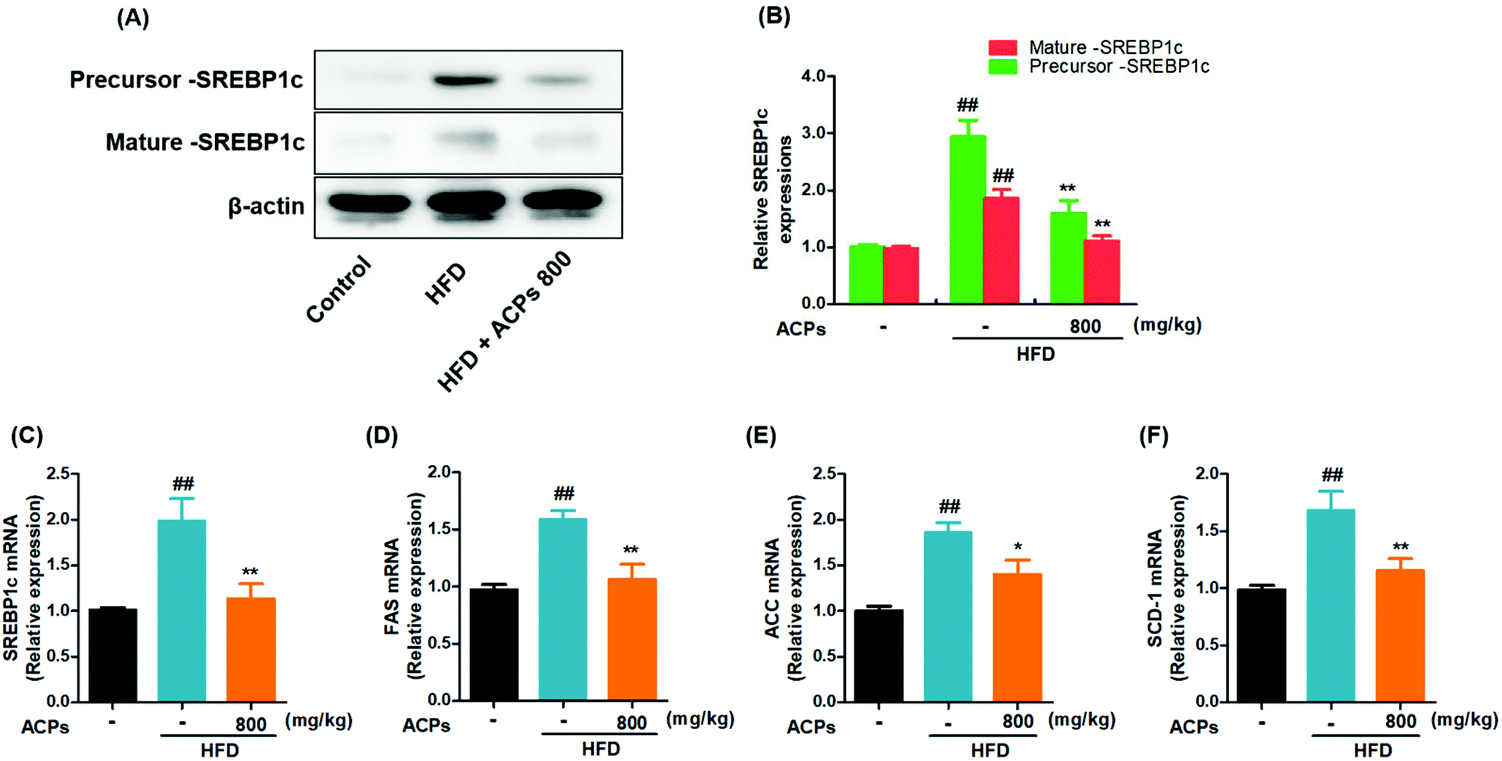 | ||
| Fig. 3 Effects of HFD and ACPs on the SREBP1c pathway in mice fed HFD for 9 continuous weeks. (A) Representative western blot images of the precursor and mature expressions of SREBP1c. (B) Densitometric quantification of SREBP1c levels as a percentage of the control conditions. Gene expressions of (C) SREBP1c, (D) FAS, (E) ACC, and (F) SCD-1. β-Actin was used as an internal control. Data are expressed as means ± SEM for 8 mice in each group. #P < 0.05, ##P < 0.01, vs. the normal control group. *P < 0.05, **P < 0.01, vs. the HFD group. | ||
3.6. ACPs reduced the inflammatory response in HFD-fed mice
Inflammation, a hallmark of nonalcoholic steatohepatitis, is strongly related to the progression of NAFLD.25 In the current study, the HFD significantly increased the CRP, a liver-secreted cytokine in response to inflammatory (Fig. 4A).26 Besides, serum levels of pro-inflammatory cytokines, such as TNF-α, IL-6 and IL-1β, were also elevated (Fig. 4B–D). However, administration of ACPs successfully attenuated the TNF-α, IL-6 and IL-1β levels. In addition, adiponectin, a known anti-inflammatory adipokine, which exerts cytoprotective effects on alcoholic liver injury,27 was markedly decreased in the HFD-fed mice, while ACPs did not significantly prevent this decline (Fig. 4E).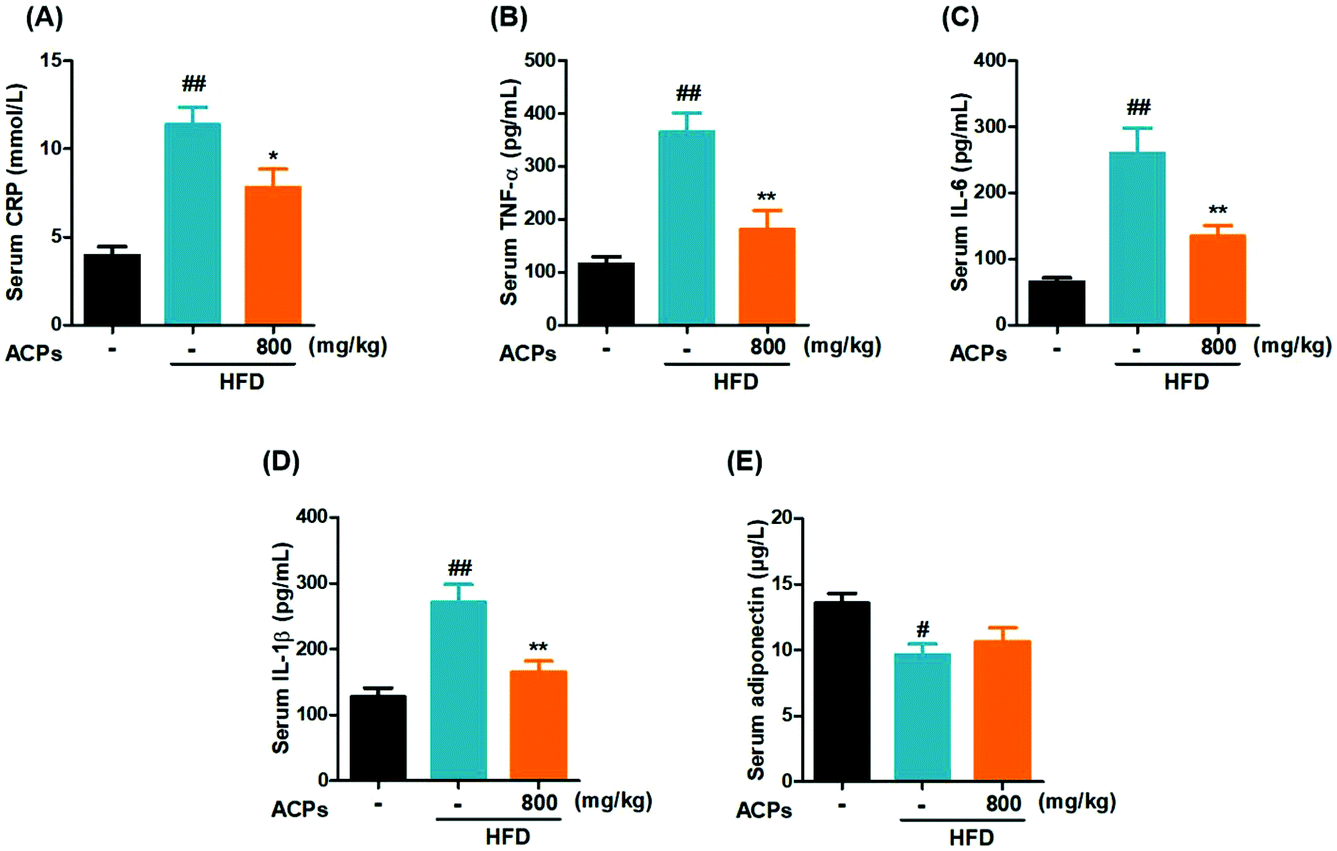 | ||
| Fig. 4 Effects of the HFD and ACPs on the liver inflammatory response in mice fed HFD for 9 continuous weeks. (A) Serum CRP, (B) TNF-α, (C) IL-6, (D) IL-1β, and (E) adiponectin levels. Data are expressed as means ± SEM for 8 mice in each group. #P < 0.05, ##P < 0.01, vs. the normal control group. *P < 0.05, **P < 0.01, vs. the HFD group. | ||
3.7. ACPs ameliorated HFD-induced oxidative stress in mice
Oxidative stress has been well established as a contributor to the pathogenesis of NAFLD.28 Consistent with that previously described, continuous HFD feeding in mice caused severe hepatic oxidative damage, as reflected by significant elevation of MDA, and reductions in SOD, and CAT levels, as well as GSH/GSSG levels (Fig. 5A–D). However, results displayed that ACPs effectively improved the oxidation and deoxidization status in HFD-fed mice by decreasing hepatic MDA formation, enhancing the GSH/GSSG ratio and antioxidant enzymic activities of SOD and CAT (Fig. 5A–D). Since Nrf2, an important factor against oxidative stress, is essential in the modulation of the antioxidant defense system,28 the expressions of Nrf2 as well as its target genes HO-1 and NQO-1 were further examined. As indicated in Fig. 5E and F, HFD feeding significantly downregulated the Nrf2, HO-1, and NQO-1 expressions, whereas co-treatment with ACPs effectively prevented the impairment of the Nrf2 pathway.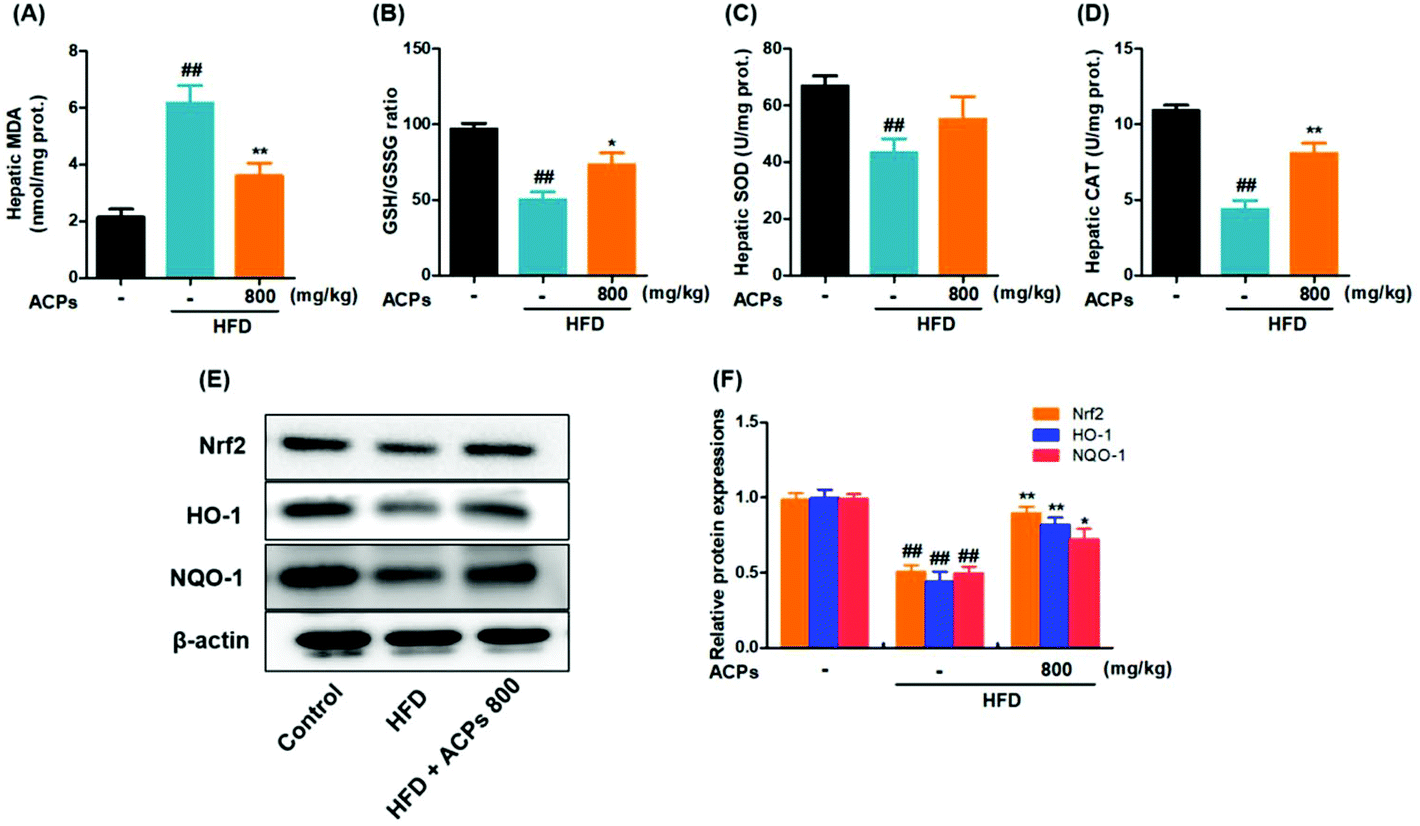 | ||
| Fig. 5 Effects of the HFD and ACPs on liver oxidative status in mice fed HFD for 9 continuous weeks. (A) Hepatic MDA levels. (B) GSH/GSSG ratio. (C) Hepatic SOD and (D) CAT levels. (E) Representative western blot images of Nrf2, HO-1 and NQO-1. (F) Densitometric quantification of Nrf2, HO-1 and NQO-1 levels as a percentage of the control conditions. β-Actin was used as an internal control. Data are expressed as means ± SEM for 8 mice in each group. #P < 0.05, ##P < 0.01, vs. the normal control group. *P < 0.05, **P < 0.01, vs. the HFD group. | ||
3.8. ACPs improved mitochondrial status in HFD mice
To investigate the involvement of mitochondria in HFD induced oxidative stress and NAFLD, the hepatic ATP homeostasis was firstly analyzed. As shown in Fig. 6A, significant ATP depletion was triggered by HFD feeding, while ACP supplementation efficiently improved the ATP levels. To determine whether the reduction of ATP was dependent on an impairment in mitochondrial ATP synthesis, the expressions of complex V (ATP-synthase) were measured, and results showed that the HFD markedly inhibited the complex V expression, whereas co-treatment with ACPs significantly reversed the complex V expression (Fig. 6B and C). UCP2, a negative regulator in the mitochondrial function, could promote proton leakage and result in depletion of cellular ATP reserves.28 Therefore, the effects of HFD and ACPs on UCP2 expressions were determined in mice. As expected, an increased UCP2 expression was observed in HFD-fed mice. However, ACPs markedly restrained the UCP2 expression (Fig. 6B and C).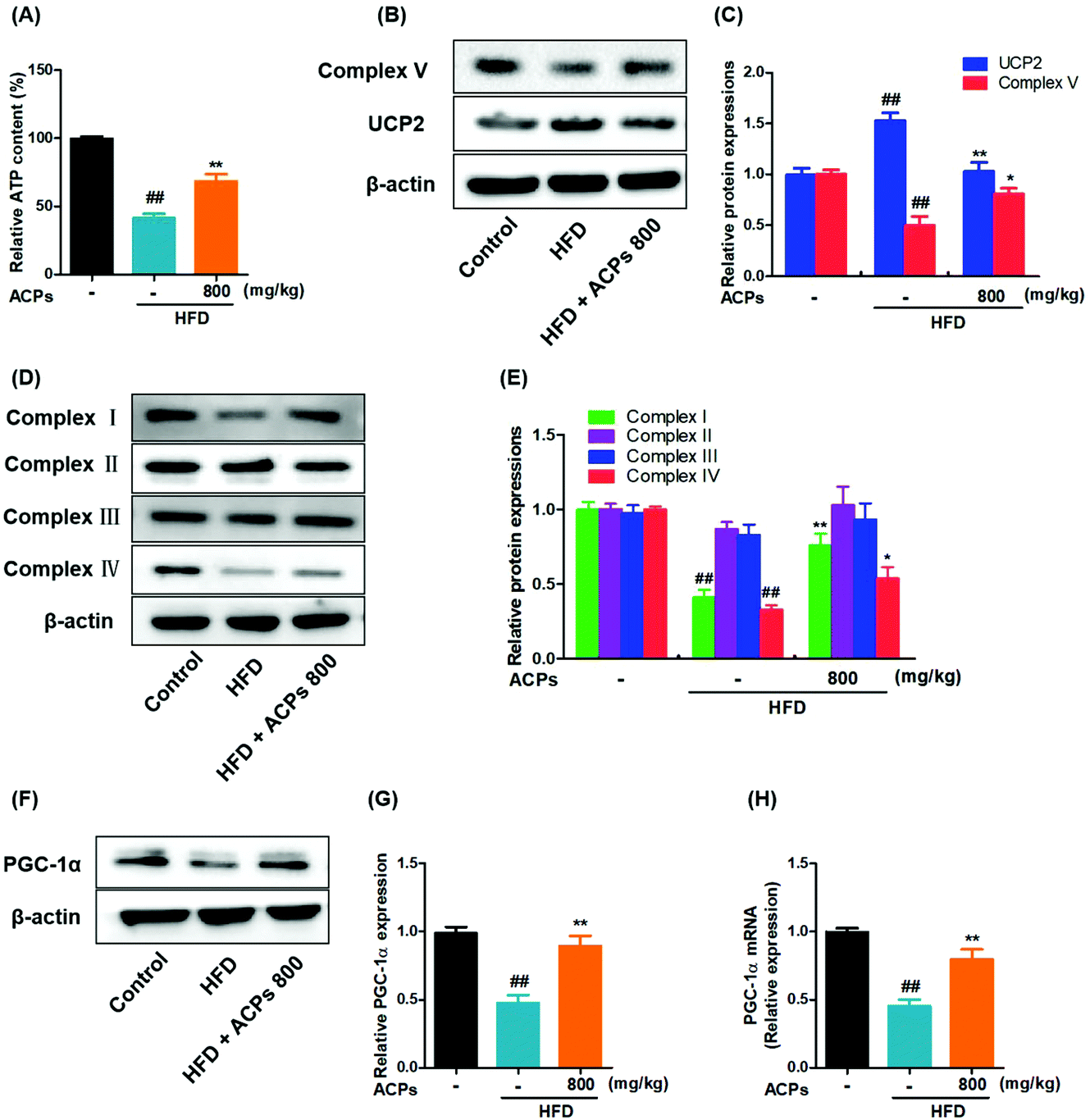 | ||
| Fig. 6 Effects of the HFD and ACPs on mitochondrial status in mice fed HFD for 9 continuous weeks. (A) Hepatic ATP levels. (B) Representative western blot images of complex V and UCP2. (C) Densitometric quantification of complex V and UCP2 levels as a percentage of the control conditions. (D) Representative western blot images of complex I, II, III, and IV. (E) Densitometric quantification of complex I, II, III, and IV levels as a percentage of the control conditions. (F) Representative western blot images of PGC-1α. (G) Densitometric quantification of PGC-1α levels as a percentage of the control conditions. (H) The mRNA levels of PGC-1α. β-Actin was used as an internal control. Data are expressed as means ± SEM for 8 mice in each group. #P < 0.05, ##P < 0.01, vs. the normal control group. *P < 0.05, **P < 0.01, vs. the HFD group. | ||
The mitochondrial respiratory chain is an important source of ROS at the subcellular level and hence a potential contributing factor to NAFLD.29 In our study, the expressions of mitochondrial complexes I and IV were down-regulated in the HFD, suggesting a severe reduction of respiratory chain proteins. However, ACP supplementation restored the mitochondrial complex I and IV activity. Complexes II and III were unaffected by either HFD or ACP treatments (Fig. 6D and E). Furthermore, the effects of HFD and ACPs on mitochondrial β-oxidation were investigated. PGC-1α is a well-known marker of mitochondrial oxidative metabolism, which plays a crucial role in regulating fatty acid β-oxidation.30 We found that ACPs significantly increased the mRNA and protein levels of PGC-1α compared with the HFD group (Fig. 6F–H). Taken together, ACP administration effectively ameliorated the HFD-induced mitochondrial dysfunction in mice.
4. Discussion
NAFLD is the most common chronic liver disease globally, and it is also considered as an independent risk factor for cardiovascular disease, which leads to increased all-cause and liver-related mortality.31 A number of predisposing factors, such as obesity, diabetes, dyslipidemia, drugs and parenteral nutrition, have been related to the prevalence of NAFLD.32 The outcomes of current therapeutic methods, involved in weight loss and the reversal of other components of metabolic pathologies, are not satisfactory.1 In recent years, natural active substances, such as polysaccharides, have been reported to be effective in HFD-induced metabolic disorders, including hyperglycemia, hyperlipidemia, and fatty liver.33 Here, we found that ACPs, hetero-polysaccharides derived from acerola, could have beneficial effects on HFD induced obesity, lipid disorders, and fatty liver by inhibiting lipogenesis, reducing oxidative stress and inflammation, and promoting the mitochondrial function in the HFD-fed C57BL/6 mice.Acerola not only contains high vitamin C and abundant phenolic compounds, but also contains a large amount of polysaccharides.12 Previous studies have demonstrated that intake of acerola juice could help in providing antioxidant defense against oxidative stress,34 reducing inflammation, and regulating obesity-associated defects in the lipolytic processes.35 The present study suggested that ACPs might be one of the key components that play a functional role. In a high-fat induced obesity-mouse model, administration of ACPs inhibited the development of obesity, restored hyperglycemia, and reduced dyslipidemia without affecting the normal diet intake. In this regard, our data indicated that ACPs were a promising candidate for the prevention of HFD-induced metabolic disorders. Since the high-dose of ACPs was found to be more effective in regulating the progression of obesity, and the HFD-induced high level of serum ALT was significantly inhibited only by the high-dose of ACPs, therefore, the high-dose of ACPs was used for the further mechanistic study.
Results of histology staining in livers further confirmed that the high-dose of ACPs significantly reduced accumulation of triglycerides in hepatocytes, so we hypothesized that ACPs may regulate the expression of genes related to fatty acid synthesis. In line with this, Dias et al. have reported that acerola juice supplementation in mice ameliorated obesity-associated defects in the lipolytic processes.36 With regard to lipid biosynthesis, activated SREBP1c can switch on downstream targets of lipogenic genes, including fatty acid synthase (ACC and FAS), and fatty acid converting enzymes (SCD-1), and promote triglyceride synthesis and lipid deposition, which results in NAFLD.24,37 Several studies demonstrated that the gene expressions of hepatic SREBP1c, FAS, ACC, and SCD-1 in obese mice were higher than those in normal control mice.37–39 Consistent with previous studies, the present study showed that HFD feeding significantly increased the levels of SREBP1c, ACC, FAS, and SCD-1, and ACPs markedly suppressed the HFD-activated SREBP1c pathway, which may be related to the lower levels of plasma TG and liver lipids. Overall, these results demonstrated that ACPs may have hepatoprotective effects on NAFLD through downregulating SREBP1c mediated lipid metabolism and lipogenesis.
Except for the activation of lipogenesis, the pathogenesis of NAFLD is usually characterized by inflammation and oxidative stress.40 A previous study reported that intake of acerola juice increased the IL-10/TNF-α ratio and phosphorylation of IκB (inhibitor of NF-κB)-α levels, and also decreased the TNF-α protein levels in mice fed a cafeteria diet.36 In agreement with them, the current study found that ACP treatment completely reversed the abnormal elevations of inflammatory markers in HFD-fed mice, including CRP, TNF-α, IL-6, and IL-1β, clearly indicating that ACPs could efficiently protect hepatocytes against the HFD-induced inflammatory response. Additionally, ACPs diminished the HFD-induced oxidative stress through decreasing MDA levels, and elevating antioxidant enzyme activities (CAT and SOD) and the GSH/GSSG ratio, suggesting that ACPs exert good beneficial effects on HFD-induced hepatic oxidative injury. Nrf2, the most important transcription factor of Phase II enzymes, is a crucial protective protein against the contribution of oxidative stress through the transcriptional activation of antioxidant-responsive element-dependent expression of gene encoding enzymes, such as HO-1.38,41 Nrf-2 activation-mediated hepatic metabolism from lipid storage to lipid oxidation has been demonstrated in mouse and human models.38 In this study, further experiments showed that ACPs significantly activated the Nrf2/HO-1 pathway, indicating an improvement of phase II enzymes. In this regard, previous studies suggested that acerola had good antioxidant effects. For example, a pectic polysaccharide fraction extracted from the acerola fruit was reported to decrease the hydrogen peroxide-induced cytotoxic effects and ROS levels in a murine fibroblast cell line.12 Acerola juice was reported to prevent the diet-induced lipid and protein oxidative damage in the hippocampus, liver, kidney and heart of rats.34 Meanwhile, Alvarez-Suarez et al. found that acerola fruits increased the antioxidant enzyme (SOD, GPx and CAT) expression and also exhibit significant improvement in mitochondrial functionality.42 In addition to these studies, our results suggested that ACP treatment effectively alleviated the HFD-induced inflammation and oxidative stress, showing good hepatoprotective effects.
As a major source of ROS production, hepatic mitochondrial dysfunction is inevitable in the development of NAFLD.43 Increasingly, studies have raised the possibility that NAFLD might be a mitochondrial disease.28 Mitochondrial dysfunction not only impairs fat homeostasis in liver, causing fat accumulation, but also leads to an overproduction of ROS and cytokines, which further exacerbate oxidative stress and induce hepatic inflammation and lethal hepatocyte injury associated with NAFLD.32 Therefore, the involvement of mitochondria in HFD-induced NAFLD was necessary to be evaluated in our study of ACPs. In the current study, results showed that HFD induced mitochondria dysfunction and impaired mitochondria β-oxidation (Fig. 6). Patients with NAFLD have decreased ATP production, and low activity of respiratory proteins in the fat, muscle and liver.44,45 Dysfunction of the mitochondrial respiratory-chain would induce the generation of mitochondrial ROS, and further activates UCP2 expression, which results in the reduction of ATP synthesis.46,47 Consistently, the present study showed that HFD feeding significantly reduced the expressions of complexes I, IV and V, and increased the UCP2 expression, and simultaneously decreased the ATP content in the livers. Interestingly, supplementary ACPs restored all these abnormal changes, showing observably protective effects on mitochondrial function (Fig. 6). In this regard, many types of polysaccharides from different sources have been reported to possess significant protective effects on mitochondria, such as from Astragalus,48Pleurotus Abalonus,49Ganoderma,50Panax Ginseng,51Cordyceps Militaris,52 and Dictyophora Indusiata.53 Under normal conditions, fatty acid β-oxidation in mitochondria plays a key role in energy homoeostasis, while mitochondrial dysfunction can result in a reduction of fatty acid β-oxidation, which is a critical feature of NAFLD.30,54 In terms of this, HFD feeding has been linked to decreased pools of mitochondrial quinine and profoundly altered mitochondrial lipid composition, which leads to the inhibition of fatty acid oxidation.28 PGC-1α not only plays a crucial role in regulating genes related to fatty acid β-oxidation, but also is responsible for mitochondrial biogenesis and oxidative metabolism.30 The results of this study clearly showed that administration of ACPs significantly stimulated the mRNA and protein levels of PGC-1α, suggesting an improvement of mitochondrial β-oxidation. From this, we inferred that mitochondria might be the primary target of ACPs and this protection of the liver mitochondrial function was related to the amelioration of HFD induced NAFLD.
In conclusion, this study for the first time demonstrated that repeated administration of the natural biomacromolecule ACPs could protect against HFD-induced NAFLD through inhibiting lipogenesis, reducing inflammation and oxidative stress, and promoting the mitochondrial function. These results suggested that ACPs might be used as a food supplement for ameliorating NAFLD in populations who consume high fat food. Further exploration of human clinical trials is needed to verify this finding.
Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
This work was financially supported by “The National Natural Science Foundation of China (31871759)”, the “National Key R&D Program of China (2018YFD0901002)”, the “National Key R&D Program of China (2018YFC0311205)”, and the “Project of Distinguished Professor of Liaoning Province (2015-153)”.References
- N. N. Than and P. N. Newsome, A concise review of non-alcoholic fatty liver disease, Atherosclerosis, 2015, 239, 192–202 CrossRef CAS.
- H. Tilg and A. R. Moschen, Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis, Hepatology, 2010, 52, 1836–1846 CrossRef CAS.
- B. W. Smith and L. A. Adams, Non-alcoholic fatty liver disease, Crit. Rev. Clin. Lab. Sci., 2011, 48, 97–113 CrossRef CAS.
- C. P. Day and O. F. James, Steatohepatitis: a tale of two “hits”?, Elsevier, 1998 Search PubMed.
- J. C. Cohen, J. D. Horton and H. H. Hobbs, Human fatty liver disease: old questions and new insights, Science, 2011, 332, 1519–1523 CrossRef CAS.
- M. Carmiel-Haggai, A. I. Cederbaum and N. Nieto, A high-fat diet leads to the progression of non-alcoholic fatty liver disease in obese rats, FASEB J., 2005, 19, 136–138 CrossRef CAS.
- P. Angulo, Nonalcoholic fatty liver disease, N. Engl. J. Med., 2002, 346, 1221–1231 CrossRef CAS PubMed.
- M. E. Rinella, Nonalcoholic fatty liver disease: a systematic review, J. Am. Med. Assoc., 2015, 313, 2263–2273 CrossRef CAS.
- M. Zhang, S. Cui, P. Cheung and Q. Wang, Antitumor polysaccharides from mushrooms: a review on their isolation process, structural characteristics and antitumor activity, Trends Food Sci. Technol., 2007, 18, 4–19 CrossRef CAS.
- K. Wang, P. Cao, H. Wang, Z. Tang, N. Wang, J. Wang and Y. Zhang, Chronic administration of Angelica sinensis polysaccharide effectively improves fatty liver and glucose homeostasis in high-fat diet-fed mice, Sci. Rep., 2016, 6, 26229 CrossRef CAS.
- T. Belwal, H. P. Devkota, H. A. Hassan, S. Ahluwalia, M. F. Ramadan, A. Mocan and A. G. Atanasov, Phytopharmacology of Acerola (Malpighia spp.) and its potential as functional food, Trends Food Sci. Technol., 2018, 74, 99–106 CrossRef CAS.
- R. R. Klosterhoff, J. M. Bark, N. M. Glänzel, M. Iacomini, G. R. Martinez, S. M. Winnischofer and L. M. Cordeiro, Structure and intracellular antioxidant activity of pectic polysaccharide from acerola (Malpighia emarginata), Int. J. Biol. Macromol., 2018, 106, 473–480 CrossRef CAS.
- R. R. Klosterhoff, L. K. Kanazawa, A. L. Furlanetto, J. V. Peixoto, C. R. Corso, E. R. Adami, M. Iacomini, R. T. Fogaça, A. Acco and S. M. Cadena, Anti-fatigue activity of an arabinan-rich pectin from acerola (Malpighia emarginata), Int. J. Biol. Macromol., 2018, 109, 1147–1153 CrossRef CAS PubMed.
- M. Dubois, K. A. Gilles, J. K. Hamilton, P. T. Rebers and F. Smith, Colorimetric method for determination of sugars and related substances, Anal. Chem., 1956, 28, 350–356 CrossRef CAS.
- T. Bitter, A modified uronic acid carbazole reaction, Anal. Biochem., 1962, 4, 330–334 CrossRef CAS.
- M. M. Bradford, A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- L. Tian, X. Shi, L. Yu, J. Zhu, R. Ma and X. Yang, Chemical composition and hepatoprotective effects of polyphenol-rich extract from Houttuynia cordata tea, J. Agric. Food Chem., 2012, 60, 4641–4648 CrossRef CAS.
- N. He, X. Shi, Y. Zhao, L. Tian, D. Wang and X. Yang, Inhibitory effects and molecular mechanisms of selenium-containing tea polysaccharides on human breast cancer MCF-7 cells, J. Agric. Food Chem., 2013, 61, 579–588 CrossRef CAS.
- L. Sun, W. Shen, Z. Liu, S. Guan, J. Liu and S. Ding, Endurance exercise causes mitochondrial and oxidative stress in rat liver: effects of a combination of mitochondrial targeting nutrients, Life Sci., 2010, 86, 39–44 CrossRef CAS PubMed.
- Z. Hou, Y. Hu, X. Yang and W. Chen, Antihypertensive effects of Tartary buckwheat flavonoids by improvement of vascular insulin sensitivity in spontaneously hypertensive rats, Food Funct., 2017, 8, 4217–4228 RSC.
- M. Montgomery, N. Hallahan, S. Brown, M. Liu, T. Mitchell, G. Cooney and N. Turner, Mouse strain-dependent variation in obesity and glucose homeostasis in response to high-fat feeding, Diabetologia, 2013, 56, 1129–1139 CrossRef CAS.
- W. N. Cong, R. Y. Tao, J. Y. Tian, G. T. Liu and F. Ye, The establishment of a novel non-alcoholic steatohepatitis model accompanied by obesity and insulin resistance in mice, Life Sci., 2008, 82, 983–990 CrossRef CAS.
- H. M′kada, M. Munteanu, H. Perazzo, Y. Ngo, N. Ramanujam, F. Imbert-Bismut, V. Ratziu, D. Bonnefont-Rousselot, B. Souberbielle and I. Schuppe-Koistinen, What are the best reference values for a normal serum alanine transaminase activity (ALT)? Impact on the presumed prevalence of drug induced liver injury (DILI), Regul. Toxicol. Pharmacol., 2011, 60, 290–295 CrossRef.
- M. Kohjima, N. Higuchi, M. Kato, K. Kotoh, T. Yoshimoto, T. Fujino, M. Yada, R. Yada, N. Harada and M. Enjoji, SREBP-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease, Int. J. Mol. Med., 2008, 21, 507–511 CAS.
- G. S. Hotamisligil, Inflammation and metabolic disorders, Nature, 2006, 444, 860 CrossRef CAS.
- P. H. Wirtz and R. von Känel, Psychological stress, inflammation, and coronary heart disease, Curr. Cardiol. Rep., 2017, 19, 111 CrossRef.
- A. Shehzad, W. Iqbal, O. Shehzad and Y. S. Lee, Adiponectin: regulation of its production and its role in human diseases, Hormones, 2012, 11, 8–20 CrossRef.
- X. Zou, C. Yan, Y. Shi, K. Cao, J. Xu, X. Wang, C. Chen, C. Luo, Y. Li and J. Gao, Mitochondrial dysfunction in obesity-associated nonalcoholic fatty liver disease: the protective effects of pomegranate with its active component punicalagin, Antioxid. Redox Signaling, 2014, 21, 1557–1570 CrossRef CAS.
- S. Orrenius, Reactive oxygen species in mitochondria-mediated cell death, Drug Metab. Rev., 2007, 39, 443–455 CrossRef CAS.
- J. H. Ye, J. Chao, M. L. Chang, W. H. Peng, H. Y. Cheng, J. W. Liao and L. H. Pao, Pentoxifylline ameliorates non-alcoholic fatty liver disease in hyperglycaemic and dyslipidaemic mice by upregulating fatty acid β-oxidation, Sci. Rep., 2016, 6, 33102 CrossRef CAS.
- C. D. Byrne and G. Targher, NAFLD: a multisystem disease, J. Hepatol., 2015, 62, S47–S64 CrossRef.
- G. Paradies, V. Paradies, F. M. Ruggiero and G. Petrosillo, Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease, World J. Gastroenterol., 2014, 39, 14205–14218 CrossRef.
- Y. Yu, M. Shen, Q. Song and J. Xie, Biological activities and pharmaceutical applications of polysaccharide from natural resources: A review, Carbohydr. Polym., 2018, 183, 91–101 CrossRef CAS.
- D. D. Leffa, G. T. Rezin, F. Daumann, L. M. Longaretti, A. L. F. Dajori, L. M. Gomes, M. C. Silva, E. L. Streck and V. M. de Andrade, Effects of Acerola (Malpighia emarginata DC.) juice intake on brain energy metabolism of mice fed a cafeteria diet, Mol. Neurobiol., 2017, 54, 954–963 CrossRef CAS.
- D. D. Leffa, J. da Silva, F. C. Petronilho, M. S. Biélla, A. Lopes, A. R. Binatti, F. Daumann, P. F. Schuck and V. M. Andrade, Acerola (Malpighia emarginata DC.) juice intake protects against oxidative damage in mice fed by cafeteria diet, Food Res. Int., 2015, 77, 649–656 CrossRef CAS.
- F. M. Dias, D. D. Leffa, F. Daumann, S. D. O. Marques, T. F. Luciano, J. C. Possato, A. A. D. Santana, R. X. Neves, J. C. Rosa and L. M. Oyama, Acerola (Malpighia emarginata DC.) juice intake protects against alterations to proteins involved in inflammatory and lipolysis pathways in the adipose tissue of obese mice fed a cafeteria diet, Lipids Health Dis., 2014, 13, 24–24 CrossRef.
- X. Li, Z. Xu, S. Wang, H. Guo, S. Dong, T. Wang, L. Zhang and Z. Jiang, Emodin ameliorates hepatic steatosis through endoplasmic reticulum–stress sterol regulatory element-binding protein 1c pathway in liquid fructose-feeding rats, Hepatol. Res., 2016, 46, E105–E117 CrossRef CAS PubMed.
- Y. T. Chen, Y. C. Lin, J. S. Lin, N. S. Yang and M. J. Chen, Sugary Kefir Strain Lactobacillus mali APS1 Ameliorated Hepatic Steatosis by Regulation of SIRT-1/Nrf-2 and Gut Microbiota in Rats, Mol. Nutr. Food Res., 2018, 62, 1700903 CrossRef.
- C. Y. Song, X. Zeng, S. W. Chen, P. F. Hu, Z. W. Zheng, B. F. Ning, J. Shi, W. F. Xie and Y. X. Chen, Sophocarpine alleviates non-alcoholic steatohepatitis in rats, J. Gastroenterol. Hepatol., 2011, 26, 765–774 CrossRef CAS.
- R. Gambino, G. Musso and M. Cassader, Redox balance in the pathogenesis of nonalcoholic fatty liver disease: mechanisms and therapeutic opportunities, Antioxid. Redox Signaling, 2011, 15, 1325–1365 CrossRef CAS.
- M. S. Brennan, H. Patel, N. Allaire, A. Thai, P. Cullen, S. Ryan, M. Lukashev, P. Bista, R. Huang and K. J. Rhodes, Pharmacodynamics of dimethyl fumarate are tissue specific and involve NRF2-dependent and-independent mechanisms, Antioxid. Redox Signaling, 2016, 24, 1058–1071 CrossRef CAS.
- J. M. Alvarez-Suarez, F. Giampieri, M. Gasparrini, L. Mazzoni, C. Santos-Buelga, A. M. González-Paramás, T. Y. Forbes-Hernández, S. Afrin, T. Páez-Watson and J. L. Quiles, The protective effect of acerola (Malpighia emarginata) against oxidative damage in human dermal fibroblasts through the improvement of antioxidant enzyme activity and mitochondrial functionality, Food Funct., 2017, 8, 3250–3258 RSC.
- Y. Guo, M. Darshi, Y. Ma, G. A. Perkins, Z. Shen, K. J. Haushalter, R. Saito, A. Chen, Y. S. Lee, H. H. Patel, S. P. Briggs, M. H. Ellisman, J. M. Olefsky and S. S. Taylor, Quantitative proteomic and functional analysis of liver mitochondria from high fat diet (HFD) diabetic mice, Mol. Cell. Proteomics, 2013, 12, 3744–3758 CrossRef CAS PubMed.
- Y. Z. Wei, R. S. Rector, J. P. Thyfault and J. A. Ibdah, Nonalcoholic fatty liver disease and mitochondrial dysfunction, World J. Gastroenterol., 2008, 14, 193–199 CrossRef CAS.
- M. Perez-Carreras, P. Del Hoyo, M. A. Martin, J. C. Rubio, A. Martin, G. Castellano, F. Colina, J. Arenas and J. A. Solis-Herruzo, Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis, Hepatology, 2003, 38, 999–1007 CrossRef CAS.
- J. Bo, S. Xie, Y. Guo, C. Zhang, Y. Guan, C. Li, J. Lu and Q. H. Meng, Methylglyoxal impairs insulin secretion of pancreatic β-cells through increased production of ROS and mitochondrial dysfunction mediated by upregulation of UCP2 and MAPKs, J. Diabetes Res., 2016, 2016, 1–14 CrossRef.
- A. Lafargue, C. Degorre, I. Corre, M. C. Alves-Guerra, M. H. Gaugler, F. Vallette, C. Pecqueur and F. Paris, Ionizing radiation induces long-term senescence in endothelial cells through mitochondrial respiratory complex II dysfunction and superoxide generation, Free Radical Biol. Med., 2017, 108, 750–759 CrossRef CAS.
- Y. F. Huang, L. Lu, D. J. Zhu, M. Wang, Y. Yin, D. X. Chen and L. B. Wei, Effects of astragalus polysaccharides on dysfunction of mitochondrial dynamics induced by oxidative stress, Oxid. Med. Cell. Longevity, 2016, 2016, 1–13 Search PubMed.
- X. Shi, Y. Zhao, Y. Jiao, T. Shi and X. Yang, Ros-dependent mitochondria molecular mechanisms underlying antitumor activity of pleurotus abalonus acidic polysaccharides in human breast cancer mcf-7 cells, PLoS One, 2013, 8, e64266 CrossRef CAS.
- W. J. Li, S. Nie, Y. F. Yao, X. Z. Liu and M. Y. Xie, Ganoderma atrum polysaccharide ameliorates hyperglycemia-induced endothelial cell death via a mitochondria-ros pathway, J. Agric. Food Chem., 2015, 37, 8182–8191 CrossRef.
- X. T. Li, R. Chen, L. M. Jin and H. Y. Chen, Regulation on energy metabolism and protection on mitochondria of panax ginseng polysaccharide, Am. J. Chin. Med., 2009, 6, 1139–1152 CrossRef.
- X. T. Li, H. C. Li, C. B. Li, D. Q. Dou and M. B. Gao, Protective effects on mitochondria and anti-aging activity of polysaccharides from cultivated fruiting bodies of cordyceps militaris, Am. J. Chin. Med., 2010, 6, 1093–1106 CrossRef.
- W. Liao, Z. Yu, Z. Lin, Z. Lei, Z. Ning, J. M. Regenstein, J. G. Yang and J. Y. Ren, Biofunctionalization of selenium nanoparticle with dictyophora indusiata polysaccharide and its antiproliferative activity through death-receptor and mitochondria-mediated apoptotic pathways, Sci. Rep., 2015, 5, 18629 CrossRef CAS.
- F. Bellanti, R. Villani, R. Tamborra, M. Blonda, G. Iannelli, G. D. Bello, A. Facciorusso, G. Poli, L. Iuliano and C. Avolio, Synergistic interaction of fatty acids and oxysterols impairs mitochondrial function and limits liver adaptation during nafld progression, Redox Biol., 2018, 15, 86–96 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |