
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Point source characterization of per- and polyfluoroalkyl substances (PFASs) and extractable organofluorine (EOF) in freshwater and aquatic invertebrates†
Alina
Koch
*a,
Anna
Kärrman
a,
Leo W. Y.
Yeung
a,
Micael
Jonsson
b,
Lutz
Ahrens
 c and
Thanh
Wang
*a
c and
Thanh
Wang
*a
aMan-Technology-Environment Research Centre (MTM), School of Science and Technology, Örebro University, SE-70182 Örebro, Sweden. E-mail: thanh.wang@oru.se; alina.koch@oru.se
bDepartment of Ecology and Environmental Science, Umeå University, SE-901 87 Umeå, Sweden
cSection for Organic Environmental Chemistry and Ecotoxicology, Department of Aquatic Sciences and Assessment, Swedish University of Agricultural Sciences (SLU), SE-750 07 Uppsala, Sweden
First published on 25th September 2019
Abstract
Major point sources of per- and polyfluoroalkyl substances (PFASs) cause ubiquitous spread of PFASs in the environment. In this study, surface water and aquatic invertebrates at three Swedish sites impacted by PFAS point sources were characterized, using homologue, isomer and extractable organofluorine (EOF) profiling as well as estimation of bioaccumulation factors (BAFs) and mass discharge. Two sites were impacted by fire training (sites A and R) and the third by industrial runoff (site K). Mean Σ25PFASs concentration in water was 1920 ng L−1 at site R (n = 3), which was more than 20- and 10-fold higher than those from sites A and K, respectively. PFOS was the most predominant PFAS in all waters samples, constituting 29–79% of Σ25PFAS concentrations. Several branched isomers were detected and they substantially contributed to concentrations in surface water (e.g. 49–78% of ΣPFOS) and aquatic invertebrates (e.g. 15–28% of ΣPFOS). BAFs in the aquatic invertebrates indicated higher bioaccumulation for long chain PFASs and lower bioaccumulation for branched PFOS isomers compared to linear PFOS. EOF mass balance showed that Σ25target PFASs in water could explain up to 55% of EOF at site R. However, larger proportions of EOF (>92%) remained unknown in water from sites A and K. Mass discharges were for the first time estimated for EOF and revealed that high amounts of EOF (e.g. 8.2 g F day−1 at site A) could be transported by water to recipient water bodies relative to Σ25PFASs (e.g. 0.15 g day−1 at site A). Overall, we showed that composition profiling, BAFs and EOF mass balance can improve the characterization of PFASs around point sources.
Environmental significanceThe majority of emissions of per- and polyfluoroalkyl substances (PFASs) are released into the aquatic environment mainly from point sources such as manufacturing plants and firefighting activities using PFAS-containing foams. However, there is still limited knowledge on the characterization of PFASs around point sources and their fate in the aquatic environment. This study demonstrates that PFAS homologue and branched isomer profiling can be useful to characterize sources, and that several PFASs showed bioaccumulation potential in aquatic invertebrates. Furthermore, extractable organofluorine (EOF) analysis combined with target analysis enabled mass balance calculations, which revealed the presence of large proportions of unknown organofluorine substances in water. This study shows that a combination of different analysis techniques together with diagnostic analysis is useful in characterizing PFAS point sources. |
Introduction
Per- and polyfluoroalkyl substances (PFASs) are a large group of synthetic highly fluorinated organic substances, that have a broad range of applications (e.g. as fire-fighting foams, industrial surfactants, surface coatings) because of their unique properties such as thermal and chemical stability and amphiphilic property.1,2 Although PFASs provide many important functions in modern society, their unique properties also lead to undesired environmental consequences such as resistance to degradation, bioaccumulation, and potential health risks to wildlife and humans.3–6 Two main manufacturing processes of PFASs are electrochemical fluorination (ECF) and telomerization. Telomerisation produces only linear and even number carbon chain length PFASs depending on the starting material and is currently the major manufacturing process.7 In contrast, ECF produces around 20–30% branched isomers and was mainly used in the past, therefore detection of branched isomers is indicative of legacy contamination.5,8 Major point sources of PFASs are discharges from manufacturing plants, fire-fighting training sites, sewage treatment plant effluents or landfill leachates.9–12 It has been estimated that >95% of perfluorooctylsulfonyl fluoride (PFOSF)-based perfluoroalkyl carboxylic acids (PFCAs) and perfluoroalkyl sulfonic acids (PFSAs) emissions have been released into aquatic environments.5,13 Due to high water solubility and amphiphilic characteristics, various PFCAs and PFSAs have been found to bioaccumulate at various trophic levels of aquatic food webs.6,14,15 Uptake rates and half-lives in organisms generally increase with increasing PFAS carbon chain length.6 Branched PFASs and shorter chain PFASs, have a faster elimination rate.16–18 Still, sources and pathways in freshwater ecosystems are not completely characterized. Further, most studies analyzed only linear PFASs and their concentrations at higher trophic levels such as fish, and little information is known about isomer profiles in aquatic invertebrates.Recently, it has been shown that at least 4700 PFASs are available on the commercial market.19 However, development of targeted analytical methods for this vast number of PFASs is not feasible. Therefore, an alternative analytical approach is to measure extractable organofluorine (EOF).20–23 This enables a mass-balance analysis, where the difference between EOF and the sum of quantified PFASs (converted into fluorine equivalents) indicate the presence of unquantifiable fluorinated substances in a sample.24 Several studies found that target PFASs contributed to only a small fraction of EOF measured in different matrices, indicating the presence of unknown/unquantifiable organofluorines.21,25–27
In this study, surface water and composite aquatic invertebrate samples were collected at three small Swedish catchments that are impacted by PFAS point sources. The aim of this study was to investigate the fate of 25 target PFASs including branched isomers in surface water and aquatic invertebrates, and to conduct EOF mass balance in surface water. The combination of homologue and isomer profiles, bioaccumulation factors (BAFs), organofluorine mass balance analysis and mass discharges will allow for a comprehensive characterization and implications of the contamination of PFAS around point sources.
Materials and methods
Sampling sites
Samples were taken from three Swedish sites that have previously been reported to be contaminated by PFASs.28–31 Site A is the catchment of Märsta River at Stockholm Arlanda Airport, site R is a small catchment near Ronneby Airport, and site K is located at the industrial area of Kvarntorp (Fig. 1). Sites A and R are located close to airports with fire-fighting training facilities, where PFAS containing AFFFs were extensively used in the past.28,31,32 Since 2011, Stockholm Arlanda Airport use only fluorine-free alternative fire-fighting foams, whereas Ronneby Airport is part of the Swedish Armed Forces, which replaced PFAS-containing foams to fluorotelomer-based fire-fighting foams.32–34 At site K, samples were taken from lake Söderhavet, which is an artificial pit lake impacted by various industrial activities from the surrounding Kvarntorp area. These include the only hazardous waste management facility in Sweden, municipal sludge deposits, a 100 m high slag heap, and paper and pigment industries. Detailed descriptions of the sampling sites can be found in the ESI.†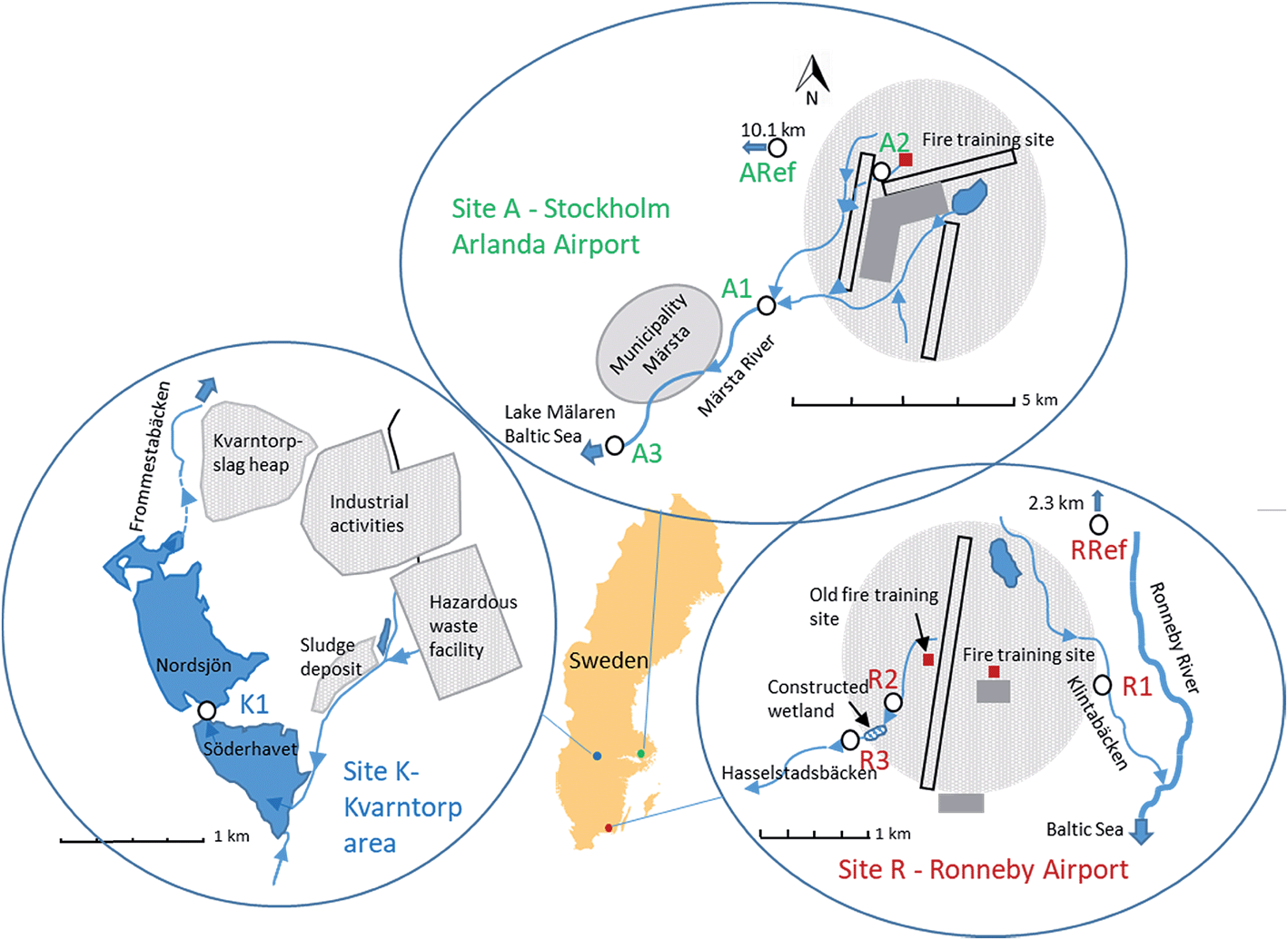 | ||
| Fig. 1 Sampling site maps, site K – Kvarntorp area (left map), site A – Stockholm Arlanda Airport (top map), and site R – Ronneby Airport (right map). Sampling spots are marked as white circles. | ||
Sample collection
Surface water and aquatic invertebrates were collected in September 2016 at: site A Stockholm Arlanda Airport (A1, A2, A3, ARef), site R Ronneby Airport (R1, R2, R3, RRef), and site K the Kvarntorp area (K1) (Fig. 1). Sampling sites A1 and A3 were located at Märsta River approximately 3 and 9 km downstream from the fire-fighting training area at Stockholm Arlanda Airport, with no other suspected PFASs point sources between the sites. Sampling site A2 is a small ditch located a few hundred meter from the cemented fire-fighting training area. Märsta River flows into Lake Mälaren, which is the 3rd largest lake and the largest drinking water source in Sweden. Reference samples were taken at a stream similar to the Märsta River about 10 km west from Stockholm Arlanda Airport. Ronneby Airport has two fire-fighting training areas, an old one located west of the runway which has not been used since the 1990s, and the current training area situated east of the runway. Sampling site R1 was located approximately 1 km east of the current training area at Kallinge creek, whereas R2 and R3 were located about 1–2 km west of the airport at a small creek (Hasselstadsbäcken) found to be impacted by the old training area. Hasselstadsbäcken flows through a constructed wetland (ca. 1.5 ha large) situated between R2 and R3. Both creeks flow into Ronneby River and finally into the Baltic Sea after a few kilometers. The reference site was located 2.3 km upstream of the airport, at the Ronneby River. In Kvarntorp, samples were taken at site K1 located at the outlet of the pit lake named Söderhavet downstream of the industrial area Kvarntorp.At each site, samples were collected within a 50 m stretch of the creeks/lake. Water parameters (pH, alkalinity, water temperature, dissolved organic carbon (DOC), total organic carbon (TOC), width and depth) were measured at A1, R1 and K1 (Table S1 in ESI†). Surface water samples (1 L) were collected a few centimeter (if possible >10 cm) below the surface in pre-rinsed polypropylene (PP) bottles. The bottles were rinsed three times with the surface water prior to sampling. After sampling, the samples were kept cool and then stored at 4 °C until analysis. For quality assurance, field blanks (1 L PP bottles filled with ultra pure MilliQ-water) were deployed with the lid removed for the duration of sampling and water samples were taken in triplicates at A1 and R1 and in duplicates at K1. The standard deviation of the replicates were low and are listed in Tables S4 and S5 in ESI.†
Benthic invertebrates were caught with a kick-net sampling method. Briefly, 10 locations for sampling were randomly chosen within the 50 m stretch, and within each of these locations, the sediment was disturbed for 60 s within an approximate area of 1 m2, while a hand-held net collected the invertebrates brought by the current. The collected invertebrates were kept in 1 L PP bottles for 24 h, which were filled with water from the sampling site, in order to clear their gut contents.35 Afterwards, their number was counted, the taxa were determined, and the samples were then frozen until analysis (Table S2 in ESI†). Due to the low amount of biomass, all invertebrates were pooled into one composite sample at each sampling site. Overall, the composite invertebrates samples contained mainly small crustaceans (i.e. amphipods and waterlouses) or aquatic insect larvae (i.e. mayfly, caddisfly or dragonfly larvae, Table S2 in ESI†). Adequate invertebrate biomass for PFAS analysis (>100 mg) was only found at A1 (containing 80% amphipods), R1 (containing 100% amphipods), and K1 (containing 18% mayfly larvae, with the rest being other aquatic insects and amphipods).
DOC and TOC analysis of water samples was performed at the department of soil and environment at the Swedish University of Agricultural Science. Daily and annual average flow data for the watershed of Hässelstad (R2 and R3) and Märsta River (A1 and A3) were obtained from the Swedish Meteorological and Hydrological Institute (SMHI)36 and from Edvisions30 for R1. Bioaccumulation factors (BAF) were calculated by the whole body PFAS concentration of the invertebrate sample (converted to wet weight) divided by the PFAS concentration in water.
Chemicals
A total of 25 target compounds were analyzed, including PFCAs with perfluorocarbon chain length of C3–C13, C15, and C17 (PFBA, PFPeA, PFHxA, PFHpA, PFOA, PFNA, PFDA, PFUnDA, PFDoDA, PFTrDA, PFTeDA, PFHxDA, PFOcDA); PFSAs with perfluorocarbon chain length of C4–C10 and C12 (PFBS, PFPeS, PFHxS, PFHpS, PFOS, PFNS, PFDS, PFDoDS); and precursors, such as fluorotelomer sulfonates (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 FTSA, 6:2 FTSA and 8:2 FTSA) and perfluorooctane sulfonamide (FOSA). The mass-labeled internal standard (IS) included 13C-PFBA, 13C-PFPeA, 13C-PFHxA, 13C-PFOA, 13C-PFNA, 13C-PFDA, 13C-PFUnDA, 13C-PFDoDA, 13C-PFTeDA, 13C-PFHxDA, 18O-PFHxS, 13C-PFOS, 13C-4:2 FTSA, 13C-6:2 FTSA, 13C-8:2 FTSA, and 13C-FOSA. Furthermore, a reference branched PFOS isomer standard was used, containing 1m-PFOS, 6/2m-PFOS, 3/4/5m-PFOS, and 4.4/4.5/5.5-m2-PFOS (brPFOSK0113). All reference compounds were purchased from Wellington Laboratories (Guelph, Canada). More details about the target PFASs can be found in Table S3 in ESI.† For the EOF analysis, a PFOS standard from Sigma-Aldrich was used.
:2 FTSA, 6:2 FTSA and 8:2 FTSA) and perfluorooctane sulfonamide (FOSA). The mass-labeled internal standard (IS) included 13C-PFBA, 13C-PFPeA, 13C-PFHxA, 13C-PFOA, 13C-PFNA, 13C-PFDA, 13C-PFUnDA, 13C-PFDoDA, 13C-PFTeDA, 13C-PFHxDA, 18O-PFHxS, 13C-PFOS, 13C-4:2 FTSA, 13C-6:2 FTSA, 13C-8:2 FTSA, and 13C-FOSA. Furthermore, a reference branched PFOS isomer standard was used, containing 1m-PFOS, 6/2m-PFOS, 3/4/5m-PFOS, and 4.4/4.5/5.5-m2-PFOS (brPFOSK0113). All reference compounds were purchased from Wellington Laboratories (Guelph, Canada). More details about the target PFASs can be found in Table S3 in ESI.† For the EOF analysis, a PFOS standard from Sigma-Aldrich was used.
PFAS analysis
Water samples were filtered through pre-baked (450 °C for 3 h) glass microfiber filters (Whatman GF/C; diameter 47 mm, 1.2 μm pore size) to remove particulate matter. The particulate matter on the filter was extracted, but the mass of particulate matter was too low (<100 mg), hence the measured levels were uncertain and excluded. Therefore, some PFASs such as the long chain homologues might be slightly underestimated in the surface water due to potential absorption to the filter and in the particles. Each water sample (500 mL) was spiked with 1 ng of IS followed by solid phase extraction (SPE) according to the ISO/DIS 25101:2009 method using Oasis weak anion exchange (WAX) cartridges (Waters 150 mg, 6 mL, 30 μm).37 The samples were eluted in two fractions; the first, the “neutral fraction” was eluted with 4 mL methanol containing mainly neutral PFASs (e.g. FOSA) and the second fraction, the “anionic fraction”, was eluted with 4 mL of 0.1% NH4OH in methanol consisting mainly of anionic PFASs (e.g. PFCAs and PFSAs).The invertebrate samples were first freeze dried, homogenized with mortar and pestle, and weighed into polypropylene (PP)-tubes (120–150 mg per sample). Extracts were then spiked with 1 ng IS, followed by alkaline digestion (0.2 M NaOH in methanol) and solid–liquid extraction (methanol), based on Powley et al.38 Afterwards, the extracts were diluted with MilliQ-water (1:9) and subjected to SPE clean-up following the same procedure as with the water samples. The neutral and anionic fractions were separately eluted, spiked with injection standard and then concentrated in LC-vials to 200 μL under a gentle nitrogen stream. The extracts from the neutral and anionic fractions were then separately analyzed for target PFASs by ultra-performance liquid chromatography coupled to a tandem mass spectrometer (UPLC-MS/MS Waters Xevo TQ-S, MA, USA). Negative ionization mode and gradient separation with a 100 mm C18 BEH column (1.7 μm, 2.1 mm) was used (for more information see ESI text and Table S3 in ESI†).
EOF analysis
A separate analysis to determine EOF in water samples was conducted. The same filtered water samples were extracted according to the target analysis using SPE. A small adjustment was added in order to remove inorganic fluorine in accordance to Miyake et al.21 After samples were loaded, the cartridges were washed in turn with 20 mL 0.01% NH4OH/MilliQ-water, 30 mL MilliQ-water and 4 mL ammonium acetate buffer. Samples were eluted in the two fractions as in the target analysis. Extracts were concentrated to 300 μL and split for the two separate instrumental analysis. EOF was determined using combustion ion chromatography (CIC), with a combustion unit from Analytik Jena (Germany), a 920 Absorber Module and 930 Compact IC Flex ion chromatograph from Metrohm (Switzerland). In brief, the extract (100 μL) was placed on a quartz boat and combusted in the furnace at 1000–1050 °C to convert organofluorine into HF, which was then adsorbed in MilliQ-water. The separation of anions was performed using an ion exchange column (Metrosep A Supp 5 – 150/4) with a carbonate buffer (64 mmol L−1 sodium carbonate and 20 mmol L−1 sodium bicarbonate) as eluent in isocratic elution. The concentration of F− was determined by a conductivity detector.Quality control and assurance and data analysis
For quantification of the PFASs, the isotope dilution method was used. For some PFASs that did not have isotope labeled standards, the internal standard closest in retention time within the same functional group was used for quantification (Table S3 in ESI†). Target PFASs were calculated against a nine-point calibration curve, whereas branched isomers of PFOS were quantified using a six-point calibration curve. The following branched isomer groups were quantified; 1m-PFOS, 6/2m-PFOS, 3/4/5m-PFOS, and 4.4/4.5/5.5-m2-PFOS. Chromatograms of the branched PFOS isomer standard, surface water (R1) and aquatic invertebrates (R1) are shown in Fig. S1 in ESI.† Branched isomers of PFHxS, PFHpS and PFOA were determined by the integration of all isomer peaks, and then semi-quantified against their respective linear isomer (assuming same response per mol for all isomers). The sum of linear and branched isomer concentration using this quantification method was referred to as e.g. T-PFOA in this study. Since PFOS branched isomer standard was available, they were quantified against a branched isomer calibration curve, and the total concentrations for branched and linear PFOS was therefore summed up and referred to ΣPFOS in this study. MilliQ-water was used for procedure blanks in the water analysis. For invertebrate analysis, in-house reference lake sediment was used as a procedure blank matrix. The MDLs and MQLs were respectively calculated as 3 times or 10 times the standard deviation (SD) plus the average concentration of all blanks (n = 3–6) used per matrix. If the target PFAS was not detected in the blanks, the lowest point of calibration (10 pg) was used to calculate the MDL and MQL. MDLs ranged between 0.004 and 0.81 ng L−1 for water samples and between 0.03 and 0.37 ng g−1 for aquatic invertebrate samples. MQLs ranged between 0.013 and 2.7 ng L−1 for water samples 0.09 and 1.25 ng g−1 for aquatic invertebrate samples. In the calculations, measured values below MQL but above MDL were reported as they were without censoring, while values below MDL were replaced by MDL/2 (Table S4 in ESI†). Recoveries ranged for C5–C11 PFCAs and PFSAs from 50–145% for water and 60–105% for invertebrate analysis (for details and standard deviations see Table S6 in ESI†).For EOF analysis, the samples were quantified by a six-point external calibration curve using a PFOS standard solution (linearity with R2 = 0.998), after the mean of the combustion blanks (n = 3) was subtracted from the peak area of the samples. For quality control a spiked (1 ng of IS) sample with MilliQ-water (QC sample) was extracted in the same batch, which was analyzed with UPLC-MS/MS. The recovery was compared to the QC values from the target analysis to ensure that the method performance was acceptable. The comparison showed the similar recovery values for the EOF and PFASs target analysis (RSD < 3.5% for C5–C11 PFCAs and PFSAs). The instrument limit of detection (LOD) was calculated by the mean of F in the combustion blanks (n = 3) plus 3× standard deviation (SD) and was 16 ng F. The MDLs were calculated from the procedural blanks and corrected for sample volumes, they ranged from 257–782 ng F L−1 in the neutral fraction and 71–217 ng F L−1 in the anionic fraction. Since EOF concentrations in samples of the neutral fraction were all below the estimated MDL, this study focuses only on the anionic fraction. Since the recovery of EOF cannot be determined, the quantified target PFAS concentrations without recovery-correction were used for mass balance analysis. For data comparison, the PFAS concentrations were convert to fluoride equivalents by the following equation:
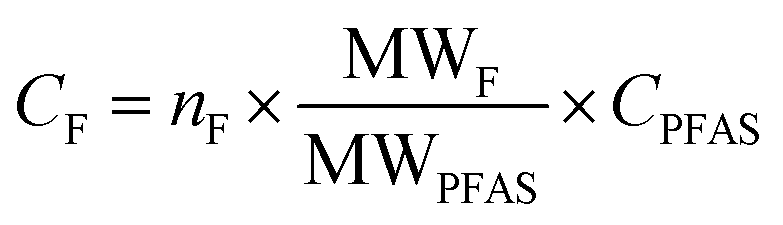
Results and discussion
PFAS concentrations in surface water
All 25 target PFASs were detected with the exception of PFDS, PFDoDS, PFOcDA, PFHxDA and 4:2 FTSA (Table S4 in ESI†). Thirteen PFASs were detected in all samples, while PFDoDA, PFTeDA, PFTrDA and 8:2 FTSA were detected in two samples and PFUnDA, PFNS and 6:2 FTSA in three to six samples. PFOS and PFHxS were the predominant PFASs in all waters samples. All samples (including reference samples) exceeded the annual average (AA)-EQS of the EU Water Framework Directive of 0.65 ng L−1 for total PFOS (sum of linear and branched isomers).
The Σ25PFAS concentrations at site A were 31 ng L−1 (A1), 94 ng L−1 (A2) and 89 ng L−1 (A3). The reference site ARef had a Σ25PFASs concentration of 14 ng L−1 (Table S4 in ESI†). Previously, PFAS concentrations were found to decrease with distance from the point source to the delta of Märsta River by a factor of 10–50.28 In this study, we found that Σ25PFAS concentrations at site A2 (closest to the point source) were much lower (94 ng L−1) compared to around 4000 ng L−1 for Σ11PFAS measured at the same location in 2011.28 One explanation for the sharp decrease at A2 could be that carbon filters have recently been installed to treat groundwater from the fire-training site.
At site R, Σ25PFAS concentrations were 282 ng L−1 (R1), 2890 ng L−1 (R2), 2580 ng L−1 (R3) and 2.0 ng L−1 (RRef). The mean Σ25PFAS concentration (±SD) at site R (1920 ± 1160 ng L−1) was more than 25 times higher than site A (71 ± 28 ng L−1) and more than 10 times higher than site K (156 ng L−1). PFOS and PFHxS constituted 57–79% and 9–25% of the Σ25PFASs, respectively. The PFAS concentrations in surface water at Ronneby Airport were comparable to levels found at other airport sites contaminated by PFAS-containing AFFFs ranging from hundreds to several thousand nanogram per liter.39–43 However, the concentrations were generally lower (about 10–30 times in R1 and 3–4 times lower in R2/R3) than reported for Σ15PFAS concentrations two years earlier (2014), just after the contamination was discovered in 2013.31
The Σ25PFAS concentration in surface water in Söderhavet (K1) was 156 ng L−1. This supports a previous report which found very high levels of PFOS in fish (mean 750 ng gww−1) from the same lake.29 The levels found at sites R and K were higher than the mean of 110 ng L−1 (Σ26PFAS) measured in 285 Swedish surface water samples from a national screening study,44 however the median concentration was low (3.9 ng L−1) indicating high variability between sites in Sweden.
PFAS concentrations in aquatic invertebrates
Twenty-two out of 24 target PFASs were detected in the benthic invertebrate samples (Table S5 in ESI†). FOSA could not be analysed in biota and is therefore excluded. Individual PFAS concentrations ranged from 0.14–44 ng gdw−1 at site A, 0.13–571 ng gdw−1 at site R, and 0.19–230 ng gdw−1 at site K, whereas the Σ24PFAS concentrations were 82, 769 and 292 ng gdw−1, respectively. To enable comparison with other studies, the concentration in dry weight was converted to wet weight (ww) using a conversion factor of 0.1, which was the mean water content (90% ± 2.3%) determined for all invertebrates in this study. PFOS was the dominant PFAS with concentrations of at least an order of magnitude higher than those of other target PFASs. The measured PFOS concentrations at all three sites (4.4 to 63 ng gww−1) were similar to concentrations measured in two amphipod species (Mysis relicta and Diporeia hoyi, 13 to 280 ng gww−1) in Lake Ontario, Canada.45 PFOS concentrations at site A1 (4.4 ng gww−1) and site K1 (25 ng gww−1) were similar to levels found in amphipods (Gammarids and D. Villosus) and damselfly larvae (Zygoptera) collected 40 km downstream of a PFAS manufacture plant from Rhône River in France (6.5–12 and 3.6–7 ng gww−1 PFOS).14 Interestingly, concentrations of long chain PFASs in this study (C9–C15 PFCAs up to 0.71 ng gww−1) were approximately two orders of magnitudes lower than those in the Rhône River (C9–C15 PFCAs maximum values of 102 ng gww−1). Amphipods from site R1, as expected from the water concentrations, had the highest Σ24PFAS concentration (769 ng gdw−1) compared to site A and K. The PFOS concentrations at R2 and R3 (2890 and 2580 ng L−1 in water) were above the Estimated No-Effects Value (ENEV) of 491 ng L−1 for aquatic organisms (used by Environment Canada46 based on the No Observed Effect Concentration (NOEC) of 49100 ng L−1 for Chironomus tentans47), indicating high exposure and potential adverse effects for invertebrates. At site A, the PFOS concentration in the invertebrates were more than 10 times lower (A1) than in amphipods collected from Lake Mälaren, which is the recipient lake of site A.48 However, the previous study collected samples in 2004, before the complete ban of PFAS-containing AFFFs at Stockholm Arlanda Airport, which may explain the lower PFAS concentrations in this study.
Isomer profiles
Chromatograms of the isomer analysis in water revealed one quantifiable-branched isomer for PFHxA, one for PFHpA, two for PFPeS, three for PFHxS, two for PFOA, three for PFNA (only in samples from site R) and six for PFOS. The proportion of the quantified branched isomers in surface water ranged from 8.6–17% for PFOA, 11–23% for PFHxS, 2.1–32% for PFHpS and 49–78% for PFOS (Fig. 2A and Table S7 in ESI†). Especially high concentrations of branched PFOS isomers were found at site R, where their contribution to the total PFOS was even higher than the linear. For example, at R1 the relative proportion to the sum of PFOS was for linear PFOS 31%, for 1-PFOS 6.2%, for 6/2-PFOS 21%, for 3/4/5-PFOS 40% and for dimethyl-PFOS 2.8% (Fig. 2B, Table S7 and Fig. S1 in ESI†). The mean contribution of branched isomers in water from this study were higher (e.g. br-PFHxS 16% and br-PFOS 66%) than the mean values found in Swedish surface waters (n = 285, e.g. br-PFHxS 7% and br-PFOS 20%).49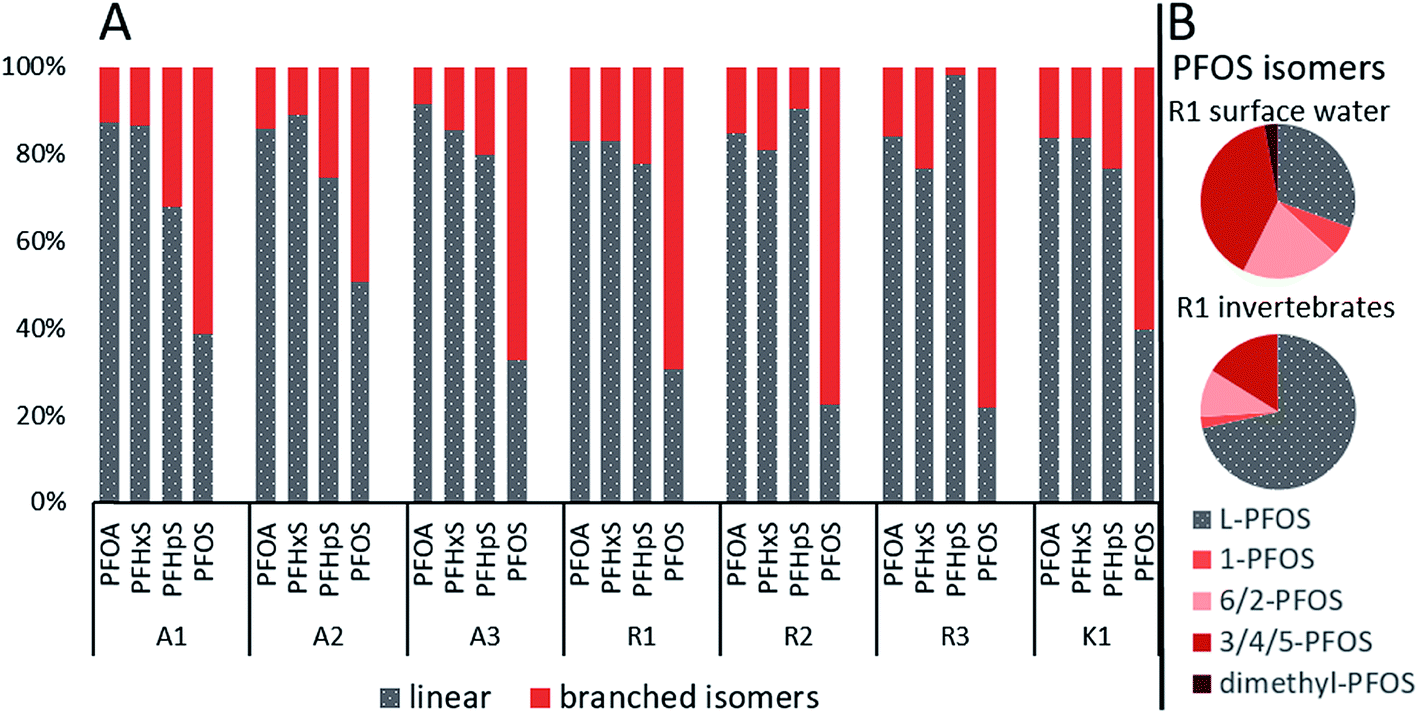 | ||
| Fig. 2 (A) Percentage of sum branched isomers vs. the linear PFASs in surface water for all sampling sites and (B) profiles of PFOS isomers in surface water and aquatic invertebrate from site R1. | ||
The branched isomer contributions in invertebrate samples were lower compared to surface water, ranging from 0.0–6.6% for PFOA, 0.0–18% for PFHpS and 15–28% for PFOS (br-PFHxS could not be quantified). This was expected since branched isomers are more water-soluble than the linear compound and have been suggested to have a lower uptake, faster elimination and/or selective retention in biota.15,41 Similar branched isomer pattern for PFOS and PFOA was found in the food web of Lake Taihu in China.50 Additionally, PFOS isomer pattern at site A, R and K in surface water (17–24% of 6/2-PFOS) and aquatic invertebrates (6.1–10% of 6/2-PFOS) were comparable to those in Lake Ontario, 22–28% of 6/2-PFOS in Lake Ontario water and 3.9–5.2% for 6/2-PFOS in amphipods (Mysis relicta).15 Without the branched isomer analysis, PFAS levels would have been substantially underestimated, e.g., br-PFOS contributed to 69% of the ΣPFOS in water at R1.
Homologue profiles of PFASs
Homologue profiles differed between the sites and between water and aquatic invertebrates, but all samples showed a dominance of PFSAs. In water, the mean (±SD) contribution of PFSAs was 65 ± 7.2% at site A, 94 ± 1.2% at site R, and 78% at site K (Fig. 3). A similar contribution of PFSAs was found in invertebrates, with 58% in sample A1, 92% in R1, and 90% in K1. The fraction of FTSAs (6:2 FTSA and 8:2 FTSA) to the overall homologue profile ranged from 0.01 to 4.3%. The percentage of the long chain PFASs (number of carbons for PFCAs ≥ 7 and PFSAs ≥ 6) was higher in the aquatic invertebrates (average 94%) than in surface water (average 81%).
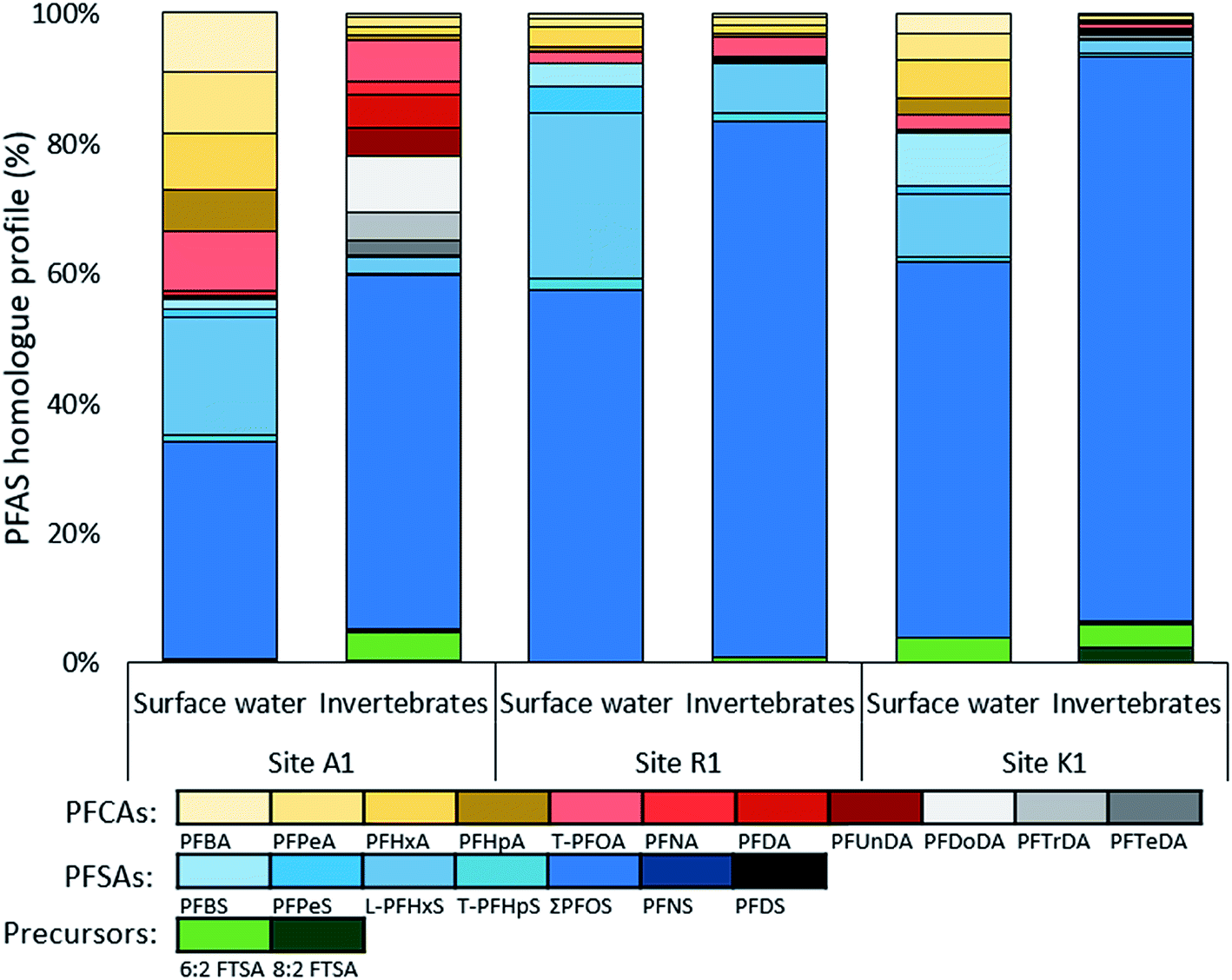 | ||
| Fig. 3 PFAS homologue profiles in surface water and aquatic invertebrates from sites A1, R1 and K1. | ||
At site A, the water profiles showed major contribution from PFOS (34–58%), PFHxS (12–18%) and short chain PFCAs (C4–C7, 19–35%). In sample A2, collected closest to the fire training area, up to 4.3% FTSAs were detected. A different profile can be seen in the aquatic invertebrates from A1 were long chain PFCAs and PFASs contributed to 92%, as well as 4.6% 6:2 FTSA. The low abundance of short chain PFASs reflects the different bioaccumulation potentials of PFASs in biota. Both profiles combined show a broad range of PFASs and indicate contamination of a mixture of historical AFFF products and products that were used in the transition time (2003–2011) until fluorine-containing foams were banned at the airport. The high proportion of PFHxS and PFOS and their branched isomers in water verify the historical use of PFOS–AFFF from 1980–2003 (pre 2003 STHMEX-AFFF 3%, Dr Richard Sthamer GmbH & Co. KG, Germany).28,32 Short-chain PFCAs and FTSAs might be used in new generation foams and therefore likely ingredients in PFOS-free AFFFs applied at site A until 2011 (Moussol APS-P from Dr Richard Sthamer GmbH & Co. KG, Germany). One explanation for the broad PFASs mixture would be that the influent creek, Kättstabäcken (A2), receives groundwater with historical PFAS contamination and surface and subsurface-water runoff from recent PFASs compositions.
The homologue profile at site R reveals that PFOS and PFHxS account for 58–79% and 10–26%, respectively, in the surface water. The remaining proportion consist of other PFSAs (i.e. PFBS, PFPeS and PFHpS) and PFCAs, mainly PFHxA. In invertebrates, the PFOS fraction is even higher, accounting for 82%, indicating a high uptake and bioaccumulation of PFOS. The contribution of PFHxS was comparatively lower at 7.7%. At Ronneby airport the military used STHMEX-AFFF 3% (pre 2003) from 1980 until early 2000, the same foam that was used at site A.32 Homologue profiles were very similar to those described for AFFF formulations (1988–2001) from the major producer 3M in the US,51 suggesting similarities in the formulation of these products. Compared to site A, homologue profiles differ since the PFCA fraction was much higher in A, suggesting that site R is mainly impacted by historical AFFFs. In fact, R2 and R3 were situated close to the old fire training area. R1 on the other hand was situated close to the recent training site (implemented in 1990) and a slightly higher contribution of PFCAs (7.6%) was measured there, indicating potential impact from new generation foams.
Major fractions in water from K1 were PFOS (58%), PFHxS (10%) and PFBS (8.3%). PFCAs (22%) contributed with C4–C8 in similar proportions and 6:2 FTSA was 3.8% of total PFASs. In the invertebrates, PFOS made up 87% of the profile and 8:2 FTSA had the highest abundance of 2.4% compared to all other samples. High proportions of PFOS and PFHxS as well as the detection of 6:2 and 8:2 FTSAs implied impacts from AFFF products in both surface water and aquatic invertebrates. 6:2 FTSA was found to be associated to fire training areas, but not in urban areas,49 strengthening the theory that site K was indirectly impacted by AFFF products. As mentioned previously, the only hazardous waste facility in Sweden is located at site K, which disposes AFFF foams by incineration and might also handle AFFF contaminated soils in landfills. Another possible source could be from the sludge deposit located along the stream that flows into Söderhavet. Furthermore, leachate from landfill could be expected from site K and besides the fact that the proportion of PFSAs were predominant in the homologue profile of K, it also had similarities to those found in Swedish landfill/waste disposal leachates.49 In general, Gobelius et al.49 found that source categories landfill, urban area, and unspecific industry had similar PFAS homologue profiles.
Log transformed concentrations of PFHxS and PFOS were found to be positively correlated in all water samples (r = 0.97, n = 7) as well as in aquatic invertebrates (r = 0.93 although the number of samples were low, i.e. n = 3) (Fig. S2†). Awad et al.39 also found a similar correlation and suggested a common origin, in their case it was AFFF impacted water. Furthermore, PFHxS and PFOS were found to be the most common PFASs at 40AFFF impacted sites in various matrices across the US.52 It is not known whether PFHxS was an intentional ingredient in PFOS products (e.g. AFFF 3M formulations) or if its occurrence in the environment is due to impurities in the production of PFOS or environmental transformation of precursors. Houtz et al.51 found evidence for transformation by showing that C6 homologue precursors in AFFF foam formulation by 3M most likely transformed into PFHxS via oxidation using a total oxidizable precursor assay. They suggested that increased PFHxS-to-PFOS ratio in environmental samples compared to those determined in AFFF (e.g. 3M AFFF had a ratio of 0.1) implies that precursors are transformed by biological or chemical transformation (e.g. aerobic oxidation by bacteria) into PFHxS. In this study, PFHxS-to-PFOS ratios in surface water at site A were 0.2–0.54, which were higher compared with the 3M formulations investigated by Houtz et al.51 indicating potential contribution from transformation of C6 precursors. The ratio was lower for K1 with 0.17 and at 0.12 for R2 and R3. However, for the surface water at R1 the ratio was 0.44, which was substantially higher than at R2 and R3. This might be explained by the fact that only R1 is impacted by the recent fire training site. In fact, it is known that the Swedish Armed Forces replaced PFOS–AFFFs by fluorotelomer-based fire-fighting foams in 2011, which would be in accordance with the PFHxS-to-PFOS ratio at R1 indicating the presence of C6 precursors. Therefore, the PFHxS-to-PFOS ratio might be a suitable indicator for different AFFF products used at fire training sites.
Bioaccumulation factors
The BAFs (based on converted wet weights) were determined for 15 PFASs and branched PFOS isomers for the aquatic invertebrates (log transformed in Fig. 4, and Table S8 in ESI†). In general, high BAF values were found for long chain PFCAs at A1, whereas at R1 short chain PFASs were higher than those in A1 and K1. Differences may be due to different compositions of invertebrates among the samples and dissimilar environmental conditions between the sites. According to regulatory criteria, PFASs with BAFs between 1000–5000 L kg−1 are usually considered as bioaccumulative.53 In the aquatic invertebrates from A1, R1 and K1, C9–C15 PFCAs and C9 PFSA (PFNS) had BAFs >1000 L kg−1, indicating bioaccumulation potential for those compounds. Furthermore, the bioaccumulation was directly related to the perfluorocarbon chain length of PFASs, which has also been described in other studies (Fig. 4).53 When looking at the per unit CF2-moiety increase, the BAFs of short chain PFASs (C4 to C7) increased by a factor of 1.3 per CF2-moiety for PFCAs and 3.2 for PFSAs. The CF2-moiety unit increase for long chain PFASs (C7 to C10) is steeper by a factor of 3.7 for PFCAs and 4 for PFSAs making the long chain PFAS even more bioaccumulative. For example, the BAF for PFNA (C8) was 866 and increased to 4280 for PFDA (C9) in A1. Branched PFOS isomers showed the lowest BAFs for dimethyl PFOS (0–14 L kg−1), and similar factors for 3/4/5-PFOS, 6/2-PFOS and 1-PFOS in the range of 85–186 L kg−1, while BAFs of linear PFOS were highest with 534–942 L kg−1. The substantial different BAFs of branched vs. linear PFOS, may be due to the higher solubility of branched isomers in water15 and the faster elimination rates for branched isomers in biota.15 In other studies, higher BAFs have been reported in aquatic food webs than in this study.15,39,54 These differences could be due to the differences in environmental conditions between sites and whether steady-state has been reached between PFAS concentrations in water and organisms.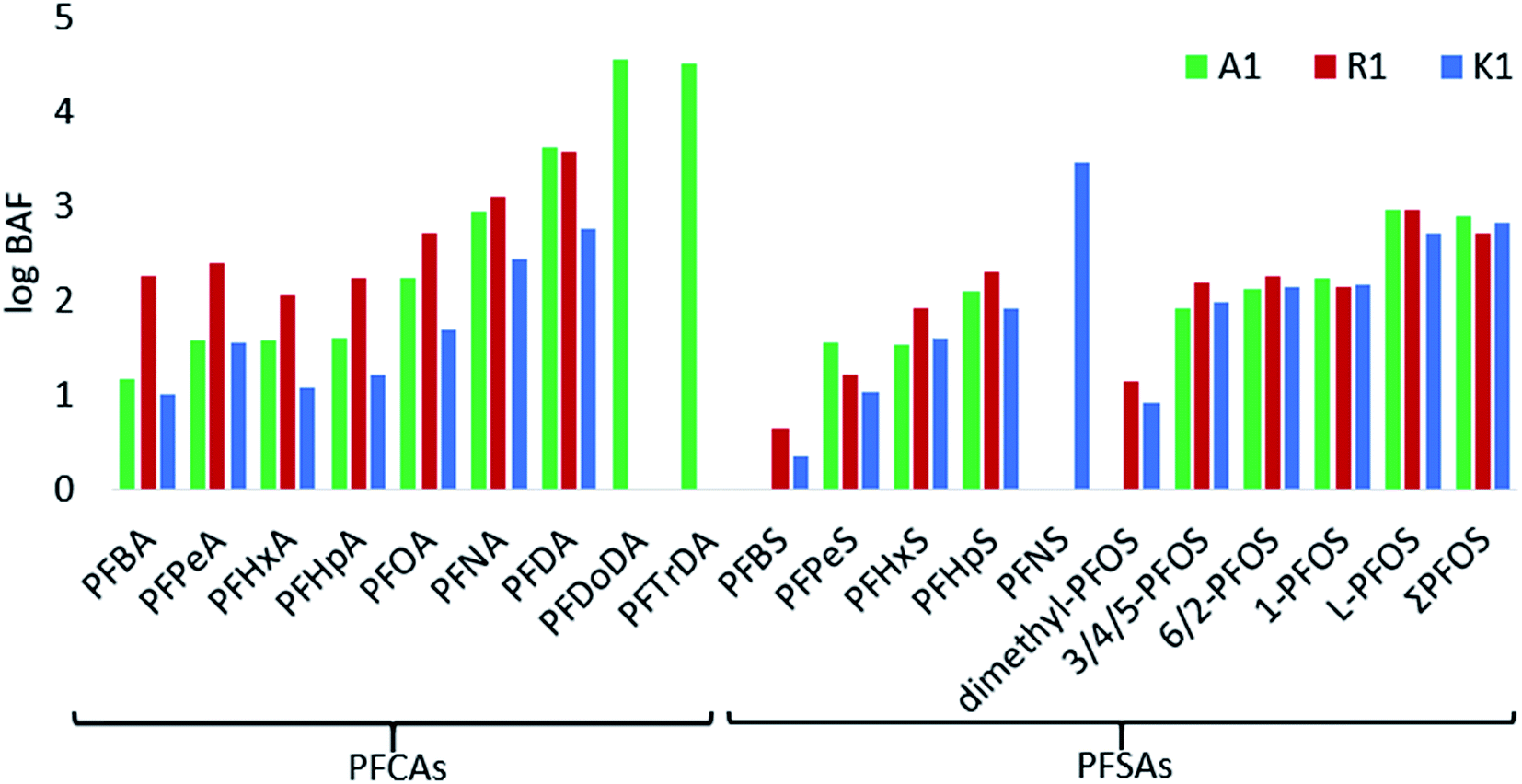 | ||
| Fig. 4 The logarithmic bioaccumulation factor (BAF) on a wet weight basis (L kg−1) of target PFCAs and PFSAs (including four branched PFOS isomers, linear PFOS and ΣPFOS) for sites A1, R1 and K1. The BAFs could not be determined for some PFASs and sites due to values below MDL. | ||
Extractable organofluorine in surface water
EOF was determined for all water samples and the mass balance analysis (the proportions between unquantified and quantified EOF) are shown in Fig. 5. At site R, the highest EOF concentrations were found for R2 and R3 with 3180 and 3930 ng F L−1, respectively, whereas R1 was 408 ng F L−1. Target PFASs (predominantly PFOS) accounted for 55%, 39% and 44% (R2, R3 and R1, respectively), indicating that the analyzed PFASs substantially contributed to the organofluorine found in the surface waters at site R. Concentrations at site A were 320, 2480, 2170 ng F L−1 at A1, A2, and A3, respectively, whereas Σ25PFASs made up only a small proportion of EOF ranging between 2.1 and 5.4%. Similarly, at site K, the EOF was 1110 ng F L−1 and Σ25PFASs represented 7.8% of the EOF. Both reference sites RRef and ARef contained 96% and 98% unquantified EOF. Comparing the three sites, the mass balance analysis showed different profiles and concentrations of EOF, indicating variation of organofluorine sources other than PFASs such as agricultural sources. For example at site R, where Σ25PFASs substantially contributed to the EOF, the sampling sites were situated in a forested area in close proximity to the airport, whereas A1 and A3 were located in an agricultural and urban landscape. At sites A and K quantified PFASs made up only a small proportion of EOF indicating other existing organofluorine sources. In general, EOF measured for R2–3, A2–3 and K1 were higher than those found in other studies, such as seawater with an average concentration of 562 ng F L−1 (ref. 21) and lake water in the range of 100–1000 ng F L−1 measured at an AFFF impacted site in Hokkaido, Japan.55 The higher EOF concentration in this study might partly be explained by the hydrological characteristics of the sampling sites, since the surface water samples from sites R and A were taken from small creeks/ditches close to the fire-fighting training area (proximity <9 km) and therefore they might have been less diluted compared to lake and seawater samples. The high portion of unquantified EOF indicates that there was a significant amount of unknown organofluorine in these samples. Their identities are currently not known but could be e.g. other PFASs, fluorinated pharmaceuticals, herbicides or other agrochemicals.56,57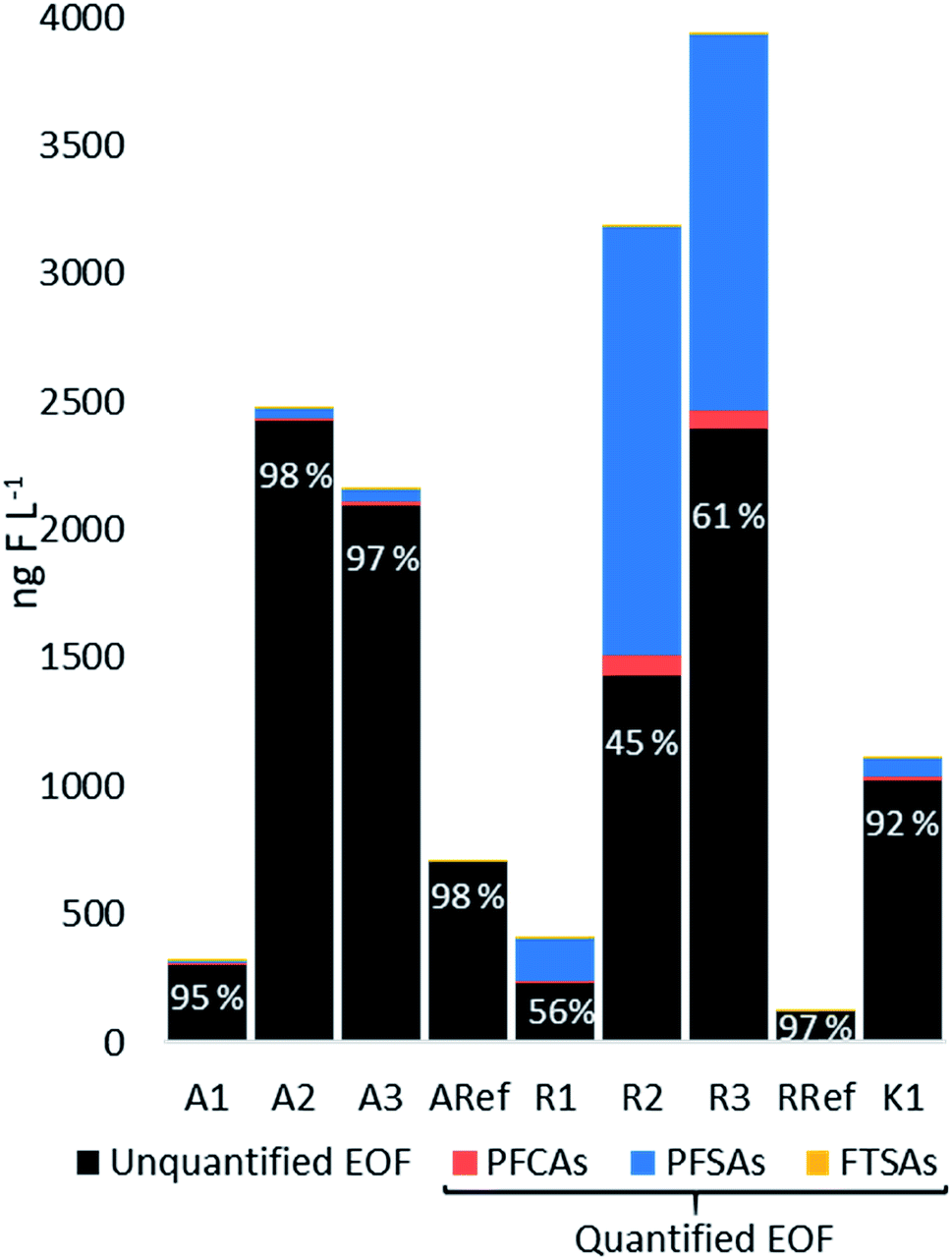 | ||
| Fig. 5 The concentrations of extractable organofluorine (EOF) measured in the anionic fraction of water samples at all three sites. Bars show the fraction of unquantified EOF (dark grey and its percentage), PFCAs (red), PFSAs (blue) and FTSAs (yellow). | ||
Mass discharge from surface waters
Mass discharges were estimated for sites A and R for Σ25PFASs, PFOS and EOF in gram per day (Table 1). At site A, the mass discharge was estimated for Märsta River (A1), located south of Stockholm Arlanda Airport, with 0.036 g day−1 for Σ25PFASs and 1.7 g F day−1 for EOF. At the mouth of the river (A3), 6 km downstream from A1, the discharge was higher with 0.15 g day−1 for Σ25PFASs and 8.2 g F day−1 for EOF. The high mass discharge of EOF at site A3 could be from the introduction of other organofluorine sources between A1 and A3 from Märsta city. For comparison with previous studies, an estimation of yearly mass discharges has been calculated based on the assumption that the PFAS concentrations have been constant over the whole year. For Märsta River and Hasselstadsbäcken, the yearly average flow rates were taken from SMHI, while for Klintabäcken a single field measurement in April 2015 was used (for more detail see Table S9 in ESI†). At site A3, a gradual decrease of the yearly mass discharge can be observed over time with 5.3 kg per year in 2011,28 a mean of 3.7 kg per year for Σ12PFAS between 2013 (ref. 3) and 2014,58 and 1.4 kg per year for Σ25PFAS in 2016 (this study). This trend is in good agreement with model predictions, where the highest PFOS amounts, 27 kg in soil and water at site A, were estimated in the 1990s and since then a yearly decrease of 0.5–1 kg was predicted.59 The model also predicted by 2060 PFOS levels in soil will drop to <5 kg and <0.3 kg in ground- and surface water at site A. Furthermore, no ecotoxicological effects on local fish as well as no human health effects from intake of fish and water (based on TDI level of 150 ng kg−1 body weight) were found in that study.59 However, due to the continued PFASs contamination even many years after the usage of PFAS–AFFFs, remediation is planned by the owner of Stockholm Arlanda Airport, and a pilot study was completed in 2018.60 Since remediation measures will be implemented in the future, it would be of interest to also monitor and identify the EOF at site A.| Sampling site | Flow rate [L day−1] | PFOS [g day−1] | Σ 25PFASs [g day−1] | EOF [g F day−1] |
|---|---|---|---|---|
| a Daily flow rate at the Märsta river (A3) on the 20th September 2016.36 b Daily flow rate measured on 14–17th of April 2015.30 c Daily flow rate of the Hasselstad watershed on the 14th of September 2016.36 | ||||
| Märsta River (A1) | 3456000a |
0.036 | 0.12 | 1.7 |
| Märsta River outlet (A3) | 3456000a |
0.15 | 0.31 | 8.2 |
| Klintabäcken (R1) | 6480000b |
1.1 | 1.7 | 3.8 |
| Hasselstadsbäcken (avg. R2/R3) | 2592000c |
5.6 ± 0.34 | 6.9 ± 0.4 | 11 ± 1.1 |
| Sum discharge from R1 and R2/3 | 6.6 | 8.7 | 14 | |
Estimated mass discharge for Klintabäcken (R1) was 1.7 g day−1 for Σ25PFASs and for Hasselstadsbäcken 6.9 g day−1 for Σ25PFASs. Both discharges combined resulted in 8.7 g day−1 for Σ25PFASs (80% was PFOS) and 14 g F day−1 for EOF that were released into Ronneby River. The yearly mass discharge of PFOS was 0.38 kg per year for Klintabäcken and 15 kg per year for Hasselstadsbäcken (Table S9 in ESI†). Mass discharge of PFOS previously estimated for Klintabäcken in 2014 was higher with 20 kg per year PFOS, while the same extent was assumed for Hasselstadsbäcken.31 However, the contribution of groundwater was included in the mass discharge estimation and PFOS concentrations measured in 2014 were higher in the surface water (3200–4000 ng L−1). This indicates that the PFOS concentration decreased by more than 10-fold from 2014 to 2016 in Klintabäcken, whereas for Hasselstadsbäcken the mass discharge remained relatively constant at high levels.
In general, the PFAS mass discharges determined for Märsta and Ronneby River were considerably lower than those discharges found for major European rivers,61 such as River Rhine with an estimate of around 6 tonnes per year for Σ4PFCAs.62 This was expected considering that the catchments of Märsta River and Ronneby Airport are relatively small with low flow rates. Also, a comparison between different sites are influenced by different factors such as input from point sources, flow rates, hydrology and seasonality. For example, PFAS concentrations were found to be varied by a factor of 1.5 between spring and fall at Märsta River (A1).58 In addition, other factors that may influence the fate of PFASs in the receiving water, such as environmental fractionation, uptake, and sorption to organic matter/sediments, were not considered.
Conclusion
Overall, we showed that homologue profiles (including PFHxS–PFOS ratio), branched isomer profiles, BAFs and EOF mass balance could improve our understanding and characterization of PFAS point sources. All three point sources showed different PFAS concentration levels, e.g. AFFF impacted site R had the highest contamination, as well as different PFAS, isomer and EOF profiles. This shows that even if point sources were from the same source category, such as AFFF impacted site A and R, contamination is still different from site to site. Site K, which is impacted by industries such as hazardous waste facilities, showed more complex mixtures of PFASs and should be considered as relevant source to the nearby environment. Aquatic invertebrates were impacted by PFASs at all three sites and the BAFs indicate potential transfer of PFASs along the food chain mediated by invertebrates. Branched isomer analysis revealed that individual PFASs would have been underestimated if only quantifying linear isomers. In this study, the concentration of PFOS in R1 would have been 69% lower without the quantification of branched isomers. Homologue profiles and PFHxS-to-PFOS ratios can help to identify type of sources and products used, e.g. PFOS vs. fluorotelomer based AFFFs. Organofluorine mass balance indicated that the majority of EOF were unidentified and unknown organofluorine substances. Furthermore, mass discharge estimations can further assist in understanding the environmental transport and fate of organofluorines and PFASs in receiving waters. Here, we found that EOF discharges were significant higher relative to discharges of Σ25PFASs. Therefore, for in depth understanding and identification of overlooked and novel PFASs as well as other organofluorine substances, additional approaches such as total oxidizable precursor assay, suspect screening and non-target screening analysis could be applied.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the Swedish Research Council Formas (project numbers: 2015-00320 and 2016-01158) and the Knowledge Foundation (KKS) of Sweden for funding the project within the Enforce Research Project (20160019). Thank you Dr Josefin Persson and Dr Petr Kukučka for the helping hands while sampling.References
- 3M, Fluorochemical use, distribution and release overview, report AR226-0550, USA, 1999 Search PubMed.
- F. M. Hekster, R. W. Laane and P. de Voogt, Environmental and toxicity effects of perfluoroalkylated substances, Rev. Environ. Contam. Toxicol., 2003, 179, 99–121 CrossRef CAS.
- L. Ahrens, Polyfluoroalkyl compounds in the aquatic environment: a review of their occurrence and fate, J. Environ. Monit., 2011, 13, 20–31 RSC.
- J. P. Giesy and K. Kannan, Global Distribution of Perfluorooctane Sulfonate in Wildlife, Environ. Sci. Technol., 2001, 35, 1339–1342 CrossRef CAS PubMed.
- K. Prevedouros, I. T. Cousins, R. C. Buck and S. H. Korzeniowski, Sources, fate and transport of perfluorocarboxylates, Environ. Sci. Technol., 2006, 40, 32–44 CrossRef CAS PubMed.
- J. W. Martin, S. A. Mabury, K. R. Solomon and D. D. Muir, Dietary accumulation of perfluorinated acids in juvenile rainbow trout (Oncorhynchus mykiss), Environ. Toxicol. Chem., 2003, 22, 189–195 CrossRef CAS PubMed.
- C. A. Moody and J. A. Field, Perfluorinated surfactants and the environmental implications of their use in fire-fighting foams, Environ. Sci. Technol., 2000, 34, 3864–3870 CrossRef CAS.
- E. Kissa, Fluorinated surfactants and repellents, CRC Press, 2001 Search PubMed.
- C. A. Huset, M. A. Barlaz, D. F. Barofsky and J. A. Field, Quantitative determination of fluorochemicals in municipal landfill leachates, Chemosphere, 2011, 82, 1380–1386 CrossRef CAS PubMed.
- J. Busch, L. Ahrens, R. Sturm and R. Ebinghaus, Polyfluoroalkyl compounds in landfill leachates, Environ. Pollut., 2010, 158, 1467–1471 CrossRef CAS PubMed.
- KemI, Occurrence and use of highly 717 fluorinated substances and alternatives, Swedish Chemicals Agency, 2015 Search PubMed.
- C. A. Moody, J. W. Martin, W. Chi Kwan, D. C. G. Muir and S. A. Mabury, Monitoring Perfluorinated Surfactants in Biota and Surface Water Samples Following an Accidental Release of Fire-Fighting Foam into Etobicoke Creek, Environ. Sci. Technol., 2002, 36, 545–551 CrossRef CAS PubMed.
- A. G. Paul, K. C. Jones and A. J. Sweetman, A First Global Production, Emission, And Environmental Inventory For Perfluorooctane Sulfonate, Environ. Sci. Technol., 2009, 43, 386–392 CrossRef CAS PubMed.
- M. Babut, P. Labadie, C. Simonnet-Laprade, G. Munoz, M.-C. Roger, B. J. D. Ferrari, H. Budzinski and E. Sivade, Per- and poly-fluoroalkyl compounds in freshwater fish from the Rhône River: Influence of fish size, diet, prey contamination and biotransformation, Sci. Total Environ., 2017, 605–606, 38–47 CrossRef CAS PubMed.
- M. Houde, G. Czub, J. M. Small, S. Backus, X. Wang, M. Alaee and D. D. Muir, Fractionation and bioaccumulation of perfluorooctane sulfonate (PFOS) isomers in a Lake Ontario food web, Environ. Sci. Technol., 2008, 42, 9397–9403 CrossRef CAS PubMed.
- A. O. De Silva, P. J. Tseng and S. A. J. E. T. Mabury, Toxicokinetics of perfluorocarboxylate isomers in rainbow trout, Environ. Toxicol. Chem., 2009, 28, 330–337 CrossRef CAS PubMed.
- J. W. Martin, S. A. Mabury, K. R. Solomon and D. C. Muir, Bioconcentration and tissue distribution of perfluorinated acids in rainbow trout (Oncorhynchus mykiss), Environ. Toxicol. Chem., 2003, 22, 196–204 CrossRef CAS PubMed.
- J. P. Benskin, A. O. De Silva, L. J. Martin, G. Arsenault, R. McCrindle, N. Riddell, S. A. Mabury and J. W. Martin, Disposition of perfluorinated acid isomers in sprague-dawley rats; Part 1: Single dose, Environ. Toxicol. Chem., 2009, 28, 542–554 CrossRef CAS PubMed.
- OECD, Towards a new comprehensive global database of per- and polyfluoroalkyl substances (PFAS): Summary report on updating the OECD 2007 list of per- and polyfluoroalkyl substances (PFAS), 2018 Search PubMed.
- N. L. A. Jamari, J. F. Dohmann, A. Raab, E. M. Krupp and J. Feldmann, Novel non-targeted analysis of perfluorinated compounds using fluorine-specific detection regardless of their ionisability (HPLC-ICPMS/MS-ESI-MS), Anal. Chim. Acta, 2019, 1053, 22–31 CrossRef CAS PubMed.
- Y. Miyake, N. Yamashita, P. Rostkowski, M. K. So, S. Taniyasu, P. K. Lam and K. Kannan, Determination of trace levels of total fluorine in water using combustion ion chromatography for fluorine: a mass balance approach to determine individual perfluorinated chemicals in water, J. Chromatogr. A, 2007, 1143, 98–104 CrossRef CAS PubMed.
- E. E. Ritter, M. E. Dickinson, J. P. Harron, D. M. Lunderberg, P. A. DeYoung, A. E. Robel, J. A. Field and G. F. Peaslee, PIGE as a screening tool for Per- and polyfluorinated substances in papers and textiles, Nucl. Instrum. Methods Phys. Res., Sect. B, 2017, 407, 47–54 CrossRef CAS.
- A. K. Tokranov, N. Nishizawa, C. A. Amadei, J. E. Zenobio, H. M. Pickard, J. G. Allen, C. D. Vecitis and E. M. Sunderland, How Do We Measure Poly- and Perfluoroalkyl Substances (PFASs) at the Surface of Consumer Products?, Environ. Sci. Technol. Lett., 2019, 6(1), 38–43 CrossRef CAS.
- A. Koch, R. Aro, T. Wang and L. W. Y. Yeung, Towards a comprehensive analytical workflow for the chemical characterisation of organofluorine in consumer products and environmental samples, TrAC, Trends Anal. Chem., 2019 DOI:10.1016/j.trac.2019.02.024.
- B. Weiner, L. W. Y. Yeung, E. B. Marchington, L. A. D'Agostino and S. A. Mabury, Organic fluorine content in aqueous film forming foams (AFFFs) and biodegradation of the foam component 6:2 fluorotelomermercaptoalkylamido sulfonate (6:2 FTSAS), Environ. Chem., 2013, 10, 486–493 CrossRef CAS.
- L. W. Yeung, Y. Miyake, S. Taniyasu, Y. Wang, H. Yu, M. K. So, G. Jiang, Y. Wu, J. Li, J. P. Giesy, N. Yamashita and P. K. Lam, Perfluorinated compounds and total and extractable organic fluorine in human blood samples from China, Environ. Sci. Technol., 2008, 42, 8140–8145 CrossRef CAS PubMed.
- Nordic Council of Ministers, A. Kärrman, T. Wang, R. Kallenborn, A. M. Langseter, S. M. Grønhovd, E. M. Ræder, J. L. Lyche, L. W. Y. Yeung, F. Chen, U. Eriksson, R. Aro and F. Fredriksson, PFASs in the Nordic environment: Screening of Poly- and Perfluoroalkyl Substances (PFASs) and Extractable Organic Fluorine (EOF) in the Nordic Environment, Nordic Council of Ministers, 2019 Search PubMed.
- L. Ahrens, K. Norstrom, T. Viktor, A. P. Cousins and S. Josefsson, Stockholm Arlanda Airport as a source of per- and polyfluoroalkyl substances to water, sediment and fish, Chemosphere, 2015, 129, 33–38 CrossRef CAS PubMed.
- D. Cederborg, S. Nordquist and S. Keiter, Slutrapport fiskundersökning Söderhavet 2015, report 1346013000, Sweco Environment AB, Karlstad, Sweden, 2016 Search PubMed.
- J. Edvinsson, MSc thesis, Stockholm Universitet, 2015.
- Swedish Armed Forces, Perfluorerade ämnen vid Blekinge flygflottilj, Ronneby kommun. Delrapport 3. Bedömning av spridningsplym, 2016 Search PubMed.
- Swedish Chemicals Agency, Survey of fire-fighting foam, Kemikalieinspektionen, 2015 Search PubMed.
- R. Berglind, J. Hellden, N. Johansson and J. Sjöström, Perfluorerade ämnen i jord, grundvatten och ytvatten – Riskbild och åtgårdsstrategier, Swedish Armed Forces, 2013 Search PubMed.
- Swedish Chemicals Agency, Occurrence and use of highly 717 fluorinated substances and alternatives, Kemikalieinspektionen, 2015 Search PubMed.
- L. T. Brooke, G. T. Ankley, D. J. Call and P. M. Cook, Gut content weight and clearance rate for three species of freshwater invertebrates, Environ. Toxicol. Chem., 1996, 15, 223–228 CrossRef CAS.
- SMHI, Vattenwebb- Hydrologiskt nuläge, http://vattenweb.smhi.se/hydronu/, accessed April, 2019 Search PubMed.
- ISO_25101:2009, Water quality – Determination of perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA) – Method for unfiltered samples using solid phase extraction and liquid chromatography/mass spectrometry, International Organization for Standardization Search PubMed.
- C. R. Powley, S. W. George, T. W. Ryan and R. C. Buck, Matrix Effect-Free Analytical Methods for Determination of Perfluorinated Carboxylic Acids in Environmental Matrixes, Anal. Chem., 2005, 77, 6353–6358 CrossRef CAS PubMed.
- E. Awad, X. Zhang, S. P. Bhavsar, S. Petro, P. W. Crozier, E. J. Reiner, R. Fletcher, S. A. Tittlemier and E. Braekevelt, Long-Term Environmental Fate of Perfluorinated Compounds after Accidental Release at Toronto Airport, Environ. Sci. Technol., 2011, 45, 8081–8089 CrossRef CAS PubMed.
- S. R. de Solla, A. O. De Silva and R. J. Letcher, Highly elevated levels of perfluorooctane sulfonate and other perfluorinated acids found in biota and surface water downstream of an international airport, Hamilton, Ontario, Canada, Environ. Int., 2012, 39, 19–26 CrossRef CAS PubMed.
- A. Kärrman, K. Elgh-Dalgren, C. Lafossas and T. Møskeland, Environmental levels and distribution of structural isomers of perfluoroalkyl acids after aqueous fire-fighting foam (AFFF) contamination, Environ. Chem., 2011, 8, 372 CrossRef.
- C. A. Moody, W. C. Kwan, J. W. Martin, D. C. G. Muir and S. A. Mabury, Determination of Perfluorinated Surfactants in Surface Water Samples by Two Independent Analytical Techniques: Liquid Chromatography/Tandem Mass Spectrometry and 19F NMR, Anal. Chem., 2001, 73, 2200–2206 CrossRef CAS PubMed.
- K. D. Oakes, J. P. Benskin, J. W. Martin, J. S. Ings, J. Y. Heinrichs, D. G. Dixon and M. R. Servos, Biomonitoring of perfluorochemicals and toxicity to the downstream fish community of Etobicoke Creek following deployment of aqueous film-forming foam, Aquat. Toxicol., 2010, 98, 120–129 CrossRef CAS PubMed.
- L. Ahrens, J. Hedlund, W. Dürig, R. Tröger and K. Wiberg, Screening of PFASs in groundwater and surface water, Institutionen för vatten och miljö, Uppsala, Sweden, 2016 Search PubMed.
- J. W. Martin, D. M. Whittle, D. C. G. Muir and S. A. Mabury, Perfluoroalkyl Contaminants in a Food Web from Lake Ontario, Environ. Sci. Technol., 2004, 38, 5379–5385 CrossRef CAS PubMed.
- Environment Canada, Canadian Environmental Protection Act, 1999 (CEPA 1999): Ecological Screening Assessment Report on Perfluorooctane Sulfonate, Its Salts and Its Precursors that Contain the 8F17SO2 or C8F17SO3, or C8F17SO2N Moiety, 2006 Search PubMed.
- M. M. MacDonald, A. L. Warne, N. L. Stock, S. A. Mabury, K. R. Solomon and P. K. Sibley, Toxicity of perfluorooctane sulfonic acid and perfluorooctanoic acid to Chironomus tentans, Environ. Toxicol. Chem., 2004, 23, 2116–2123 CrossRef CAS PubMed.
- T. Jacobson, K. Holmström, G. Yang, A. T. Ford, U. Berger and B. Sundelin, Perfluorooctane sulfonate accumulation and parasite infestation in a field population of the amphipod Monoporeia affinis after microcosm exposure, Aquat. Toxicol., 2010, 98, 99–106 CrossRef CAS PubMed.
- L. Gobelius, J. Hedlund, W. Dürig, R. Tröger, K. Lilja, K. Wiberg and L. Ahrens, Per- and Polyfluoroalkyl Substances in Swedish Groundwater and Surface Water: Implications for Environmental Quality Standards and Drinking Water Guidelines, Environ. Sci. Technol., 2018, 52, 4340–4349 CrossRef CAS PubMed.
- S. Fang, X. Chen, S. Zhao, Y. Zhang, W. Jiang, L. Yang and L. Zhu, Trophic magnification and isomer fractionation of perfluoroalkyl substances in the food web of Taihu Lake, China, Environ. Sci. Technol., 2014, 48, 2173–2182 CrossRef CAS PubMed.
- E. F. Houtz, C. P. Higgins, J. A. Field and D. L. Sedlak, Persistence of Perfluoroalkyl Acid Precursors in AFFF-Impacted Groundwater and Soil, Environ. Sci. Technol., 2013, 47, 8187–8195 CrossRef CAS PubMed.
- R. H. Anderson, G. C. Long, R. C. Porter and J. K. Anderson, Occurrence of select perfluoroalkyl substances at U.S. Air Force aqueous film-forming foam release sites other than fire-training areas: Field-validation of critical fate and transport properties, Chemosphere, 2016, 150, 678–685 CrossRef CAS PubMed.
- J. M. Conder, R. A. Hoke, W. d. Wolf, M. H. Russell and R. C. Buck, Are PFCAs Bioaccumulative? A Critical Review and Comparison with Regulatory Criteria and Persistent Lipophilic Compounds, Environ. Sci. Technol., 2008, 42, 995–1003 CrossRef CAS PubMed.
- K. Kannan, L. Tao, E. Sinclair, S. D. Pastva, D. J. Jude and J. P. Giesy, Perfluorinated Compounds in Aquatic Organisms at Various Trophic Levels in a Great Lakes Food Chain, Arch. Environ. Contam. Toxicol., 2005, 48, 559–566 CrossRef CAS PubMed.
- S. Taniyasu, N. Yamashita, E. Yamazaki, P. Rostkowski, L. W. Y. Yeung, S. K. Kurunthachalam, K. Kannan and B. G. Loganathan, in Water Challenges and Solutions on a Global Scale, ed. S. Ahuja, J. B. DeAndrade, D. D. Dionysiou, K. D. Hristovski and B. G. Loganathan, 2015, vol. 1206, pp. 221–244 Search PubMed.
- A. M. Thayer, Fabulous Fluorine – Having fluorine in life sciences molecules brings desirable benefits, but the trick is getting it in place and making sought-after building blocks, http://pubs.acs.org/email/cen/html/060606071341.html?print%EF%BF%BD%C3%9C, accessed October, 2018 Search PubMed.
- T. Fujiwara and D. O'Hagan, Successful fluorine-containing herbicide agrochemicals, J. Fluorine Chem., 2014, 167, 16–29 CrossRef CAS.
- M. A. Nguyen, K. Norström, K. Wiberg, J. Gustavsson, S. Josefsson and L. Ahrens, unpublished work.
- K. Norström, T. Viktor, A. Palm Cousins and M. Rahmberg, RE-PATH: Risks and Effects of the dispersion of PFAS on Aquatic, Terrestrial and Human populations in the vicinity of International Airports, Report B 2232, IVL Swedish Environmental Research Institute, Stockholm, Sweden, 2015 Search PubMed.
- A. B. Swedavia,Miljörapport 2018 Stockholm Arlanda Airport, 2018 Search PubMed.
- M. S. McLachlan, K. E. Holmström, M. Reth and U. Berger, Riverine discharge of perfluorinated carboxylates from the European continent, Environ. Sci. Technol., 2007, 41, 7260–7265 CrossRef CAS PubMed.
- A. Möller, L. Ahrens, R. Surm, J. Westerveld, F. van der Wielen, R. Ebinghaus and P. de Voogt, Distribution and sources of polyfluoroalkyl substances (PFAS) in the River Rhine watershed, Environ. Pollut., 2010, 158, 3243–3250 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9em00281b |
| This journal is © The Royal Society of Chemistry 2019 |