 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Kinetic analysis of bioorthogonal reaction mechanisms using Raman microscopy†
William J.
Tipping
 a,
Martin
Lee
b,
Valerie G.
Brunton
b,
Guy C.
Lloyd-Jones
a and
Alison N.
Hulme
*a
a,
Martin
Lee
b,
Valerie G.
Brunton
b,
Guy C.
Lloyd-Jones
a and
Alison N.
Hulme
*a
aEaStCHEM School of Chemistry, The University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: Alison.Hulme@ed.ac.uk
bEdinburgh Cancer Research Centre, MRC Institute of Genetics and Molecular Medicine, The University of Edinburgh, Crewe Road South, Edinburgh, EH4 2XR, UK
First published on 18th June 2019
Abstract
Raman spectroscopy is well-suited to the study of bioorthogonal reaction processes because it is a non-destructive technique, which employs relatively low energy laser irradiation, and water is only very weakly scattered in the Raman spectrum enabling live cell imaging. In addition, Raman spectroscopy allows species-specific label-free visualisation; chemical contrast may be achieved when imaging a cell in its native environment without fixatives or stains. Combined with the rapid advances in the field of Raman imaging over the last decade, particularly in stimulated Raman spectroscopy (SRS), this technique has the potential to revolutionise our mechanistic understanding of the biochemical and medicinal chemistry applications of bioorthogonal reactions. Current approaches to the kinetic analysis of bioorthogonal reactions (including heat flow calorimetry, UV-vis spectroscopy, fluorescence, IR, NMR and MS) have a number of practical shortcomings for intracellular applications. We highlight the advantages offered by Raman microscopy for reaction analysis in the context of both established and emerging bioorthogonal reactions, including the copper(I) catalysed azide–alkyne cycloaddition (CuAAC) click reaction and Glaser–Hay coupling.
Introduction
Bioorthogonal chemistries have been rapidly adopted both in traditional synthetic chemistry and as a means to site-specifically label biomolecules inside cells.1–5 More recently their use has been proposed as a means to both construct and de-cage complex drug molecules in vivo as a new therapeutic modality.6,7 Critical to the success of reactants that couple under bioorthogonal conditions are that they are mutually reactive but do not cross-react, or interact, with other biological functionalities or reactions in a cell; that they are stable and non-toxic in physiological settings; and that their mutual reaction is highly specific and fast.1 Rapid reaction rates are required particularly to monitor fast intracellular processes and also where the intracellular abundance of the reacting species is comparatively low.2 However, despite many studies of these reactions in solution (illustrated for the bioorthogonal reactions of alkynes in Fig. 1),2,4,5 current approaches to their kinetic analysis are limited in their intracellular application by the need for coupling to a specific substrate (for fluorescence),8,9 the low spatial resolution (for IR10 and MS11), and the concentrations required (for NMR).12 Differences arising from intracellular macromolecular crowding,13 variations in pH across cell populations and within individual cells,14 and the sequestration of substrates into cellular structures or organelles15 are poorly accounted for by bulk in vitro solution phase analysis, necessitating the urgent development of techniques which might be applicable in an in cellulo or in vivo microenvironment.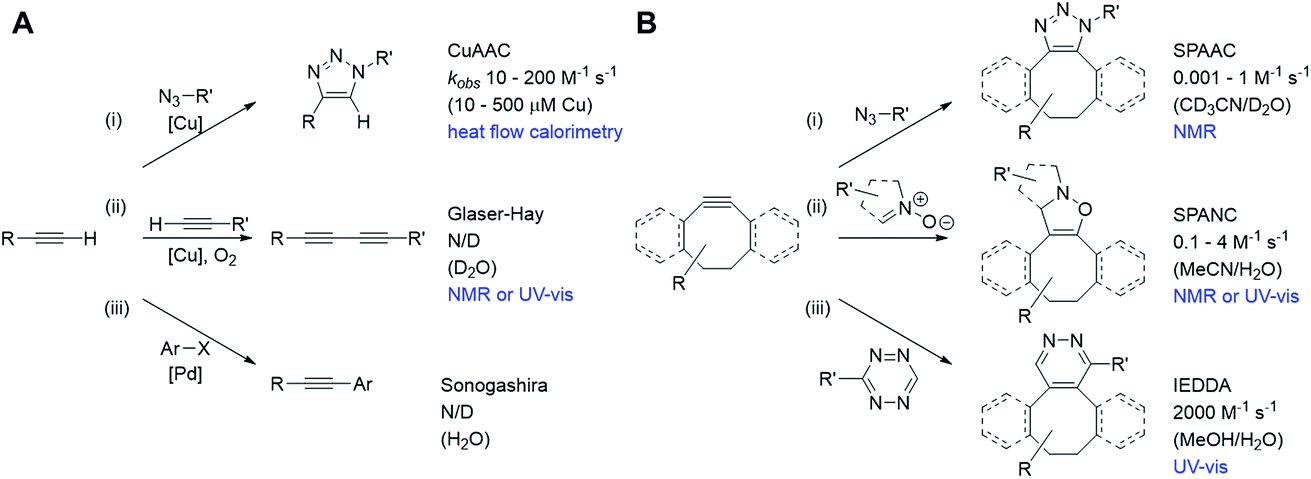 | ||
| Fig. 1 Bioorthogonal reactions of terminal alkynes (A) and strained alkynes (B), with approximate rate constants in aqueous based media (N/D = not determined). (A) (i) Copper(I) catalysed azide–alkyne cycloaddition (CuAAC);23 (ii) Glaser–Hay reaction;24 (iii) copper-free Sonogashira reaction.25 (B) (i) Strain-promoted azide–alkyne cycloaddition (SPAAC);26 (ii) strain-promoted alkyne nitrone cycloaddition (SPANC);26 (iii) inverse electron demand Diels–Alder (IEDDA).27 | ||
Raman is well-suited to the study of bioorthogonal reaction processes because it is a non-destructive technique, which employs relatively low energy laser irradiation, and water is only very weakly scattered in the Raman spectrum enabling live cell imaging.16 In addition, Raman spectroscopy allows species-specific label-free visualisation; chemical contrast may be achieved when imaging a cell in its native environment without fixatives or stains. Combined with the rapid advances in the field of Raman imaging over the last decade,17 most notably in coherent techniques such as stimulated Raman scattering (SRS), this has the potential to revolutionise our mechanistic understanding of the biochemical and medicinal chemistry applications of bioorthogonal reactions.
We chose to focus our study on the reactions of alkynes, as they are widely used in bioorthogonal chemistry2–5 and have been used to probe biological processes, such as DNA, RNA and protein synthesis, effectively using Raman spectroscopy and SRS microscopy.18 Two notable bioorthogonal reactions of alkynes are: (a) the copper(I) catalysed azide–alkyne cycloaddition (CuAAC) click reaction (Fig. 1A(i));19,20 and (b) the Glaser–Hay reaction (Fig. 1A(ii)).21,22 These two copper-catalysed reactions provide complementary reaction pathways from a terminal alkyne, and are thought to occur (in solution) at comparable, or slower, rates than other bioorthogonal reactions of terminal alkynes and the strain-promoted cycloaddition reactions of cyclooctyne derivatives with dipoles or tetrazines (Fig. 1B).23–27
The CuAAC reaction has emerged as a leading example of ‘click chemistry’, a term developed by Sharpless et al. in 2001 to describe near-perfect bond-forming reactions which are useful for rapid assembly of molecules with desired function.28 The use of Cu(I) salts, or a Cu(II) precursor in combination with a reducing agent to provide catalytically-active copper species in situ has been shown to mediate azide–alkyne cycloaddition with high rates of reaction. Current mechanistic understanding of the CuAAC reaction (Fig. 2A) supports a model with two chemically equivalent copper atoms working together to bring about the regioselective formation of 1,4-substituted 1,2,3-triazoles.29 Formation of the copper acetylide is turnover rate-limiting, thus strategies which aid alkyne deprotonation accelerate the CuAAC reaction.30 When particularly acidic alkynes are used, a switch in the rate limiting step to azide ligation/migratory insertion is observed.31 Heat flow calorimetry has been used extensively in detailed mechanistic studies of the CuAAC reaction, which clearly does not translate to an in cellulo or in vivo environment.
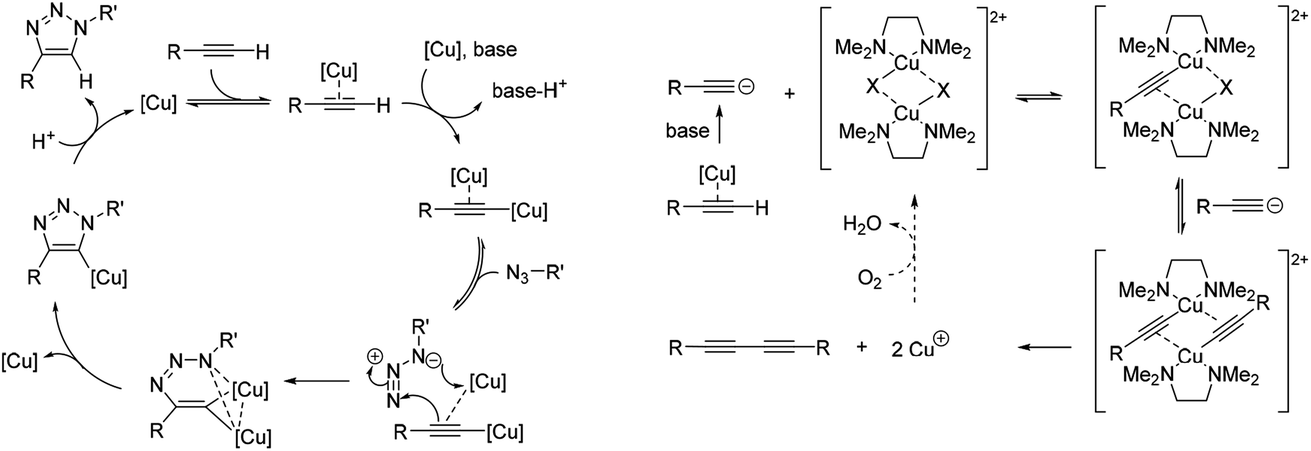 | ||
| Fig. 2 Simplified mechanisms for the catalytic cycle of (A) the CuAAC reaction,29a and (B) the Glaser–Hay reaction.34 | ||
Oxidative homocoupling of copper(I)-phenylacetylide upon exposure to air was first observed in 1869 by Carl Glaser,32 and consequently the reaction bears his name. Modifications to the initial Glaser coupling conditions sought to form the copper(I) acetylide in situ;33 in the Hay modification, oxidative acetylinic coupling was demonstrated in an oxygen environment in the presence of catalytic amounts of CuCl and the bidentate ligand, TMEDA.21 The Glaser–Hay reaction is emerging as a bioorthogonal reaction which is particularly useful for producing bioconjugates where the reduced steric demands of a bisalkyne linker are critical. The mechanism of the Glaser–Hay reaction has been studied, using a variety of analytical techniques.24,34,35 An early report by Bohlmann et al. examining the homocoupling of terminal acetylenes in MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (80:20) at pH 3 (HCl) identified the formation of a dicopper(II)-diacetylide complex as the rate-limiting step in the observed second-order kinetics using UV absorption spectroscopy.34 The proposed mechanism involved reductive elimination of this species to yield the 1,3-diyne product (Fig. 2B). A more recent DFT study broadly supports this mechanism, but suggests that (as for the CuAAC reaction) the turnover rate-limiting step corresponds to the Cu-coordinated alkyne deprotonation.36 In contrast, studies combining NMR and UV spectroscopy to probe the reaction of terminal acetylenes in D2O or CH2Cl2 have suggested that the mechanism involves a mono-nuclear Cu2+ intermediate.24,35 Again, the analytical techniques used to study the Glaser–Hay reaction to date do not readily translate to a more biological setting. Thus this provided a second reaction against which we could test our Raman spectroscopic approach.
:H2O (80:20) at pH 3 (HCl) identified the formation of a dicopper(II)-diacetylide complex as the rate-limiting step in the observed second-order kinetics using UV absorption spectroscopy.34 The proposed mechanism involved reductive elimination of this species to yield the 1,3-diyne product (Fig. 2B). A more recent DFT study broadly supports this mechanism, but suggests that (as for the CuAAC reaction) the turnover rate-limiting step corresponds to the Cu-coordinated alkyne deprotonation.36 In contrast, studies combining NMR and UV spectroscopy to probe the reaction of terminal acetylenes in D2O or CH2Cl2 have suggested that the mechanism involves a mono-nuclear Cu2+ intermediate.24,35 Again, the analytical techniques used to study the Glaser–Hay reaction to date do not readily translate to a more biological setting. Thus this provided a second reaction against which we could test our Raman spectroscopic approach.
Results and discussion
Establishing Raman spectroscopy as a method to follow bioorthogonal reactions
Real-time chemical reaction monitoring has been reported using both infrared (IR) and Raman spectroscopy. For a molecule to be IR active, there must be an accompanying change in dipole moment of the chemical bond as a result of absorption of the incident IR radiation. The CuAAC reaction has been studied previously using IR spectroscopy in an organic solvent (DMF) following the decay of the azide peak at 2096 cm−1.10 Here, we show that the CuAAC reaction can be followed by IR spectroscopy in an aqueous solvent mixture (Fig. S1†), but not one that is compatible with biological cells. In general, functional groups which generate a strong peak in an IR spectrum tend to be weaker in Raman and vice versa. Raman active shifts are a result of a change in bond polarisation upon interaction of monochromatic light. Raman scattering, therefore, is detected as an inelastic scattered photon, but only 1 in 108 photons undergo Raman scattering.37 Despite this, even low intensity vibrations (e.g. C–H) can provide signals for imaging purposes if visualised at high enough concentrations. Spectroscopically bioorthogonal functional groups, including alkyne (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), nitrile (CN) and deuterated groups (C–D), produce peaks in the cellular-silent region of the Raman spectrum, between 1800–2800 cm−1.17,18 Of these, the alkyne-containing thymidine analogue, EdU (Fig. 3, 1), is the most widely studied small molecule for Raman imaging applications. EdU is used as a probe for DNA synthesis in proliferating cells, using CuAAC conjugation with a fluorescent probe38 or biotin group for pull-down experiments,39 and more recently, inherent visualisation of the alkyne group present within EdU at 2120 cm−1 has been achieved in a single step using Raman and SRS imaging approaches (Fig. 3).18 SRS imaging provides fast acquisition rates (up to video rate speeds) and can generate images based on the inherent chemical contrast of the cell. Fig. 3B shows SRS images of the CH3 (2939 cm−1, proteins) and CH2 (2844 cm−1, lipids) signal in HeLa cells. Additionally, the incorporation of EdU is detected in the nucleus of the cells at 2120 cm−1.
C), nitrile (CN) and deuterated groups (C–D), produce peaks in the cellular-silent region of the Raman spectrum, between 1800–2800 cm−1.17,18 Of these, the alkyne-containing thymidine analogue, EdU (Fig. 3, 1), is the most widely studied small molecule for Raman imaging applications. EdU is used as a probe for DNA synthesis in proliferating cells, using CuAAC conjugation with a fluorescent probe38 or biotin group for pull-down experiments,39 and more recently, inherent visualisation of the alkyne group present within EdU at 2120 cm−1 has been achieved in a single step using Raman and SRS imaging approaches (Fig. 3).18 SRS imaging provides fast acquisition rates (up to video rate speeds) and can generate images based on the inherent chemical contrast of the cell. Fig. 3B shows SRS images of the CH3 (2939 cm−1, proteins) and CH2 (2844 cm−1, lipids) signal in HeLa cells. Additionally, the incorporation of EdU is detected in the nucleus of the cells at 2120 cm−1.
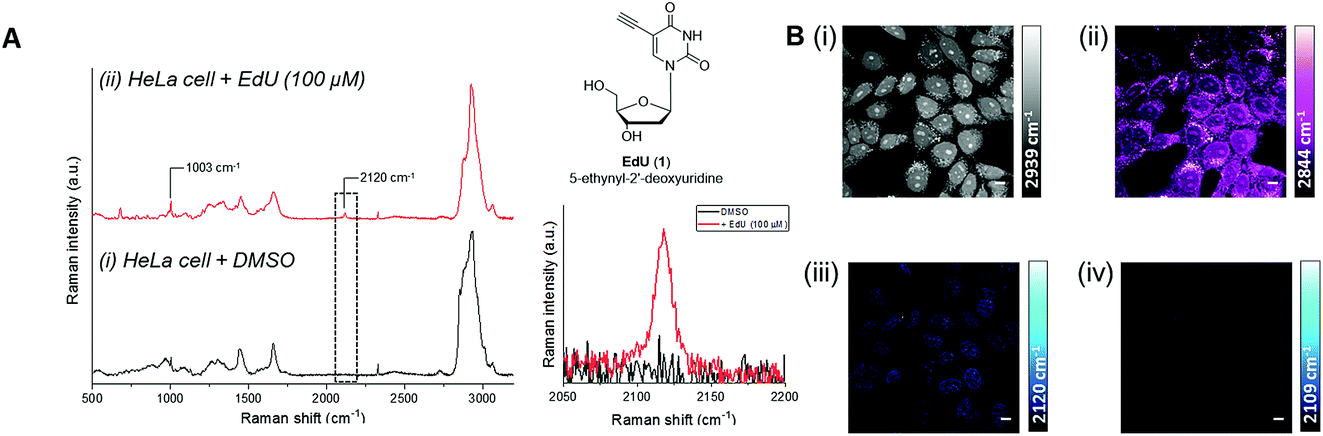 | ||
| Fig. 3 Detection of alkyne-labelled DNA using EdU (1) and Raman imaging. (A) Spontaneous Raman spectroscopy of single fixed HeLa cells treated with (i) DMSO and (ii) EdU (100 µM, 18 h).18 The spectra were normalised to the intensity of the peak at 1003 cm−1 (phenylalanine ring breathing mode), scaled between 1000–20000 counts and offset for clarity. Peak annotations: 1003 cm−1 (phenylalanine ring breathing mode); 2120 cm−1 (CC, EdU). Raman spectra acquired using λex = 532 nm using a 50× objective lens and 30 s integration time. Inset: chemical structure of EdU (top) and expansion of the EdU alkyne region (2050–2220 cm−1 indicated by the dashed box; bottom). (B) SRS imaging of HeLa cells treated with EdU (100 µM, 18 h).18 SRS images were acquired at (i) 2939 cm−1 (CH3), (ii) 2844 cm−1 (CH2), (iii) 2120 cm−1 (CC, EdU) and (iv) 2109 cm−1 (cell-silent region). SRS images were acquired using 1024 × 1024 pixels and 20 µs pixel dwell time. False colours were applied to different detection wavenumbers. Scale bars: 10 µm. | ||
Reaction time-course experiments were first measured in solution-phase using Raman spectroscopy. To assess the feasibility of using a Raman spectral approach to monitor the reaction progress of bioorthogonal transformations, initial investigations focussed upon the CuAAC reaction between EdU (1) and biotin-PEG3-azide (2) in the presence of CuSO4 and ascorbic acid in DMSO(aq.). The intense Raman signal of the EdU alkyne (at 2120 cm−1) was preferred for reaction monitoring, over the weak Raman signal associated with the azide (at 2096 cm−1) (Fig. S2†). The time-course dataset was normalised to the intensity of the Raman peak of the DMSO reaction solvent at 1418 cm−1 (DMSO CH def.) which enabled direct comparison of the relative peak intensities and the peak positions in Raman spectra at each individual timepoint. The spectra are presented in Fig. 4A. In the course of the reaction, the intensity of the Raman peak at 2120 cm−1 (EdU, CC) decreases to the baseline level. Additionally, a shift in the peak at 1624 cm−1 to 1646 cm−1 indicates a change in the local bonding environment surrounding the amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of EdU upon formation of the triazole product. Integration of the alkyne peak at 2120 cm−1 indicates that the reaction reaches completion within 40 min, whilst in the absence of Cu catalyst, there is only very marginal reduction in the alkyne signal over 60 min (Fig. 4B). An advantage of the Raman spectral approach detailed here over NMR-based reaction monitoring methods is that extremely small reaction volumes can be employed; here the reactions were performed using 25 µL total volume at t = 0 min.
O of EdU upon formation of the triazole product. Integration of the alkyne peak at 2120 cm−1 indicates that the reaction reaches completion within 40 min, whilst in the absence of Cu catalyst, there is only very marginal reduction in the alkyne signal over 60 min (Fig. 4B). An advantage of the Raman spectral approach detailed here over NMR-based reaction monitoring methods is that extremely small reaction volumes can be employed; here the reactions were performed using 25 µL total volume at t = 0 min.
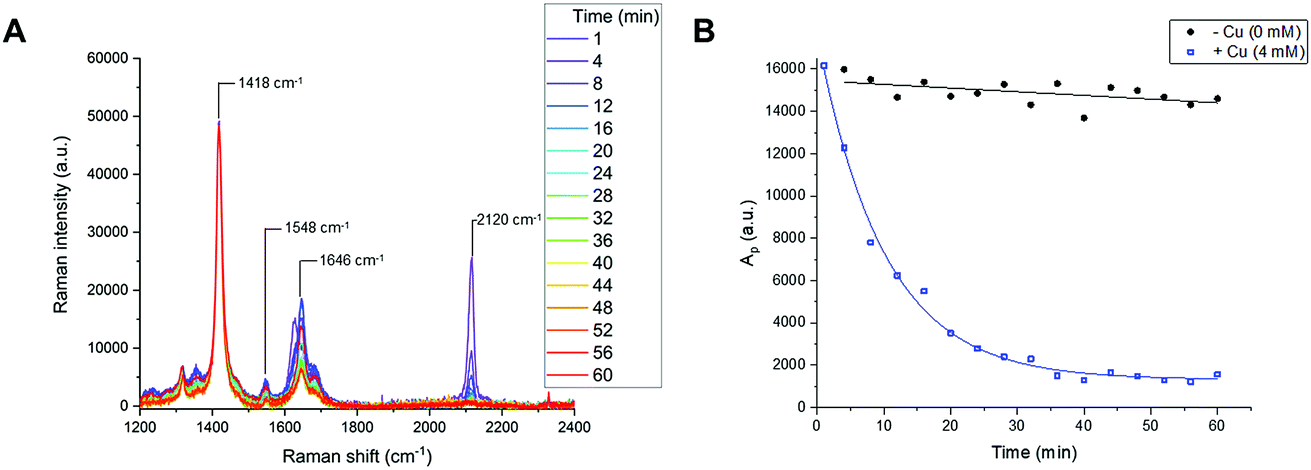 | ||
| Fig. 4 Reaction monitoring of the CuAAC reaction between EdU (1) and biotin-PEG3-azide (2) using Raman spectroscopy. (A) Raman spectra of a reaction mixture comprising EdU (50 mM), biotin-PEG3-azide (50 mM), CuSO4 (4 mM) and ascorbic acid (20 mM) in DMSO(aq.). An aliquot of the reaction mixture was removed at each timepoint and the individual Raman spectra presented between 0–60 min. Spectra were acquired using λex = 532 nm for 20 s using a 20× objective lens. The Raman spectra were normalised to the intensity of the peak at 1418 cm−1 (DMSO CH def.). (B) Integration analysis of the normalised Raman peak at 2120 cm−1 (CC, EdU) for the reaction presented in (A) (blue squares, +Cu (4 mM) with exponential fitting applied) and a control reaction (black circles, −Cu, linear fitting) where [Cu] = 0 mM. Ap = peak area @ 2120 cm−1. | ||
Kinetic analysis of the CuAAC click reaction
For more demanding applications, including CuAAC reactions in the cellular environment, the use of Cu-coordinating ligands has been found to accelerate the CuAAC reaction even further, up to several thousand times more than the ligand-free process.23 The requirement for Cu-coordinating ligands was particularly evident for bioconjugation reactions involving biological molecules, which impose specific demands in that they must be chemoselective, biocompatible, operate in aqueous environments at low reactant concentrations and induce fast catalyst turnover.40 A number of Cu-coordinating ligands have been developed for CuAAC reactions in cells, including THPTA (Fig. 5) and TBTA (Fig. 5); these are known to significantly accelerate the CuAAC reaction and stabilize the Cu(I) oxidation state in aqueous mixtures.41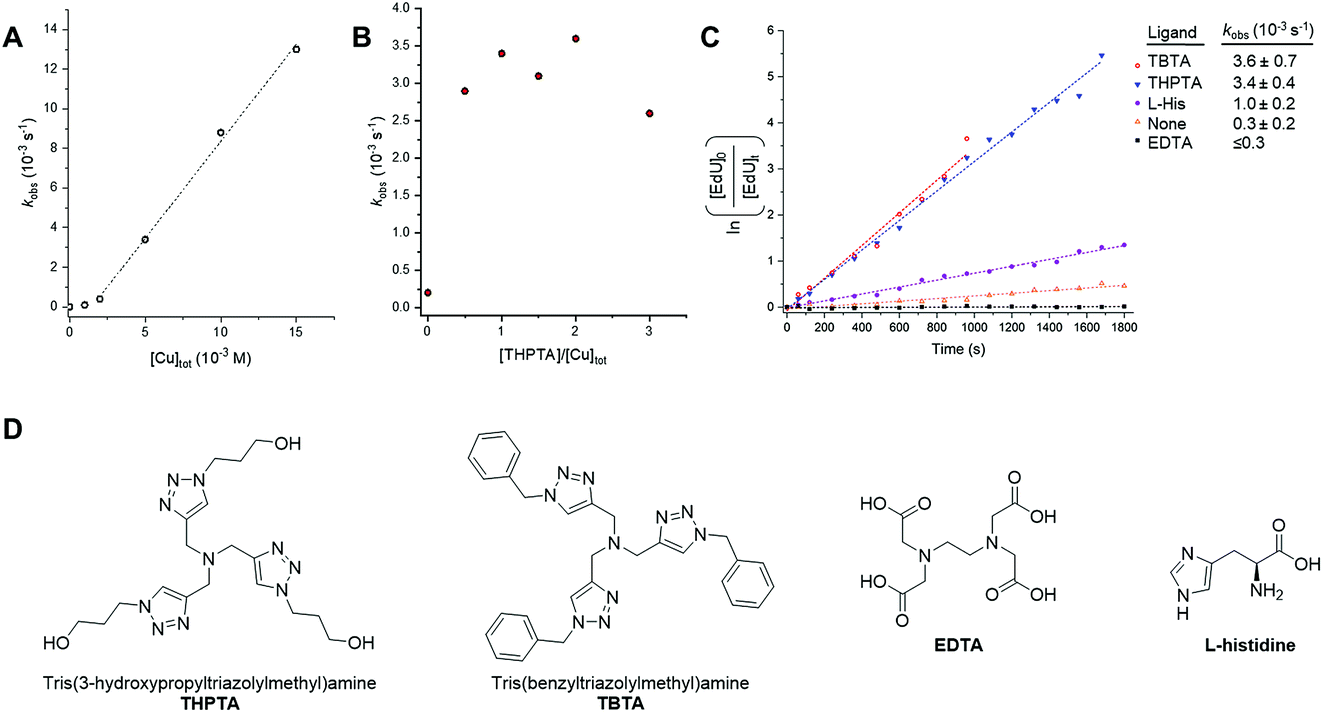 | ||
| Fig. 5 Investigating catalyst composition effects on the kinetics of the CuAAC reaction between EdU and biotin-PEG3-azide in DMSO/H2O 3:2 v/v. (A) Plot of kobs as a function of [Cu]tot, showing a first-order dependence on [Cu]tot. A linear fitting is only applied between [Cu]tot = 2–15 mM, due to the suspected presence of low concentrations of inhibitors in the reactants. Experimental conditions are described in entry 1, Table 1. Tabulated data with kobs ± 0.5 RMSE, see Table S1.† (B) Plot of kobs as a function of [THPTA]/[Cu]tot ratio. Experimental conditions are described in entry 2, Table 1. Tabulated data with kobs ± 0.5 RMSE, see Table S2.† (C) Plot of the CuAAC reaction with different CuAAC ligands with kobs ± 0.5 RMSE. Experimental conditions are described in entry 3, Table 1. (D) Chemical structure of the Cu(I) ligands, THPTA, TBTA, EDTA and L-histidine. | ||
Having established Raman spectroscopy as a suitable method for following the progress of the ligand-free CuAAC reaction, we next sought to perform a kinetic analysis of the ligand-accelerated CuAAC reaction. Firstly, the initial [Cu]tot was varied between 0–15 mM (with equimolar [THPTA]) using a fixed concentration of both the EdU (100 mM, alkyne) and biotin-PEG3-azide (100 mM, azide) reagents. A plot of kobs as a function of [Cu]tot showed a first-order dependence on [Cu]tot (Fig. 5A). This observation is consistent with the recently reported empirical rate law for the Cu-catalysed reaction of phenylacetylene with benzyl azide determined using heat-flow calorimetry.29b Additionally, a non-linear correlation at [Cu] ≤2 mM indicated the presence of low concentrations of inhibitors in the reactants. Next, we investigated the specific activity for the CuAAC reaction between EdU and biotin-PEG3-azide accelerated by THPTA. A plot of kobs as a function of the [THPTA]/[Cu] ratio is provided in Fig. 5B. In the absence of THPTA ligand, the reaction rate is markedly reduced, and a maximum rate-enhancing effect of the THPTA ligand was found at [THPTA]: [Cu] ≥ 0.5.23 Finally, the nature of the Cu-coordinating ligand was varied (Fig. 5C). The rate-enhancement by different ligands followed the order THPTA ≈ TBTA ≫ L-histidine, with no reaction in the presence of EDTA.42 Taken together, these results indicate that Raman spectroscopy is a simple and effective technique for profiling the bioorthogonal ligand-assisted CuAAC reaction in solution-phase.
Kinetic analysis of the Glaser–Hay reaction
Like the CuAAC reaction, the Glaser–Hay reaction has also been used in bioconjugation applications. In a seminal report, the Glaser–Hay reaction was used to generate protein bioconjugates under aqueous conditions, through site-specific incorporation of an alkyne-containing unnatural amino acid in an expressed protein and coupling under modified Glaser–Hay conditions.43 Additional studies have used Glaser–Hay bioconjugation for protein immobilisation44 and the synthesis of natural products using solid-supports.45 Given the increasing interest in the Glaser–Hay reaction for bioconjugation, we attempted to study aspects of its reaction kinetics using Raman spectroscopy to enable further optimisation of the reaction for biological applications. We chose to study the homodimerisation of propargyl choline (3) as a first step towards this goal (Fig. 6A). Propargyl choline is an alkyne-containing choline metabolite that was developed for the imaging of choline-containing phospholipids in vivo using CuAAC conjugation.46 More recently, however, it has been detected in living cells and tissues using SRS microscopy, which localises the metabolically-incorporated alkyne directly without the need for a second chemical reaction.18 The Raman spectrum of propargyl choline (as its bromide salt) shows an intense peak at 2131 cm−1 associated with the alkyne group (Fig. S3†). Glaser–Hay reaction time-course experiments in DMSO solution (Fig. 6B) indicated that in the presence of 7.5 mol% of CuI/TMEDA catalyst, the formation of the homodimer butadiyne product (4) was observed as a discrete peak at 2269 cm−1 (with the starting material detected at 2122 cm−1). Integration of the relative peak areas at 2122 cm−1 and 2269 cm−1 shows a significant increase in the intensity of the product as a function of reaction time, whilst the intensity of the starting material is seen to decrease (Fig. 6C). The relative intensity differences for the two alkyne signals reflects the observation that bis-alkynes have an overall higher Raman scattering cross-section compared to single, terminal alkynes (Fig. S4†). This experiment indicated that Raman spectroscopy could be used to follow the progress of the Glaser–Hay reaction in solution. The detection of the butadiyne product at a discrete Raman shift is a major advantage of this approach, as the relative intensities of both the starting material consumption and product formation could be assessed quantitatively in the same spectral acquisition. Furthermore, the ability to detect both alkyne components within the cellular-silent region of the Raman spectrum may facilitate optimisation of Glaser–Hay bioconjugations; for example, in determining the reaction yield.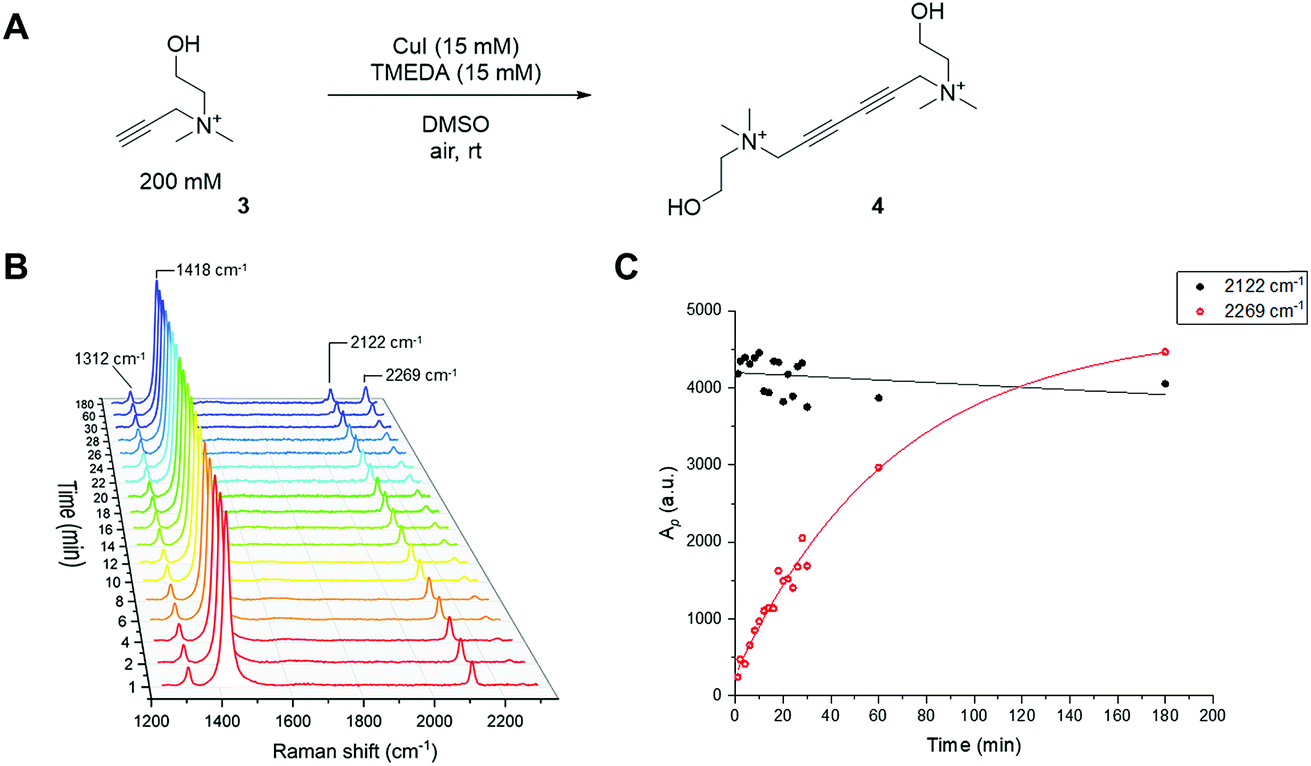 | ||
| Fig. 6 Reaction monitoring of the Glaser–Hay homocoupling of propargyl choline using Raman spectroscopy. (A) Glaser–Hay reaction of propargyl choline (200 mM) using CuI (15 mM, 7.5 mol%) and TMEDA (15 mM, 7.5 mol%) in DMSO. (B) Raman spectra of the reaction mixture in (A) acquired at the indicated timepoints following addition of the CuI/TMEDA catalyst. An aliquot of the reaction mixture was removed at each timepoint and the individual Raman spectra presented between 0–180 min. Spectra were acquired using λex = 532 nm for 20 s using a 20× objective lens. The Raman spectra were normalised to the intensity of the peak at 1418 cm−1 (DMSO CH def.). (C) Integration analysis of the Raman peaks at 2122 cm−1 (propargyl choline) and 2269 cm−1 (dimer) presented in (B). Ap = peak area @ 2122 cm−1/2269 cm−1. The lines through the data are merely a guide to the eye, not kinetic fitting. | ||
We next sought to study the kinetics of the Glaser–Hay reaction and used the homodimerisation of phenylacetylene (PA) as a model reaction. We chose PA because it has been widely used in this context, most recently in a DFT study of the reaction.36 The study was performed on a 20 µL scale, with air constantly bubbling through the reaction mixture. Initially, phenylacetylene (100 mM) was reacted in the presence of CuI (10 mM) and TMEDA (10 mM) in DMSO. In the course of the reaction, the intensity of the PA alkyne signal at 2102 cm−1 is seen to decrease, whilst the intensity of the product signal at 2218 cm−1 greatly increases (Fig. S5A†).47 Additionally, new peaks are generated at 1341 cm−1 and 1496 cm−1, whilst the intensity of the peak at 1597 cm−1 also increases. These observations are indicative of the formation of the 1,4-bisphenyl-1,3-butadiyne product (Fig. S5B†). Using the normalised peak area for the Raman signals of PA (at 2102 cm−1) and the butadiyne product (at 2218 cm−1), the kinetics of the Glaser–Hay reaction were assessed in response to varying [Cu]tot, [PA], Cu source and Cu-coordinating ligand (Fig. S6†). Firstly, the [PA] was fixed at 100 mM and [Cu]tot varied between 0–20 mM. Empirical second-order rate constants, kobs, were estimated by linear correlation of 1/[PA] versus time; i.e. rate ≈ kobs[PA]2. A plot of kobsversus [Cu]tot2 resulted in a linear relationship, i.e. kobs ≈ k [Cu]tot2 (Fig. 7A). Additionally, the [PA] was varied between 50–150 mM, with [Cu] and [TMEDA] fixed at 10 mM, yielding consistent kobs values over the concentration range tested (Fig. S7†). The effect of varying the ratio [TMEDA]/[Cu] was also investigated (Fig. 7B) and it was found that excess TMEDA causes inhibition of the reaction. The empirical rate equation is thus described by:
| Rate ≈ kobs[PA]2; where kobs ≈ 0.5[Cu]tot2 M−1 s−1 |
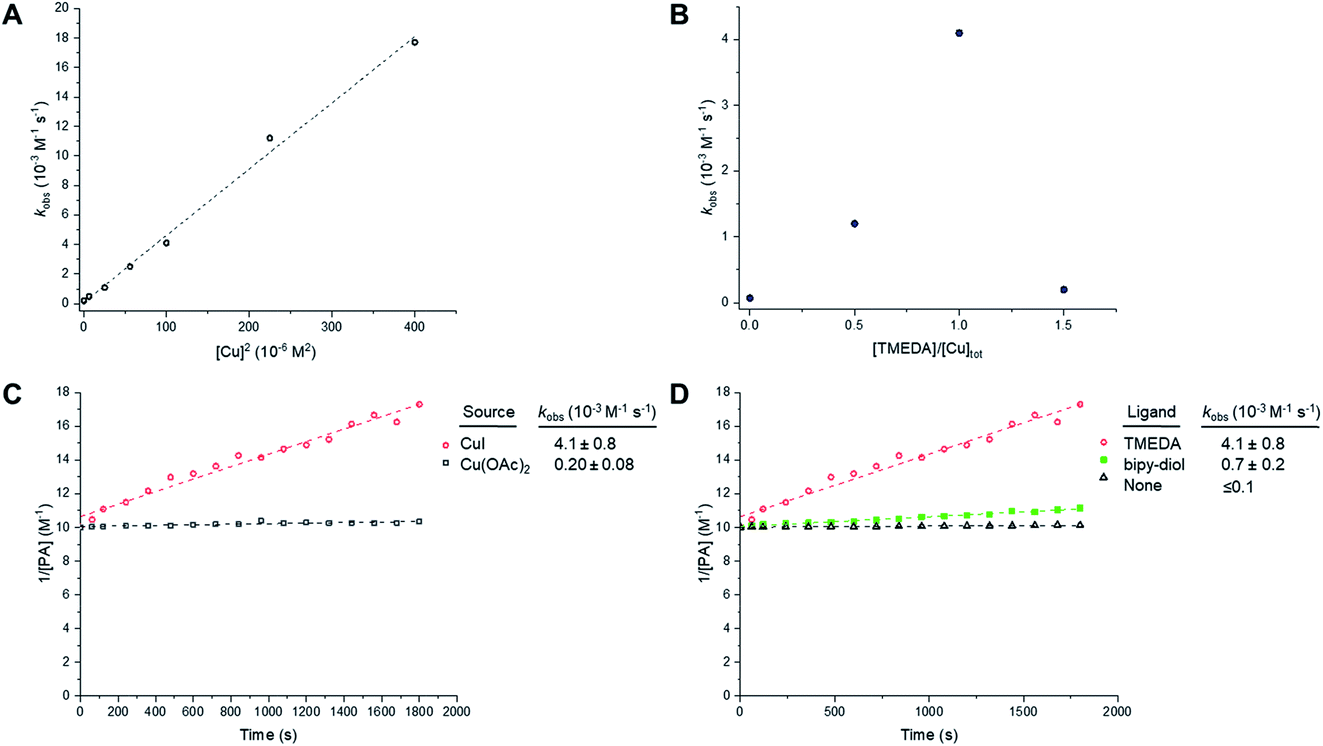 | ||
| Fig. 7 Empirical correlations for the kinetics of the Glaser–Hay homocoupling of phenylacetylene (PA). Experimental conditions are tabulated in Table S3.† (A) Correlation of the observed second-order rate constant with total [Cu]. Experimental conditions are described in entry 1, Table S3.† Tabulated data with kobs ± 0.5 RMSE, see Table S4.† (B) Effect of Cu:TMEDA ratio on observed second-order rate constant. Experimental conditions are described in entry 2, Table S3.† Tabulated data with kobs ± 0.5 RMSE, see Table S5.† Empirical second-order kinetic analysis of Glaser–Hay reactions with various Cu-sources (C), and ligand (D) with kobs ± 0.5 RMSE. Experimental conditions are described in entries 3 and 4, Table S3.† | ||
These results indicate a second-order dependence in both the copper and PA. Further studies investigated the catalyst composition effects; CuI showed an overall higher rate of reaction than Cu(OAc)2 (Fig. 7C), whilst TMEDA gave a higher rate of reaction than bipy-diol (Fig. 7D). The implications of these findings are summarised in Fig. 8. In terms of mechanism, the data are consistent with an equilibrium between (TMEDA)CuX and PA, to afford a complex [Cu·PA] in which there is one TMEDA, or in which the TMEDA is lost as [TMEDA·H]+X−, coupled to a second equilibrium involving two [Cu·PA], presumably generating one [Cu2·PA2] intermediate, and then a turnover rate-limiting irreversible reaction of [Cu2·PA2] that directly, or indirectly, generates product and regenerates CuX or (TMEDA)CuX. This scenario is broadly consistent with the mechanism proposed by Maseras et al. using DFT analysis (Fig. 2).36 Taken together, these data indicate that Raman spectroscopy is a useful method for profiling the Glaser–Hay reaction.
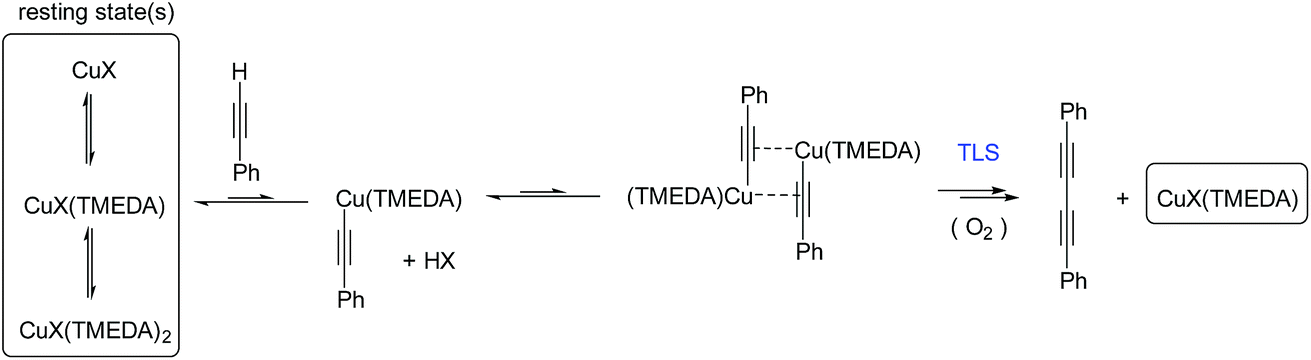 | ||
| Fig. 8 Mechanism for the Glaser–Hay reaction consistent with kinetic data obtained by Raman spectral analysis. TLS = turnover rate-limiting step. | ||
Conclusions
The studies described here indicate that Raman spectroscopy is ideally suited to analysing the kinetics of alkyne-based reactions. The intense alkyne signal in the Raman spectrum is well resolved from other solvent peaks in the fingerprint region (<1700 cm−1), and can thus be used to monitor the consumption of a starting material (i.e. EdU in the CuAAC reaction) or the generation of a product (i.e. butadiyne in the Glaser–Hay reaction). With the emergence of portable and in-line Raman instrumentation, Raman spectroscopy is expected to serve as a useful technique for profiling chemical reactions at scale. For the purposes of studying reactions at the microscopic level, the small sample volumes required for Raman analysis and the ability to use advanced Raman imaging systems (e.g. SRS microscopy) to provide detailed analysis of cellular features will enable rigorous kinetic investigations at the sub-cellular level. This approach would thus enhance current understanding of bioconjugation reactions in the cellular environment, which would enable a thorough understanding of both the intracellular chemistry and biology as a result.Materials and methods
Infra-red spectroscopy
FT infra-red (FTIR) spectra were recorded on a Shimadzu IRAffinity-1 spectrophotometer with a Pike Technologies horizontal ATR attachment, ZnSe lens single reflection ATR plate and Happ Genzel apodization. The recorded spectral range was 550–4000 cm−1. Data acquisition was performed over 10 scans. Spectral resolution for the system is 0.5 cm−1.Raman spectroscopy
The spontaneous Raman spectra were acquired using a confocal Raman spectrometer (inVia Raman microscope, Renishaw) at room temperature. A 297 mW (206 mW after objective) 785 nm diode laser or a 30 mW 532 nm laser excitation source was used to excite the sample through a 50×, N.A. 0.75 objective, a 20×, N.A. 0.40 objective or a 5×, N.A. 0.12 objective (Leica Biosystems). The recorded spectral range for grating 1200 g mm−1 was 100–200 cm−1. The total data acquisition was performed for 60 s for spectra recorded at λex = 785 nm or 10 s for spectra recorded at λex = 532 nm using the WiRE software. Spectral resolution for the system is 0.5 cm−1. The spectra were baseline corrected using the Baseline Subtract function available on WiRE™ software. Peak normalisation was performed using Origin Lab Data Analysis and Graphing software. Peak integration was performed on Origin using the Integrate function.SRS microscopy
Images were acquired using a custom-built multi-modal microscope setup. A picoEmerald (APE, Berlin, Germany) laser provided both a tunable pump laser (720–990 nm, 7 ps, 80 MHz repetition rate) and a spatially and temporally overlapped Stokes laser (1064 nm, 5–6 ps, 80 MHz repetition rate). The output beams were inserted into the scanning unit of an Olympus FV1000MPE microscope using a series of dielectric mirrors and a 2 lens based beam-expanding module. The resulting 2.4 mm beams were expanded by a further 3.6 lens within the microscope and directed into an Olympus XLPL25XWMP N.A. 1.05 objective lens using a short-pass 690 nm dichroic mirror (Olympus). The objective was under-filled to achieve higher power transmissions through the microscope, which were shown to be essential to detecting the SRS signal. Backscattered emission signals from two-photon fluorescence were separated from any backscattered excitation light using a short-pass 690 nm dichroic mirror and IR cut filter (Olympus). A series of filters and dichroic mirrors were then used to deconvolve the different emission signals onto one of 4 available photo-multiplier tubes (PMT).For SRS measurements, the Stokes beam was modulated with a 20 MHz EoM built into the picoEmerald. Forward scattered light was collected by a 20 Olympus XLUMPLFLN N.A. 1.00 objective lens and Stokes light was removed by filtering with an ET890/220m filter (Chroma). A telescope focused the light onto an APE silicon photodiode connected to an APE lock in amplifier with the time constant set to 20 µs. The lock in amplifier signal was fed into an Olympus FV10-Analog unit. Laser powers after the objective were measured up to 40–70 mW for the pump laser and up to 70 mW for the Stokes laser. All images were recorded at 512 × 512 or 1024 × 1024 pixels with a pixel dwell time between 2 and 20 µs, using FluoView FV10-ASW scanning software (Olympus). Images of the EdU distribution were recorded at 2120 cm−1 and are presented following removal of background signal using the Image Calculate function on ImageJ (2120–2109 cm−1). False colour LUTs and scale bars were applied to images using ImageJ software.
Chemicals and reagents
All chemicals were received from commercial suppliers unless otherwise noted, and were used as received without further purification. EdU (Thermo Fisher Scientific), CuSO4·5H2O, Tris-buffered saline (TBS), Tris–HCl, ascorbic acid, sodium ascorbate and biotin-PEG3-azide were all received from Sigma Aldrich.Kinetic measurements
:THPTA (5 mM, 1 µL from a pre-mixture comprising 5 µL CuSO4 (100 mM stock in water) + 1 µL THPTA (500 mM stock in DMSO) + 4 µL water) and sodium ascorbate (100 mM, 2 µL from 0.5 M stock in water), added last to the mixture. The 10 µL mixture was mixed by gentle agitation and vortex mixing before a sample (0.25 µL) was removed every 2 minutes and analysed using spontaneous Raman spectroscopy. Control reaction mixtures were prepared by varying the biotin-PEG3-azide content (replacing with 2 µL DMSO) or by varying the CuSO4 content (replacing CuSO4 in the pre-mixture with 5 µL water). Where increased biotin-PEG3-azide/EdU/CuSO4 concentrations were used, the volume of DMSO was reduced to ensure the total sample volume was consistent and contained a final concentration of DMSO:water of 3:2 v/v.
Kinetic analysis
The spectra were baseline corrected using the Baseline subtract function available on WiRE™ software. The spectra were then normalised to the area of the peak at 1420 cm−1 (CH def. DMSO, area: 1380–1460 cm−1; Raman), or the intensity of the peak at 2920 cm−1 (C–H, DMSO, IR). After this, the spectra were either overlaid for comparison or the peak area at 2096 cm−1 (N3, CuAAC, area (biotin azide, 2): 2050–2150 cm−1), 2120 cm−1 (CC, CuAAC, area (EdU, 1): 2075–2140 cm−1), 2122 cm−1 (CC, Glaser–Hay, area (PC): 2090–2150 cm−1), 2269 cm−1 (CC, Glaser–Hay, area (PC product, 4): 2230–2290 cm−1), 2102 cm−1 (CC, Glaser–Hay, area (PA): 2080–2130 cm−1) and 2218 cm−1 (CC, Glaser–Hay, area (PA product, 5): 2180–2240 cm−1) was determined using integration analysis available on Origin Software. Data were plotted and fitted using Origin graphic analysis package. Temporal concentrations/mol-fraction data for the CuAAC reaction were fitted to a standard exponential decay using the solver function in Excel, with initial concentration and first-order rate constants as unrestricted variables. Reciprocal temporal concentrations of [PA] were plotted to estimate kobs for the Glaser–Hay reaction. Errors in rate determinations were estimated as the root-mean square of the difference between kobs calculated from the analytical rate equation for each datapoint and kobs obtained by linear-regression of the entire dataset.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results received funding from the BBSRC (Grant Ref: BB/N021614/1), Cancer Research UK (Grant ref: C157/A25140 and C157/A15703), and the European Research Council under the European Union’s Seventh Framework Programme (FP7/2007–2013)/ERC Grant Agreement 340163. We thank Dr Colin Campbell for use of the spontaneous Raman microscope [UK Regenerative Medicine Platform Niche Hub, MR/K026666/1].Notes and references
- (a) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed; (b) J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13–21 CrossRef CAS PubMed.
- C. S. McKay and M. G. Finn, Chem. Biol., 2014, 21, 1075–1101 CrossRef CAS PubMed.
- N. Krall, F. P. da Cruz, O. Boutureira and G. J. L. Bernardes, Nat. Chem., 2015, 8, 103–113 CrossRef PubMed.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS PubMed.
- K. Lang and J. Chin, ACS Chem. Biol., 2014, 9, 16–20 CrossRef CAS PubMed.
- (a) K. Neumann, A. Gambardella, A. Lilienkampf and M. Bradley, Chem. Sci., 2018, 9, 7198–7203 RSC; (b) J. Clavadetscher, E. Indrigo, S. V. Chankeshwara, A. Lilienkampf and M. Bradley, Angew. Chem., Int. Ed., 2017, 56, 6864–6868 CrossRef CAS PubMed; (c) J. Clavadetscher, S. Hoffmann, A. Lilienkampf, L. Mackay, R. M. Yusop, S. A. Rider, J. J. Mullins and M. Bradley, Angew. Chem., Int. Ed., 2016, 55, 15662–15666 CrossRef CAS PubMed.
- (a) N. K. Devaraj, ACS Cent. Sci., 2018, 4, 952–959 CrossRef CAS PubMed; (b) J. G. Rebelein and T. R. Ward, Curr. Opin. Biotechnol., 2018, 53, 106–114 CrossRef CAS PubMed.
- A. Nadler and C. Schultz, Angew. Chem., Int. Ed., 2013, 52, 2408–2410 CrossRef CAS PubMed.
- K. Sivakumar, F. Xie, B. M. Cash, S. Long, H. N. Barnhill and Q. Wang, Org. Lett., 2004, 6, 4603–4606 CrossRef CAS PubMed.
- S. Sun and P. Wu, J. Phys. Chem. A, 2010, 114, 8331–8336 CrossRef CAS PubMed.
- A. R. Buchberger, K. DeLaney, J. Johnson and L. Li, Anal. Chem., 2018, 90, 240–265 CrossRef CAS PubMed.
- M. V. Gomez and A. de la Hoz, Beilstein J. Org. Chem., 2017, 13, 285–300 CrossRef CAS PubMed.
- (a) R. Grima, J. Chem. Phys., 2010, 132, 185102 CrossRef; (b) H.-X. Zhou, G. Rivas and A. P. Minton, Annu. Rev. Biophys., 2008, 37, 375–397 CrossRef CAS PubMed.
- (a) C. Corbet and O. Feron, Nat. Rev. Cancer, 2017, 17, 577–593 CrossRef CAS PubMed; (b) S. K. Parks, J. Chiche and J. Pouysségur, Nat. Rev. Cancer, 2013, 13, 611–623 CrossRef CAS PubMed.
- (a) Q. Li, T. Zhou, F. Wu, N. Li, R. Wang, Q. Zhao, Y.-M. Ma, J.-Q. Zhang and B.-L. Ma, Drug Metab. Rev., 2018, 50, 430–447 CrossRef CAS PubMed; (b) X. Chu, K. Korzekwa, R. Elsby, K. Fenner, A. Galetin, Y. Lai, P. Matsson, A. Moss, S. Nagar, G. R. Rosania, J. P. F. Bai, J. W. Polli, Y. Sugiyama and K. L. R. Brouwer, Clin. Pharmacol. Ther., 2013, 94, 126–141 CrossRef CAS PubMed.
- (a) B. G. Saar, C. W. Freudiger, J. Reichman, C. M. Stanley, G. R. Holtom and X. S. Xie, Science, 2010, 330, 1368–1370 CrossRef CAS PubMed; (b) B. G. Saar, Y. Zeng, C. W. Freudiger, Y.-S. Liu, M. E. Himmel, X. S. Xie and S.-Y. Ding, Angew. Chem., Int. Ed., 2010, 49, 5476–5479 CrossRef CAS PubMed.
- (a) S. Vanden-Hehir, W. J. Tipping, M. Lee, V. G. Brunton, A. Williams and A. N. Hulme, Nanomaterials, 2019, 9, 341 CrossRef CAS PubMed; (b) R. C. Prince, R. R. Frontiera and E. O. Potma, Chem. Rev., 2017, 117, 5070–5094 CrossRef CAS PubMed; (c) L. Wei, F. Hu, Z. Chen, Y. Shen, L. Zhang and W. Min, Acc. Chem. Res., 2016, 49, 1494–1502 CrossRef CAS PubMed; (d) C. Krafft, I. W. Schie, T. Meyer, M. Schmitt and J. Popp, Chem. Soc. Rev., 2016, 45, 1819–1849 RSC; (e) W. J. Tipping, M. Lee, A. Serrels, V. G. Brunton and A. N. Hulme, Chem. Soc. Rev., 2016, 45, 2075–2089 RSC.
- (a) W. J. Tipping, M. Lee, A. Serrels, V. G. Brunton and A. N. Hulme, Chem. Sci., 2017, 8, 5606–5615 RSC; (b) F. Hu, M. R. Lamprecht, L. Wei, B. Morrison and W. Min, Sci. Rep., 2016, 6, 39660 CrossRef CAS PubMed; (c) S. Hong, T. Chen, Y. Zhu, A. Li, Y. Huang and X. Chen, Angew. Chem., Int. Ed., 2014, 53, 5827–5833 CrossRef CAS PubMed; (d) Z. Chen, D. W. Paley, L. Wei, A. L. Weisman, R. A. Friesner, C. Nuckolls and W. Min, J. Am. Chem. Soc., 2014, 136, 8027–8033 CrossRef CAS PubMed; (e) L. Wei, F. Hu, Y. Shen, Z. Chen, Y. Yu, C.-C. Lin, M. C. Wang and W. Min, Nat. Methods, 2014, 11, 410–414 CrossRef CAS PubMed.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (b) C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS PubMed.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS PubMed.
- A. S. Hay, J. Org. Chem., 1962, 27, 3320–3321 CrossRef CAS.
- Z. M. Nimmo, J. F. Halonski, L. E. Chatkewitz and D. D. Young, Bioorg. Chem., 2018, 76, 326–331 CrossRef CAS PubMed.
- S. I. Presolski, V. Hong, S.-H. Cho and M. G. Finn, J. Am. Chem. Soc., 2010, 132, 14570–14576 CrossRef CAS PubMed.
- C. R. Travis, L. E. Mazur, E. M. Peairs, G. H. Gaunt and D. D. Young, Org. Biomol. Chem., 2019, 17, 3396–3402 RSC.
- N. Li, K. Reyna, V. Lim, S. Edwardraja and Q. Lin, J. Am. Chem. Soc., 2011, 133, 15316–15319 CrossRef CAS PubMed.
- M. F. Debets, S. S. Van Berkel, J. Dommerholt, A. J. Dirks, F. P. J. T. Rutjes and F. L. Van Delft, Acc. Chem. Res., 2011, 44, 805–815 CrossRef CAS PubMed.
- (a) K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 10317–10320 CrossRef CAS PubMed; (b) W. Chen, D. Wang, C. Dai, D. Hamelberg and B. Wang, Chem. Commun., 2012, 48, 1736–1738 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- (a) B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed; (b) R. Chung, A. Vo, V. V. Fokin and J. E. Hein, ACS Catal., 2018, 8, 7889–7897 CrossRef CAS.
- L. Zhu, C. J. Brassard, X. Zhang, P. M. Guha and R. J. Clark, Chem. Rec., 2016, 16, 1501–1517 CrossRef CAS PubMed.
- C. P. Seath, G. A. Burley and A. J. B. Watson, Angew. Chem., Int. Ed., 2017, 56, 3314–3318 CrossRef CAS PubMed.
- C. Glaser, Ber. Dtsch. Chem. Ges., 1869, 2, 422–424 CrossRef.
- P. Siemsen, R. C. Livingston and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 2632–2657 CrossRef CAS PubMed.
- F. Bohlmann, H. Schönowsky, E. Inhoffen and G. Grau, Chem. Ber., 1964, 97, 794–800 CrossRef CAS.
- M. H. Vilhelmsen, J. Jensen, C. G. Tortzen and M. B. Nielsen, Eur. J. Org. Chem., 2013, 701–711 CrossRef CAS.
- J. Jover, P. Spuhler, L. Zhao, C. McArdle and F. Maseras, Catal. Sci. Technol., 2014, 4, 4200–4209 RSC.
- H. J. Butler, L. Ashton, B. Bird, G. Cinque, K. Curtis, J. Dorney, K. Esmonde-White, N. J. Fullwood, B. Gardner, P. L. Martin-Hirsch, M. J. Walsh, M. R. McAinsh, N. Stone and F. L. Martin, Nat. Protoc., 2016, 11, 664–687 CrossRef CAS PubMed.
- A. Salic and T. J. Mitchison, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2415–2420 CrossRef CAS PubMed.
- A. E. Kliszczak, M. D. Rainey, B. Harhen, F. M. Boisvert and C. Santocanale, Sci. Rep., 2011, 1, 95 CrossRef PubMed.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- V. O. Rodinov, S. I. Presolski, D. Diaz Diaz, V. V. Fokin and M. G. Finn, J. Am. Chem. Soc., 2007, 129, 12705–12712 CrossRef PubMed.
- D. C. Kennedy, C. S. McKay, M. C. B. Legault, D. C. Danielson, J. A. Blake, A. F. Pegoraro, A. Stolow, Z. Mester and J. P. Pezacki, J. Am. Chem. Soc., 2011, 133, 17993–18001 CrossRef CAS PubMed.
- J. S. Lampkowski, J. K. Villa, T. S. Young and D. D. Young, Angew. Chem., Int. Ed., 2015, 54, 9343–9346 CrossRef CAS PubMed.
- J. C. Maza, J. R. McKenna, B. K. Raliki, M. T. Freedman and D. D. Young, Bioconjugate Chem., 2015, 26, 1884–1889 CrossRef CAS PubMed.
- J. S. Lampkowski, D. M. Uthappa, J. C. Maza and D. D. Young, J. Org. Chem., 2016, 81, 12520–12524 CrossRef CAS PubMed.
- C. Y. Jao, M. Roth, R. Welti and A. Salic, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15332–15337 CrossRef CAS PubMed.
- R. Bai, G. Zhang, H. Yi, Z. Huang, X. Qi, C. Liu, J. T. Miller, A. J. Kropf, E. E. Bunel, Y. Lan and A. Lei, J. Am. Chem. Soc., 2014, 136, 16760–16763 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S7 and Tables S1–S4. See DOI: 10.1039/c9fd00057g. Primary data files can be found at https://doi.org/10.7488/ds/2576 |
| This journal is © The Royal Society of Chemistry 2019 |