
Glycyrrhizic acid promotes neural repair by directly driving functional remyelination†
Jing
Tian‡
a,
Xing
Li‡
a,
Li
Zhao‡
a,
Peixin
Shen
a,
Zhezhi
Wang
a,
Lin
Zhu
b,
Cuiqin
Li
 a,
Chun
Su
a and
Yuan
Zhang
*a
a,
Chun
Su
a and
Yuan
Zhang
*a
aNational Engineering Laboratory for Resource Development of Endangered Crude Drugs in Northwest China, The Key Laboratory of Medicinal Resources and Natural Pharmaceutical Chemistry, The Ministry of Education, College of Life Sciences, Shaanxi Normal University, Xi'an, Shaanxi 710119, China. E-mail: yuanzhang_bio@126.com
bDepartment of Pharmacy, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan 450052, China
First published on 28th November 2019
Abstract
Natural compounds are a rich source of effective candidate drugs for the treatment of neurological disorders. Glycyrrhizic acid (GA), the major water-soluble ingredient isolated from Glycyrrhiza glabra, is reported to show anti-inflammatory and immunomodulatory activities. However, its effect on CNS demyelinating disease is unclear. In this study, we showed that GA ameliorated the clinical disease severity of experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis (MS), especially at the chronic stage of clinical EAE. Histological evaluation demonstrated that, in the prophylactic treatment regimen, GA significantly inhibited inflammatory demyelination in the CNS. During the chronic stage when myelin and axon damage has already occurred, GA induced oligodendrocyte progenitor cell (OPC) differentiation into mature oligodendrocytes, thus effectively accelerating remyelination. Evidence from the cuprizone-induced mouse model of de- and remyelination, ex vivo organotypic slice cultures, and in vitro OPC maturation experiments indicated that the observed efficacy of this compound resulted directly from enhanced remyelination rather than immune suppression. Furthermore, we found that GA promoted oligodendrocyte maturation through modulating GSK-3β signaling pathways. Our data led to the conclusion that GA could be used as a potential therapeutic candidate for the treatment of demyelinating diseases such as MS, which remains refractory to available treatments.
1. Introduction
Multiple sclerosis (MS) and its animal model, experimental autoimmune encephalomyelitis (EAE), are chronic autoimmune diseases of the central nervous system (CNS), accompanied by remarkable lesions of the spinal cord and inflammation, axonal loss and demyelination.1 The disease process can be broadly divided into neuroinflammatory reactions and demyelinating diseases. The prevalence of MS varies from region to region but its causative factors are unknown. It is estimated to affect over two million people worldwide, resulting in a high disability rate especially in young adults.2 EAE is commonly induced by stimulating an immune response with myelin antigens emulsified in an adjuvant.3 As with MS, the disease process can be broadly divided into two phases, acute neuroinflammatory reactions and chronic demyelinating plaques.4 In the initial phase of the disease, myelin-reactive T cells, together with other immunocytes in the periphery, cross the disrupted blood–brain barrier (BBB) and infiltrate into the CNS. Subsequently, the infiltrating inflammatory cells are reactivated, secrete cytokines and chemokines that recruit microglia, and promote cell- and antibody-mediated immunity.5,6 At the early stage of MS/EAE, disease progression is associated with demyelination, axonal damage and neuronal loss.7 In order to prevent neural degeneration and subsequent demyelination, remyelination must be promoted by the generation of new OPCs. However, although OPCs are abundant in the demyelination areas of MS and EAE, they fail to differentiate or recruit in lesions.8 Failure of remyelination might be attributed to insufficient OPC migration to lesions (recruitment), a lack of OPC maturation, poor oligodendrocyte survival within the inflammatory milieu, and a lack of neurotrophic factors in MS/EAE lesions.9Currently, several immunosuppressive and immunomodulatory therapies are broadly available for the treatment of acute MS.10,11 However, treatment for chronic diseases, which is mainly chronic-progressive (CP) and relapsing–remitting (RR) MS, remains a significant challenge in this field, because immunomodulatory therapies alone lack the neuroregenerative capacity to repair already damaged CNS tissue, e.g., demyelination, axonal damage and neuron loss, and the failure of spontaneous neuroregeneration.12 A therapeutic strategy that has both immunomodulatory and neuroregenerative effects is therefore required to overcome this deficiency.13
It is worth noting that in recent years many natural compounds and their derivatives have shown significant effects in the treatment of EAE. Glycyrrhizic acid (GA), a triterpenoid saponin, is a natural component obtained from the roots and rhizomes of Glycyrrhiza glabra. Owing to their wide spectrum of pharmacological activities, such as anti-inflammatory,14 anti-viral,15 and anti-tumor activities,16,17 root extracts of G. glabra have been widely used as a sweetener or thickener in various foods (such as bread, candy, candied fruit, chewing gum, beer, soft drinks, etc.) for thousands of years in China, Japan, and India.18–21 It has been reported that GA is effective in several inflammatory diseases or related experimental models, including allergic reaction22 and asthma.23 Although these findings suggest that GA may also be beneficial for autoimmune diseases such as MS, important questions regarding its pharmacological activity, cytological basis, and molecular mechanisms of action remain to be answered.
In the current study, we demonstrate that GA treatment significantly decreases the clinical severity of EAE mice. Findings from an in vivo cuprizone-induced model of demyelination, CNS organotypic slice culture, and in vitro OPC differentiation experiments indicated that the observed efficacy of this drug results directly from enhanced remyelination rather than immunomodulation. Finally, we showed that GA functions by a mechanism that significantly inhibits the phosphorylation of GSK-3β in OPCs. Our data demonstrate the novel potential of GA as an anti-inflammatory and neural repair agent, especially at the chronic stage, which is a more challenging goal and more relevant to human MS.
2. Materials and methods
2.1 EAE induction and treatment
Eight-week-old female C57BL/6 mice were ordered from the Fourth Military University (Xi'an, China). The experimental protocol was approved by the Committee on the Ethics of Animal Experiments of Shaanxi Normal University (No. ECES-2015-0247). The mice were immunized subcutaneously on the back for active induction of EAE as described.24 Briefly, to induce active EAE, the mice were subcutaneously immunized with 200 μg of myelin oligodendrocyte glycoprotein (MOG35–55) peptide (Genescript, Piscataway, NJ) emulsified in CFA (Difco Lab, Lawrence, KS) containing 5 mg mL−1Mycobacterium tuberculosis H37Ra (Difco Lab). 200 ng of pertussis toxin (Sigma-Aldrich, St Louis, MO) were administered intraperitoneally on days 0 and 2 post-immunization (p.i.). Daily clinical scores and body weights were recorded blindly by two researchers on a 0–5 scale as described previously.25 In brief: 0, no clinical signs; 0.5, stiff tail; 1, limp tail; 1.5, limp tail and wadding gait; 2, paralysis of one limb; 2.5, paralysis of one limb and weakness of another limb; 3, complete paralysis of both hind limbs; 4, moribund; 5, death. Glycyrrhizic acid (purity of 97%) was obtained from Sigma-Aldrich and dissolved in dimethyl sulfoxide (Millipore, Billerica, MA) to obtain a stock solution and diluted with PBS before administration at the final concentration. In vivo dosage was determined based on previous studies.22,26 GA (50 mg kg−1) or PBS (the same concentration of dimethyl sulfoxide as that in the GA-treated group, similarly hereinafter) was administered daily, starting on the day of immunization, or day 30 p.i.2.2 Cuprizone-induced demyelination and treatment
For the cuprizone model, 8 week-old male C57BL/6 mice (purchased from the Fourth Military University (Xi'an, China)) were fed with the standard rodent diet, mixed with 0.2% copper chelator cuprizone, which inhibits mitochondrial function and causes CNS demyelination, purchased from Envigo's Teklad business unit.27 For demyelination experiments, the mice were fed with cuprizone in standard chow for 6 weeks, and treated with GA (50 mg kg−1) or PBS every day. Besides, it was also compared with a pre-treatment of GA (50 mg kg−1) or PBS every day for 2 weeks before inducing demyelination using cuprizone for 6 weeks. For induced remyelination experiments, the mice were exposed to cuprizone for 4 weeks to achieve complete demyelination, after which cuprizone was withdrawn. GA (50 mg kg−1) or PBS was administered daily between weeks 4 and 6 to induce remyelination. All the animals were euthanized and their brains were fixed with 4% PFA, and the corpus callosum was obtained for western blotting analysis. To investigate whether GA affects de- or remyelination, the brains were further embedded in paraffin, sectioned and stained for histopathological and immunohistochemistry analyses.2.3 Remyelination effect of GA in organotypic cerebellar slice culture
Postnatal day 3 (P3) C57BL/6 mice were sacrificed and their brains were used to prepare cerebellar slices. The culture conditions were based on previously described procedures.25 Briefly, the brain and hippocampus were dissected into 350 μm slices with scissors, spatulas, etc. and kept cold and wet, and 4–5 slices were grown on each cell culture insert (Millipore) in 6 well plates. The time schedule and culture medium were the same as described previously.25 Briefly, after 2 days of recovery, the slices were treated with 0.5 mg mL−1 lysophosphatidylcholine (LPC) for 18 h to induce demyelination. The slices were then washed and treated with PBS and GA for the next 14 days. APC and MBP expression was measured to evaluate maturation OLG numbers and the remyelination effect.2.4 Mouse primary OPC generation and differentiation
Primary OPCs were isolated from 3 day-old newborn mouse brains, separated using a Neural Tissue Dissociation Kit (Miltenyi Biotec, San Diego, CA), and then concentrated with anti-A2B5 microbeads (Miltenyi Biotec). OPCs were cultured in OPC proliferation medium: DMEM/F12 (Thermo Fisher Scientific, Waltham, MA) supplemented with 2% B27, 1% N2, 2 mM GlutaMAX-I and penicillin–streptomycin (Life Technology), 20 ng mL−1 bFGF, and 20 ng mL−1 PDGF-BB (Thermo Fisher Scientific) at 37 °C with 5% CO2.In order to induce OPC differentiation into OLG in vitro, the cell culture medium was switched to OPC differentiation medium: DMEM/F12 supplemented with 2% B27—without vitamin A, 20 μg mL−1 triiodothyronine (T3; TCI, Shanghai, China), 20 μg mL−1 noggin (Life Technology), 10 μg mL−1 Shh (Life Technology) and 2 mM penicillin–streptomycin at 37 °C with 5% CO2. Differentiated OLGs were analyzed by the immunofluorescence staining of APC and MBP. After fixation, permeabilization and blocking, the cells were labeled with CNPase (Abcam), followed by incubation with Alexa Fluor ®488 or 594 conjugated secondary antibodies (Abcam). Nuclei were visualized by DAPI staining.
To determine the mechanism of GA-induced effects, primary OPCs were obtained using the method described above and then treated with GA (10 μM) or PBS for 48 h in OPC differentiation medium. In order to perform experiments to evaluate the pharmacological inhibition of kinase, primary OPCs were pre-treated with the GSK-3β inhibitor (AR-A014418, 20 μM (TargetMol, Shanghai, China)) for 48 h in OPC differentiation medium, followed by western blotting analysis.
2.5 Histological analysis
EAE mice were sacrificed at 30 or 60 d.p.i. and perfused with cold PBS transcardially. Lumbosacral enlargement of lumbar spinal cords was harvested, fixed with 4% PFA, and further embedded in paraffin, cut into 5 μm sections, for pathological assessment, and stained with hematoxylin and eosin (H&E) to assess inflammation and with Luxol Fast Blue (LFB) to assess demyelination. Slides were evaluated and scored in a blinded manner for inflammation followed by previous procedures.28 For demyelination quantification, the total white matter was manually outlined, and the area (%) of demyelination was calculated using Image-Pro Plus software.For toluidine staining, procedures were followed as described previously.13 The lumbar spinal cord tissues were dissected into 1 μm thick semi-thin slices and fixed in 4% paraformaldehyde overnight. The incubated slices were immersed in 2% osmium tetroxide, dehydrated in a graded series of acetone and then embedded into EPON. One μm thick semi-thin sections were incubated with 1% toluidine blue solution and images were photographed by light microscopy. For analysis purpose, 10 micrographs per mouse that cover almost all the white matter in the lumbar spinal cord were selected, and 5 mice per group were estimated. To count myelinated axons in the area of ventral of the lumbar spinal cord, a line-sampling method was performed as described previously.25
For immunofluorescence staining, spinal cord tissues were fixed with 4% PFA for 24 h and subsequently cryoprotected with 30% sucrose solution over 48 h. The tissues were embedded in OCT solution (Tissue-Tek, Sakura Finetek, Japan) for preparing frozen sections and then cut coronally into 8 μm sections. Transverse sections of the tissues were cut and stained with specific antibodies. Immunofluorescence controls were routinely generated by omitting primary antibodies. The results were visualized by fluorescence microscopy (Nikon Eclipse E600; Nikon, Melville, NY) or confocal microscopy (Zeiss LSM 510; Carl Zeiss, Thornwood, NY). For the quantification of H&E, LFB, MBP and APC, ten areas in the white matter at the lumbar spinal cord were selected (Fig. S3†).
2.6 Real-time quantitative PCR
RNA extract, reverse transcription and RT-PCR were performed according to a previously described procedure.29 Total RNA was extracted from the cells using the RNAprep Pure Cell kit (TIANGEN Biotech, (Beijing) Co., Ltd) according to the manufacturer's instructions. Reverse transcription was carried out using the Prime Script® RT Master Mix (TakaRa). Real-time PCR was conducted using the ChamQTM SYBR® qPCR Master Mix (TakaRa), and the detection was performed using Roche Molecular Biochemicals Light Cycler Software Version 3.5 (Roche, Nutley, NJ). GAPDH was chosen as an internal control, and the relative expression was calculated by using the 2−ΔCt method. Primers used in this study are listed in ESI Table 1.†2.7 Western blotting analysis
Protein was isolated from OPCs/tissues using PMSF (Invitrogen, Gaithersburg, MD) and protein lysis buffer. The protein concentration in the supernatant of the cell/tissue extract was determined using a BCA protein assay kit (Zoman Biotechnology, Beijing). Twenty ng of proteins were loaded on SDS-polyacrylamide stacking and separating gels. After electrophoresis, the proteins obtained in the gel were transferred to PVDF membranes (Life Technology), and the blots were then probed with primary antibodies including JNK, phospho-JNK, GSK-3β, phospho-GSK-3β, and β-actin (all from Cell Signal Technology), followed by incubation with HRP-conjugated secondary antibodies and chemiluminescent enhancement by using Tanon 4600 (Shanghai, China). Normalization of the results was ensured by performing parallel western blotting analysis with β-actin antibody (Invitrogen). Optical density was quantified using an image processing and analysis program (Image J, Ederick, MA).2.8 Statistical analysis
All results in this paper obtained from the various quantifications are presented with standard deviations (SD). Statistical differences between two or multiple groups were implemented using the unpaired Student's t-test or ANOVA. Differences with a P value of less than 0.05 were considered statistically significant. All statistical analyses were performed using GraphPad Prism 6 (GraphPad, La Jolla, CA).3. Results
3.1 Prophylactic efficacy of GA in the MOG-induced EAE model
To determine whether GA has the therapeutic potential to alleviate clinical severity in C57BL/6 EAE mice, we initially examined disease development in PBS-treated (control) and GA-treated MOG35–55-immunized EAE mice. In the prophylactic treatment regimen, GA and PBS administration starting from day 0 post-immunization (p.i.), with a daily dosage of 50 mg kg−1 in EAE animals, led to significantly delayed development and decreased disease severity in the GA-treated group compared with the PBS-treated group (Fig. 1a). At the peak (day 20 p.i.) stage of EAE disease, the mean clinical score of the PBS-treated group is 2.85. However, in the GA-treated group, the mean clinical score is 1.1. Furthermore, the decrease in the body weight of the EAE mice was also prevented under the treatment of GA, which is consistent with the change in the clinical signs (Fig. 1b).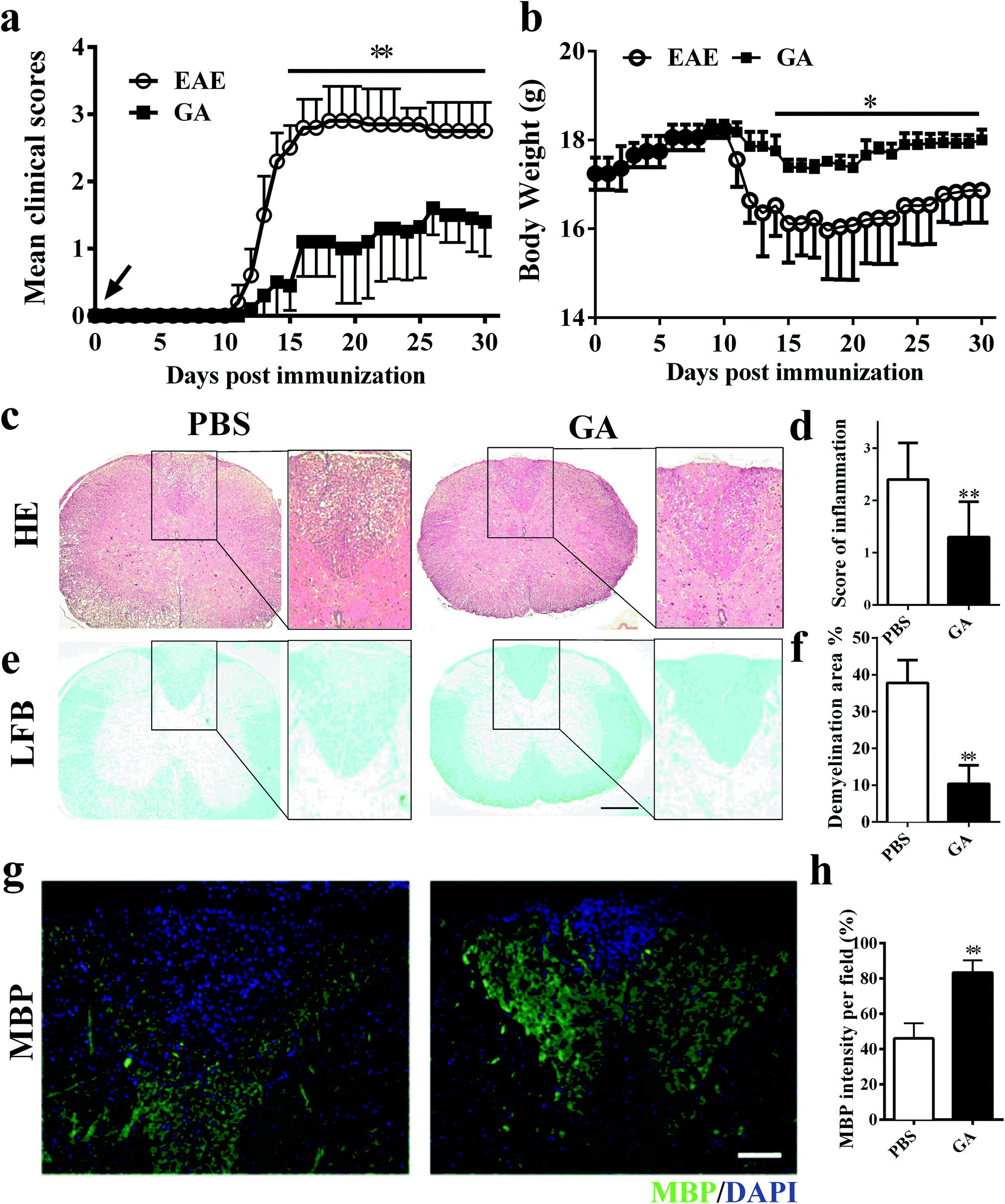 | ||
| Fig. 1 GA reduced CNS inflammation and myelin damage in EAE. Clinical scores (a) and body weight (b) of EAE mice that were administered daily with GA (50 mg kg−1) or PBS control from day 0 p. i. (n = 5, preventive treatment). (c, e) Transverse sections of spinal cords from control EAE mice and GA-treated mice were stained with H&E or Luxol fast blue on day 30 p.i. to assess inflammatory infiltration and demyelination. (d, f) Histopathology score of CNS inflammatory infiltration, and demyelination was quantified by manually outlining with Image-Pro Plus software. (g) Immunolabeling for myelin basic protein (MBP) was performed to determine the percentage of myelinated areas within a lesion (defined in the DAPI channel as nuclei). (h) MBP intensity was measured and quantified in the lesion areas in the white matter of the lumbar spinal cord using Image-Pro. Data are mean ± SD (n = 5 each group). Scale bar = 500 μm in (c, e). Scale bar = 100 μm in (g). *p < 0.05 and **p < 0.01 as determined by two-way ANOVA or the nonparametric t test. One representative of three independent experiments is shown. | ||
To further evaluate the pathological changes in the GA-treated EAE mice, we performed histological analyses in lumbar spinal cords to examine CNS inflammatory infiltration and demyelination at day 30 post immunization. As shown in Fig. 1c and d, more inflammatory infiltration was evident in the spinal cord of the PBS-treated mice rather than the GA-treated EAE mice. In Fig. 1e and f, demyelinated lesions were detected by reduced LFB intensity in the white matter of PBS-treated, but not the GA-treated, EAE mice. These results indicated that the prophylactic dosing of GA is effective at inhibiting inflammatory infiltration and demyelination of clinical EAE. Furthermore, demyelinated lesions were determined via myelin basic protein (MBP) expression in the dorsal column of the lumbar spinal cord. A significantly increased proportional area of intact myelin (MBP+) was detected in the GA-treated mice compared with that in the PBS group (Fig. 1g and h), indicating that GA treatment may inhibit demyelination and/or enhance remyelination in CNS lesions.
3.2 GA effectively alleviated the disease process when treatment was started at the chronic stage of EAE
To further explore the therapeutic effects of GA, the chronic EAE model was used in this study. The mice were treated starting from day 30 p.i. (at the stable stage when CNS demyelination and damage have already occurred and remyelination has started).30 During the next 30 days of treatment, as shown in Fig. 2a, the GA-treated mice showed gradual and remarkably improved amelioration of clinical disease compared to the PBS-treated group. The weight change corresponds perfectly to the clinical scores during the 60 day treatment of the EAE mice (Fig. 2b). These results were further confirmed by LFB staining (Fig. 2c and f), which demonstrated that the demyelinated areas were significantly decreased after GA treatment compared with the PBS group (all the mice were sacrificed at day 60 p.i.). These data suggest that GA treatment plays a primary role in the chronic stage of EAE. We then determined the effects of GA on remyelination and oligodendrocyte maturity within spinal cord lesions using anti-MBP (myelin marker) and anti-APC (mature oligodendrocyte marker) (Fig. 2d and e). As shown in Fig. 1g and h, in the PBS-treated group, severe demyelination (∼45% of MBP intensity/field) had occurred before treatment at day 30 p.i. and day 60 p.i. (∼55% of MBP intensity/field). At day 60 p.i., the GA-treated mice showed a higher MBP intensity compared to the PBS-treated mice (∼70% of MBP intensity/field, p < 0.01, Fig. 2d and g). Importantly, the GA-treated group showed significantly increased MBP intensity compared to the PBS group at day 30 p.i., indicating that GA treatment not only reduced further myelin damage, but also promoted myelin recovery. Furthermore, significantly increased APC + oligodendrocyte numbers were detected in the GA-treated mice (Fig. 2e and h), suggesting that GA induced OPC maturation in CNS lesions. In order to further investigate the effect of GA on remyelination in EAE lesions, histological sections were stained with toluidine blue staining. The well-preserved original myelin sheaths were thicker than those of the newly formed ones (remyelination). As shown in Fig. 2i and k, GA treatment not only significantly increased the number of myelinated axons, but also reduced the G-ratio of the remyelinated axons, indicating a better recovery from demyelination.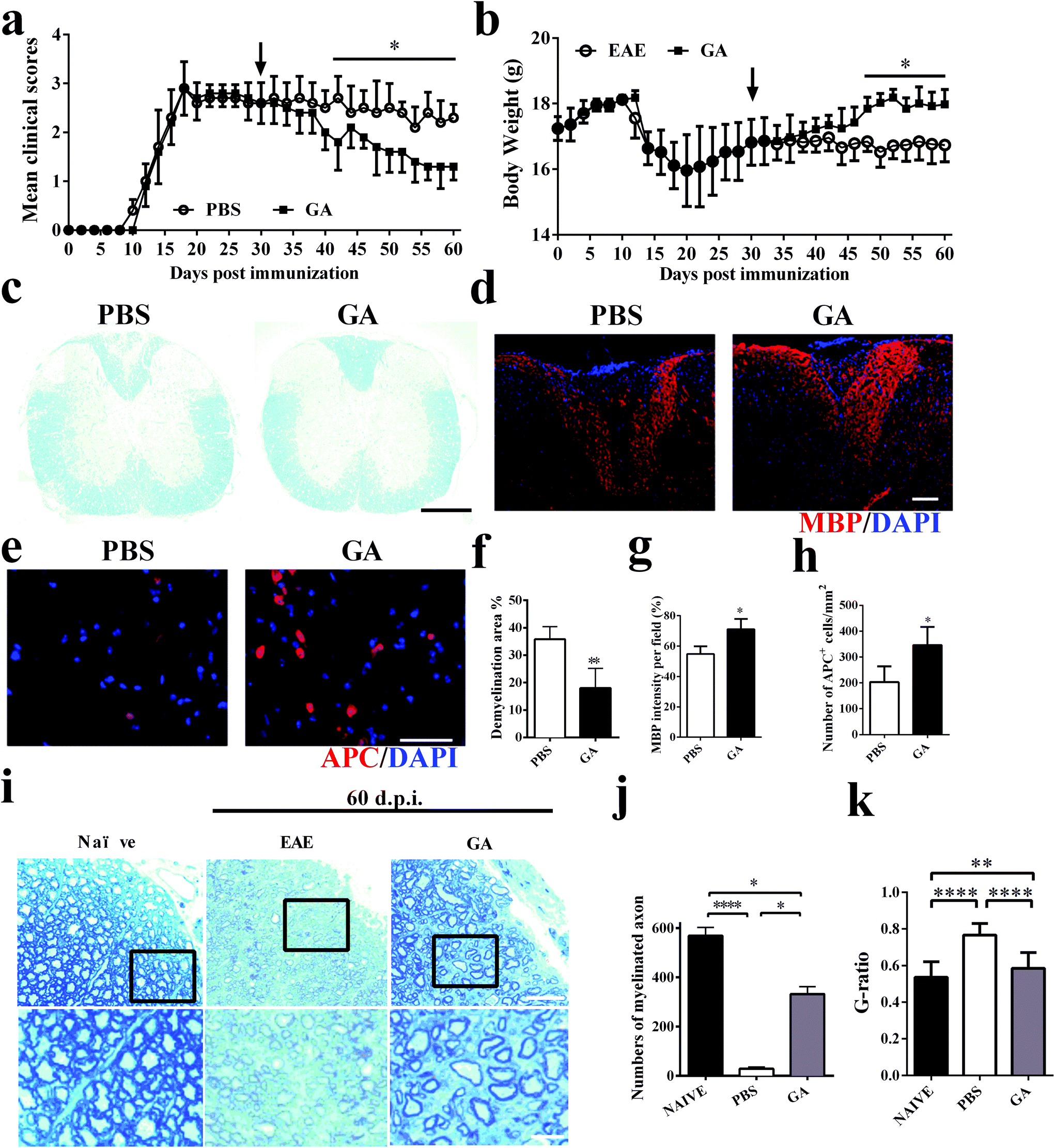 | ||
| Fig. 2 GA improved the clinical signs in chronic EAE and promoted remyelination of myelin at the chronic stage of EAE. Clinical scores (a) and body weight (b) of EAE mice that were administered daily with GA (50 mg kg−1 day−1) or vehicle control from day 30 p.i. (n = 5, therapeutic treatment). (c) Mice were sacrificed on day 60 p.i. (n = 5 each group), and spinal cords were harvested from control EAE mice, and GA-treated mice were stained with LFB. (d) MBP staining of spinal cord sections for the detection of MBP expression. (e) The effect of GA on the number of mature oligodendrocytes in the spinal cord compared with PBS-treated mice. APC and DAPI co-immunolabeling was evaluated to determine the number of mature oligodendrocytes. (f) Demyelinated area was measured by manually outlining the total white matter area with Image-Pro Plus software. The demyelinated area is expressed as a percentage of the total white matter area. (g) MBP intensity was measured and quantified in the lesion areas in the white matter of lumbar spinal cord. (h) Quantification of total APC + cells. (i) Histological sections stained with toluidine blue show the extent of remyelination and occurrence of remyelination in the lesions of naïve, PBS-treated and GA-treated animals sacrificed at day 60 p.i. (j) Quantification of the percentage of myelinated axons among total axons as shown. (k) Mean G ratio (axon diameter divided by the entire myelinated fiber diameter) was determined using Image-Pro Plus software. Data are mean ± SD (n = 5 each group). Scale bar = 1 mm in (c). Scale bar = 10 μm in (d, e). Scale bar = 25 μm and 5 μm in (i).*p < 0.05, **p < 0.01, and ****p < 0.0001 as determined by two-way ANOVA. One representative of three experiments is shown. | ||
3.3 GA enhanced remyelination in the cuprizone-induced demyelinated model
In the EAE model, the beneficial effects of GA on remyelination may be partly due to its immunomodulatory effects on the immune system. To further investigate the direct effects of GA on OPC maturation and remyelination in vivo, we therefore used the toxic cuprizone-induced demyelinated model under minimal inflammatory and T-cell-independent microenvironments. As shown in Fig. 3, the mice responded to cuprizone treatment as expected, showing progressive weight loss and obvious demyelination of the corpus callosum during the 6 weeks of cuprizone exposure. However, during the demyelination phase, cuprizone-mediated weight loss (Fig. 3a) and demyelination (Fig. 3b) were not altered by GA supplementation. Similar results are also shown in Fig. S1,† and even if GA was pre-administered for 2 weeks, it could not alter the weight loss (Fig. S1a†) and demyelination damage (Fig. S1b and d†) caused by cuprizone. To determine the effects of GA during remyelination, the mice were exposed to cuprizone for 4 weeks to achieve complete demyelination, and then administered with GA (50 mg kg−1) or PBS every day for 2 weeks after cuprizone withdrawal. At week 6, there was a significant change in weight and a slight remyelination was observed in the PBS-treated group (Fig. 3c and d). Remarkably, GA supplementation increased the body weight and enhanced remyelination as assessed by histochemical (LFB) and immunohistochemical staining (myelin) (Fig. 3d). To determine whether GA could stimulate oligodendrocyte maturation in vivo, immunohistochemical staining using OPC/OLG specific antibodies was performed at week 6. Co-staining of APC/DAPI and NG2/DAPI in the corpus callosum revealed that GA promoted OPC maturation into OLGs (Fig. 3e). Hence, GA supplementation directly stimulates oligodendrocyte maturation and promotes remyelination following the creation of a cuprizone-induced demyelination environment. | ||
| Fig. 3 GA enhanced remyelination in the cuprizone-induced demyelinated model. For the demyelination model, 8-week old naïve male C57BL/6 mice were fed daily with standard rodent diets, and cuprizone model mice were treated with PBS and GA (50 mg kg−1) for 6 weeks, respectively. Body weights (a) of mice were recorded every week. (b) LFB and FluoroMyelin stains of the corpus callosum. For the remyelination model, 8-week old naïve male C57BL/6 mice were fed daily with cuprizone for 4 weeks; PBS or GA (50 mg kg−1) was administered daily between weeks 4 and 6. Quantitative analysis of myelinated and fluoromyelinated areas measured at random areas in the white matter of spinal cords using Image Pro software. (c) Body weights of mice were recorded every week. (d) LFB and myelin stains of the corpus callosum. Quantitative analysis of myelinated and fluoromyelinated areas measured at random areas in the white matter of spinal cords using Image Pro. (e) Immunofluorescence stains of the corpus callosum for APC+ OLGs and NG2+ OPCs. Quantification analysis of the percentage of APC+ cells and NG2+ cells measured at random areas using Image Pro. Data are mean ± SD (n = 5 each group). Scale bar = 80 μm in (b, d). Scale bar = 200 μm in (e). *p < 0.05, **p < 0.01, and ***p < 0.001 as determined by two-way ANOVA. One representative of three experiments is shown. | ||
3.4 GA promoted remyelination in organotypic slice culture
To determine the direct effects of GA on remyelination with minimal involvement of the immune response, an organotypic slice culture of brain tissues was generated. Demyelination of the brain slices from newborn mice was induced with lysophosphatidylcholine (LPC) as described previously31 and then they were treated with GA or PBS for the next 14 days. Compared with the GA treatment, significantly fewer APC + mature oligodendrocytes were observed in slices treated with PBS only (Fig. 4a and b). Meanwhile, although LPC induced a remarkable decrease in the MBP expression of the brain tissues, addition of GA caused a significant increase in the MBP intensity compared with the PBS treatment (Fig. 4c and d). These results indicated that GA effectively increased the number of mature oligodendrocytes and promoted myelin protein production in LPC-induced demyelination in organotypic slice culture.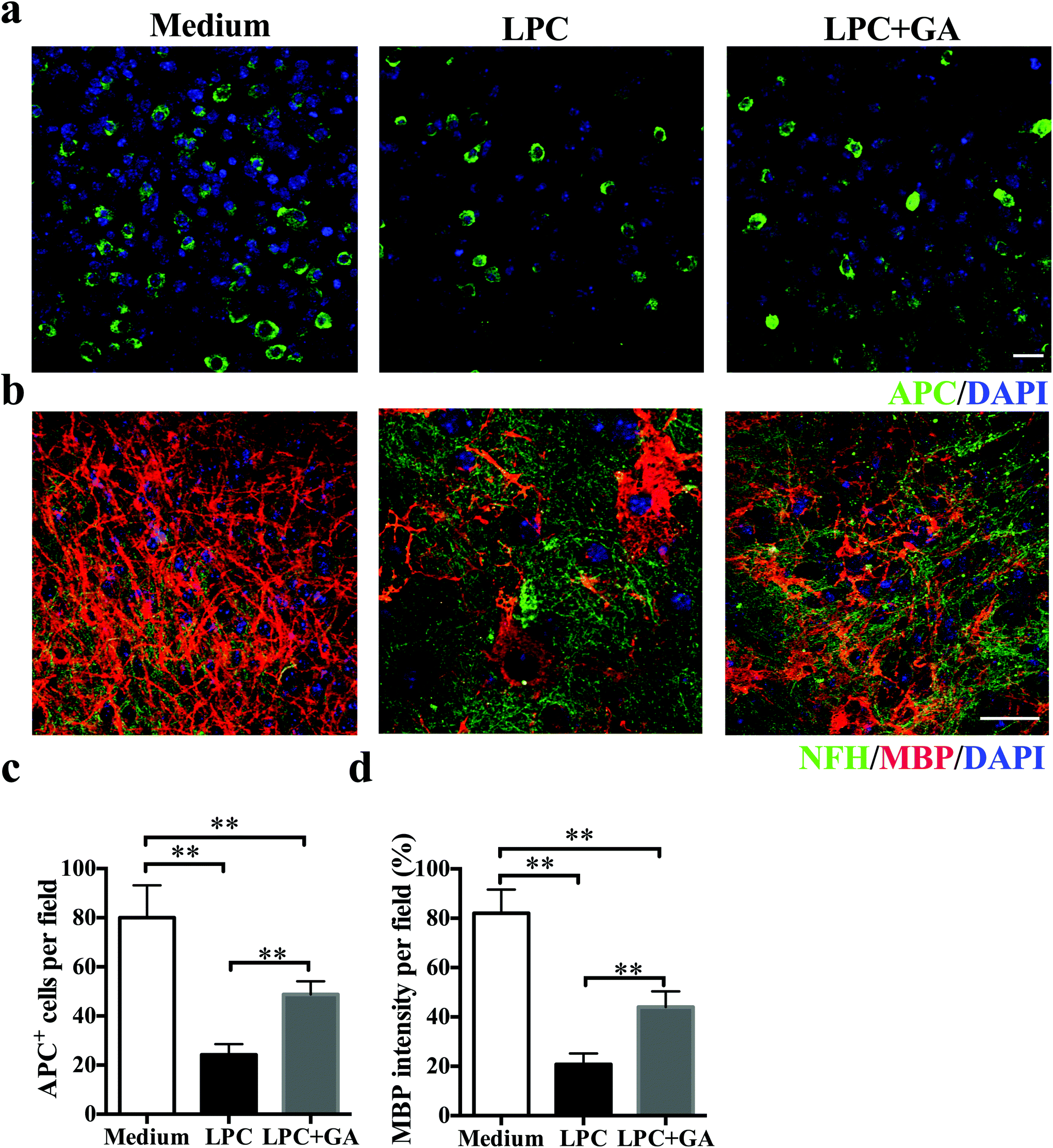 | ||
| Fig. 4 GA induced OLG maturation and improved CNS remyelination ex vivo. (a) Confocal images of mouse cerebellar slices treated with GA or PBS for 14 days (original myelination without LPC treatment as the control) and stained for APC (green) and DAPI (blue). (b) Double immunofluorescence staining of MBP (red) and NFH (green) in cerebellar slices. (c) Quantitative analysis was performed for APC + cells for mature OLGs. (d) Quantitative data of MBP intensity in NFH labeled axons in the GA (10 μM) treatment group compared to the PBS group. Data are shown as mean values ± SD (n = 5 slices each group). Scale bar = 10 μm in (a). Scale bar = 20 μm in (b). **p < 0.01, as determined by Student's t-test. One representative of three experiments is shown. | ||
3.5 GA promoted OPC maturation in vitro
To investigate the direct impact of GA on oligodendrocyte maturation, dissociated single OPCs cultured in differentiation medium were treated for 7 days with PBS or GA (10 μM) alone, followed by immunocytochemical staining. Mature oligodendrocytes were stained with CNPase, whereas proliferating OPCs were labelled with Ki67. GA-treated OPCs exhibited a more extended complex branched morphology after 2 weeks of culture (Fig. 5a), and the total branch length of oligodendrocytes increased more than fourfold in the GA-treated group compared with the PBS control group (Fig. 5b). Furthermore, the percentage of Ki67 + proliferative OPCs under differentiation conditions was significantly decreased in the GA-treated OPCs (Fig. 5c and d), as the cell cycle progression is generally inhibited at the stages of cellular maturation and differentiation. Consistent with the immunofluorescence findings, GA treatment up-regulated the transcription levels of the oligodendrocyte-specific transcription factors (e.g., Olig2, Sox10, and Sox 17) and oligodendrocyte-specific markers MBP, as well as the expression of the neurotrophic factor CNTF (Fig. 5e). Together, these results indicated that GA acts as a potent pro-myelinating compound, directly inducing OPC maturation in vitro.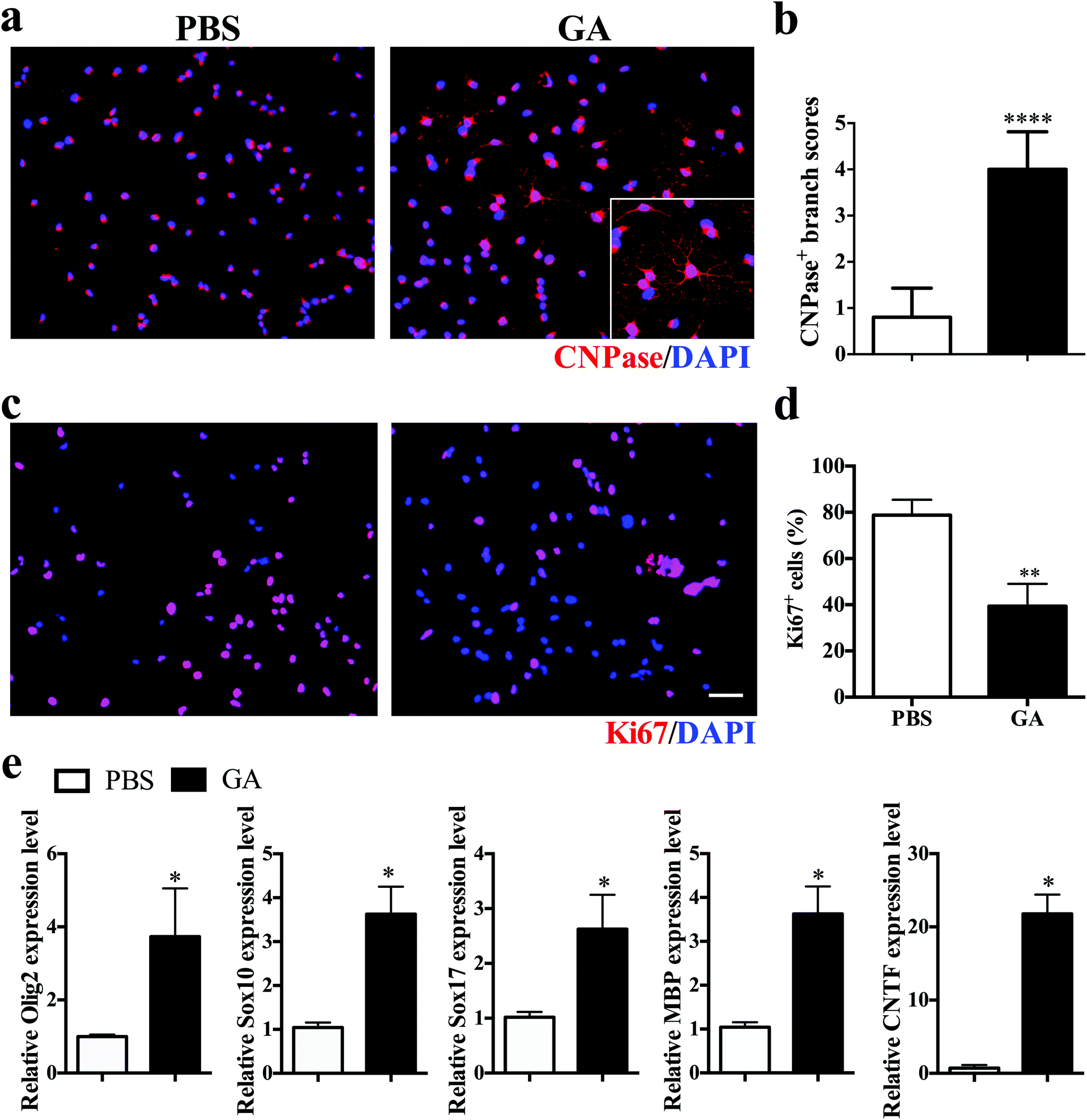 | ||
| Fig. 5 GA promoted OPC differentiation and myelination in vitro. OPCs were prepared from newborn (postnatal day 3) C57BL/6 mice and treated with GA (10 μM) for one week in the OPC differentiation medium. (a) Cells were immunostained for CNPase (red) and DAPI (blue). (b) Quantitative analysis was performed to calculate the branch score of differentiated OLGs. (c) Cells were then immunostained for Ki67 (red) and DAPI (blue). (d) Quantification of the percentage of Ki67 + cells. (e) Real-time quantitative PCR was performed to test Olig2, Sox10, Sox17, MBP, and CNTF in GA- and PBS-treated groups. Data are mean ± SD (n = 5 each group). Scale bar = 50 μm in (a, c). *p < 0.05, **p < 0.01, and ****p < 0.0001 as determined by two-way ANOVA. | ||
3.6 GA modulated remyelination through regulation of GSK-3β signaling pathways
Previous studies have reported that phosphorylation of JNK and GSK-3β plays a critical role in the process of OPC proliferation and differentiation.32,33 To determine the mechanism by which GA triggers oligodendrocyte maturation, we next investigated whether this compound could modulate the JNK or GSK-3β signaling pathway. After GA treatment for 48 h, the expression levels of total phosphorylation of JNK and GSK-3β were tested in OPCs cultured under oligodendrocyte differentiation conditions (Fig. 6a). As shown in Fig. 6b, compared with total JNK, no significant differences in two phospho-JNK isoforms, phospho-JNK54 and phosphor-JNK46, were observed between the GA- and PBS-treated groups. These results indicated that the GA-induced OPC differentiation is independent of the JNK signaling pathway. However, the expression of phospho-GSK-3β, compared with total GSK-3β and β-actin, was significantly reduced in the GA-treated group (decreased by 54.14% and 39.58% of the PBS-treated group, respectively) (Fig. 6c). To further confirm the pharmacological inhibition of GA on GSK-3β phosphorylation, AR-A014418, a GSK-3β inhibitor, was used. The results show that both GA and AR-A014418 treatment decreased the expression of phosphor-GSK-3β significantly, compared with total GSK-3β and β-actin (Fig. S2a–c†). The state of GSK-3β was also determined in the cuprizone-induced demyelination model. As shown in Fig. 6d and e, the expression of phospho-GSK-3β, compared with total GSK-3β and β-actin, was significantly reduced in the corpus callosum of the GA-treated group (decreased by 69.14% and 45.31% of the PBS-treated group, respectively), which is consistent with the in vitro results. Altogether, our data suggest that GA promoted OPC differentiation through the regulation of GSK-3β signaling pathways.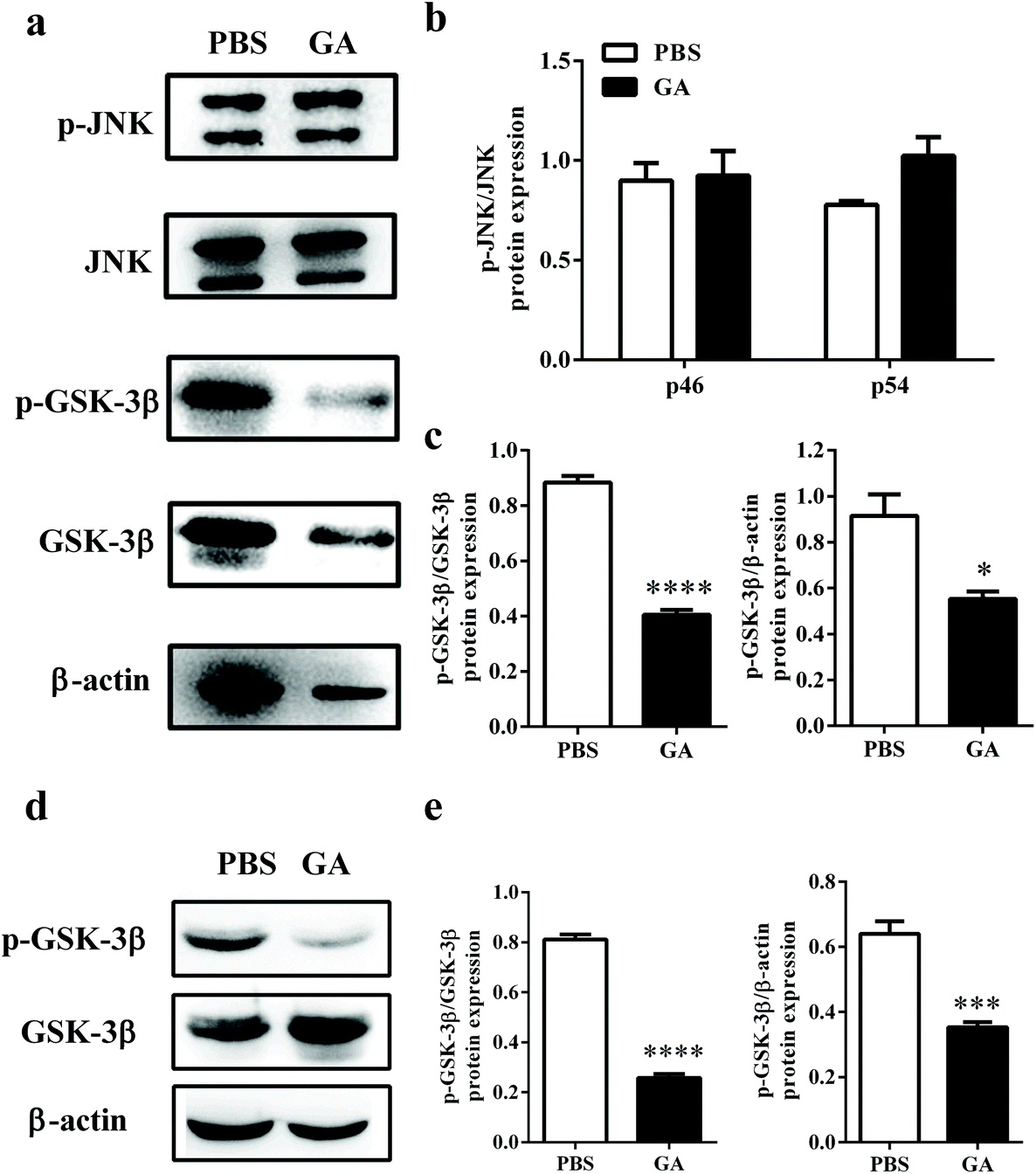 | ||
| Fig. 6 GA promoted OPC maturation through the regulation of the GSK-3β signaling pathway. (a) Primary OPCs isolated from the brains of newborn C57BL/6 mice (postnatal day 3) were treated with GA (10 μM) and vehicle in OPC differentiation medium for 48 h, and then probed by the western blotting of total JNK and GSK-3β and their phosphorylation counterparts, and β-actin. (b, c) Densitometry was determined using Image J and data were analyzed using Graphpad Prism 6. (d) For the remyelination model, 8-week old naïve male C57BL/6 mice were fed daily with cuprizone for 4 weeks; PBS or GA (50 mg kg−1) was administered daily between weeks 4 and 6 and the corpus callosum was collected for western blotting analysis. (e) Densitometry was determined using Image J and data were analyzed using Graphpad Prism 6. Data are mean ± SD (n = 3 each group). *p < 0.05, ***p < 0.001, and ****p < 0.0001, as determined by the nonparametric test. | ||
4. Discussion
Given the inflammation-induced tissue damage in the CNS at the chronic stage of MS/EAE, an ideal therapeutic strategy would be both anti-inflammatory and would enhance neural repair with minimal side effects. As reported in previous studies, the anti-inflammatory effects of GA and its derivative, 18-glycyrrhetinic acid, have been widely studied in animal models of liver damage34 and Parkinson's disease.35 In addition, 18-glycyrrhetinic acid was reported to show unique therapeutic effects in the EAE model, leading to microglia activation in the CNS in the EAE mice.36 In the current study, we demonstrated that, for the first time, GA effectively alleviated the clinical signs of the EAE mice and inhibited the infiltration of CNS inflammation and inflammation-induced demyelination, especially targeting the chronic stage which remains refractory to the available treatments. In MS and EAE when myelin damage occurred, OPCs activate and recruit to the lesion site by proliferation and migration, and then differentiate into new oligodendrocytes to achieve the regeneration of the myelin sheath.37 The remission of pathogenicity at the chronic stage of MS mainly depends on remyelination. Pathological studies have demonstrated that there are a large number of OPC clusters in the chronic lesions of MS patients, suggesting that the ability of OPCs to proliferate, survive, and migrate to the lesion site is not a major cause of myelin regeneration failure. However, the failure of normal differentiation and maturation is a limiting factor in the regeneration of the myelin sheath, and thus for the treatment of demyelinating diseases.38 Therefore, promoting OPC maturation into myelinating oligodendrocytes is important for remyelination and tissue repair.In this study, the potential of GA as a pro-myelinating therapy is clearly highlighted by several lines of evidence. Firstly, GA treatment not only significantly inhibited ongoing demyelination, but also promoted MBP production and remyelination at the chronic stage of EAE, when axonal damage and neuron loss have already occurred. Secondly, we further demonstrated the direct effects of GA to enhance remyelination in vivo, using the cuprizone-induced demyelination model. Although GA cannot prevent the subsequent demyelination induced by cuprizone, it improved remyelination effectively when cuprizone was withdrawn. Thirdly, GA treatment enhanced remyelination and increased the number and membrane extension of mature oligodendrocytes following LPC-induced demyelination of organotypic cerebellar slices. Last but not least, exposure of primary OPCs to GA, under the given conditions in vitro, decreased OPC proliferation and promoted their differentiation, providing supporting proof that GA stimulated the differentiation of OPCs. These encouraging findings resulting from different assays add weight to the hypothesis that GA played a dominant role in OPC differentiation into immature OLGs during the remyelination period. Thus, together with the anti-inflammatory effect, our data strongly suggest that GA, with its anti-inflammatory and remyelination capability, is a potential candidate drug for MS, especially at the intractable chronic stage.
Mechanically, we provide evidence indicating that GA directly induced OPC maturation and myelin repair via GSK-3β signaling, but not the phosphorylation of JNK. Luo et al. reported that phosphorylation of GSK-3β promoted the Notch 1 signaling pathway, which inhibited OPC differentiation and myelination.39 Azim et al. also proved that GSK-3β negatively regulated OPC differentiation.32 In addition, VP3.15 acting as a small molecule inhibitor of GSK-3β ameliorated the clinical disease severity of EAE and promoted remyelination.40 These findings are consistent with our results. Contradictorily, Zhou et al. reported that GSK-3β is a positive modulator of OPC differentiation.41 We speculated that the promising reasons of contradictory results are most probably due to dynamic and multilevel regulations. It might be attractive in the therapy of neurodegenerative diseases, such as multiple sclerosis, because GSK-3β may act as a pharmacological checkpoint in myelin-related diseases.
The direct receptor of GA is not clear up to now. Han et al.42 reported that the AKT/GSK-3β/snail axis is important in modulating the epithelial–mesenchymal transition (EMT), and miR-215 inhibits AKT phosphorylation mediation, and subsequently suppresses GSK-3β phosphorylation. Wang et al.43 indicted that activated AKT, as an important upstream regulator of GSK-3β, inhibits the activation of GSK-3β by promoting the phosphorylation of GSK-3β. Therefore, we speculated that GA may suppress the action of AKT and then impair the phosphorylation of GSK-3β indirectly, and eventually promote OPC differentiation and remyelination. Besides, Sitia et al.44 indicated that GA is an inhibitor of HMGB1, and it may directly bind to HMGB1, consequently inhibiting the phosphorylation and secretion of HMGB1. Nicaise et al.45 reported that HMGB1 can be secreted by neural progenitor cells, activated astrocytes and microglia in lesion areas, and was found to be a senescence-associated inhibitor of OPC differentiation. In MS patients, endogenous progenitor cells exhibit higher levels of HMGB1 and suppress OPC maturation, which may explain why immature oligodendroglia within lesions fail to remyelinate in MS.45 Therefore, HMGB1 may be another potent target regulated by GA for OPC differentiation and remyelination. With these clues, we will focus on the underlying mechanism of GA on OPC maturation and myelination in our further studies.
Taken together, our findings in the present study showed that GA alleviated the pathophysiology of demyelination diseases by both reducing inflammation and directly promoting remyelination (Fig. 7). These studies, together with the wide spectrum of pharmacological activities, the abundant sources and the low cost, pave the way for clinical applications in demyelination diseases in the CNS.
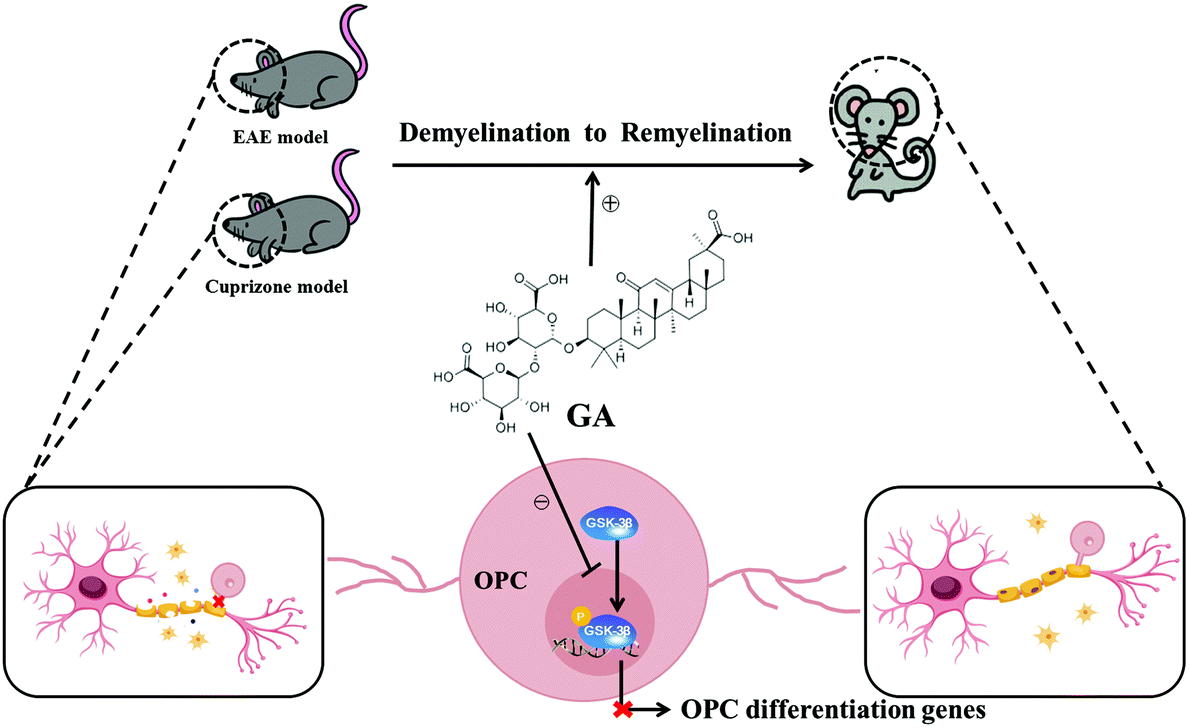 | ||
| Fig. 7 Model of GA-mediated repair by directly driving functional remyelination. GA promoted the maturation and differentiation of OPCs by inhibiting the phosphorylation of GSK-3β, accelerated remyelination in the CNS, and alleviated the clinical symptoms of EAE/cuprizone mice. | ||
Author contributions
Y. Z. and X. L. conceived and designed the experiments. J. T., L. Z. and P. X. S. carried out the experiments. C. Q. L., C. S. and L. Z. analyzed the data and wrote the manuscript. Z. Z. W. and Y. Z. co-supervised the study and revised the paper. All the authors read and approved the final manuscript.Funding sources
This study was supported by the National Key R&D Program of China (2018YFC1706500), the Chinese National Natural Science Foundation (Grant No. 81771345, 31970771), the Natural Science Foundation of Shaanxi Province, China (Grant No. 2018JZ3001, 2019KJXX-022, 2018JQ8033), the Fundamental Research Funds for the Central Universities (Grant No. GK20182010, GK201903062, GK201701009, 2018CSLZ018, 2019CSLZ016), and the Open Fund of Shanxi Key Laboratory of Inflammatory Neurodegenerative Diseases, Shanxi Datong University (Grant No. KF2019001, KF2019006).Conflicts of interest
The authors declare no competing interest.Acknowledgements
We thank Katherine Regan for editorial assistance.References
- H. Lassmann and M. Bradl, Multiple sclerosis: experimental models and reality, Acta Neuropathol., 2017, 133, 223–244 CrossRef CAS PubMed.
- R. Ehling, T. Berger and M. Reindl, Multiple sclerosis - established and novel therapeutic approaches, Cent. Nerv. Syst. Agents Med. Chem., 2010, 10, 3–15 CrossRef CAS PubMed.
- D. H. Rodrigues, B. P. Leles, V. V. Costa, A. S. Miranda, D. Cisalpino, D. A. Gomes, D. G. de Souza and A. L. Teixeira, IL-1beta Is Involved with the Generation of Pain in Experimental Autoimmune Encephalomyelitis, Mol. Neurobiol., 2016, 53, 6540–6547 CrossRef CAS PubMed.
- J. van Horssen, M. E. Witte, G. Schreibelt and H. E. de Vries, Radical changes in multiple sclerosis pathogenesis, Biochim. Biophys. Acta, 2011, 1812, 141–150 CrossRef CAS PubMed.
- I. M. Stromnes and J. M. Goverman, Active induction of experimental allergic encephalomyelitis, Nat. Protoc., 2006, 1, 1810–1819 CrossRef CAS PubMed.
- Q. Yang, C. Zheng, J. Cao, G. Cao and P. Shou, Spermidine alleviates experimental autoimmune encephalomyelitis through inducing inhibitory macrophages, Cell Death Differ., 2016, 23, 1850–1861 CrossRef CAS PubMed.
- Y. Bando, Y. Hagiwara, Y. Suzuki, K. Yoshida, Y. Aburakawa, T. Kimura, C. Murakami, M. Ono, T. Tanaka, Y. P. Jiang, B. Mitrovi, H. Bochimoto, O. Yahara and S. Yoshida, Kallikrein 6 secreted by oligodendrocytes regulates the progression of experimental autoimmune encephalomyelitis, Glia, 2018, 66, 359–378 CrossRef PubMed.
- J. Zhang, J. Chen, Y. Li, X. Cui, X. Zheng, C. Roberts, M. Lu, S. B. Elias and M. Chopp, Niaspan treatment improves neurological functional recovery in experimental autoimmune encephalomyelitis mice, Neurobiol. Dis., 2008, 32, 273–280 CrossRef CAS.
- M. Kipp, Remyelination strategies in multiple sclerosis: a critical reflection, Expert Rev. Neurother., 2016, 16, 1–3 CrossRef CAS.
- D. Bates, Treatment effects of immunomodulatory therapies at different stages of multiple sclerosis in short-term trials, Neurology, 2011, 76, S14–S25 CrossRef CAS.
- M. Kabrine and F. Laraba-Djebari, Immunomodulatory and protective properties of tacrolimus in experimental scorpion envenomation, Int. J. Immunopathol. Pharmacol., 2014, 27, 69–78 CrossRef CAS.
- O. Aktas, B. Kieseier and H. P. Hartung, Neuroprotection, regeneration and immunomodulation: broadening the therapeutic repertoire in multiple sclerosis, Trends Neurosci., 2010, 33, 140–152 CrossRef CAS.
- F. J. Najm, M. Madhavan, A. Zaremba, E. Shick, R. T. Karl, D. C. Factor, T. E. Miller, Z. S. Nevin, C. Kantor, A. Sargent, K. L. Quick, D. M. Schlatzer, H. Tang, R. Papoian, K. R. Brimacombe, M. Shen, M. B. Boxer, A. Jadhav, A. P. Robinson, J. R. Podojil, S. D. Miller, R. H. Miller and P. J. Tesar, Drug-based modulation of endogenous stem cells promotes functional remyelination in vivo, Nature, 2015, 522, 216–220 CrossRef CAS PubMed.
- C. Marianecci, F. Rinaldi, L. Di Marzio, M. Mastriota, S. Pieretti, C. Celia, D. Paolino, M. Iannone, M. Fresta and M. Carafa, Ammonium glycyrrhizinate-loaded niosomes as a potential nanotherapeutic system for anti-inflammatory activity in murine models, Int. J. Nanomed., 2014, 9, 635–651 CrossRef PubMed.
- G. Hoever, L. Baltina, M. Michaelis, R. Kondratenko, L. Baltina, G. A. Tolstikov, H. W. Doerr and J. Cinatl Jr., Antiviral activity of glycyrrhizic acid derivatives against SARS-coronavirus, J. Med. Chem., 2005, 48, 1256–1259 CrossRef CAS.
- Y. Guo, W. Zhang, Y. Y. Yan, C. G. Ma, X. Wang, C. Wang and J. L. Zhao, Triterpenoid pristimerin induced HepG2 cells apoptosis through ROS-mediated mitochondrial dysfunction, J. BUON, 2013, 18, 477–485 Search PubMed.
- T. G. van Rossum, A. G. Vulto, R. A. de Man, J. T. Brouwer and S. W. Schalm, Review article: glycyrrhizin as a potential treatment for chronic hepatitis C, Aliment. Pharmacol. Ther., 1998, 12, 199–205 CrossRef CAS.
- R. A. Isbrucker and G. A. Burdock, Risk and safety assessment on the consumption of Licorice root (Glycyrrhiza sp.), its extract and powder as a food ingredient, with emphasis on the pharmacology and toxicology of glycyrrhizin, Regul. Toxicol. Pharmacol., 2006, 46, 167–192 CrossRef CAS.
- M. N. Asl and H. Hosseinzadeh, Review of pharmacological effects of Glycyrrhiza sp. and its bioactive compounds, Phytother. Res., 2008, 22, 709–724 CrossRef CAS.
- X. Feng, L. Ding and F. Qiu, Potential drug interactions associated with glycyrrhizin and glycyrrhetinic acid, Drug Metab. Rev., 2015, 47, 229–238 CrossRef CAS PubMed.
- X. Gao, W. Wang, S. Wei and W. Li, [Review of pharmacological effects of Glycyrrhiza radix and its bioactive compounds], Zhongguo Zhongyao Zazhi, 2009, 34, 2695–2700 CAS.
- S. Han, L. Sun, F. He and H. Che, Anti-allergic activity of glycyrrhizic acid on IgE-mediated allergic reaction by regulation of allergy-related immune cells, Sci. Rep., 2017, 7, 7222 CrossRef.
- Q. Wu, Y. Tang, X. Hu, Q. Wang, W. Lei, L. Zhou and J. Huang, Regulation of Th1/Th2 balance through OX40/OX40L signalling by glycyrrhizic acid in a murine model of asthma, Respirology, 2016, 21, 102–111 CrossRef.
- Y. Zhang, X. Li, B. Ciric, C. G. Ma, B. Gran, A. Rostami and G. X. Zhang, Therapeutic effect of baicalin on experimental autoimmune encephalomyelitis is mediated by SOCS3 regulatory pathway, Sci. Rep., 2015, 5, 17407 CrossRef CAS.
- X. Li, Y. Zhang, Y. Yan, B. Ciric, C. G. Ma, J. Chin, M. Curtis, A. Rostami and G. X. Zhang, LINGO-1-Fc-Transduced Neural Stem Cells Are Effective Therapy for Chronic Stage Experimental Autoimmune Encephalomyelitis, Mol. Neurobiol., 2017, 54, 4365–4378 CrossRef CAS.
- N. F. Abo El-Magd, M. El-Mesery, A. El-Karef and M. M. El-Shishtawy, Glycyrrhizin ameliorates high fat diet-induced obesity in rats by activating NrF2 pathway, Life Sci., 2018, 193, 159–170 CrossRef CAS PubMed.
- L. Liu, A. Belkadi, L. Darnall, T. Hu, C. Drescher, A. C. Cotleur, D. Padovani-Claudio, T. He, K. Choi, T. E. Lane, R. H. Miller and R. M. Ransohoff, CXCR2-positive neutrophils are essential for cuprizone-induced demyelination: relevance to multiple sclerosis, Nat. Neurosci., 2010, 13, 319–326 CrossRef CAS.
- X. Li, Y. Zhang, Y. Yan, B. Ciric, C. G. Ma, B. Gran, M. Curtis, A. Rostami and G. X. Zhang, Neural Stem Cells Engineered to Express Three Therapeutic Factors Mediate Recovery from Chronic Stage CNS Autoimmunity, Mol. Ther., 2016, 24, 1456–1469 CrossRef CAS.
- Y. Zhang, J. J. Han, X. Y. Liang, L. Zhao, F. Zhang, J. Rasouli, Z. Z. Wang, G. X. Zhang and X. Li, miR-23b Suppresses Leukocyte Migration and Pathogenesis of Experimental Autoimmune Encephalomyelitis by Targeting CCL7, Mol. Ther., 2018, 26, 582–592 CrossRef CAS.
- B. Blanchard, T. Heurtaux, C. Garcia, N. M. Moll, C. Caillava, L. Grandbarbe, A. Klosptein, C. Kerninon, M. Frah, D. Coowar, A. Baron-Van Evercooren, E. Morga, P. Heuschling and B. Nait Oumesmar, Tocopherol derivative TFA-12 promotes myelin repair in experimental models of multiple sclerosis, J. Neurosci., 2013, 33, 11633–11642 CrossRef CAS.
- Y. Dombrowski, T. O'Hagan, M. Dittmer, R. Penalva, S. R. Mayoral, P. Bankhead, S. Fleville, G. Eleftheriadis and C. Zhao, Regulatory T cells promote myelin regeneration in the central nervous system, Nat. Neurosci., 2017, 20, 674–680 CrossRef CAS.
- K. Azim and A. M. Butt, GSK3beta negatively regulates oligodendrocyte differentiation and myelination in vivo, Glia, 2011, 59, 540–553 CrossRef.
- J. X. Zhang, Y. F. Feng, Q. Qi, L. Shen, R. Wang, J. S. Zhou, H. Z. Lu and J. G. Hu, JNK is necessary for oligodendrocyte precursor cell proliferation induced by the conditioned medium from B104 neuroblastoma cells, J. Mol. Neurosci., 2014, 52, 269–276 CrossRef CAS.
- J. Y. Yu, J. Y. Ha, K. M. Kim, Y. S. Jung, J. C. Jung and S. Oh, Anti-Inflammatory activities of licorice extract and its active compounds, glycyrrhizic acid, liquiritin and liquiritigenin, in BV2 cells and mice liver, Molecules, 2015, 20, 13041–13054 CrossRef CAS.
- S. Ojha, H. Javed, S. Azimullah, S. B. Abul Khair and M. E. Haque, Glycyrrhizic acid Attenuates Neuroinflammation and Oxidative Stress in Rotenone Model of Parkinson's Disease, Neurotoxic. Res., 2016, 29, 275–287 CrossRef CAS.
- J. Zhou, W. Cai, M. Jin, J. Xu, Y. Wang, Y. Xiao, L. Hao, B. Wang, Y. Zhang, J. Han and R. Huang, 18beta-glycyrrhetinic acid suppresses experimental autoimmune encephalomyelitis through inhibition of microglia activation and promotion of remyelination, Sci. Rep., 2015, 5, 13713 CrossRef.
- R. J. Franklin and C. Ffrench-Constant, Remyelination in the CNS: from biology to therapy, Nat. Rev. Neurosci., 2008, 9, 839–855 CrossRef CAS.
- D. Kremer, O. Aktas, H. P. Hartung and P. Kury, The complex world of oligodendroglial differentiation inhibitors, Ann. Neurol., 2011, 69, 602–618 CrossRef CAS.
- F. Luo, K. Burke, C. Kantor, R. H. Miller and Y. Yang, Cyclin-dependent kinase 5 mediates adult OPC maturation and myelin repair through modulation of Akt and GSK-3beta signaling, J. Neurosci., 2014, 34, 10415–10429 CrossRef CAS.
- E. M. Medina-Rodriguez, A. Bribian, A. Boyd, V. Palomo, J. Pastor, A. Lagares, C. Gil, A. Martinez, A. Williams and F. de Castro, Promoting in vivo remyelination with small molecules: a neuroreparative pharmacological treatment for Multiple Sclerosis, Sci. Rep., 2017, 7, 43545 CrossRef.
- L. Zhou, C. Y. Shao, S. M. Xu, J. Ma, Y. J. Xie, L. Zhou, P. Teng, Y. Wang, M. Qiu and Y. Shen, GSK3beta promotes the differentiation of oligodendrocyte precursor cells via beta-catenin-mediated transcriptional regulation, Mol. Neurobiol., 2014, 50, 507–519 CrossRef CAS.
- J. Han, M. Zhang, C. Nie, J. Jia, F. Wang, J. Yu, W. Bi, B. Liu, R. Sheng, G. He, L. Kong, L. Zheng, R. Pang, Z. Ding, L. Chen, Q. Guan, S. Pan, X. Meng, J. Xu, L. Liu and J. Zhang, miR-215 suppresses papillary thyroid cancer proliferation, migration, and invasion through the AKT/GSK-3beta/Snail signaling by targeting ARFGEF1, Cell Death Dis., 2019, 10, 195 CrossRef.
- H. Wang, H. Sui, Y. Zheng, Y. Jiang, Y. Shi, J. Liang and L. Zhao, Curcumin-primed exosomes potently ameliorate cognitive function in AD mice by inhibiting hyperphosphorylation of the Tau protein through the AKT/GSK-3beta pathway, Nanoscale, 2019, 11, 7481–7496 RSC.
- G. Sitia, M. Iannacone, S. Muller, M. E. Bianchi and L. G. Guidotti, Treatment with HMGB1 inhibitors diminishes CTL-induced liver disease in HBV transgenic mice, J. Leukocyte Biol., 2007, 81, 100–107 CrossRef CAS PubMed.
- A. M. Nicaise, L. J. Wagstaff, C. M. Willis, C. Paisie, H. Chandok and P. Robson, Cellular senescence in progenitor cells contributes to diminished remyelination potential in progressive multiple sclerosis, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9030–9039 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9fo01459d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |