
AMPK activation is involved in hypoglycemic and hypolipidemic activities of mogroside-rich extract from Siraitia grosvenorii (Swingle) fruits on high-fat diet/streptozotocin-induced diabetic mice†
Hesheng
Liu
ab,
Xiangyang
Qi
 *a,
Keke
Yu
a,
Anjie
Lu
a,
Kaifeng
Lin
a,
Jiajing
Zhu
a,
Min
Zhang
ac and
Zhida
Sun
b
*a,
Keke
Yu
a,
Anjie
Lu
a,
Kaifeng
Lin
a,
Jiajing
Zhu
a,
Min
Zhang
ac and
Zhida
Sun
b
aBeijing Advanced Innovation Center for Food Nutrition and Human Health, College of Biological and Environmental Sciences, Zhejiang Wanli University, Ningbo 315100, P. R. China. E-mail: qixiangyang85@sina.com; Tel: +86 574 88222461
bCollege of Food Science and Technology, Huazhong Agricultural University, Wuhan 430070, P. R. China
cThe School of Food and Chemical Engineering, Beijing Technology & Business University (BTBU), Beijing 100048, P. R. China
First published on 15th November 2018
Abstract
Previous studies suggested the anti-diabetic effect of mogrosides in type 1 diabetes. To evaluate the potential effect of mogrosides in type 2 diabetes, we herein investigated the hypoglycemic and hypolipidemic effects and the underlying mechanism of mogroside-rich extract (MGE) using a high-fat diet in combination with streptozotocin (STZ)-induced diabetic model. MGE feeding for 5 weeks did not result in any obvious impact on the body weight and energy intake, but caused a moderate decrease of organ index in diabetic mice. MGE-supplemented diabetic mice showed a notable reduction of fasting blood glucose (FBG), glycated serum protein (GSP), serum insulin, homeostasis model assessment-insulin resistance (HOMA-IR), and serum atherogenic lipid profiles in a dose-dependent manner, whereas significant increases in the anti-atherogenic lipid profile, insulin sensitivity, glucose and insulin tolerance capacity with high dose (300 mg kg−1) MGE were observed (P < 0.01). Besides, hepatocyte polymorphism, lipid accumulation and steatosis were ameliorated and restored to near normal at the high dose. Furthermore, hepatic 5′AMP-activated protein kinase (AMPK) signaling was dose-dependently activated. Accordingly, the mRNA levels of hepatic gluconeogenic and lipogenic genes were downregulated and fat oxidation-associated genes were upregulated. These findings suggest that the hypoglycemic and hypolipidemic activities of MGE are probably attributed to the attenuation of insulin resistance and activation of hepatic AMPK signaling.
1. Introduction
Diabetes is a complex metabolic syndrome characterized as persistent hyperglycemia and abnormal lipid metabolism. If treatments fail to tightly control these symptoms, it will eventually progress to serious complications, such as atherosclerosis, coronary heart disease, stroke and renal failure.1–3 Various commercially available drugs, including α-glucosidase inhibitors, biguanides, sulfonylureas, thiazolidinediones and dipeptidyl peptidase inhibitors, have been used as therapeutic agents for diabetes. Unfortunately, some of these drugs are restricted and intolerant to diabetic individuals. Furthermore, long-term administration of these drugs causes several side effects.4,5 Hence, alternative therapeutic agents with no or low toxicity, such as bioactive compounds of dietary or plant sources, have gained increasing interest.6,7AMPK, a phylogenetically conserved serine/threonine kinase, senses intracellular energy conditions. It is activated by elevated adenosine monophosphate (AMP)/adenosine triphosphate (ATP) ratios and subsequently inhibits energy-consuming anabolic pathways such as glycogen, lipid and protein biosynthesis, while promoting ATP-generating catabolic pathways such as glycolysis and fatty acid oxidation.8,9 It has been well documented that AMPK activation elicits beneficial effects on metabolic disorders, including obesity, dyslipidemia and type 2 diabetes. Once activated, AMPK improves insulin resistance, hepatic gluconeogenesis and lipogenesis, and enhances fat oxidation.10 Thus, AMPK is a potential therapeutic target for treatments of diabetes and other metabolic diseases.
The fruit of Siraitia grosvenorii (Swingle) is an edible traditional Chinese medicine and has been used as a folk medicine for sore throat, dry cough, thirst and constipation for a long history in China.11 Its major bioactive substances are cucurbitane-type tetracyclic triterpene glycosides (termed mogrosides),12 with extreme sweetness (approximately 300 times sweeter than sucrose), low-calories, non-nutritive value and low toxicity.13,14 Recently, mogroside extract has been approved as a generally recognized as safe (GRAS) substance by the U.S. Food and Drug Administration (FDA) and a food additive in China and Japan.14,15 Pharmacological studies revealed the anti-cancer, antioxidant, anti-fibrotic, anti-inflammatory and anti-diabetic activities of mogrosides.11,16,17 Our previous work showed that mogroside extract markedly suppressed hyperglycemia, and improved immune balance and serum lipid profiles in alloxan-induced diabetic mice,18 but the underlying mechanisms of hypoglycemic and hypolipidemic effects were not defined. Recently, mogrosides and their metabolites have been found to be capable of activating AMPK in Hep G2 cells19 or cell-free kinase activity in vitro experiments.20 Harada et al. (2016) also reported that mogrol (the aglycone of mogrosides) suppressed triglyceride accumulation in 3T3-L1 adipocytes by inhibiting the transcriptional activity of cAMP response element-binding protein (CREB) and activating AMPK signaling.21 Although the AMPK-activating effect of mogrosides has been proved in vitro, whether MGE activates AMPK and exerts anti-diabetic effects through AMPK signaling or not in vivo is still unknown. Therefore, the present study aimed at investigating the anti-hyperglycemic and anti-hyperlipidemic effects of mogrosides on type 2 diabetes through the activation of AMPK signaling.
2. Materials and methods
2.1. Chemicals
MGE from Siraitia grosvenorii (Swingle) was provided by Guilin Layn Natural Ingredients Corp. Its chemical constituents and main individual mogrosides were analyzed in our previous report,22 consisting of moisture 8.69%, total phenolics 1.43%, total flavonoids 0.38%, total mogrosides 80.14% (colorimetric method), mogroside V 44.52%, 11-O-mogroside V 7.34%, mogroside IV 0.97%, mogroside VI 4.58%, mogroside III 0.58% and mogroside IIA2 0.32%, using a high performance liquid chromatography method. A pelletized high-fat diet (HFD, 45% kcal fat) was purchased from Jiangsu Medicine Biomedical Co., Ltd (Yangzhou, China) and its ingredient formula is listed in the ESI (Table S1†). The antibodies AMPK (#5831), phosphor-AMPK (Thr172) (#2535), acetyl-CoA carboxylase (ACC) (#3676), phosphor-ACC (Ser79) (#11818), fatty acid synthase (FAS) (#3180), horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (#7074) and HRP-conjugated anti-mouse IgG (#7076) were purchased from Cell Signaling Technology (Danvers, MA, USA), and β-actin monoclonal antibody from TransGen Biotech Co., Ltd (Beijing, China). STZ was purchased from Sigma-Aldrich (St Louis, MO, USA). A rat/mouse insulin enzyme-linked immunosorbent assay (ELISA) kit was obtained from Millipore Corporation (Billerica, MA, USA). Recombinant human insulin was obtained from ProSpec-Tany TechnoGene Ltd (Ness Ziona, Israel). Unless otherwise indicated, all other chemicals used in the current study were of analytical grade.2.2. Animal treatments
Four-week-old male C57BL/6 mice (SCXK (hu) 2012-0002) were purchased from Slaccas Laboratory Animal Co., Ltd (Shanghai, China). The mice received diets and water ad libitum and were maintained in a temperature- and relative humidity-controlled animal room (23 ± 3 °C and 40–70%, respectively), and on a 12 h light (7:00 a.m. light)/12 h dark cycle. All animal experiments were performed in accordance with the Regulations of Laboratory Animal Management in China (National Science and Technology Commission, 2013) and guide for Laboratory Animal Care and Use of Huazhong Agricultural University and approved by the Ethics Committee of Laboratory Animal Use and Care of Ningbo University (approval No. SYXK 2016-0103). Animal welfare was maximized to minimize the suffering as much as possible during animal operations.After acclimatization for 1 week, mice were randomly divided into 2 groups according to their body weight. Group 1 (NC, 10 mice) was fed with regular pelletized chow (13.8% kcal fat) from Shanghai Puluteng Biotechnology Co., Ltd (Shanghai, China) throughout the experimental period. Group 2 (80 mice) was fed with a HFD throughout the feeding period and at the end of the 8th week, the mice were intraperitoneally injected with 110 mg per kg bw STZ after fasting for 14 h. STZ was dissolved in 0.1 mol L−1 citrate buffer, pH 4.5, and freshly prepared before injection. Mice in the NC group were injected with an equal amount of a citrate buffer vehicle. One week after STZ injection, mice with fasting blood glucose (FBG) above 7.8 mmol L−1 together with the symptoms of polyuria, polydipsia, polyphagia and body weight loss were denoted as type 2 diabetic animals. Established diabetic animals (40 mice, appropriately 55% mice were successfully induced to diabetes) were randomly divided into 4 groups based on FBG, 10 mice each group. One group (DM) without MGE or any other drug treatment was given only distilled water intragastrically and the other three groups were given low (150 mg per kg bw, DM + LMGE) and high (300 mg per kg bw, DM + HMGE) doses of MGE, and 200 mg per kg bw metformin (DM + Met), respectively. The treatments were maintained for another five weeks and mice in all groups except NC feeding regular rodent diets received the HFD during the following experimental periods. Food intake in each cage was monitored every day and the body weight and FBG were measured every week. At the end of the 14th week, mice were fasted overnight and blood was collected by cardiac puncture after anesthesia. Serum was obtained by centrifugation at 4 °C, 1500g for 15 min and stored at −80 °C for biochemical analysis. All mice were anaesthetized using pentobarbital sodium (70 mg per kg bw) and sacrificed by cervical dislocation. Liver and epididymal white adipose tissue (eWAT) were harvested and weighed promptly after sacrifice and the organ index was expressed as 100× organ weight/body weight. A small portion of the left lobe of the liver was immersed in neutral formaldehyde for histopathological analysis, and the remaining liver was sharply frozen in liquid nitrogen and stored at −80 °C before use.
2.3. Intraperitoneal glucose tolerance test (IPGTT) and insulin tolerance test (IPITT)
As described by Yu et al.,23 IPGTT and IPITT were performed with minor modification. Briefly, in the last week, mice in all groups were injected intraperitoneally with glucose at a dose of 1 g per kg bw after a 14 h food deprival. Blood drops were collected by clipping a very small portion of the tail and the glucose concentration was determined using a hand-held glucometer (Accu-Chek, Roche Diagnostics, Mannheim, Germany) by a glucose oxidase method at 0, 15, 30, 60 and 120 min after glucose ingestion. Specifically, the value at 0 min is referred to as the concentration before glucose injection. With regard to the IPITT, 3 days before the end of the feeding period, mice in all groups were intraperitoneally injected with recombinant human insulin (0.8 U per kg bw) after fasting for 6 h. Similar to the IPGTT, the glucose concentration was also measured at 0, 30, 60, 90 and 120 min after insulin injection.2.4. Serum biomarker analysis
The levels of glycated serum protein (GSP), total triglyceride (TG), total cholesterol (TC), low density lipoprotein cholesterol (LDL-C) and high density lipoprotein cholesterol (HDL-C) were assayed using commercially available kits from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). The FBG was measured with a commercial strip using a hand-held Accu-Chek active glucometer (Roche Diagnostics). The serum insulin level was analyzed using an ELISA kit according to the manufacturer's instructions. HOMA-IR was calculated using the formula fasting plasma glucose concentration (mmol L−1) × fasting plasma insulin level (FINS, μU mL−1) divided by 22.5,24 and the insulin sensitivity index (ISI)25 was calculated using the formula Ln (FINS × FBG)−1.2.5. Liver histopathology analysis
Liver tissue fixed in 4% PBS-buffered neutral formaldehyde for 48 h was embedded in paraffin and cut into a section with 5 μm thickness, and then was stained by a hematoxylin and eosin (H&E) method. Histopathological changes of the section were examined and imaged by a bright-field microscopy (Olympus CX41, Japan).2.6. Gene expression analysis
Total RNA from liver was isolated and purified using a commercial reagent. Briefly, 30–50 mg frozen liver was fully homogenized with an appropriate amount of the RNAiso Plus reagent (TaKaRa, Dalian, China). Isolation and purification procedures were carried out according to the manufacturer's instructions and finally dissolved with RNase-free deionized water. The RNA quality was checked from the ratio of OD260 nm/OD280 nm and by 1% agarose gel electrophoresis and its concentration was determined by using an ultra-micro spectrophotometer (Nanodrop 2000, Thermo Fisher, MA, USA). Subsequently, 1 μg total RNA with a ratio of 1.8 to 2.0 was reverse transcribed to single-stranded cDNA using a PrimeScript™ II 1st strand cDNA synthesis kit (TaKaRa, Dalian, China) at 42 °C for 45 min. An appropriate amount (500 ng) of cDNA with a SYBR Premix DimerEraser (TaKaRa, Dalian, China) was used to amplify on a LightCycler 480 real-time thermo cycler (Roche Diagnostics, Switzerland). Specific primers (ESI, listed in Table S2†) were designed using an online tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast)26 or according to reported literature studies,27,28 and synthesized by Sangon Biotech Co., Ltd (Shanghai, China). A high-resolution melting curve was obtained after amplification for primer specificity analysis. The data were obtained from 5 mice in two replicates. The relative level of mRNA was calculated by the 2−ΔΔCt method, which was normalized to the mRNA level of β-actin.2.7. Western blotting
About 30 mg of liver tissue was thoroughly homogenized in pre-cooled Western and IP cell lysis buffer (Beyotime, Nantong, China) on ice. 1 mM phenylmethylsulfonyl fluoride, protease and phosphatase inhibitor cocktail (Roche, Mannheim, Germany) were added just prior to homogenization. The lysate was centrifuged at 4 °C, 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 30 min to collect the supernatant and its protein concentration was quantified by a bicinchoninic acid assay (BCA) method. An appropriate amount of the total protein sample (30–50 μg) was separated by 8–10% polyacrylamide gel electrophoresis (Bio-Rad) and electrically transferred onto a 0.45 μm pore size nitrocellulose membrane (GE Healthcare, Wisconsin, USA). After blocking in 5% (w/v) skimmed milk dissolved in tris-buffered saline solution containing 0.1% Tween-20 for 1 h at ambient temperature, the membrane was incubated at 4 °C overnight with the primary antibodies as follows: AMPK (1:1000), phospho-AMPK (p-AMPK, 1:1000), ACC (1:1000), phospho-ACC (p-ACC, 1:1000), FAS (1:1000) and β-actin (1:3000). After incubation with HRP-conjugated secondary antibodies for 1 h at ambient temperature, the signal was detected using an enhanced chemiluminescence-based reagent (GE Healthcare, Wisconsin, USA). The relative expression level of protein was calculated from the gray intensity of the band and normalized using the protein level of β-actin.
000g for 30 min to collect the supernatant and its protein concentration was quantified by a bicinchoninic acid assay (BCA) method. An appropriate amount of the total protein sample (30–50 μg) was separated by 8–10% polyacrylamide gel electrophoresis (Bio-Rad) and electrically transferred onto a 0.45 μm pore size nitrocellulose membrane (GE Healthcare, Wisconsin, USA). After blocking in 5% (w/v) skimmed milk dissolved in tris-buffered saline solution containing 0.1% Tween-20 for 1 h at ambient temperature, the membrane was incubated at 4 °C overnight with the primary antibodies as follows: AMPK (1:1000), phospho-AMPK (p-AMPK, 1:1000), ACC (1:1000), phospho-ACC (p-ACC, 1:1000), FAS (1:1000) and β-actin (1:3000). After incubation with HRP-conjugated secondary antibodies for 1 h at ambient temperature, the signal was detected using an enhanced chemiluminescence-based reagent (GE Healthcare, Wisconsin, USA). The relative expression level of protein was calculated from the gray intensity of the band and normalized using the protein level of β-actin.
2.8. Statistical analysis
Data are presented as mean ± standard error of the mean (SEM). Normality was evaluated by Shapiro-Wilk's test and the homogeneity of variances was tested by Levene's test. When two groups are compared, unpaired Student's t test with or without Welch's correction was employed. For comparisons of three or more groups, one-way analysis of variance (ANOVA) followed by Dunnett's multiple comparisons was applied to equal variances, while one-way ANOVA with Welch's correction followed Games–Howell's multiple comparisons to unequal variances. P < 0.05 denotes statistical significance.3. Results
3.1. MGE treatment on the body weight, energy intake and organ index
As shown in Fig. 1A, before STZ injection, the body weight of the HFD groups increased notably compared to the regular chow-fed group, but STZ injection resulted in body weight loss dramatically. Although MGE or Met intervention did not reverse the body weight, it suppressed a sharp drop in the body weight in contrast to the continuous loss of the body weight in diabetic mice. With respect to energy intake, no significant difference was observed between regular chow-fed mice and HFD-fed mice before diabetic model establishment, while the energy intake of diabetic mice increased notably compared to non-diabetic mice, but the difference between MGE- and Met-treated diabetic mice was not significant (Fig. 1B). These results indicate that MGE or Met intervention did not affect the energy intake and appetite of diabetic mice. The data of the organ index indicated that diabetic mice had a higher liver and eWAT index, and MGE or Met ingestion reduced the elevated organ index (Fig. 1C), which suggests a lipid-lowering effect of MGE on diabetic mice.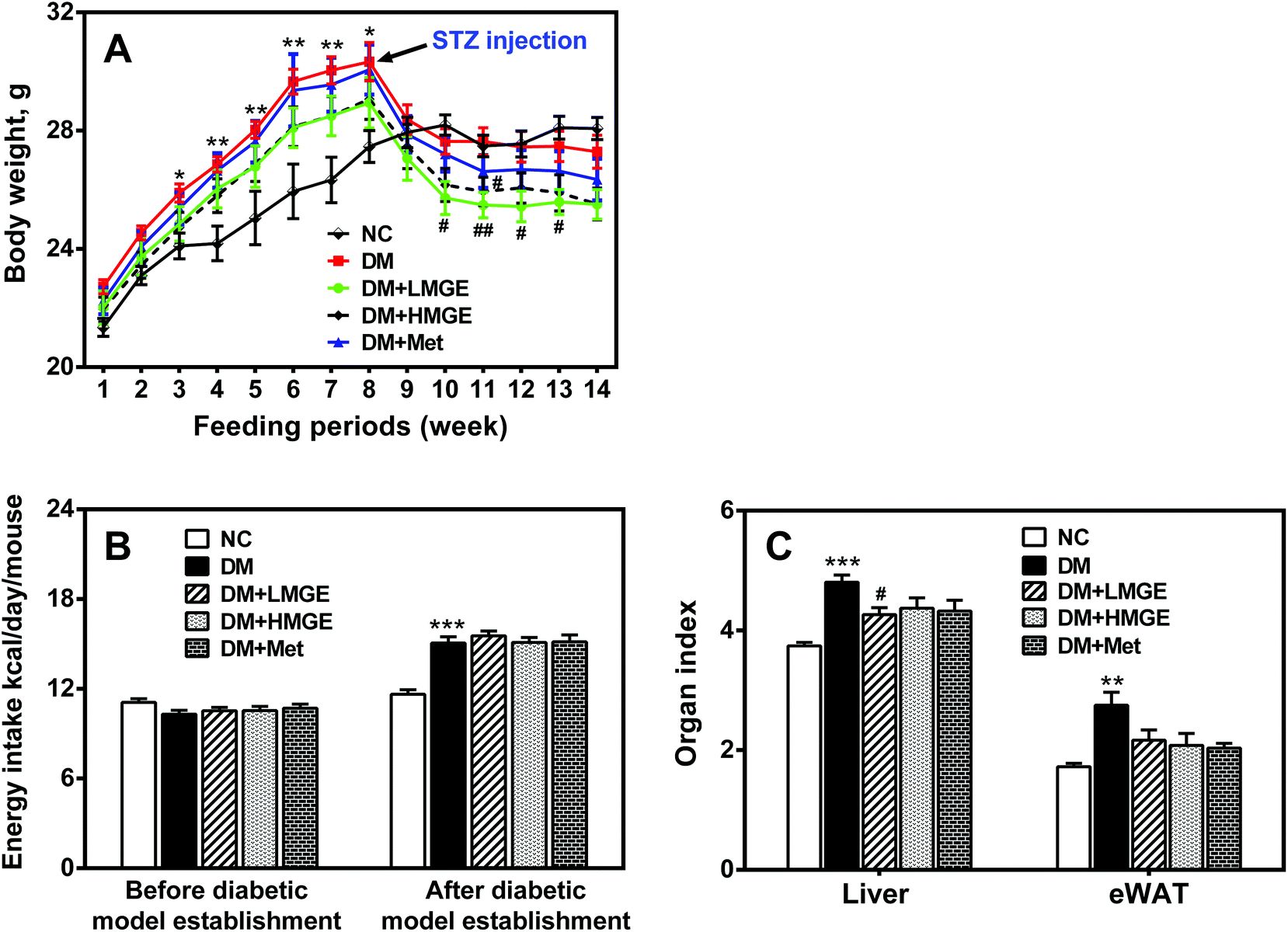 | ||
| Fig. 1 Effects of MGE treatment on the phenotype of HFD/STZ-induced diabetic mice. (A) Body weight alteration during the experimental period. (B) Energy intake before and after diabetic model establishment. (C) Organ index. Data are expressed as mean ± SEM (n = 9–10) and analyzed by one-way ANOVA followed by Dunnett's multiple comparisons for equal variances or Games–Howell multiple comparisons for unequal variances. *p < 0.05, **p < 0.01, ***p < 0.001 versus NC group, and #p < 0.05, ##p < 0.01, ###p < 0.001 versus DM group. | ||
3.2. MGE treatment on FBG and GSP
Diabetes-associated hyperglycemia induces protein glycation and causes dysfunction of the protein, which may lead to a variety of diseases. Thus, the reduction of FBG and GSP is of great importance. In the current study, despite increased FBG in diabetic and MGE- or Met-treated mice in the feeding periods, a rapid rise of FBG in diabetic mice was inhibited by MGE and Met intervention compared to diabetic mice without treatment (Fig. 2A). Similarly, the GSP level in the DM group was much higher than that in the NC group. Low and high dose MGE or MGE treatment groups showed a lower GSP level than that in the DM group, and the effect seems to be dose-dependent, in spite of no significance difference (P > 0.05) (Fig. 2B). These data suggest a beneficial effect of MGE treatment on hyperglycemia control and the inhibition of elevated GSP mediated by diabetes.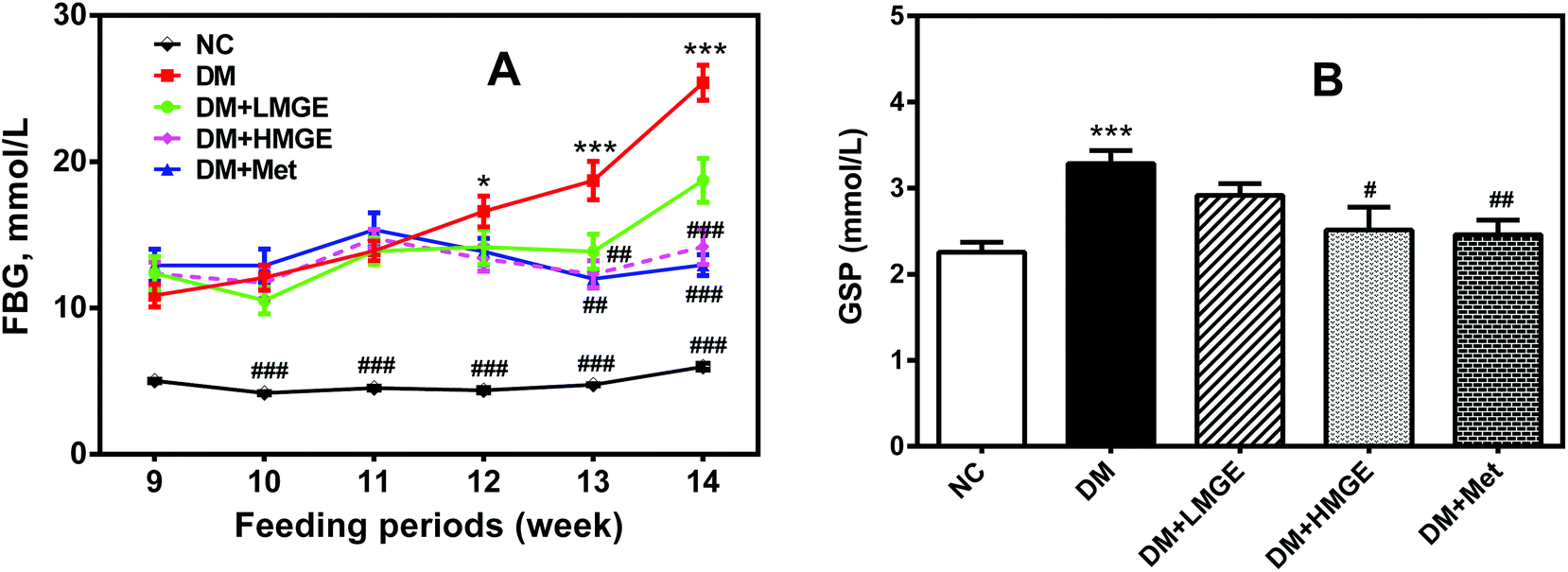 | ||
| Fig. 2 Effects of MGE treatment on the (A) FBG and (B) GSP levels. Significance among groups compared with the NC or DM group was analyzed using one-way ANOVA followed by Dunnett's multiple comparisons. Data are expressed as mean ± SEM (n = 9–10). *p < 0.05, **p < 0.01, ***p < 0.001 versus NC group, and #p < 0.05, ##p < 0.01, ###p < 0.001 versus DM group. | ||
3.3. MGE treatment on insulin, HOMA-IR, ISI, IPGTT and IPITT
In order to investigate the effect of MGE intake on insulin action and the regulation of glucose metabolism, we assayed the serum insulin level, HOMA-IR, ISI, IPGTT and IPITT. As illustrated in Fig. 3A, compared with the NC group, a remarkable increase (by 104.3%, P < 0.01) of insulin was observed in mice from the DM group, while MGE dose dependently lowered the insulin level of DM mice. Diabetic mice supplemented with 300 mg mg−1 MGE caused a 46.0% reduction of the insulin level compared to that of untreated diabetic mice, which was comparable to those of the positive and normal controls. Similarly, a sharp increase of HOMA-IR and a large reduction of the ISI were observed in diabetic mice (P < 0.01), whereas diabetic mice treatment with low and high dose MGE or Met notably decreased serum HOMA-IR by 46.1%, 70.5% and 72.4% and increased the ISI by 9.8%, 19.4% and 20.2%, respectively (Fig. 3B and C). These beneficial effects of high dose MGE were comparable to that of Met treatment. In a study of the IPGTT, the glucose concentration reached the highest value at 15 min, and then gradually decreased. Mice in the NC group were more tolerant to the glucose load and the glucose concentration of normal mice reduced rapidly close to the initial value, but that of the DM group dropped quite slowly. A moderate improvement in glucose tolerance was observed in diabetic mice fed with 300 mg kg−1 MGE or Met (P < 0.05), despite no obvious change in 150 mg kg−1 MGE-fed diabetic mice (P > 0.05). The results of the IPITT revealed that MGE in different dosage groups was capable of enhancing the potency of insulin-stimulated glucose uptake, and the beneficial effects of 300 mg kg−1 MGE were comparable to that of Met, although there was a large gap between HMGE and Met or NC groups.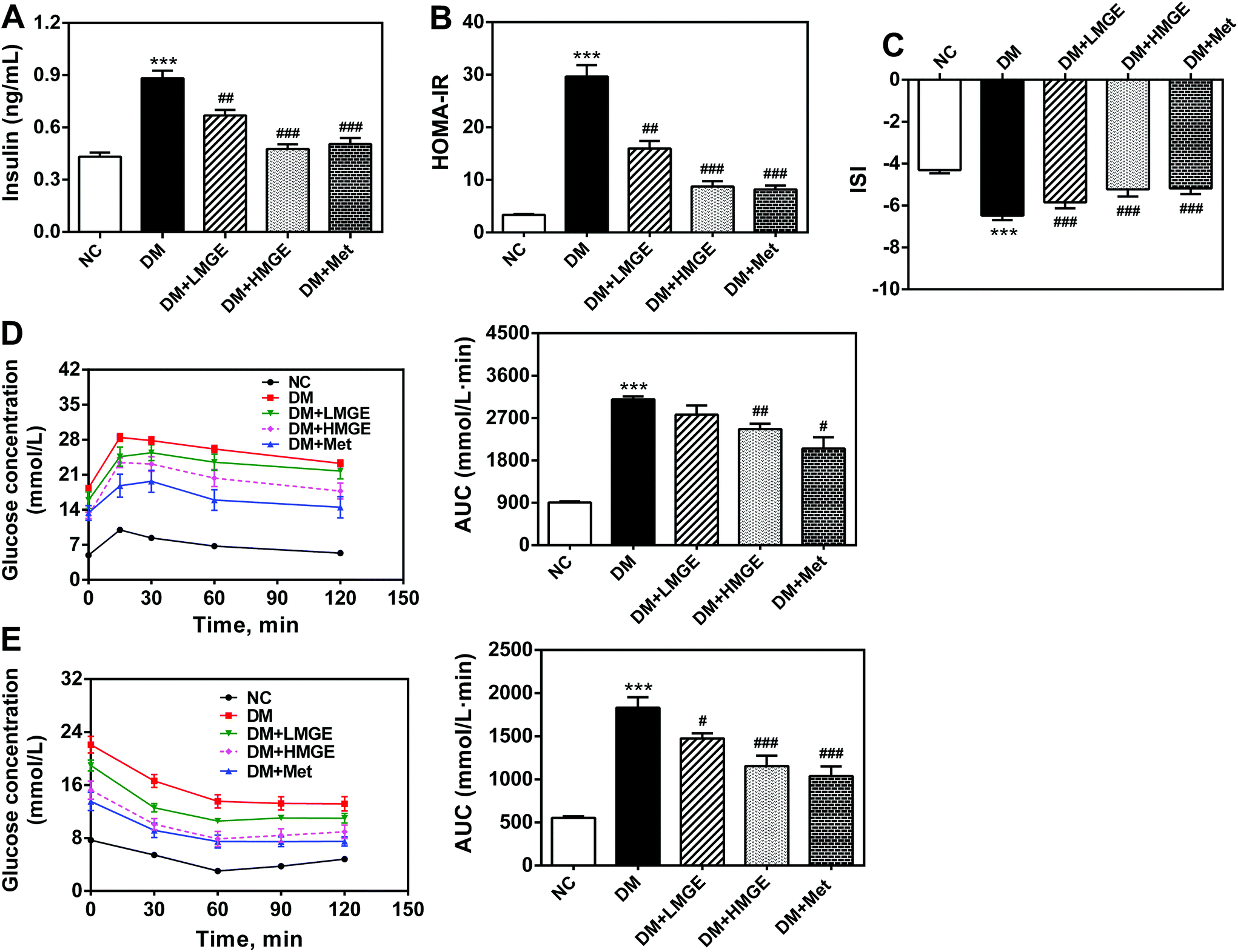 | ||
| Fig. 3 Effects of MGE treatment on the serum (A) insulin level, (B) HOMA-IR, (C) ISI, (D) IPGTT plus AUC and (E) IPITT plus AUC. Data are expressed as mean ± SEM (n = 8–10) and the differences between groups compared with the NC or DM group were analyzed using one-way ANOVA followed by Dunnett's multiple comparisons for equal variances or Games–Howell's multiple comparisons for unequal variances. *p < 0.05, **p < 0.01, ***p < 0.001 versus NC group, and #p < 0.05, ##p < 0.01, ###p < 0.001 versus DM group. | ||
These data collectively demonstrate that MGE ingestion, especially in a high dose, resulted in reduced insulin resistance, enhanced insulin sensitivity, and improved glucose and insulin tolerance.
3.4. MGE treatment on serum lipid profiles
To study the possible ameliorative effect of MGE feeding on serum lipid profiles in diabetic animals, the serum TG, TC, LDL-C and HDL-C levels of mice with different treatments were determined. Fig. 4 shows that compared to the NC group, TG, TC and LDL-C increased notably in the DM group, while HDL-C reduced slightly in the DM group compared to the NC group. However, MGE supplementation reduced TG, TC and LDL-C in a dose-dependent manner, despite no significant reduction in the TC and LDL-C levels. Interestingly, MGE elicited a noticeable increase of HDL-C and 300 mg kg−1 MGE induced an acute rise by 65.6% compared to the DM group (P < 0.05). These results indicate that MGE treatment may alleviate dyslipidemia resulting from diabetes.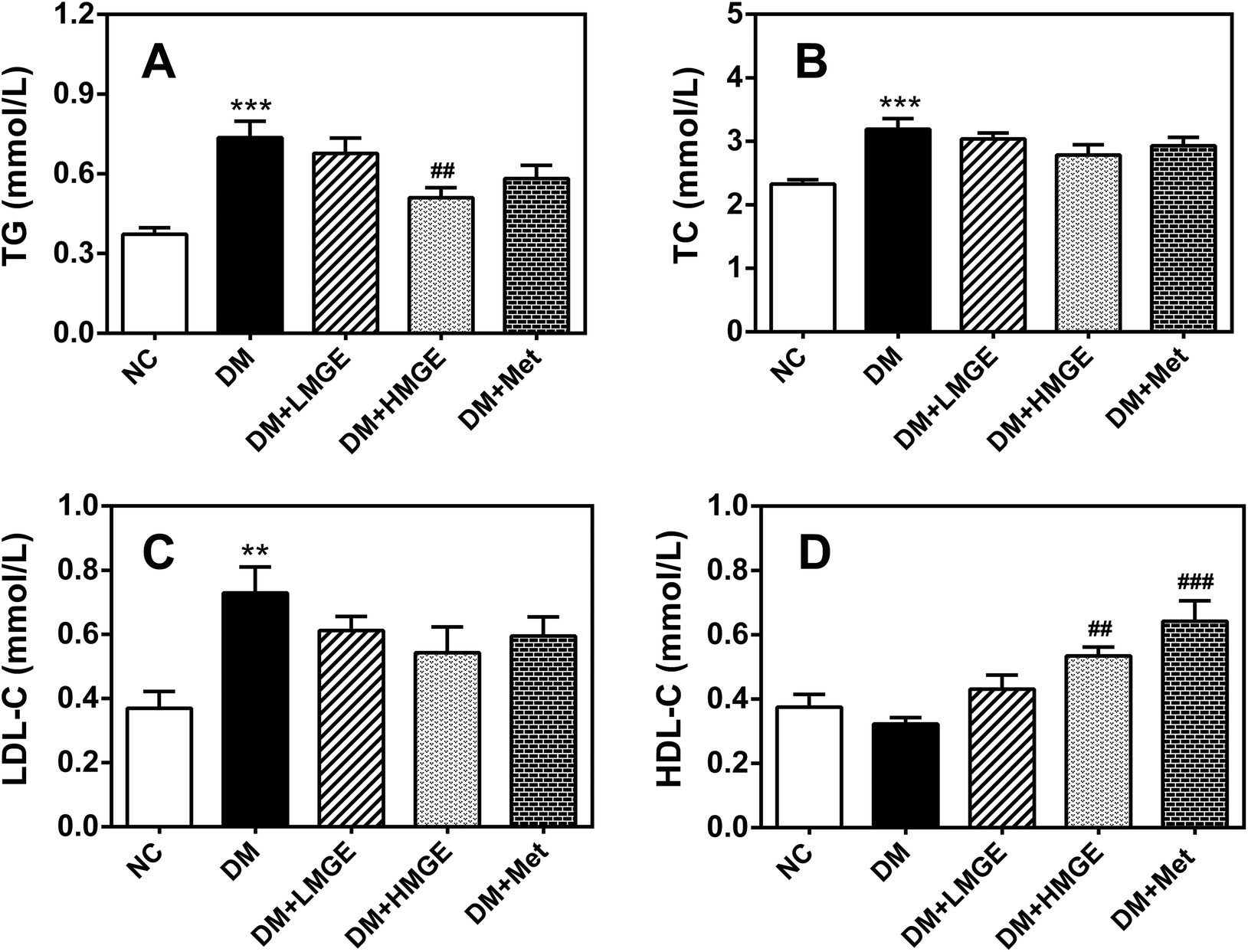 | ||
| Fig. 4 Effects of MGE treatment on serum (A) TG, (B) TC, (C) LDL-C and (D) HDL-C. Data are expressed as mean ± SEM (n = 9–10) and differences between groups compared with the NC or DM group were analyzed using one-way ANOVA followed by Dunnett's multiple comparisons for equal variances or Games–Howell's multiple comparisons for unequal variances. *p < 0.05, **p < 0.01, ***p < 0.001 versus NC group, and ##p < 0.01, ###p < 0.001 versus DM group. | ||
3.5. MGE treatment on liver histopathology
Liver histopathological examination (Fig. 5) showed that no abnormality of the hepatocyte architecture and morphology was observed in normal mice. However, the liver of the diabetic model exhibited severe hepatocyte necrosis and macrovesicular steatosis, as well as the infiltration of inflammatory cells, whereas MGE feeding for 5 weeks exhibited a beneficial effect on diabetes-mediated pathological changes, with marked attenuation of macrovesicular steatosis, liver inflammation and focal necrosis. In addition, the hepatocyte morphology of diabetic mice fed with high dose MGE was restored to near normal, suggesting the improvement of hepatic disorders related to lipid metabolism after MGE ingestion.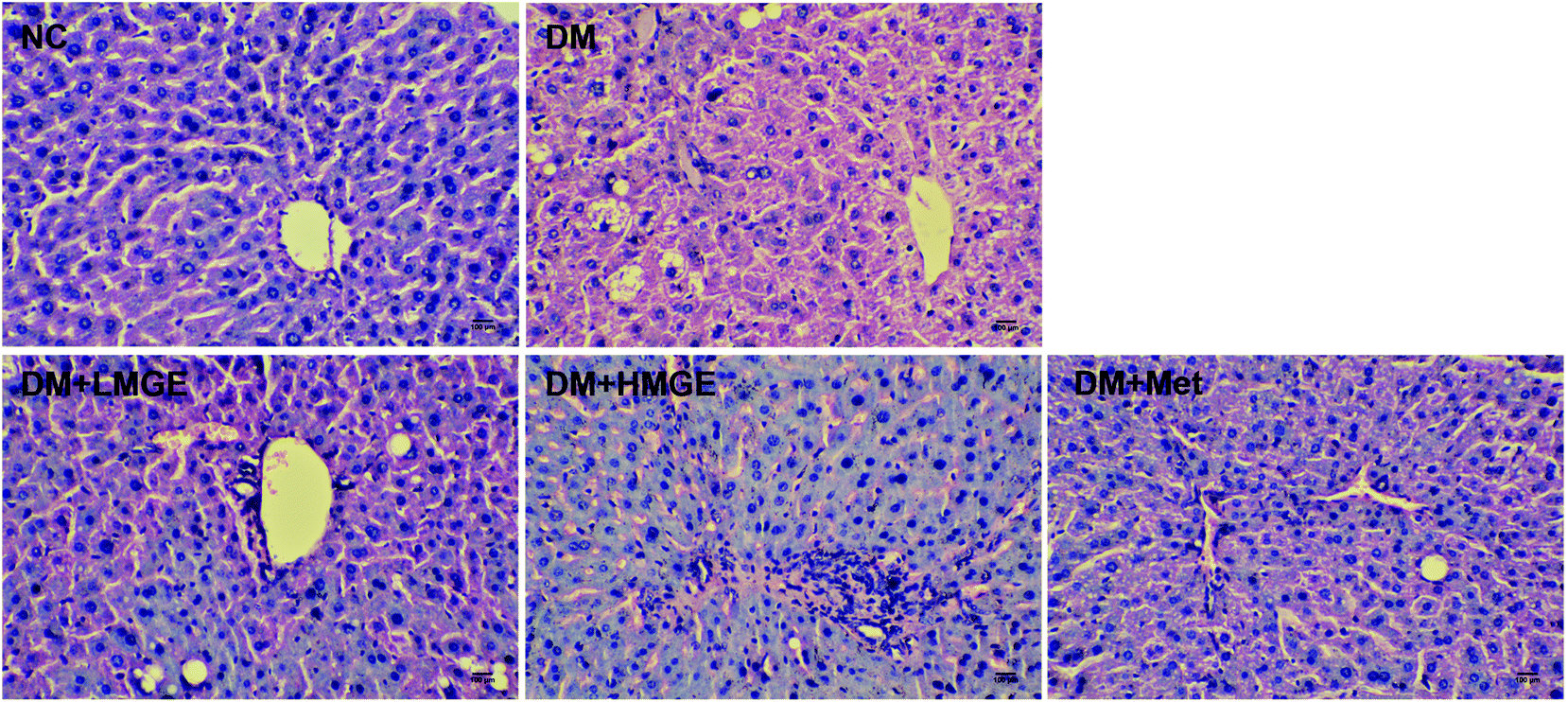 | ||
| Fig. 5 Effects of MGE treatment on hepatocyte polymorphism and steatosis using H&E staining. The marked scale bar is 100 μm. | ||
3.6. MGE treatment on the activation of the AMPK pathway
To understand the potential mechanism of MGE in hypoglycemic and hypolipidemic activities, western blotting was employed to determine the protein levels of phosphorylated AMPKα (Thr172) and ACC (Ser79) as well as FAS. As shown in Fig. 6, livers of diabetic mice presented a noticeable reduction in AMPKα and ACC phosphorylation and a large increase in the FAS protein level. Met and low- or high-dose MGE treatment stimulated the phosphorylation of AMPKα and ACC, and a marked decrease of the FAS level, but there was no obvious impact on the total AMPKα and ACC protein levels. These data indicate that MGE treatment activates the hepatic AMPK pathway in diabetic mice.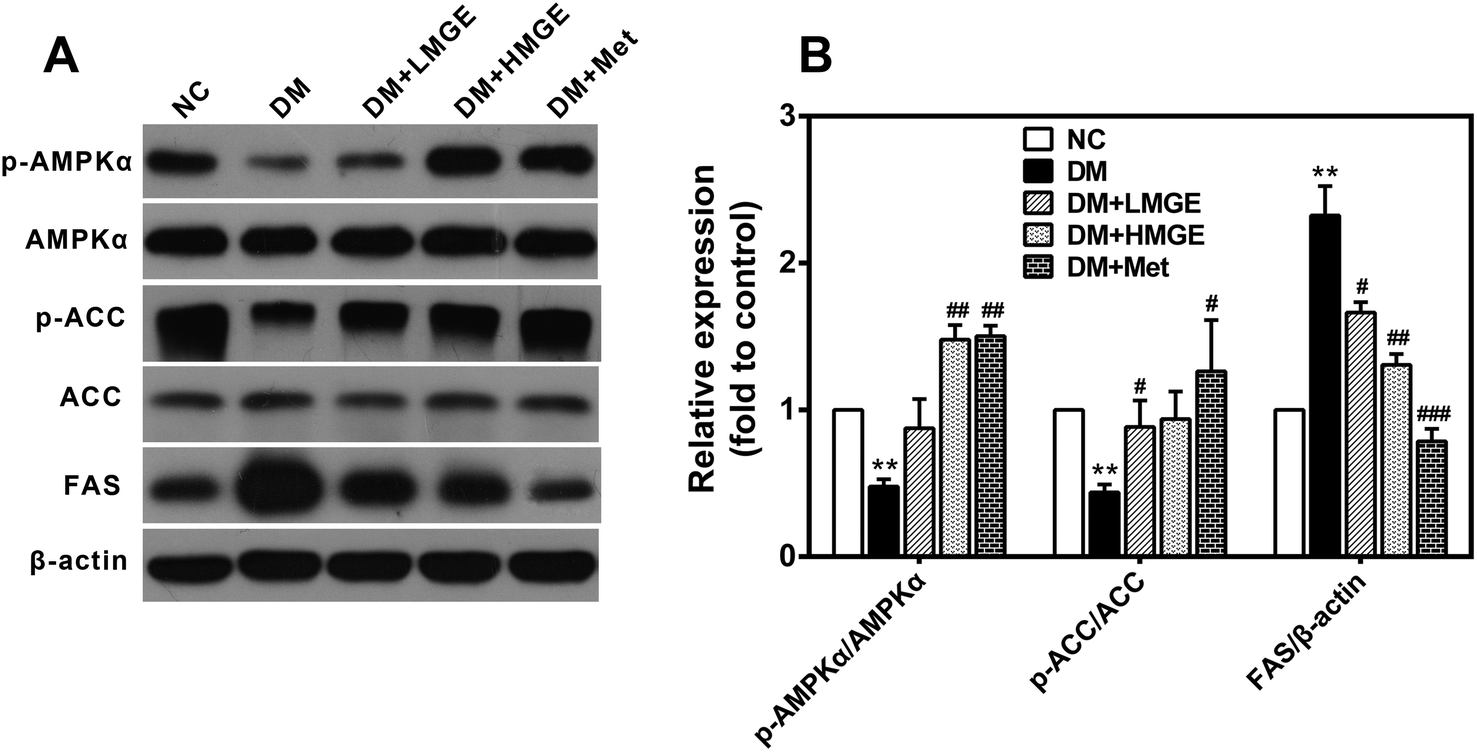 | ||
| Fig. 6 Effects of MGE treatment on hepatic AMPK signaling. (A) Western blotting photographs of p-AMPK, total AMPK, p-ACC, total ACC, FAS, and housekeeping β-actin. (B) Relative expression levels of the protein involved in the AMPK pathway. Data are expressed as mean ± SEM (n = 5) and differences between groups compared with the NC or DM group were analyzed using two-tailed unpaired Student's t test with Welch's correction. *p < 0.05, **p < 0.01, ***p < 0.001 versus NC group, and #p < 0.05, ##p < 0.01, ###p < 0.001 versus DM group. | ||
3.7. MGE treatment on liver gene expressions
To explore the potential effect of MGE administration on the expression levels of key genes related to liver glucose and lipid metabolism, a real-time quantitative polymerase chain reaction (RT-qPCR) was used to analyze the relative levels of mRNA. In the current study, diabetes induced a large increase in the mRNA levels of glucose 6-phosphatase (G6Pase), phosphoenolpyruvate carboxykinase (PEPCK), sterol regulatory element-binding protein 1 (SREBP1), stearoyl-CoA desaturase-1 (SCD-1) and diglyceride acyltransferase 2 (DGAT2), and a dramatic reduction in glycogen synthase (GS) and carnitine palmitoyltransferase 1a (CPT1a), a modest decrease in peroxisome proliferator-activated receptor α (PPARα) and almost no change in the low density lipoprotein receptor (LDLR) (Fig. 7). Compared with diabetic mice, the mRNA levels of G6Pase, PEPCK, SREBP1, SCD-1 and DGAT2 reduced markedly after a 5-week feeding of low- or high-dose MGE or Met, and this effect appeared to be dose-dependent, but the levels of PPARα and CPT1a increased slightly without dose dependence. There was almost no change in the GS mRNA level after MGE supplementation compared to diabetic mice without treatment. However, Met-supplemented diabetic mice showed an increased GS mRNA level in the liver (P < 0.05, Fig. 7). Collectively, these findings suggest that MGE ingestion inhibits gluconeogenesis and lipogenesis and promotes fatty acid oxidation in diabetic mice.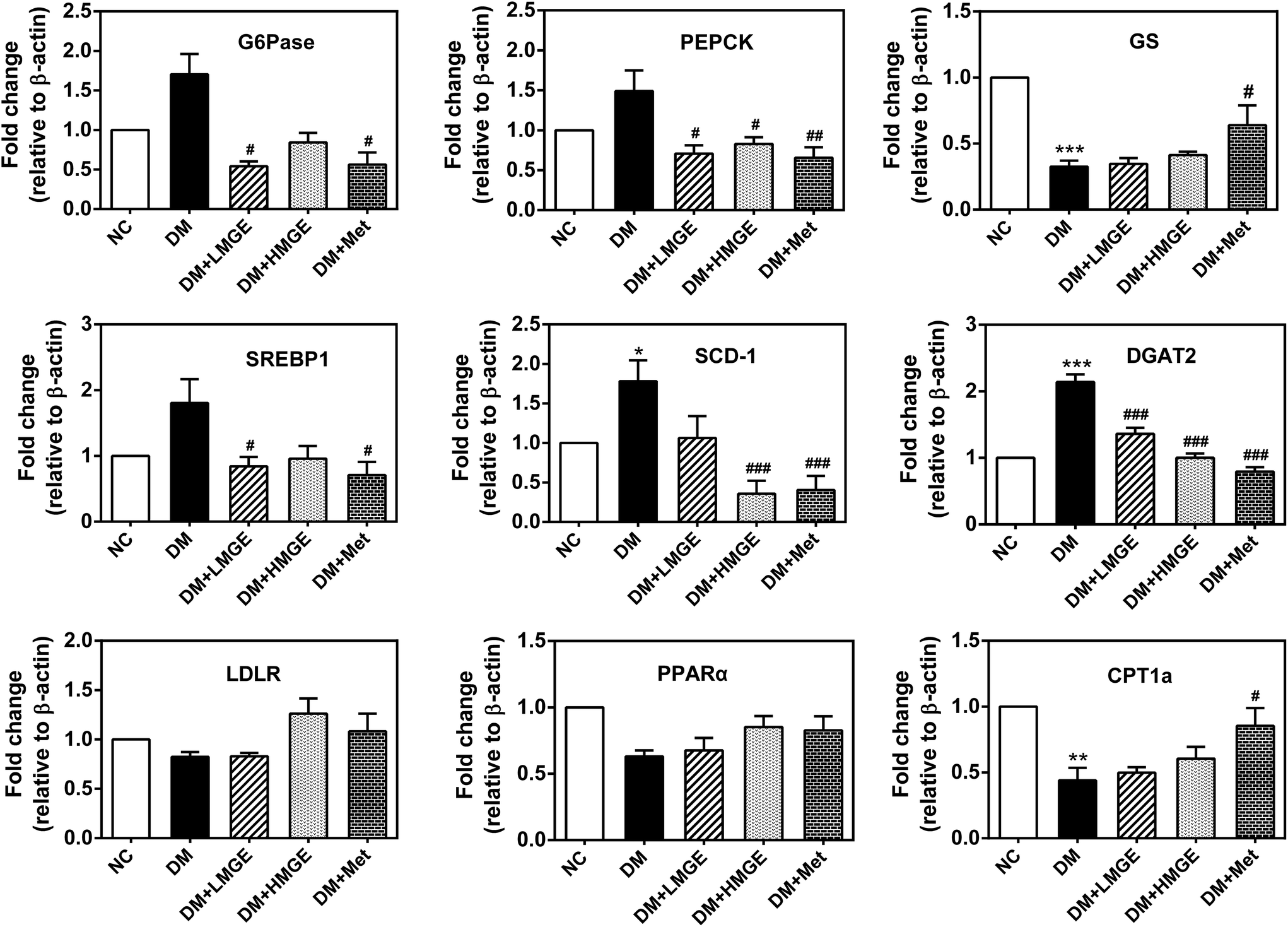 | ||
| Fig. 7 Effects of MGE treatment on the mRNA levels of the genes associated with gluconeogenesis, glycogen synthesis, de novo lipogenesis and fatty acid oxidation. Data are expressed as mean ± SEM (n = 4–5) and differences between groups compared with the NC or DM group were analyzed using two-tailed unpaired Student's t test with Welch's correction. *p < 0.05, **p < 0.01, ***p < 0.001 versus NC group, and #p < 0.05, ##p < 0.01, ###p < 0.001 versus DM group. | ||
4. Discussion
Although the pathogenesis of type 2 diabetes is complex and remains to be further explored, it is now clear that the insulin secretion deficiency of pancreatic β cells and insulin insensitivity (insulin resistance) in liver and peripheral tissues are the major causes.2 Liver plays a crucial role in modulating glucose homeostasis mainly through the regulation of hepatic glucose production, gluconeogenesis and glycogen synthesis.29 Under physiological conditions, postprandially elevated circulating glucose is transported into hepatocytes via glucose transporter 2 (GLUT2) through the insulin-stimulated glucose uptake pathway and subsequently used for glycogen synthesis or glycolysis. Simultaneously, the gluconeogenic pathway is suppressed in insulin signaling.30 While in the state of insulin resistance, hepatic glucose production increases largely due to the aberrant activation of gluconeogenesis and glycogen synthesis pathways.31 In the present study, diabetic mice showed much higher FBG than normal mice (Fig. 2A), and elevated mRNA levels of the G6Pase and PEPCK (Fig. 7), two rate-limiting enzymes in gluconeogenesis.32 In conformity with our previous studies,18,33 in the current study, MGE feeding decreased the elevated FBG and mRNA levels of G6Pase and PEPCK induced by STZ. These results suggest that the hypoglycemic effect of MGE on type 2 diabetes is, at least partially, attributed to the inhibition of gluconeogenesis. In diabetes, the impaired insulin pathway activates glycogen synthase kinase 3 (GSK3) which subsequently phosphorylates and inactivates glycogen synthase, leading to the reduction of glycogen synthesis.34,35 It is noteworthy that in spite of the large reduction of the GS mRNA level in diabetic mice, the level of MGE-fed diabetic mice showed almost no change (Fig. 7). Abundant evidence shows that, apart from insulin signaling, hepatic gluconeogenesis and glycogen synthesis are also improved through an insulin-independent AMPK pathway.36–39 Our present study showed significant activation of AMPK in diabetic mice fed with MGE. Although no direct evidence was provided in the present study, AMPK activation by MGE probably contributed to the inhibition of gluconeogenesis in diabetic mice.In addition to hyperglycemia, dyslipidemia, including hypertriglyceridemia and hypercholesterolemia, is another characteristic feature of type 2 diabetes, which is a vital risk factor for adverse cardiovascular disease.40,41 Although the precise mechanism remains unknown, previous studies revealed that insulin resistance was a main cause leading to dyslipidemia.42 Thus, therapeutic attenuation of insulin resistance in type 2 diabetes retards the progression of diabetic complications. In the present study, insulin resistance in MGE treated diabetic mice reduced significantly, which was characterized as a dramatic decrease in serum insulin and HOMA-IR and a noticeable increase in the ISI. Although the evidence that mogroside intervention directly relieved diabetes-associated insulin resistance was not confirmed previously, it was reported that mogroside IIIE blocked toll-like receptor 4 (TLR4)–c-Jun N-terminal kinase (JNK) signaling,11 and the inhibitory effect of MGE on oxidative stress was also confirmed in type 1 diabetes.33 Both TLR4-JNK and oxidative stress pathways could mediate insulin resistance. Moreover, several studies reported that AMPK activation was involved in ameliorating insulin resistance through insulin-independent pathways.8,43–45 Thus, it could be speculated that MGE-elicited AMPK activation probably contributes to the improvement of dyslipidemia via the attenuation of insulin resistance.
Additionally, dyslipidemia is also modulated by transcription factors. SREBP1 is a key transcription factor regulating lipogenesis. In insulin resistance and/or the diabetes state, the transcriptional activity of SREBP1 increases and enhances the expression of downstream lipogenic genes, including FAS, SCD-1 and DGAT2,46 eventually leading to hyperlipidemia. Previous studies showed that AMPK activation directly or indirectly inhibited the expression and transcriptional activity of SREBP1,47,48 resulting in the reduction of de novo lipogenesis and the improvement of lipid metabolism. Consistently, MGE-elicited AMPK activation reduced the mRNA levels of SREBP1, SCD-1 and DGAT2, as well as improvement of serum lipid profiles and hepatocyte steatosis (Fig. 4, 5 and 7). Accordingly, the effect of mogroside-mediated AMPK activation was also observed in vitro using a Hep G2 cell line.19 However, in 3T3-L1 preadipocytes, mogrol, an aglycone of mogrosides, rather than mogrosides activated AMPK and mediated the suppression of adipogenesis via CREB-dependent signaling.21 The discrepancies are unclear and may be caused by distinct cell types which need further study. Nonetheless, it should be noted that in the current in vivo experiment, the mogroside or its metabolites that actually exerted AMPK-activating activity was not identified. The in vivo metabolism study of mogroside should be carried out in future.
The anti-hyperlipidemic activity of MGE in diabetic mice was further evaluated by fatty acid oxidation. Normal fatty acid oxidation is suppressed due to reduced CPT1a expression in type 2 diabetes. CPT1a is an essential enzyme responsible for the transport of long-chain fatty acyl-CoA from the cytosol to the mitochondrial matrix for β-oxidation and its activity is inhibited by malonyl-CoA, a product of ACC.49 ACC is a downstream target of AMPK. Activated AMPK phosphorylated and inactivated ACC, leading to decreased malonyl-CoA and increased fatty acid oxidation.50 Besides, the critical role of PPARα in lipid metabolism regulation has been intensively elucidated.51,52 Activated PPARα enhances the expression of genes implicated in fatty acid oxidation, such as acyl-CoA oxidase and CPT1a.53 Moreover, PPARα activation-mediated fatty acid oxidation may be enhanced in an AMPK-dependent manner.54 In this study, MGE ingestion showed a marked activation of AMPK and the inhibition of ACC (phosphorylated ACC) and FAS activities (Fig. 6) as well as increased mRNA levels of PPARα and CPT1a (Fig. 7). These results indicated that the MGE-induced enhancement of AMPK and PPARα activities may be the main aspect in fatty acid oxidation. Similarly, some other tetracyclic triterpenoids also promoted fatty acid oxidation and inhibited hyperlipidemia in STZ-induced diabetic mice evidenced by the activation of AMPK and PPARα.46
In summary, MGE treatment showed multiple beneficial effects on type 2 diabetic mice, including the reduction of FBG and increase of serum insulin, the inhibition of gluconeogenesis and insulin resistance, the enhancement of hepatic fatty acid oxidation, the amelioration of serum lipid profiles and impairment of hepatic AMPK and insulin signaling. These improvements are, at least in part, attributed to the activation of hepatic AMPK signaling, although the exact metabolites of mogrosides serving as the activator of the hepatic AMPK pathway remain to be further investigated.
Abbreviations
| ACC | Acetyl-CoA carboxylase |
| AMP | Adenosine monophosphate |
| AMPK | 5′AMP-activated protein kinase |
| ANOVA | Analysis of variance |
| ATP | Adenosine triphosphate |
| G6Pase | Glucose 6-phosphatase |
| cAMP | Cyclic adenosine monophosphate |
| CPT1a | Carnitine palmitoyltransferase 1a |
| CREB | cAMP response element-binding protein |
| DGAT2 | Diglyceride acyltransferase 2 |
| DM | Diabetes mellitus |
| ELISA | Enzyme-linked immunosorbent assay |
| eWAT | Epididymal white adipose tissue |
| FAS | Fatty acid synthase |
| FBG | Fasting blood glucose |
| FINS | Fasting plasma insulin |
| GLUT2 | Glucose transporter 2 |
| GRAS | Generally recognized as safe |
| GS | Glycogen synthase |
| GSK3 | Glycogen synthase kinase 3 |
| GSP | Glycated serum protein |
| H&E | Hematoxylin and eosin |
| HDL-C | High density lipoprotein cholesterol |
| HOMA-IR | Homeostasis model assessment-insulin resistance |
| IPGTT | Intraperitoneal glucose tolerance test |
| IPITT | Intraperitoneal insulin tolerance test |
| ISI | Insulin sensitivity index |
| JNK | c-Jun N-terminal kinase |
| LDL-C | Low density lipoprotein cholesterol |
| LDLR | Low density lipoprotein receptor |
| Met | Metformin |
| MGE | Mogroside-rich extract |
| NC | Normal control |
| PBS | Phosphate-buffered saline |
| PEPCK | Phosphoenolpyruvate carboxykinase |
| PPARα | Peroxisome proliferator-activated receptor α |
| RT-qPCR | Real-time quantitative polymerase chain reaction |
| SCD-1 | Stearoyl-CoA desaturase-1 |
| SEM | Standard error of the mean |
| SREBP1 | Sterol regulatory element-binding protein 1 |
| STZ | Streptozotocin |
| T2D | Type 2 diabetes |
| TC | Total cholesterol |
| TG | Total triglyceride |
| TLR4 | Toll-like receptor 4 |
Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
We are grateful to Guilin Layn Natural Ingredients Corp. for generous provision of mogroside-rich extract. This study was funded by the National Natural Science Foundation of China (No. 31171780), Beijing Advanced Innovation Center for Food Nutrition and Human Health (No. 20171033), Zhejiang Provincial Natural Science Foundation of China (LY19C200004), Zhejiang Provincial Top Discipline of Biological Engineering (Level A) (No. ZS2018001), Zhejiang Provincial Top Key Discipline of Bioengineering (No. CX2016008) and Department of Education of Zhejiang Province (Y201738544).References
- V. Fonseca, Clinical significance of targeting postprandial and fasting hyperglycemia in managing type 2 diabetes mellitus, Curr. Med. Res. Opin., 2003, 19, 635–631 CrossRef PubMed.
- S. E. Inzucchi, Oral Antihyperglycemic Therapy for Type 2 Diabetes: Scientific Review, J. Am. Med. Assoc., 2002, 287, 360–372 CrossRef CAS PubMed.
- M. R. Taskinen and J. Borén, New insights into the pathophysiology of dyslipidemia in type 2 diabetes, Atherosclerosis, 2015, 239, 483–495 CrossRef CAS PubMed.
- D. E. Moller, New drug targets for type 2 diabetes and the metabolic syndrome, Nature, 2002, 414, 821–827 CrossRef PubMed.
- B. Satirapoj and S. G. Adler, Comprehensive approach to diabetic nephropathy, Kidney Res. Clin. Pract., 2014, 33, 121–131 CrossRef PubMed.
- J. Naveen and V. Baskaran, Antidiabetic plant-derived nutraceuticals: a critical review, Eur. J. Nutr., 2018, 57, 1275–1299 CrossRef CAS PubMed.
- Y. S. Oh and H. S. Jun, Role of Bioactive Food Components in Diabetes Prevention: Effects on Beta-Cell Function and Preservation, Nutr. Metab. Insights, 2014, 7, 51–59 Search PubMed.
- B. B. Kahn, T. Alquier, D. Carling and D. G. Hardie, AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism, Cell Metab., 2005, 1, 15–25 CrossRef CAS PubMed.
- D. G. Hardie, AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function, Genes Dev., 2011, 25, 1895–1908 CrossRef CAS PubMed.
- K. A. Coughlan, R. J. Valentine, N. B. Ruderman and A. K. Saha, AMPK activation: a therapeutic target for type 2 diabetes?, Diabetes, Metab. Syndr. Obes., 2014, 7, 241–253 Search PubMed.
- L. Tao, J. Yang, F. Cao, H. Xie, M. Zhang, Y. Gong and C. Zhang, Mogroside IIIE, a Novel Anti-Fibrotic Compound, Reduces Pulmonary Fibrosis through Toll-Like Receptor 4 Pathways, J. Pharmacol. Exp. Ther., 2017, 361, 268–279 CrossRef CAS.
- J. S. Jin and J. H. Lee, Phytochemical and pharmacological aspects of Siraitia grosvenorii, luo han kuo, Orient. Pharm. Exp. Med., 2012, 12, 233–239 CrossRef.
- T. Takemoto, S. Arihara, T. Nakajima and M. Okuhira, Studies on the constituents of fructus Momordicae. I. On the sweet principle, Yakugaku Zasshi, 1983, 103, 1151–1154 CrossRef CAS.
- F. Xu, D. P. Li, Z. C. Huang, F. L. Lu, L. Wang, Y. L. Huang, R. F. Wang, G. X. Liu, M. Y. Shang and S. Q. Cai, Exploring in vitro, in vivo metabolism of mogroside V and distribution of its metabolites in rats by HPLC-ESI-IT-TOF-MSn, J. Pharm. Biomed. Anal., 2015, 115, 418–430 CrossRef CAS.
- R. S. Pawar, A. J. Krynitsky and J. I. Rader, Sweeteners from plants—with emphasis on Stevia rebaudiana (Bertoni) and Siraitia grosvenorii (Swingle), Anal. Bioanal. Chem., 2013, 405, 4397–4407 CrossRef CAS PubMed.
- M. H. Pan, J. R. Yang, M. L. Tsai, S. Sang and C. T. Ho, Anti-inflammatory effect of Momordica grosvenori Swingle extract through suppressed LPS-induced upregulation of iNOS and COX-2 in murine macrophages, J. Funct. Foods, 2009, 1, 145–152 CrossRef.
- J. S. Jin and J. H. Lee, Phytochemical and pharmacological aspects of Siraitia grosvenorii, luo han kuo, Orient. Pharm. Exp. Med., 2012, 12, 233–239 CrossRef.
- X. Qi, W. Chen, L. Liu, P. Yao and B. Xie, Effect of a Siraitia grosvenori extract containing mogrosides on the cellular immune system of type 1 diabetes mellitus mice, Mol. Nutr. Food Res., 2006, 50, 732–738 CrossRef CAS PubMed.
- X. B. Chen, J. J. Zhuang, J. H. Liu, M. Lei, L. Ma, J. Chen, X. Shen and L. H. Hu, Potential AMPK activators of cucurbitane triterpenoids from Siraitia grosvenorii Swingle, Bioorg. Med. Chem., 2011, 19, 5776–5781 CrossRef CAS.
- Z. Luo, F. Qiu, K. Zhang, X. Qin, Y. Guo, H. Shi, L. Zhang, Z. Zhang and X. Ma, In vitro, AMPK activating effect and in vivo pharmacokinetics of mogroside V, a cucurbitane-type triterpenoid from Siraitia grosvenorii fruits, RSC Adv., 2016, 6, 7034–7041 RSC.
- N. Harada, M. Ishihara, H. Horiuchi, Y. Ito, H. Tabata, Y. A. Suzuki, Y. Nakano, R. Yamaji and H. Inui, Mogrol Derived from Siraitia grosvenorii Mogrosides Suppresses 3T3-L1 Adipocyte Differentiation by Reducing cAMP-Response Element-Binding Protein Phosphorylation and Increasing AMP-Activated Protein Kinase Phosphorylation, PLoS One, 2016, 11, e0162252 CrossRef PubMed.
- H. Liu, C. Wang, X. Qi, J. Zou and Z. Sun, Antiglycation and antioxidant activities of mogroside extract from Siraitia grosvenorii (Swingle) fruits, J. Food Sci. Technol., 2018, 55, 1880–1888 CrossRef CAS PubMed.
- L. Yu, W. Cai, Y. Zhang, L. Feng and C. Huang, Red bayberry extract prevents high-fat diet-induced metabolic disorders in C57BL/6 mice, J. Funct. Foods, 2015, 14, 278–288 CrossRef CAS.
- J. Nie, Y. Chang, Y. Li, Y. Zhou, J. Qin, Z. Sun and H. Li, Caffeic Acid Phenethyl Ester (Propolis Extract) Ameliorates Insulin Resistance by Inhibiting JNK and NF-κB Inflammatory Pathways in Diabetic Mice and HepG2 Cell Models, J. Agric. Food Chem., 2017, 65, 9041–9053 CrossRef CAS PubMed.
- Y. Gao, M. Zhang, T. Wu, M. Xu, H. Cai and Z. Zhang, Effects of D-Pinitol on Insulin Resistance through the PI3 K/Akt Signaling Pathway in Type 2 Diabetes Mellitus Rats, J. Agric. Food Chem., 2015, 63, 6019–6026 CrossRef CAS PubMed.
- J. Ye, G. Coulouris, I. Zaretskaya, I. Cutcutache, S. Rozen and T. L. Madden, Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction, BMC Bioinf., 2012, 13, 134 CrossRef CAS PubMed.
- K. H. Kim, S. Choi, Y. Zhou, E. Y. Kim, J. M. Lee, P. K. Saha, S. Anakk and D. D. Moore, Hepatic FXR/SHP axis modulates systemic glucose and fatty acid homeostasis in aged mice, Hepatology, 2017, 66, 498–509 CrossRef CAS PubMed.
- O. Akinrotimi, R. Riessen, P. VanDuyne, J. E. Park, Y. K. Lee, L. J. Wong, A. M. Zavacki, K. Schoonjans and S. Anakk, Small heterodimer partner deletion prevents hepatic steatosis and when combined with farnesoid X receptor loss protects against type 2 diabetes in mice, Hepatology, 2017, 66, 1854–1865 CrossRef CAS PubMed.
- P. J. Klover and R. A. Mooney, Hepatocytes: critical for glucose homeostasis, Int. J. Biochem. Cell Biol., 2004, 36, 753–758 CrossRef CAS PubMed.
- V. T. Samuel and G. I. Shulman, Mechanisms for Insulin Resistance: Common Threads and Missing Links, Cell, 2012, 148, 852–871 CrossRef CAS PubMed.
- G. Jayanthy and S. Subramanian, RA abrogates hepatic gluconeogenesis and insulin resistance by enhancing IRS-1 and AMPK signalling in experimental type 2 diabetes, RSC Adv., 2015, 5, 44053–44067 RSC.
- Y. Zhang, F. Y. Lee, G. Barrera, H. Lee, C. Vales, F. J. Gonzalez, T. M. Willson and P. A. Edwards, Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1006–1011 CrossRef CAS.
- X. Y. Qi, W. J. Chen, L. Q. Zhang and B. J. Xie, Mogrosides extract from Siraitia grosvenori, scavenges free radicals in vitro and lowers oxidative stress, serum glucose, and lipid levels in alloxan-induced diabetic mice, Nutr. Res., 2008, 28, 278–284 CrossRef CAS.
- Z. Chen, C. Wang, Y. Pan, X. Gao and H. Chen, Hypoglycemic and hypolipidemic effects of anthocyanins extract from black soybean seed coat in high fat diet and streptozotocin-induced diabetic mice, Food Funct., 2018, 9, 426–439 RSC.
- I. Cordero-Herrera, M. Á. Martín, L. Goya and S. Ramos, Cocoa flavonoids attenuate high glucose-induced insulin signalling blockade and modulate glucose uptake and production in human HepG2 cells, Food Chem. Toxicol., 2014, 64, 10–19 CrossRef CAS PubMed.
- Q. F. Collins, H. Y. Liu, J. Pi, Z. Liu, M. J. Quon and W. Cao, Epigallocatechin-3-gallate (EGCG), A Green Tea Polyphenol, Suppresses Hepatic Gluconeogenesis through 5′-AMP-activated Protein Kinase, J. Biol. Chem., 2007, 282, 30143–30149 CrossRef CAS PubMed.
- J. M. Lee, W. Y. Seo, K. H. Song, D. Chanda, Y. D. Kim, D. K. Kim, M. W. Lee, D. Ryu, Y. H. Kim, J. R. Noh, C. H. Lee, J. Y. L. Chiang, S. H. Koo and H. S. Choi, AMPK-dependent repression of hepatic gluconeogenesis via disruption of CREB/CRTC2 complex by orphan nuclear receptor SHP, J. Biol. Chem., 2010, 285, 32182–32191 CrossRef CAS PubMed.
- S. J. Jiang, H. Dong, J. B. Li, L. J. Xu, X. Zou, K. F. Wang, F. E. Lu and P. Yi, Berberine inhibits hepatic gluconeogenesis via the LKB1-AMPK-TORC2 signaling pathway in streptozotocin-induced diabetic rats, World J. Gastroenterol., 2015, 21, 7777–7785 CrossRef CAS PubMed.
- Z. Ren, Z. Xie, D. Cao, M. Gong, L. Yang, Z. Zhou and Y. Ou, C-Phycocyanin inhibits hepatic gluconeogenesis and increases glycogen synthesis via activating Akt and AMPK in insulin resistance hepatocytes, Food Funct., 2018, 9, 2829–2839 RSC.
- J. M. Chehade, M. Gladysz and A. D. Mooradian, Dyslipidemia in Type 2 Diabetes: Prevalence, Pathophysiology, and Management, Drugs, 2013, 73, 327–339 CrossRef CAS PubMed.
- J. A. Farmer, Diabetic dyslipidemia and atherosclerosis: Evidence from clinical trials, Curr. Diabetes Rep., 2008, 8, 71–77 CrossRef.
- H. J. Warraich and J. S. Rana, Dyslipidemia in diabetes mellitus and cardiovascular disease, Cardiovasc. Endocrinol. Metab., 2017, 6, 27–32 CrossRef CAS.
- I. Chopra, H. F. Li, H. Wang and K. A. Webster, Phosphorylation of the insulin receptor by AMP-activated protein kinase (AMPK) promotes ligand-independent activation of the insulin signalling pathway in rodent muscle, Diabetologia, 2012, 55, 783–794 CrossRef CAS PubMed.
- A. Tzatsos and K. V. Kandror, Nutrients Suppress Phosphatidylinositol 3-Kinase/Akt Signaling via Raptor-Dependent mTOR-Mediated Insulin Receptor Substrate 1 Phosphorylation, Mol. Cell. Biol., 2006, 26, 63–76 CrossRef CAS PubMed.
- T. Zheng, X. Yang, D. Wu, S. Xing, F. Bian, W. Li, J. Chi, X. Bai, G. Wu, X. Chen, Y. Zhang and S. Jin, Salidroside ameliorates insulin resistance through activation of a mitochondria-associated AMPK/PI3 K/Akt/GSK3β pathway, Br. J. Pharmacol., 2015, 172, 3284–3301 CrossRef CAS PubMed.
- Y. H. Kuo, C. H. Lin and C. C. Shih, Antidiabetic and Antihyperlipidemic Properties of a Triterpenoid Compound, Dehydroeburicoic Acid, from Antrodia camphorata in Vitro and in Streptozotocin-Induced Mice, J. Agric. Food Chem., 2015, 63, 10140–10151 CrossRef CAS.
- J. Yang, L. Craddock, S. Hong and Z. M. Liu, AMP-activated protein kinase suppresses LXR-dependent sterol regulatory element-binding protein-1c transcription in rat hepatoma McA-RH7777 cells, J. Cell. Biochem., 2009, 106, 414–426 CrossRef CAS PubMed.
- Y. Li, S. Xu, M. M. Mihaylova, B. Zheng, X. Hou, B. Jiang, O. Park, Z. Luo, E. Lefai, J. Y. J. Shyy, B. Gao, M. Wierzbicki, T. J. Verbeuren, R. J. Shaw, R. A. Cohen and M. Zang, AMPK Phosphorylates and Inhibits SREBP Activity to Attenuate Hepatic Steatosis and Atherosclerosis in Diet-Induced Insulin-Resistant Mice, Cell Metab., 2011, 13, 376–388 CrossRef CAS PubMed.
- J. D. McGarry and N. F. Brown, The Mitochondrial Carnitine Palmitoyltransferase System—From Concept to Molecular Analysis, Eur. J. Biochem., 1997, 244, 1–14 CrossRef CAS.
- B. Viollet, L. Lantier, J. Devin-Leclerc, S. Hebrard, C. Amouyal, R. Mounier, M. Foretz and F. Andreelli, Targeting the AMPK pathway for the treatment of Type 2 diabetes, Front. Biosci., 2009, 14, 3380–3400 CrossRef CAS.
- P. Ferre, The biology of peroxisome proliferator-activated receptors: relationship with lipid metabolism and insulin sensitivity, Diabetes, 2004, 53, 43–50 CrossRef.
- S. Mandard, M. Müller and S. Kersten, Peroxisome proliferator-activated receptor α target genes, Cell. Mol. Life Sci., 2004, 61, 393–416 CrossRef CAS PubMed.
- R. Kimura, N. Takahashi, K. Murota, Y. Yamada, S. Niiya, N. Kanzaki, Y. Murakami, T. Moriyama, T. Goto and T. Kawada, Activation of peroxisome proliferator-activated receptor-α (PPARα) suppresses postprandial lipidemia through fatty acid oxidation in enterocytes, Biochem. Biophys. Res. Commun., 2011, 410, 1–6 CrossRef CAS PubMed.
- M. Pawlak, P. Lefebvre and B. Staels, Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease, J. Hepatol., 2015, 62, 720–733 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8fo01486h |
| This journal is © The Royal Society of Chemistry 2019 |