
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRe-examining the role of the gut microbiota in the conversion of the lipid-lowering statin monacolin K (lovastatin) into its active β-hydroxy acid metabolite
D.
Beltrán
,
M. D.
Frutos-Lisón
,
J. C.
Espín
 and
R.
García-Villalba
*
and
R.
García-Villalba
*
Laboratory of Food & Health, Research Group on Quality, Safety and Bioactivity of Plant Foods, Department of Food Science and Technology, CEBAS-CSIC, P.O. Box 164, 30100 Campus de Espinardo, Murcia, Spain. E-mail: rgvillalba@cebas.csic.es
First published on 12th March 2019
Abstract
Monacolin K (MK, lovastatin), a naturally occurring statin, only exerts lipid-lowering effects in its active β-hydroxy acid form (MKA). This activation was thought to be mediated by the gut microbiota (GM). We report here for the first time that the GM does not convert MK into MKA (a spontaneous pH-dependent conversion) but catabolises MKA. The GM might hamper the lipid-lowering effects by degrading the active metabolite MKA.
Introduction
Monacolin K (MK) is a naturally occurring statin and widely consumed in food complements as lipid-lowering products. MK can be found in oyster mushrooms, red yeast rice, and Pu-erh tea.1 In particular, the traditional Chinese red yeast rice is a MK-rich food supplement produced by fermenting rice with the red mould species Monascus purpureus.2 The European Food Safety Authority (EFSA) approved a health claim regarding the consumption of red yeast rice as a food supplement containing MK (10 mg) for maintaining normal blood LDL-cholesterol concentrations.3 MK is identical to the pharmacological drug lovastatin, and thus the EFSA has recently raised safety concerns related to some side-effects commonly associated with the consumption of statins.4 Therefore, although identical, the term MK is used in the context of food supplements and lovastatin as a pharmaceutical drug.MK is administered as an inactive lactone (prodrug) and its lipid-lowering effects depend on its conversion into its active β-hydroxy acid form (MKA) (Fig. 1). MKA is a well-known competitive inhibitor of the 3-hydroxy-3-methylglutaryl coenzyme-A reductase, the rate-limiting enzyme in cholesterol biosynthesis.5 Liver cytochrome P450-dependent monooxygenases have been reported to be involved in the metabolism of MK, but not in the conversion from MK into MKA.6 Although Tang et al.7 described the in vivo hydrolysis of the lactone ring by plasma and liver carboxylesterases, these authors did not demonstrate this enzymatic process unequivocally.
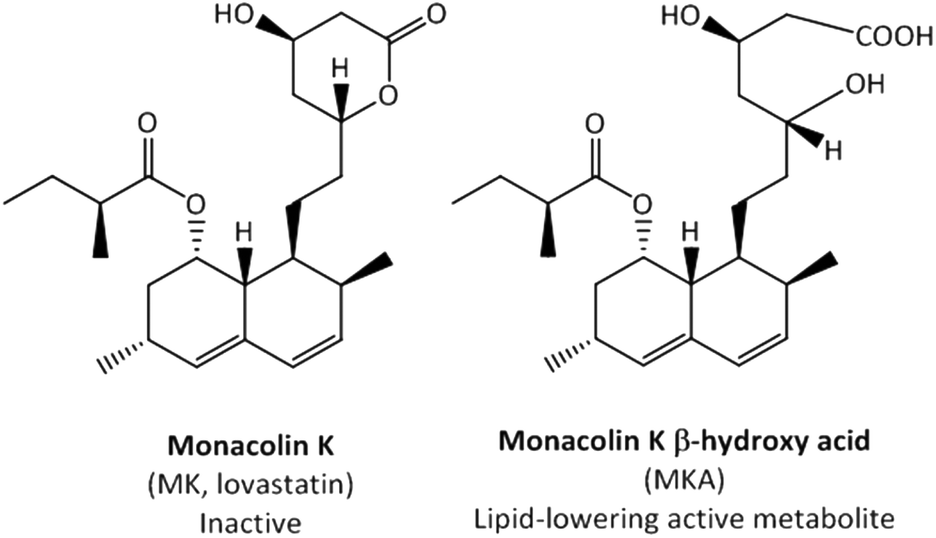 | ||
| Fig. 1 The inactive lactone prodrug monacolin K (MK) and its lipid-lowering active metabolite monacolin K β-hydroxy acid (MKA). | ||
The role of the GM in the metabolism of orally administered xenobiotics has gradually been gaining interest.8,9 In general, the microbial metabolism of statins has not been comprehensively studied so far. Aura et al.10 described some microbiota-derived simvastatin metabolites (a methylated analogue of MK). Remarkably, Yoo et al.11 reported lower plasma levels of MKA in antibiotic-treated rats and concluded that the GM was specifically involved in the activation of MK to yield MKA. Recently, the in vitro conversion of MK into MKA was also reported after MK incubation with 5 anaerobes that are common inhabitants of the human GM.12 However, these studies11,12 did not demonstrate unequivocally that the GM catalysed the conversion of MK into MKA.
We aim here to assess whether the GM is really involved in the conversion of MK into MKA and (or) in the catabolism of both compounds.
Materials and methods
Chemicals and samples
Monacolin K (MK, lovastatin) was purchased from Sigma-Aldrich (St Louis, MO, USA) and MK hydroxy acid sodium salt (MKA) from Santa Cruz Biotechnology (Santa Cruz, California, USA). Stock solutions of MK and MKA (10 mM) were prepared in propylene glycol (PPG) and water, respectively. Working solutions of a standard mixture were prepared by appropriate dilutions in methanol (MeOH). All solutions were stored at −20 °C. Acetonitrile was obtained from J. T. Baker (Deventer, The Netherlands), formic acid and hydrochloric acid from Panreac (Barcelona, Spain) and ethyl acetate from RCI Labscan (Gliwice, Poland). Milli-Q system (Millipore Corp., Bedford, MA, USA) ultrapure water was used throughout the study.MKA synthesis
MK (100 mg) was added to 50 mL of 0.1 M NaOH and kept under agitation at 30 °C for 24 hours. The samples were filtered through a 0.45 μm nylon filter and an aliquot of 5 mL was loaded onto a SPE cartridge Chromafix C18 (950 mg) (Macherey-Nagel, Düren, Germany), which was previously conditioned with 10 mL MeOH and then 10 mL water. After loading the sample, the cartridge was washed with 10 mL water for removing the excess of sodium and other water-soluble impurities. Finally, MKA was eluted with 5 mL MeOH and dried in a speed vacuum concentrator. The residue was dissolved in 5 mL water to obtain 5 mM MKA. An aliquot of the sample was diluted in water and injected into an HPLC-DAD-ESI-MS system to check the conversion rate by comparing the area with that of the commercial standard.Stability studies
MK and MKA (30 μM) were incubated in the absence of bacteria at different pH values and temperatures. MK was incubated at pH 7 (25 mM phosphate buffer), pH 12.5 (0.1 M NaOH) and pH 2 (0.1 N HCl), and at 30 and 60 °C. MKA was also used as the starting material to follow its stability and possible conversion into its lactone form (MK) and was incubated at pH 2 (0.1 N HCl). Samples were collected at 2, 4, 7 and 9 days for further analysis in the HPLC-DAD-ESI-MS system.Faecal cultures
This study was included in the Spanish National Project AGL2015-73744-JIN (MINECO, Spain). All experiments were performed in accordance with the ethical guidelines outlined in the Declaration of Helsinki, and approved by the Spanish National Research Council's Bioethics Committee (Madrid, Spain). Nine healthy volunteers (five men and four women) aged between 25 and 48 years gave their written consent before participating. Inclusion criteria were age over 18 years and good health status. Exclusion criteria were diagnosed pathology, previous gastrointestinal surgery, chronic medication as well as the intake of probiotics, prebiotics, and antibiotics for 3 months before participating.Preparation of faecal suspensions and culturing experiments were conducted as reported elsewhere.13 Aliquots of filtered faecal suspensions (50 μL) were inoculated into 5 mL of fermentation medium to grow anaerobes (anaerobe basal broth, ABB, Oxoid) containing MK or MKA at 15 μM. Triplicate samples were prepared in parallel. As controls, MK and MKA were incubated without the faecal suspension, and the faecal suspension was incubated without adding MK or MKA. Samples (5 mL), collected at 0, 3 and 6 days, were extracted with 5 mL of ethyl acetate. The mixture was vortexed for 10 min and centrifuged at 3500g for 10 min. The organic phase was evaporated under reduced pressure, redissolved in 250 μL of methanol and filtered through a 0.22 μm PVDF filter. Samples were diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 in methanol prior to the injection into the HPLC-DAD-ESI-MS system. The extraction protocol was tested by spiking 15 μM MK or MKA in the faecal medium. The recovery percentages of 97.7 ± 6.0 for MKA and 98.2 ± 4.3 for MK confirmed the suitability of the protocol.
:4 in methanol prior to the injection into the HPLC-DAD-ESI-MS system. The extraction protocol was tested by spiking 15 μM MK or MKA in the faecal medium. The recovery percentages of 97.7 ± 6.0 for MKA and 98.2 ± 4.3 for MK confirmed the suitability of the protocol.
HPLC-DAD-ESI-MS analyses
Chromatographic separation was carried out on an Agilent 1290 (Agilent Technologies, Aldbronn, Germany) equipped with a photodiode array detector and a single quadrupole (single Q) mass spectrometer detector in series (6120 Quadrupole, Agilent). A reverse phase column Poroshell 120 C18 (100 × 3 mm, 2.7 μm) (Agilent) operating at 25 °C was used. Mobile phases consisting of water acidified with 0.5% formic acid (phase A) and acetonitrile (phase B) at a flow rate of 0.5 mL min−1 were used with the following gradient: 0 min, 20% B, from 0 to 15 min, 20–50% B, from 15 to 23 min, 50–75% B, and from 23 to 28 min, 75–95% B; this percentage was maintained from 1 min and then returned to the initial condition for 5 min. Nitrogen was used as a nebuliser gas to optimise ESI-MS parameters as follows: capillary voltage: 3000 V, drying gas flow: 9 L min−1, nebuliser pressure: 40 psi, and drying temperature: 300 °C. MS spectra were acquired in positive ionisation mode and measured in selective ion monitoring (SIM) mode. The fragmentor voltage was optimised to 100 V by flow injection analysis (FIA) of each standard at 100 μM in MeOH. The mass ions 405 ([MK + H]+), 427 ([MK + Na]+), 423 ([MKA + H]+) and 445 ([MKA + Na]+) were recorded. The identification of the metabolites was carried out by a direct comparison with commercial standards and taking into account their mass, UV spectra and retention times. MK and MKA were quantified using UV spectra at 237 nm. Calibration curves from 0.5 to 100 μM were obtained for both compounds.Results
Stability of MK and MKA
The incubation of MK in the absence of faecal inocula and at a plausible colonic pH and temperature (pH 7.0 and 37 °C) resulted in the transformation of MK into MKA, which confirmed that the GM is not essential for the opening of the lactone ring to take place (Fig. 2A). A faster transformation was observed at 60 °C, although after 2 days the kinetics of the decrease was similar to that obtained at pH 7 and 37 °C, not reaching complete conversion after 9 days (data not shown).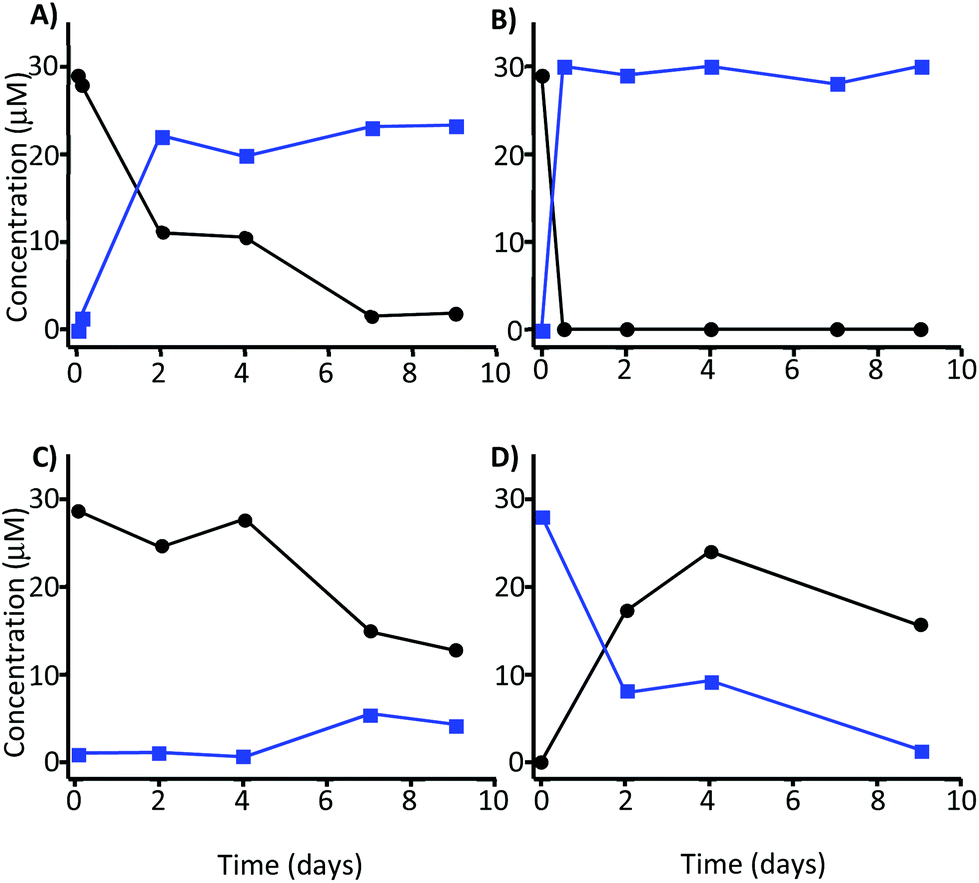 | ||
Fig. 2 Stability of 30 μM MK (●) and MKA ( ) at 37 °C and different pH values. Incubation of (A) MK at pH 7, (B) MK at pH 12.5, (C) MK at pH 2, and (D) MKA at pH 2. ) at 37 °C and different pH values. Incubation of (A) MK at pH 7, (B) MK at pH 12.5, (C) MK at pH 2, and (D) MKA at pH 2. | ||
The fast and total transformation of MK into MKA was achieved at pH 12.5 and was independent of the temperature (Fig. 2B). MKA was very stable at this pH. These alkaline conditions were applied to isolate MKA with a conversion rate of 99% for further incubation processes with the faecal microbiota. At pH 2, MK became unstable after 4 days, and approximately 60% of MK was degraded after 7 days with only a slight transformation into MKA (5 μM) (Fig. 2C). At pH 2, MKA decreased concomitantly with the increase of MK, indicating that MKA could be reversed into MK at this pH. Again the degradation of both MK and MKA became evident after 4 days (Fig. 2D). Overall, these results suggested a GM-independent conversion of MK into MKA. Therefore, we next explored the role of the GM in the metabolism of both MK and MKA.
Microbial metabolism
The faecal incubation of MK at different time points reduced its concentration (Fig. 3A), but this reduction was similar in the absence and presence of faecal inocula. MK was converted into MKA over time (Fig. 3A), but the conversion was lower in the presence than in the absence of faecal inocula after 6 days of incubation. | ||
| Fig. 3 (A) Incubation of MK (15 μM) in the absence (black) or presence (grey) of faecal inocula, and its conversion into MKA in the absence (striped dark blue) and presence (striped cyan) of inocula. (B) Incubation of MKA (15 μM) in the absence (blue) or presence (cyan) of faecal inocula. Results are expressed as mean ± SD (n = 9 volunteers in triplicate). | ||
We next incubated the previously synthesised MKA in the absence and in the presence of bacteria (Fig. 3B), confirming the stability of MKA in the growth medium at pH 7 in the absence of bacteria. However, the presence of the faecal inocula caused a significant decrease of MKA (Fig. 3B). The degradation of MK and MKA after 6 days, depending on the faecal inoculum, ranged from 73% to 91% in the case of MK, and from 58% to 79% in the case of MKA.
While the catabolism of MKA by the GM occurred unequivocally (Fig. 3B), it is difficult to confirm the involvement of the GM in the degradation of MK because similar reduction was observed in the absence or presence of faecal inocula (Fig. 3A), indicating only a pH-dependent conversion. Overall, these results showed the GM-independent conversion of MK into MKA and the catabolism of MKA by the GM, and also suggested that MK is not metabolised by the GM.
Discussion
The role of the GM in the metabolism of xenobiotics, including dietary phytochemicals and drugs, has been investigated extensively.8,9,14 However, the metabolism of statins by the GM, and in particular that of MK, is still not well understood. The first study that assigned a crucial role to the GM in the conversion of MK into MKA was that reported by Yoo et al.,11 who observed lower plasma levels of MKA in antibiotic-treated rats, and concluded that the GM was specifically involved in this activation. Afterwards, other reports have repeatedly cited this study and assumed the transformation of MK into MKA catalysed by the GM.15–17 Unfortunately, none of these reports, including that of Yoo et al.,11 showed incubation of MK under plausible physiological colonic conditions (pH 7 and 37 °C) in the absence of bacteria.In the present study, we demonstrate that the conversion of MK into the lipid-lowering active metabolite MKA occurs spontaneously at neutral and alkaline pH values, and independently of the GM. In contrast, MKA is very stable under physiological colonic pH and temperature, but it is catabolised by the GM to other unknown metabolites, whose identification requires further research. Besides, our results suggest a critical role of the β-hydroxy acid moiety in MKA to become a substrate of the GM since no apparent catabolism of lactone-containing MK by the GM occurred.
Our results could also suggest the presence of high and low MKA metabolisers with clear involvement of the GM. However, the assays were carried out with the faecal microbiota of only 9 volunteers, and some individuals yielded disparate results at different sampling periods consistent with an experimental variation rather than with a specific individual feature. Therefore, more research is needed in a larger group to establish whether the population can be stratified in high and low MKA metabolisers.
The pH-dependent degradation of MK has been previously reported in different biological fluids.18,19 We observed the complete transformation of MK into MKA at pH 12.5 in which MKA was very stable, in agreement with previous reports.20 However, strong acidic conditions (pH 2.5) only partially reversed MKA into MK since both metabolites were further degraded. Overall, the inter-conversion between MK and MKA was clearly dependent on the pH as previously reported for other statins.21,22
A key point in the present research is to find a plausible explanation for the results obtained by Yoo et al.,11 who concluded that the GM was specifically involved in the conversion of MK into MKA since they detected lower plasma levels of MKA in antibiotic-treated rats. It is known that ATP-binding cassette (ABC) and solute carrier (SLC) membrane transporters transport statins across the cell membrane.23 Besides, it is also known that some antibiotics, including erythromycin, can compete with MK-MKA for some ABC transporters such as P-glycoprotein (P-gp, MDR1).24 Yoo et al.11 administered an acute dose of an antibiotic cocktail, including erythromycin. However, these authors did not unequivocally evaluate the involvement of the GM in the conversion of MK into MKA (i.e., incubation of MK in growth medium in the presence and absence of the GM), since the effect of antibiotics on other key variables, such as statin cellular transporters, was not evaluated. Therefore, lower plasma MKA levels in antibiotic-treated rats do not necessarily imply lower MKA formation but lower MKA transport from the intestine to the bloodstream due to a possible interaction between the antibiotics and the ABC transporters involved in MKA absorption. In contrast, while antibiotics seem to hamper MKA transport to the bloodstream, we speculate that the administration of antibiotics could prevent the microbial metabolism of MKA in the colon lumen by inhibiting the GM.
Conclusions
We describe here for the first time that the transformation of monacolin K (lovastatin, MK) into its active lipid-lowering metabolite the β-hydroxy acid metabolite (MKA) is a spontaneous process that occurs at neutral pH and without the participation of the GM. Besides, our results also show for the first time that the GM catabolises MKA to other compounds unknown so far and thus, the GM could mediate the lipid-lowering effect of MKA in vivo. Further research is needed before speculating the existence of individuals who harbour a GM more or less efficient in the metabolism of MKA, which might give rise to different responses in individuals after consuming MK. Our ongoing research aims at the identification of the MKA-derived microbial metabolites, the specific microbial groups involved in MKA catabolism and the possible modulation of these groups with antibiotics and prebiotics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research reported in this article has been supported by the Project AGL2015-73744-JIN (MINECO, Spain).References
- P. Svoboda, D. Sander, K. Plachká and L. Nováková, Development of matrix effect-free MISPE-UHPLC-MS/MS method for determination of lovastatin in PU-erh tea, oyster mushroom, and red yeast rice, J. Pharm. Biomed. Anal., 2017, 140, 367–376 CrossRef CAS PubMed.
- H. N. Huang, Y. Y. Hua, G. R. Bao and L. H. Xie, The quantification of monacolin K in some red yeast rice from fujian providence and the comparison of the other product, Chem. Pharm. Bull., 2006, 54, 687–689 CrossRef CAS PubMed.
- EFSA, Scientific Opinion on the substantiation of health claims related to monacolin K from red yeast rice and maintenance of normal blood LDL cholesterol concentrations (ID 1648, 1700) pursuant to Article 13(1) of Regulation (EC) No 1924/2006, EFSA J., 2011, 9, 2304 Search PubMed.
- EFSA, Scientific opinion on the safety of monacolins in red yeast rice, EFSA J., 2018, 16, 5368 Search PubMed.
- A. W. Alberts, Lovastatin and simvastatin-inhibitors of HMG CoA reductase and cholesterol biosynthesis, Cardiology, 1990, 77, 14–21 CrossRef PubMed.
- R. W. Wang, P. H. Kari, A. Y. Lu, P. E. Thomas, F. P. Guengerich and K. P. Vyas, Biotransformation of lovastatin. IV. Identification of cytochrome P450 3A proteins as the major enzymes responsible for the oxidative metabolism of lovastatin in rat and human liver microsomes, Arch. Biochem. Biophys., 1991, 290, 355–361 CrossRef CAS PubMed.
- B. K. Tang and W. Kalow, Variable activation of lovastatin by hydrolytic enzymes in human plasma and liver, Eur. J. Clin. Pharmacol., 1995, 47, 449–451 CrossRef CAS PubMed.
- P. Spanogiannopoulos, E. N. Bess, R. N. Carmody and P. J. Turnbaugh, The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism, Nat. Rev. Microbiol., 2016, 14, 273–287 CrossRef CAS PubMed.
- J. C. Espín, A. González-Sarrías and F. A. Tomás-Barberán, The gut microbiota: a key factor in the therapeutic effects of (poly)phenols, Biochem. Pharmacol., 2017, 139, 82–93 CrossRef PubMed.
- A. M. Aura, I. Mattila, T. Hyötyläinen, P. Gopalacharyulu, C. Bounsaythip, M. Oresic and K. M. Oksman-Caldentey, Drug metabolome of the simvastatin formed by human intestinal microbiota in vitro, Mol. Biosyst., 2011, 7, 437–446 RSC.
- D. H. Yoo, I. S. Kim, T. K. Van Le, I. H. Jung, H. H. Yoo and D. H. Kim, Gut microbiota-mediated drug interactions between lovastatin and antibiotics, Drug Metab. Dispos., 2014, 42, 1508–1513 CrossRef PubMed.
- V. Demonfort-Nkamga, N. Armstrong and M. Drancourt, In vitro susceptibility of cultured human methanogens to lovastatin, Int. J. Antimicrob. Agents, 2017, 49, 176–182 CrossRef CAS PubMed.
- R. García-Villalba, D. Beltrán, J. C. Espín, M. V. Selma and F. A. Tomás-Barberán, Time course production of urolithins from ellagic acid by human gut microbiota, J. Agric. Food Chem., 2013, 61, 8797–8806 CrossRef PubMed.
- T. Sousa, R. Paterson, V. Moore, A. Carlsson, B. Abrahamsson and A. W. Basit, The gastrointestinal microbiota as a site for the biotransformation of drugs, Int. J. Pharm., 2008, 363, 1–25 CrossRef CAS PubMed.
- D. H. Kim, Gut microbiota mediated drug-antibiotic interactions, Drug Metab. Dispos., 2015, 43, 1581–1589 CrossRef CAS PubMed.
- C. D. Klaassen and J. Y. Cui, Review: mechanisms of how the intestinal microbiota alters the effects of drugs and bile acids, Drug Metab. Dispos., 2015, 43, 1505–1521 CrossRef CAS PubMed.
- H. Li, J. He and W. Jia, The influence of gut microbiota on drug metabolism and toxicity, Expert Opin. Drug Metab. Toxicol., 2016, 12, 31–40 CrossRef CAS PubMed.
- A. Lueje-Álvarez, J. Pastine, J. A. Squella and L. J. Nuñez-Vergara, Assessment of the hydrolytic degradation of lovastatin by HPLC, J. Chil. Chem. Soc., 2005, 50, 639–646 Search PubMed.
- A. Saha, H. Jangala, P. Vats, R. Thakur, A. Khuroo and T. Monif, Stability indicating LC-MS/MS method for estimation of lovastatin in human plasma: application to a bioequivalence study, J. Anal. Sci. Technol., 2015, 6, 19–30 CrossRef.
- D. J. Yang and L. S. Hwang, Study on the conversion of three natural statins from lactone forms to their corresponding hydroxy acid forms and their determination in Pu-Erh tea, J. Chromatogr. A, 2006, 1119, 277–284 CrossRef CAS PubMed.
- A. Y. Yang, L. Sun, D. G. Musson and J. J. Zhao, Application of a novel ultra-low elution volume 96-well solid-phase extraction method to the LC/MS/MS determination of simvastatin and simvastatin acid in human plasma, J. Pharm. Biomed. Anal., 2005, 38, 521–527 CrossRef CAS PubMed.
- A. S. Kearny, L. F. Crawford, S. C. Mehta and G. W. Radebaugh, The interconversion kinetics, equilibrium, and solubilities of the lactone and hydroxyacid forms of the HMG-CoA reductase inhibitor, CI-981, Pharm. Res., 1993, 10, 1461–1465 CrossRef.
- K. C. E. Rocha, B. M. V. Pereira and A. C. Rodrigues, An update on efflux and uptake transporters as determinants of statin response, Expert Opin. Drug Metab. Toxicol., 2018, 14, 613–624 CrossRef CAS PubMed.
- R. B. Kim, C. Wandel, B. Leake, M. Cvetkovic, M. F. Fromm, P. J. Dempsey, M. M. Roden, F. Belas, A. K. Chaudhary, D. M. Roden, A. J. Wood and G. R. Wilkinson, Interrelationship between substrates and inhibitors of human CYP3A and P-glycoprotein, Pharm. Res., 1999, 16, 408–414 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |