
Self-assembled Ti3C2 MXene and N-rich porous carbon hybrids as superior anodes for high-performance potassium-ion batteries†
Ruizheng
Zhao
,
Haoxiang
Di
,
Xiaobin
Hui
,
Danyang
Zhao
,
Rutao
Wang
,
Chengxiang
Wang
* and
Longwei
Yin
 *
*
Key Laboratory for Liquid–Solid Structural Evolution and Processing of Materials, Ministry of Education, School of Materials Science and Engineering, Shandong University, Jinan 250061, P. R. China. E-mail: wcxmat@sdu.edu.cn; yinlw@sdu.edu.cn; Fax: +86 531 88396970; Tel: + 86 531 88396970
First published on 20th November 2019
Abstract
Potassium-ion batteries (PIBs) are attracting increased attention because of their low cost and similar energy storage mechanism to lithium-ion batteries. Considering the low structural stability and poor electrochemical redox reaction kinetics resulting from the large size of K+ (1.38 Å), we elaborately designed novel PDDA-NPCN/Ti3C2 hybrids as PIBs anodes via an electrostatic attraction self-assembly approach, while N-rich porous carbon nanosheets (NPCNs) are derived from metal–hexamine frameworks. The coupled PDDA-NPCN/Ti3C2 hybrids with stacked structure and large specific surface area could ensure intimate contact between Ti3C2 and the NPCNs to efficiently take advantage of both components and more accessible active sites. The hybrids afford enlarged interlayer spacing and unique 3D interconnected conductive networks to accelerate the ionic/electronic transport rates. Meanwhile, the robust hybrids contribute high chemical stabilities due to favorable tolerance to volume change caused by phase transformations during the fast charge/discharge process. DFT calculations further indicate that the PDDA-NPCN/Ti3C2 hybrids efficiently reduce the adsorption energy of K+ and accelerate the reaction kinetics. The hybrids possess a remarkable synergetic effect, leading to a high reversible capacity of 358.4 mA h g−1 after 300 cycles at 0.1 A g−1 and long cycling stability of 252.2 mA h g−1 with only 0.03% degradation per cycle within 2000 cycles at 1.0 A g−1. This work paves the way for further self-assembled coupled hybrids in energy storage devices.
Broader contextNowadays, the ever-increasing energy consumption and rapid depletion of fossil fuels have driven extensive research activities on the development of green and cost-effective electrochemical energy storage and conversion technologies. Owing to lithium's rarity, uneven distribution in the Earth's crust and high cost, potassium-ion batteries (PIBs) have attracted tremendous attention recently owing to their low cost, fast ionic conductivity in electrolyte, high operating voltage and similar energy storage mechanism to lithium/sodium-ion batteries (LIBs/SIBs). However, the commercialization of PIBs anodes is greatly hindered by the low structural stability, poor electrochemical redox reaction kinetics and fast capacity fading caused by the large size of K+ (1.38 Å) as compared to 0.76 Å/1.02 Å for Li+/Na+. Herein, we develop an elaborate strategy to synthesize PDDA-NPCN/Ti3C2 hybrids as anodes for high-performance PIBs, which exhibit excellent electrochemical performance, showing long cycling stability with only 0.03% degradation per cycle within 2000 cycles at 1.0 A g−1. It is a novel structure for PIBs application, and it seems that these questions for PIBs are well resolved in this work. This ingenious strategy can effectively accelerate the ionic/electronic transport kinetics, paving the way for self-assembled hybrids in energy storage devices. |
Introduction
Potassium-ion batteries (PIBs), which possess a similar “rocking chair” mechanism to lithium/sodium-ion batteries (LIBs/SIBs), have attracted tremendous attention in large-scale energy storage applications owing to their low cost, fast ionic conductivity in electrolyte and high operating voltage.1–5 The redox standard hydrogen potential of K/K+ (−2.92 V vs. standard hydrogen electrode (SHE)) is lower than that of Na/Na+ (−2.71 V vs. SHE) and close to that of Li/Li+ (−3.04 V vs. SHE), which enables PIBs to be operated in a higher potential window and thereby provides a higher energy density. Smaller solvated K+ caused by the much weaker Lewis acidity of K+ leads to a lower desolvation energy and smaller Stokes’ radius (3.6 Å) compared with Li+ (4.8 Å) and Na+ (4.6 Å), indicating that it has a better ion mobility and ion conductivity in electrolyte.6,7 However, the poor ion diffusivity in solid graphitic carbon with confined interspace electrodes leads to sluggish reaction kinetics and large volume variations during cycling due to the larger diameter of K+ (1.38 Å vs. 0.76 Å/1.02 Å for Li+/Na+), which induces structural instability and fast capacity fading.8–10 Therefore, it is still a great challenge to develop appropriate electrode materials to accommodate huge volume changes and improve the K+ diffusivity/reaction kinetics, thus enhancing the rate capability and cycling performance.MXenes, as an emerging potential 2D material, have gained widespread interest due to their high metallic conductivity, abundant surface functional groups and controllable flexible interlayer spacing, which is beneficial for fast ion diffusion and offering more ion intercalation channels over the whole exposed surface.11–21 It is revealed that few-layer Ti3C2 nanosheets display a high conductivity of 6.76 × 105 S m−1 and a low diffusion barrier of K+ of about 0.103 eV, which indicates excellent electron transport and superior K+ diffusion kinetics.11,12 More recently, theoretical simulations and experimental measurements reveal that Ti3C2 is electrochemically redoxable, which can act as active sites for efficient potassium storage, delivering reversible capacities of 42–136 mA h g−1, and excellent rate capability.12,14,19,20 The electrochemical performance of multilayer MXene nanosheets can be further improved by exfoliating them into few-layer nanosheets. Nevertheless, the aggregation and self-restacking of few-layer MXene nanosheets are usually inevitable during the drying and electrode fabrication processes due to the strong van der Waals interactions and hydrogen bonds between adjacent nanosheets, which usually limits the accessibility to electrolyte ions and hinders the full utilization of their functional surfaces.21,22
In order to overcome this shortcoming, recently, layer-by-layer structures could offer a stable scaffold for volume expansion and a stable SEI layer for side reactions, which have been proved definitely to make full use of both characteristics of the components for superior electrochemical performance compared with other methods.23–25 Among the various 2D materials,15,26–31 metal–hexamine framework (MHF) derived N-rich porous carbon nanosheets (NPCNs) with a large amount of open pores will benefit both electron and ion transfer during cycling, as well as fully contact and store sufficient electrolyte for rechargeable batteries. Specifically, N-doping could introduce defects and more electronegative sites in carbon, which further enhances the electronic conductivity of carbon and generates more active sites with stronger attraction for ion storage in carbon.27,32,33 On the other hand, NPCNs would provide an ideal “buffer” or interlayer spacer for coupling with MXene nanosheets to effectively prevent self-restacking between the MXene adjacent nanosheets, which is desirable to ensure structural stability through accelerating the ionic/electronic transport and overcome the serious volume changes during the fast charge/discharge process. However, it is a great challenge and important to rationally design unique NPCN/Ti3C2 hybrids for high-performance PIBs. These robust hybrids are expected to make full utilization of every nanosheet, and enhance interfacial charge separation and transfer between adjacent layers, which is important to further achieve high electrochemical performance.34,35
In this paper, we elaborately design novel PDDA-NPCN/Ti3C2 hybrids via an electrostatic attraction self-assembly approach. The synergistic effect between Ti3C2 and NPCNs affords enlarged interlayer spacing and a unique 3D porous interconnected conductive network architecture, which exhibits more accessibility of the electrolyte, and shortens the ion transmission paths to facilitate rapid ionic/electronic transport, thereby contributing to enhanced performance. The robust hybrids may alleviate the serious volume changes caused by the phase transformations during the fast charge/discharge process and assist durable rate capability and cycling stability. DFT calculations further show that the PDDA-NPCN/Ti3C2 hybrids improve the interfacial K+ adsorption ability and promote the potassiation process. Benefiting from those advantages, the PDDA-NPCN/Ti3C2 anode exhibits a high reversible capacity of 358.4 mA h g−1 after 300 cycles at 0.1 A g−1 and long cycling stability of 252.2 mA h g−1 with only 0.03% degradation per cycle within 2000 cycles at 1.0 A g−1. The related mechanisms of NPCNs and Ti3C2 in the potassiation/depotassiation process are deeply investigated.
Results and discussion
Microstructure characterization
Fig. 1 schematically illustrates the overall fabrication process of the coupled PDDA-NPCN/Ti3C2 hybrids. It follows a static electronic self-assembly process between negatively charged exfoliated Ti3C2 nanosheets (ex-Ti3C2) and N-rich porous carbon nanosheets modified by positively charged polymer poly(diallyldimethylammonium chloride) (PDDA-NPCNs). The ex-Ti3C2 nanosheets would be covered by negative charge groups, such as –OH, –F or –O groups, showing a zeta potential of −33.2 mV.36 The scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of the ex-Ti3C2 nanosheets (Fig. 2a and b) clearly present a crinkled and self-restacked sheet-like morphology with an ultrathin thickness. The surface functional groups endow them with a hydrophilic nature and uniform dispersion in water, showing a clear Tyndall effect (Fig. 2a, inset).15,37 The atomic force microscopy (AFM) image of the ex-Ti3C2 nanosheets presents a uniform and flat surface with a few-layer thickness, e.g., ∼3.3 nm (Fig. S1, ESI†) in a bilayer structure.38 The as-prepared NPCNs are modified by PDDA and became positively charged with a zeta potential of +35.3 mV (Fig. S2a–g, ESI†). The TEM image (Fig. 2c) reveals that the PDDA-NPCNs have ample pores that uniformly distribute over the body, and its colloidal suspension (Fig. 2c, inset) is stable and exhibits a typical Tyndall effect.39 The PDDA-NPCNs (Fig. S2h and i, ESI†) show a uniform thickness of ∼3.7 nm after being modified with PDDA, which is in agreement with previous reports.40 The PDDA-NPCN/Ti3C2 hybrids (Fig. 2d, inset) with a zeta potential of +2.77 mV are obtained through flocculation based on a mass ratio of 2.0.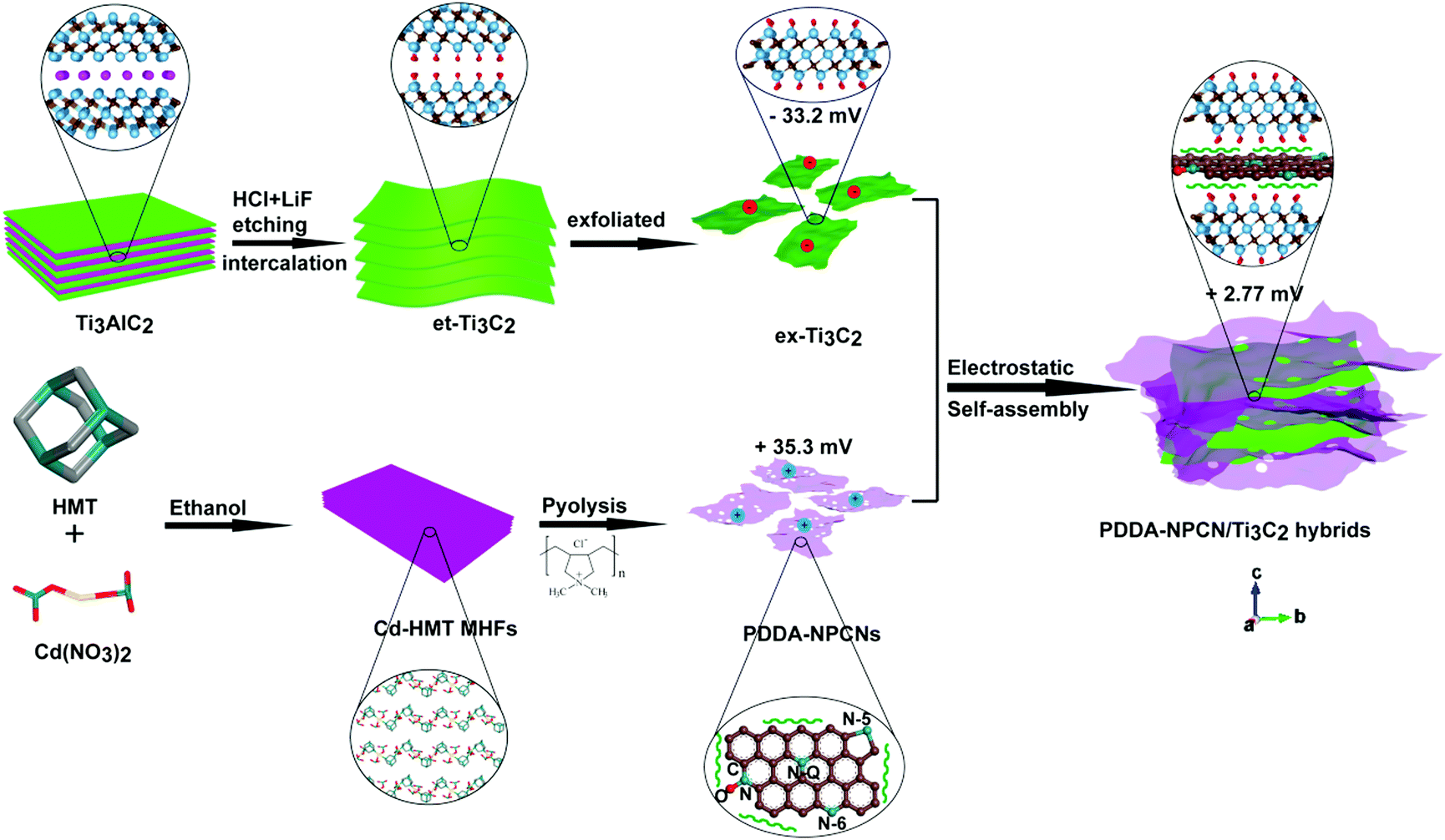 | ||
| Fig. 1 Schematic illustration of the fabrication of PDDA-NPCN/Ti3C2 hybrids. The water molecules between the layers are not shown here for clarity. | ||
 | ||
| Fig. 2 Mophology and chemical composition characterization. (a and b) SEM and TEM images of ex-Ti3C2 nanosheets. (c) TEM image of PDDA-NPCNs. The inset shows the Tyndall effect. (d) SEM, (e) TEM and (f) ED pattern of PDDA-NPCN/Ti3C2 flocculation. The inset shows flocculation. T stands for ex-Ti3C2 and C is PDDA-NPCNs. (g) EDS spectrum. (h) HAADF-STEM image of PDDA-NPCN/Ti3C2 and the corresponding elemental mapping images of Ti, C and N elements, respectively. (i) AFM image and thickness profile of PDDA-NPCN/Ti3C2. | ||
The SEM and TEM images (Fig. 2d and e) present the stacking of PDDA-NPCNs and ex-Ti3C2 nanosheets, which shows a porous interconnected conductive network structure, tremendously shortening the ion transport pathways and facilitating fast ionic/electronic transport. The clear lattice fringes of 0.34 nm and 0.15 nm under high-resolution transmission electron microscopy (HRTEM) (Fig. S3, ESI†) roughly correspond to the (002) plane of the PDDA-NPCNs and the (110) plane of the ex-Ti3C2 nanosheets. The electron diffraction (ED) patterns (Fig. 2f and Fig. S3, inset, ESI†) reveal the in-plane diffraction of the (100) and (110) planes of ex-Ti3C2 (T100, T110) and the (100) and (110) planes of the PDDA-NPCNs (C100, C110), again proving the existence of both nanosheets. More importantly, a typical energy dispersive X-ray spectroscopy (EDS) spectrum (Fig. 2g) shows that the real mass ratio of ex-Ti3C2 to PDDA-NPCNs is estimated to be ∼2.1, which is close to the calculated value. And a typical high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image and the corresponding elemental mapping images (Fig. 2h) indicate that the Ti, C and N elements homogeneously distribute over the PDDA-NPCN/Ti3C2 hybrids, revealing the stacking of ex-Ti3C2 and the PDDA-NPCNs in the lateral dimension. Typical stacking of both nanosheets is observed in the AFM observation (Fig. 2i), in which the PDDA-NPCNs (∼3.8 nm in thickness) are superimposed on the ex-Ti3C2 nanosheets (∼3.3 nm in thickness), in accordance with the TEM results.
The X-ray diffraction (XRD) pattern (Fig. 3a) of ex-Ti3C2 shows that the (001), (002) and (003) peaks are initially at 6.8°, 13.6° and 20.5° after the etching, intercalation and exfoliation process, corresponding to an interlayer spacing value of 13.0 Å. The PDDA-NPCNs only exhibit a broad peak at 21.7°, which is indexed to the (002) plane of carbon materials with a low degree of graphitization, and the interlayer spacing could be calculated to be 4.0 Å. The flocculated PDDA-NPCN/Ti3C2 material shows coexisting crystallographic phases of ex-Ti3C2 with a predominant (001) peak and well maintained PDDA-NPCNs with a weak (002) peak. In particular, the (001) plane downshifts to 4.6°, revealing an interlayer spacing of 19.2 Å. The increased spacing (6.2 Å) might be caused by the intercalation of PDDA-NPCNs into the interlayer of ex-Ti3C2 through the strong electrostatic interactions between them as well as the water content in the gallery of the flocculated products, which is in agreement with previous reports.23,24,37
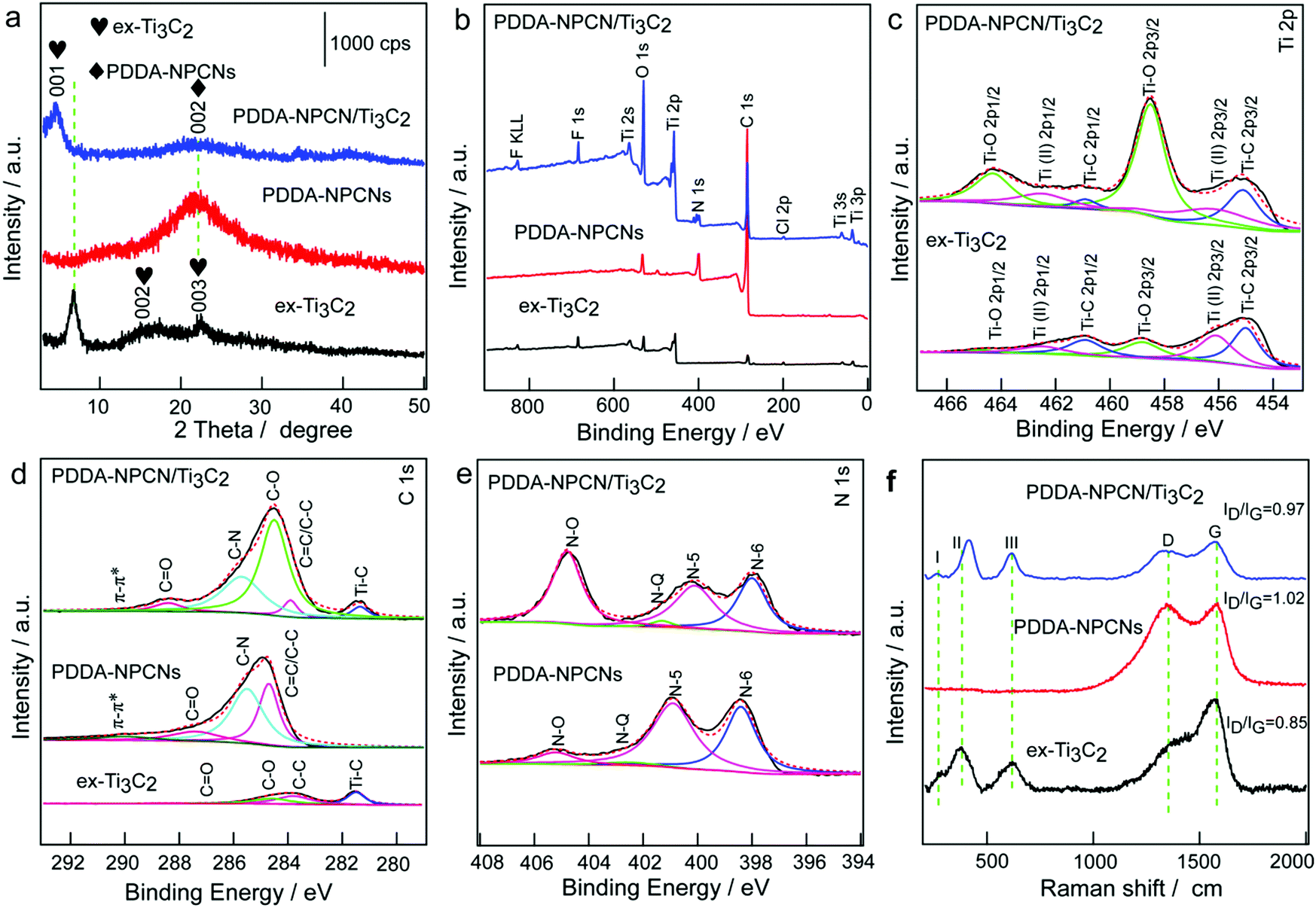 | ||
| Fig. 3 Structure and chemical composition characterization. (a) XRD patterns of ex-Ti3C2, PDDA-NPCNs and PDDA-NPCN/Ti3C2 hybrids, respectively. XPS spectra of ex-Ti3C2, PDDA-NPCNs and PDDA-NPCN/Ti3C2 hybrids, respectively. (b) Full spectra. (c–e) High-resolution spectra of Ti 2p, C 1s and N 1s. (f) Raman spectra of ex-Ti3C2, PDDA-NPCNs and PDDA-NPCN/Ti3C2 hybrids. | ||
To profoundly understand the elemental compositions and corresponding chemical bonding states of Ti, C, O, F and N species in the PDDA-NPCN/Ti3C2 hybrids, X-ray photoelectron spectroscopy (XPS) (Fig. 3b–e and Fig. S4, ESI†) is performed. The XPS spectra as shown in Fig. 3b indicate that the ex-Ti3C2 nanosheets are mainly composed of Ti, C, O and F. After the PDDA-NPCNs are introduced into the ex-Ti3C2 nanosheets, a new peak at a binding energy of 399.9 eV in the spectra for the PDDA-NPCN/Ti3C2 hybrids is assigned to N 1s. The high-resolution Ti 2p spectra of ex-Ti3C2 and the PDDA-NPCN/Ti3C2 hybrids (Fig. 3c) could be fitted with three doublets (Ti 2p3/2–Ti 2p1/2).17 The Ti 2p3/2 components of ex-Ti3C2 located at 455.0, 456.1 and 458.8 eV correspond to Ti–C, Ti(II) and Ti–O bonds, respectively. The C 1s core level of the ex-Ti3C2 nanosheets (Fig. 3d) could be fitted with four components centered at 281.5, 283.8, 284.6, and 288.5 eV, which could be assigned to Ti–C, C–C, C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, respectively.17 The C 1s spectrum of the PDDA-NPCNs displays four peaks at 289.9, 287.4, 285.5, and 284.7 eV, which could be attributed to π–π*, CO, C–N, and sp2 carbon (CC/C–C), respectively.40 The strongest peak at 284.7 eV indicates that graphitic carbon is the major species for the layered bulk MHF derived porous carbon. Moreover, deconvolution of the C 1s region for the PDDA-NPCN/Ti3C2 hybrids shows that the characteristic peaks of Ti–C, CC/C–C, C–O, C–N, CO and π–π* are still present. Correspondingly, the N 1s spectrum of the PDDA-NPCNs in Fig. 3e could be fitted with four components centered at 398.4, 400.9, 402.5, and 405.2 eV, which could be assigned to pyridinic N (N-6), pyrrolic N (N-5), graphitic N (N-Q) and N–O, indicating that N atoms are indeed inserted into the carbon lattice of the PDDA-NPCNs.41 Benefiting from the intrinsic high N content of the HMT ligand (N/C = 66.7%), the PDDA-NPCNs also possess a high N-doping level of 10.35 at% in addition to the novel structure, which can greatly improve the wettability and conductivity of the electrode and increase the active sites for potassium storage. According to previous reports, among the various types of N, pyridinic-N is more active compared with the other types of N because it can efficiently induce sufficient defects and more active sites, and is more energetically favorable towards adsorbing alkali metal ions.42,43
O bonds, respectively.17 The C 1s spectrum of the PDDA-NPCNs displays four peaks at 289.9, 287.4, 285.5, and 284.7 eV, which could be attributed to π–π*, CO, C–N, and sp2 carbon (CC/C–C), respectively.40 The strongest peak at 284.7 eV indicates that graphitic carbon is the major species for the layered bulk MHF derived porous carbon. Moreover, deconvolution of the C 1s region for the PDDA-NPCN/Ti3C2 hybrids shows that the characteristic peaks of Ti–C, CC/C–C, C–O, C–N, CO and π–π* are still present. Correspondingly, the N 1s spectrum of the PDDA-NPCNs in Fig. 3e could be fitted with four components centered at 398.4, 400.9, 402.5, and 405.2 eV, which could be assigned to pyridinic N (N-6), pyrrolic N (N-5), graphitic N (N-Q) and N–O, indicating that N atoms are indeed inserted into the carbon lattice of the PDDA-NPCNs.41 Benefiting from the intrinsic high N content of the HMT ligand (N/C = 66.7%), the PDDA-NPCNs also possess a high N-doping level of 10.35 at% in addition to the novel structure, which can greatly improve the wettability and conductivity of the electrode and increase the active sites for potassium storage. According to previous reports, among the various types of N, pyridinic-N is more active compared with the other types of N because it can efficiently induce sufficient defects and more active sites, and is more energetically favorable towards adsorbing alkali metal ions.42,43
As shown in the Raman spectra (Fig. 3f), the three I, II and III peaks around 258, 410 and 605 cm−1 correspond to the vibrations from Ti–C bonds for ex-Ti3C2, respectively.36 Besides, the two new obvious peaks at 1383 and 1570 cm−1 could be attributed to the D band (disordered/defect-induced) and G band (graphite), respectively, and the ratio of intensity between ID and IG of the ex-Ti3C2 nanosheets is 0.85, suggesting a high graphitization degree. The PDDA-NPCNs show two typical characteristic bands of the D-band (1346 cm−1) and G-band (1578 cm−1), and the ratio of intensity between ID and IG is 1.02, which could be ascribed to the higher defect concentration induced by nitrogen doping. After flocculation with PDDA-NPCNs, the I and III peaks of the PDDA-NPCN/Ti3C2 hybrids broaden and downshift. Notably, some shift for the II peak of the PDDA-NPCN/Ti3C2 hybrids is possibly due to the slightly-oxidized surface in sonication during the fabrication of the PDDA-NPCN/Ti3C2 hybrid process, in accordance with XPS results, which is consistent with a previous report.44 And the ratio of 0.97 for the intensity between ID and IG of the PDDA-NPCN/Ti3C2 hybrids could be ascribed to the combined effects of the higher defect concentration induced by the PDDA-NPCNs and the higher graphitization degree induced by ex-Ti3C2. Furthermore, there are no redox reactions occurring between ex-Ti3C2 and the PDDA-NPCNs, and the presence of surface groups in the flocculation process is further revealed by FTIR (Fig. S5, ESI†).15,36,40,45,46 The designed PDDA-NPCN/Ti3C2 hybrids (Fig. S6 and Table S1, ESI†) show a higher specific surface area of 147.87 m2 g−1 with a pore volume of 0.809 cm3 g−1 compared with the ex-Ti3C2 material, which could provide enough transport pathways for K+ insertion and expose more accessible active sites for enhanced performance.
Potassium ion storage performance
The electrochemical behaviors as PIBs anodes are shown in Fig. 4. The galvanostatic charge/discharge profiles of the PDDA-NPCN/Ti3C2 anode (Fig. 4a) are measured in the first five cycles at 0.1 A g−1. It delivers initial discharge and charge capacities of 797.3 and 583.6 mA h g−1, respectively (all the capacities are calculated based on the mass of the total electrode), corresponding to a CE of 73.2%. Such a low CE in the first cycle can be attributed to the formation of the solid electrolyte interface (SEI) layer on a large surface area, which leads to large irreversible consumption and decomposition of the electrolyte and electrode material. As the cycle number increases, the capacity gradually becomes stable, with CE up to 95.9% at the fifth cycle, corresponding to high cycling stability of the electrode. The prolonged sloping region and an indistinct plateau below 1.5 V of potassiation for the first cycle could be ascribed to different reactions, including the reactions between K+ and surface functional groups, the decomposition of the electrolyte and formation of the SEI layer, and the stepwise insertion of K+ into the Ti3C2 and NPCNs layers to form a K-intercalated compound, which agrees with the weak peaks observed between 0 and 1.5 V in the first cathodic process (Fig. S7, ESI†).12,19,29,32,47 The voltage profile of depotassiation in the first cycle monotonically increasing from 0.01 V to 3 V is ascribed to the combination of multiple charge storage mechanisms with the stepwise extraction of K+ from the K-intercalated compound and Ti3C2 layers, and the reversible reactions between K+ and surface functional groups, which is also revealed by the broad peak between 0.2 and 1.0 V in the CV profiles.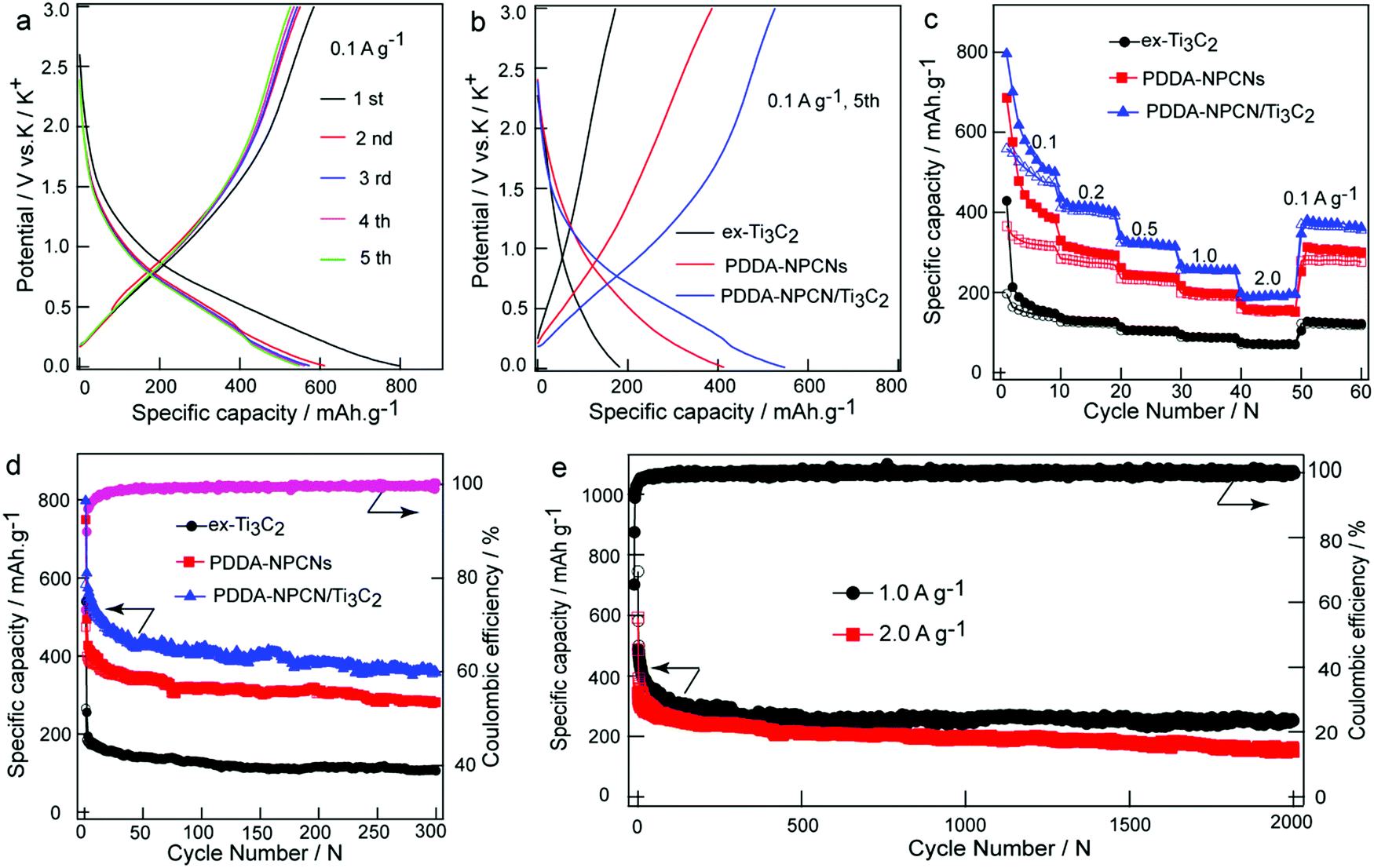 | ||
| Fig. 4 Potassium ion storage performance. (a) Initial five galvanostatic charge/discharge profiles at 0.1 A g−1 for the PDDA-NPCN/Ti3C2 anode. (b) The 5th charge/discharge profiles at 0.1 A g−1, (c) rate performance at different current densities and (d) cycling stability at 0.1 A g−1 for different anodes, respectively. (e) Long cycling performance of the PDDA-NPCN/Ti3C2 anode at 1.0 and 2.0 A g−1 for 2000 cycles. | ||
To further reveal the performance of different materials, the galvanostatic charge/discharge profiles of the fifth cycle at 0.1 A g−1 are extracted for comparison as shown in Fig. 4b. The ex-Ti3C2, PDDA-NPCNs and PDDA-NPCN/Ti3C2 anodes exhibit reversible capacities of 172.3, 387.3 and 526.6 mA h g−1, respectively, which suggests that PDDA-NPCN/Ti3C2 is more conducive to making full use of the active materials for efficient energy storage. A high-rate capability and long-term cycle performance are also important for practical applications. As shown in Fig. 4c, among the three anodes, the PDDA-NPCN/Ti3C2 anode exhibits excellent rate performance, especially under high current densities. Specifically, the PDDA-NPCN/Ti3C2 anode delivers reversible capacities of 499.8, 402.2, 319.4, 254.8 and 191.2 mA h g−1 at 0.1, 0.2, 0.5, 1.0 and 2.0 A g−1, respectively. Noticeably, it could quickly recover a capacity of 363.8 mA h g−1 when the current density is back to 0.1 A g−1. In contrast, both ex-Ti3C2 and the PDDA-NPCNs are not able to be comparable with PDDA-NPCN/Ti3C2 in capacity and rate performance. For example, they only have reversible capacities of 68.9 (ex-Ti3C2) and 152.0 mA h g−1 (PDDA-NPCNs) at 2.0 A g−1 after 50 cycles, obviously worse than that of the PDDA-NPCN/Ti3C2 anode. This is because the PDDA-NPCN/Ti3C2 anode shows a unique structure with enlarged interlayer spacing and high conductivity, which is favorable for rapid electron transfer and abundant electrolyte infiltration, thereby contributing to enhanced performance compared to the other anodes.
In addition, in order to further investigate the influence of the NPCNs content on the electrochemical performance, PDDA-NPCN/Ti3C2 hybrids with three different mass ratios of Ti3C2/NPCN are prepared and measured under identical test conditions. As shown in Fig. S8 (ESI†), the PDDA-NPCN/Ti3C2-2 anode (a mass ratio of 2.0 for Ti3C2/NPCN) shows much better electrochemical performance than the PDDA-NPCN/Ti3C2-1 and PDDA-NPCN/Ti3C2-3 anodes. The enhanced electrochemical performance of the PDDA-NPCN/Ti3C2-2 anode could be attributed to the full utilization of the high theoretical capacity of the NPCNs and the high conductivity of Ti3C2, because there is no extra self-stacking of the NPCNs and Ti3C2 to reduce self-utilization. Specifically, the initial discharge capacity increases with the NPCNs content, while the initial coulombic efficiency decreases contrarily with the NPCNs content, which is possibly because the NPCNs with a well-developed pore structure and high specific surface area consume a large amount of electrolyte for SEI layer formation. For the PDDA-NPCN/Ti3C2-1 anode with higher NPCNs content, although it shows a higher initial discharge capacity, it displays poorer rate performance than the PDDA-NPCN/Ti3C2-2 anode. For the PDDA-NPCN/Ti3C2-3 anode with a relatively lower content of NPCNs, it shows better rate performance, although it shows lower reversible capacity than the PDDA-NPCN/Ti3C2-2 and PDDA-NPCN/Ti3C2-1 anodes.
The cycling stabilities of the different anodes are further examined at 0.1 A g−1 as depicted in Fig. 4d. The PDDA-NPCN/Ti3C2 anode also exhibits the highest reversible capacity and excellent cycling performance. An excellent reversible capacity of 358.4 mA h g−1 still remains after 300 cycles, which corresponds to 61.4% of the initial reversible capacity. Except for the first few cycles, a stable CE of 99.0% is obtained in the following cycles, revealing excellent cycling stability, which is comparable or superior to those of other previously reported Ti3C2-based and carbonaceous anodes (Table S2, ESI†).12,19–22,27,29,32,48–56 The ex-Ti3C2 and PDDA-NPCNs anodes also display stable reversible capacities of 106.2 and 280.9 mA h g−1, respectively. It should be noticed that the capacity of PDDA-NPCN/Ti3C2 is not the combination of Ti3C2 and the NPCNs, but much larger than both of them. This means there is a synergetic effect between Ti3C2 and NPCNs within such hybrids, which is further verified by DFT calculations as follows. In addition, high-rate and long-term cycling performance (Fig. 4e) is also measured on the PDDA-NPCN/Ti3C2 anode. Impressively, it retains reversible capacities of 252.2 mA h g−1 with a capacity decay as low as 0.03% per cycle at 1.0 A g−1 and 151.2 mA h g−1 at 2.0 A g−1 after 2000 cycles, which is comparable or much superior to those of other previously reported anode materials (Table S3, ESI†).19,21,22,27,49–58 It evidently reveals that the PDDA-NPCN/Ti3C2 hybrid anode intrinsically has a better ability to accommodate and survive large stresses and strains caused by K+ insertion/extraction during the potassiation/depotassiation process. It could also provide multiple accessible active sites, further accelerate electron/ion transport and facilitate access of electrolyte ions to the electrode, thus offering high reversible capacity and cycling stability.
Electrochemical kinetic behaviors
Electrochemical impedance spectroscopy (EIS) is further performed to explain the electrochemical performance. As can be seen in Fig. 5a, the Nyquist plots mainly include a single depressed semicircle at the high-medium frequency region associated with the charge transfer resistance (Rct) and an inclined line in the low-frequency region related to the K+ diffusion ability defined as the Warburg impedance (W0). Specifically, Rct of the PDDA-NPCN/Ti3C2 anode is 769.5 Ω, which is obviously lower than that of ex-Ti3C2 (1257.3 Ω) and the PDDA-NPCNs (1050.1 Ω), indicating that this PDDA-NPCN/Ti3C2 anode can remarkably accelerate interfacial charge separation and transfer between adjacent layers during repeated K+ insertion/extraction processes. The K+ diffusion coefficient in the electrode could be obtained by the plots in the Warburg region based on the following equation:41| |Z| = Rct + Re + σω−1/2 | (1) |
| Dk = R2T2/(2A2n4F4Ck2σ2) | (2) |
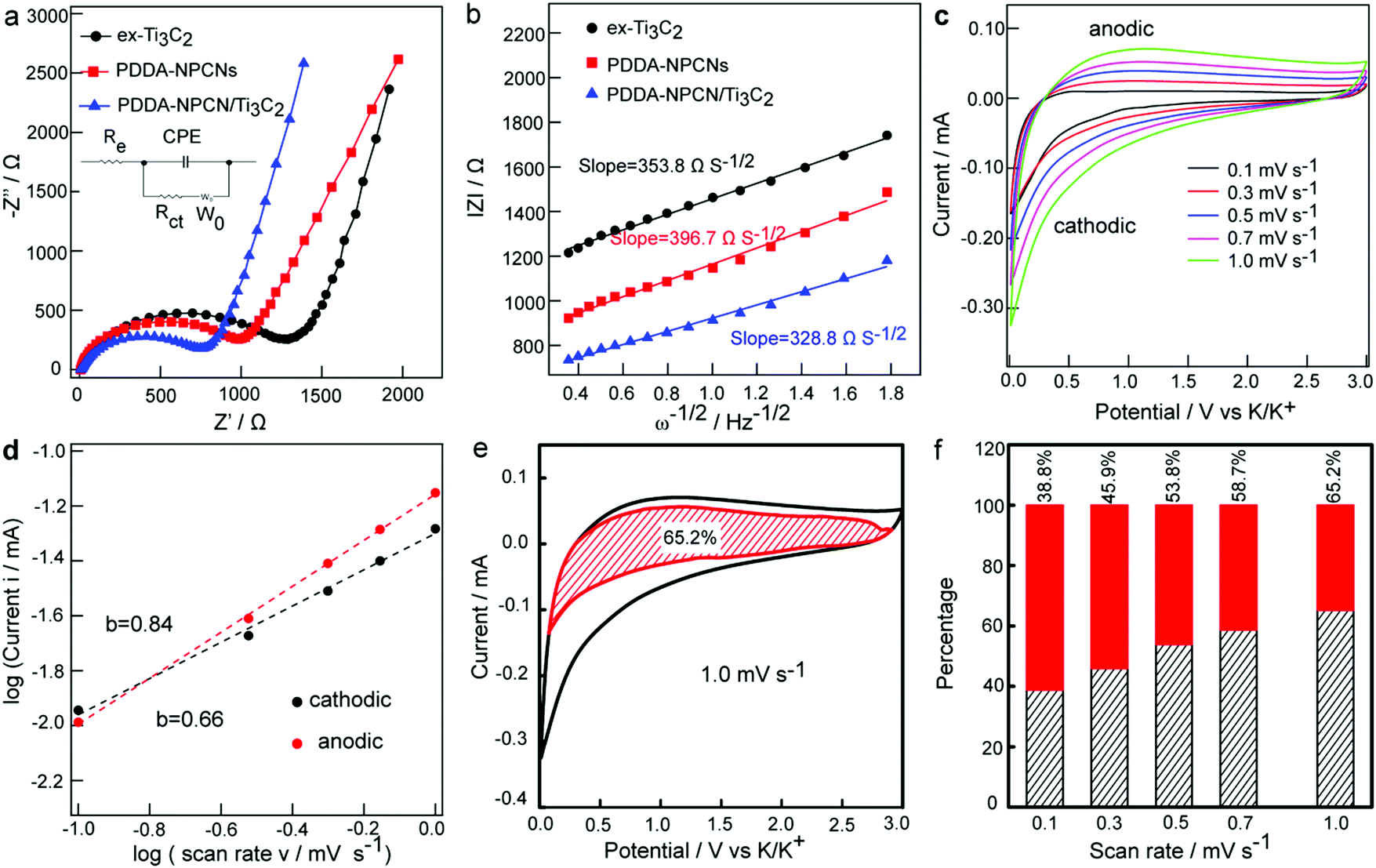 | ||
| Fig. 5 Electrochemical kinetic behaviors. (a) Nyquist plots. The inset exhibits the equivalent circuit. (b) Extracted |Z| vs. ω1/2 plot in the Warburg region for different anodes, respectively. (c) CV curves at various scan rates and (d) relationship between log(i) vs. log(ν) for the PDDA-NPCN/Ti3C2 anode. (e) Capacitive-controlled and diffusion-controlled contributions at 1.0 mV s−1 for the PDDA-NPCN/Ti3C2 anode. (f) Normalized contribution ratio of capacitive-controlled capacities for the PDDA-NPCN/Ti3C2 anode at different scan rates. | ||
To better understand the high-rate capability of the PDDA-NPCN/Ti3C2 anode, CV curves (Fig. 5c) are obtained at various scan rates from 0.1 to 1.0 mV s−1 to investigate the K+ storage kinetic behaviors. With an increasing scan rate, the measured peak current (i) at a certain potential is not proportional to the square root of the scan rate (ν), indicating that the oxidation and reduction processes are not simple ion diffusion-controlled processes.17,32 Here, the relationship between i and ν can be described by the following equations:32,47
| i = aνb, (0.5 ≤ b ≤ 1) | (3) |
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) i = blogν + loga i = blogν + loga | (4) |
i vs. logν profile. At the oxidation and reduction peak potential of 1.2 V, the b-values are 0.84 and 0.66, respectively, revealing that capacitive behaviors cannot be ignored in the PDDA-NPCN/Ti3C2 anode. The same kinetics analysis is performed on the ex-Ti3C2 (Fig. S9, ESI†) and PDDA-NPCNs anodes (Fig. S10, ESI†) as well, which demonstrates that the PDDA-NPCN/Ti3C2 and PDDA-NPCNs anodes show superior K+ diffusion kinetics and better rate capability than the ex-Ti3C2 anode. That's the reason why the PDDA-NPCN/Ti3C2 anode shows a much larger capacity than the ex-Ti3C2 anode at the same current density. In fact, the current response can be further expressed as:32,47| i(ν) = k1ν + k2ν1/2 | (5) |
| i(ν)/ν1/2 = k1ν1/2 + k2 | (6) |
Electrochemical reaction mechanisms
To better understand the superior cycling and rate performance, the voltage profile and the corresponding ex situ XRD patterns of the PDDA-NPCN/Ti3C2 anode in the initial charge/discharge cycle are investigated the structure evolution of the anode shown in Fig. 6a and b. As shown in Fig. 6b, the anode shows a clear XRD pattern, corresponding to that of the diffraction features of the hybrids. Upon potassiation, the peak of the NPCNs gradually decreases and shifts towards a lower angle, which indicates that the interlayer distance of the NPCNs increases due to the K+ insertion. Notably, the diffraction peaks of the NPCNs do not vanish until the potential reaches 0.30 V, where new peaks at 22.0 and 29.4° appear, which are attributed to KC36 (Fig. 6c).48,65 Upon further potassiation, KC36 further transforms to KC24 (Fig. 6d), corresponding to the new peaks at 20.2 and 30.6°. In the full potassiation state, new characteristic peaks at 16.2 and 33.5° are observed, corresponding to the formation of phase-pure KC8 (Fig. 6e).7 The interlayer distance of the fresh NPCNs is 4.08 Å and it increases to 5.37 Å after K+ insertion, for which the corresponding volume expansion of the NPCNs is estimated to be 31.6%. The ultimate potassiation product of KC8 leads to a plateau well above the plating potential of potassium metal, which may relieve the danger of dendrite formation. Meanwhile, the (001) peak of Ti3C2 shifts to a lower angle, suggesting a significant expansion of the interlayer distance from 19.2 Å to 24.6 Å, which corresponds to K+ insertion into the PDDA-NPCN/Ti3C2 anode. During the first depotassiation process (Fig. 6b), the XRD peaks are widened, which suggests that the extraction of K+ causes certain damage to the hybrids.48 According to the ex situ XRD analysis above, the reactions during the potassiation/depotassiation process could be denoted as the following equations:| 8C + xK+ + xe− ↔ KxC8 | (7) |
| Ti3C2Tx + yK+ + ye− ↔ KyTi3C2Tx | (8) |
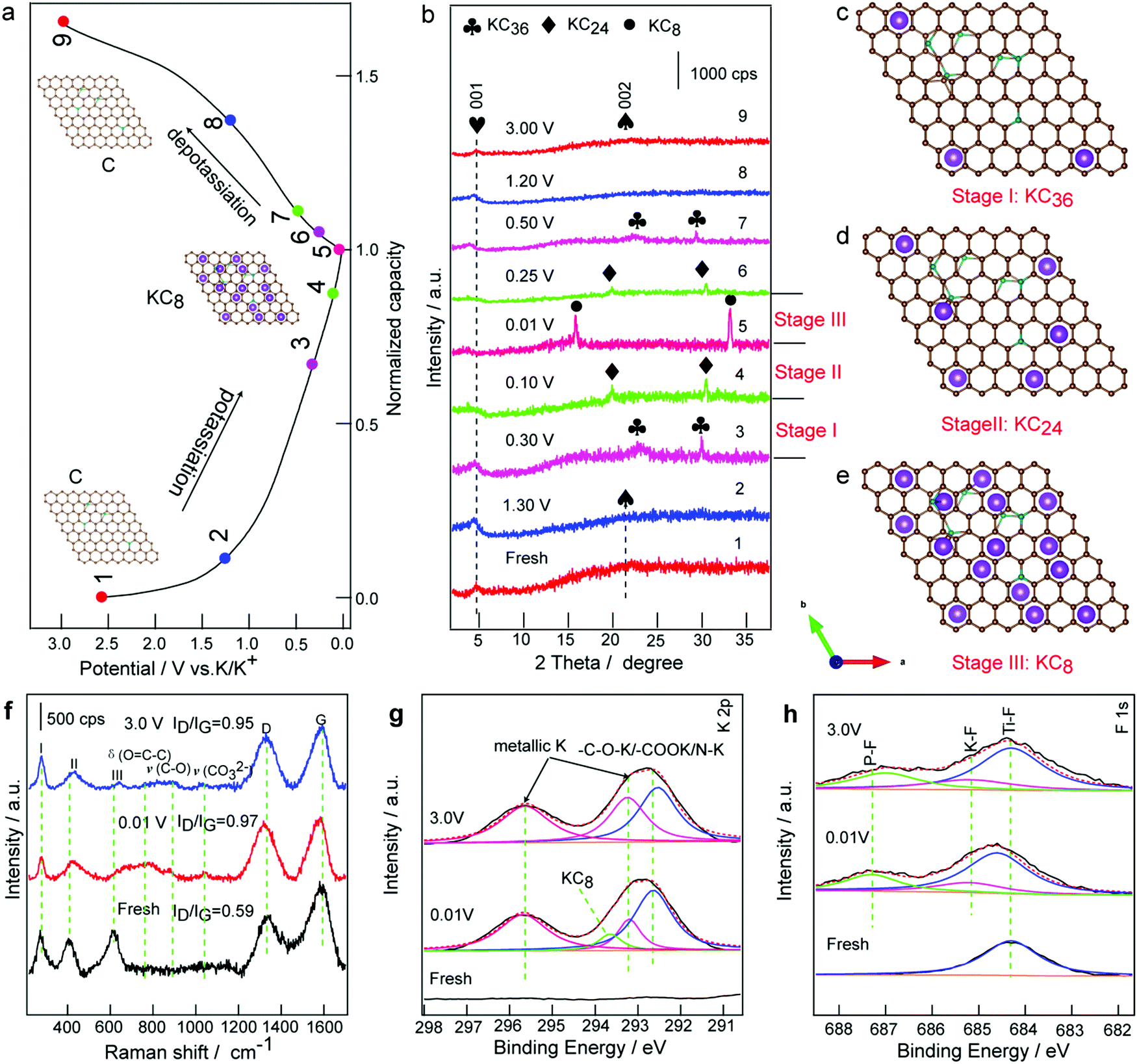 | ||
| Fig. 6 Electrochemical mechanisms. (a) Voltage profile and (b) corresponding ex situ XRD patterns for the PDDA-NPCN/Ti3C2 anode at different states during the initial potassiation/depotassiation process. Supercells of (c) KC36, (d) KC24 and (e) KC8. (f) Ex situ Raman spectra and (g and h) ex situ XPS spectra of the PDDA-NPCN/Ti3C2 anode at different potassiation/depotassiation states. | ||
To characterize the microstructures, ex situ SEM images of the ex-Ti3C2, PDDA-NPCNs and PDDA-NPCN/Ti3C2 anodes after 300 cycles are investigated in Fig. S12 (ESI†). Notably, the ex-Ti3C2 anode (Fig. S12a, ESI†) agglomerates together seriously after 300 cycles owing to pulverization, which inevitably reduced the K+ accessible surface area and impeded electrolyte penetration. However, due to the existence of the PDDA-NPCNs (Fig. S12b, ESI†), the morphology of the PDDA-NPCN/Ti3C2 anode (Fig. S12c, ESI†) is well retained compared with ex-Ti3C2, exhibiting excellent structure stability. From the ex situ TEM image, a porous interconnected conductive network and the inner void space could be well preserved for the PDDA-NPCN/Ti3C2 anode even after 300 cycles as shown by Fig. S13a (ESI†), and a thin SEI coating covers the hybrids, which further demonstrates that the cycling stability is not deteriorated upon repeated large radius K+ insertion/extraction, making it possible to build long-life PIBs. Ti, C, O, F, N and K elements are homogeneously distributed among the whole anode (Fig. S13, ESI†), further proving successful potassiation of the PDDA-NPCN/Ti3C2 anode. Ex situ Raman and ex situ XPS analysis are further carried out for the PDDA-NPCN/Ti3C2 anode to study structure changes in the charge/discharge process. For the fresh PDDA-NPCN/Ti3C2 anode (Fig. 6f), the I, II and III peaks located at 273, 409, and 613 cm−1 are assigned to the vibrational modes of Ti–C bonds, respectively.36 The two obvious peaks at 1327 and 1588 cm−1 could be attributed to the D and G bands, respectively.15 The ratio of intensity between ID and IG of the fresh PDDA-NPCN/Ti3C2 anode is 0.59, suggesting a high graphitization degree. When discharged to 0.01 V, the I and II peaks shift, while the III peak disappears, corresponding to the partial breaking of Ti–C bonds after formation of the Ti3C2TxKy phase.14 Meanwhile, a slight shift of the G band peak occurs accompanied by a decrease in the relative intensity, indicating a more disordered structure as a result of the potassiation process. When charged to 3.0 V, the III peak of the Ti–C bonds appears again but not as strong as that of the fresh anode, which is probably caused by different states of Ti–C bonds between the crystalline Ti3C2TxKy nanosheets and re-formed Ti3C2 nanosheets. Correspondingly, the ratio of intensity between ID and IG related to the disorder degree of carbon increases from 0.59 to 0.97 and then decreases to 0.95 for the fresh anode, fully discharged and fully charged, respectively, which illuminates the change of the disorder degree due to the K+ insertion/extraction, further confirming the structural sustainability and reversibility of the PDDA-NPCN/Ti3C2 anode upon the potassiation/depotassiation process. Furthermore, extra weak peaks appear at 757, 872 and 1037 cm−1, which are possibly associated with the decomposition products of the electrolytes because they are only observed for the cycled samples.66
As shown in the ex situ XPS spectra (Fig. S14, ESI†), the atomic percent of O, K and F is greatly enhanced after discharging to 0.01 V compared with that of the fresh PDDA-NPCN/Ti3C2 anode. The high-resolution XPS spectrum of K 2p (Fig. 6g) deconvolutes into four peaks: metallic K (293.2 and 295.7 eV), KC8 (293.6 eV) and –C–O–K/–COOK/NK (292.6 eV), which could be attributed to the insertion of K+, the formation of KC8 and the SEI layer.53 However, when charged to 3.0 V again, the atomic percent of O, K and F falls back. The peak at 293.6 eV of the KC8 state disappears, meanwhile the decreased percent of K could be readily considered as the reversible storage mechanism of K+. The residual K in the PDDA-NPCN/Ti3C2 anode could be ascribed to various potassium salts in the SEI layer and the trapped metallic K in the nanopores. However, the formation of the SEI layer occurs vigorously during the discharge process, which looks similar to adsorption. In this regard, the change of F 1s (Fig. 6h) on the PDDA-NPCN/Ti3C2 anode surface is distinguished during the charge/discharge process. The major component of the fresh anode is located at 684.3 eV, corresponding to the Ti–F surface functional group.15 The Ti–F peak shifts to higher binding energy 684.6 eV upon discharging to 0.01 V. Meanwhile, the K–F and P–F bonds at 685.2 and 687.3 eV appear, indicating that the SEI layer starts to form during the potassiation process.67 When charged to 3.0 V, the Ti–F peak shifts back to the initial again, demonstrating that K+ can be reversibly inserted into/extracted from within the PDDA-NPCN/Ti3C2 anode. However, the P–F bonds shift to 687.0 eV, which can explain the irreversible reactions of the SEI layer and side reactions of the surface functional groups during the initial cycling. Based on the discussions above, a schematic illustration (Fig. S15, ESI†) of the possible electrochemical mechanism is proposed. The interlayer distance changes when K+ is inserted into/extracted from within the robust PDDA-NPCN/Ti3C2 anode through mesoporous tunnels and nanopores. It is noted that the NPCNs could transform into KC8 at low voltage, and further recover to NPCNs at high voltage during the potassiation/depotassiation process, which provide an ideal “buffer” or interlayer spacer for coupling with MXene nanosheets, further enhancing the durability of high reversible capacity and long-term cycling stability.
Based on first principles, we further perform DFT calculations to investigate the adsorption abilities of K+ in detail. We construct four types of computational models, i.e., Ti3C2, NPCNs and NPCN/Ti3C2 hybrids (Fig. S16a–d, ESI†) and then calculate the corresponding adsorption energies (ΔEa). As a comparison, the ΔEa (Fig. S16e, ESI†) of the NPCN/Ti3C2 hybrids are −2.26 and −2.42 eV, much lower than those of the bare Ti3C2 (−0.43 eV) and NPCNs (−2.11 eV), which indicates NPCN/Ti3C2 hybrids with stronger K+ adsorption capability than Ti3C2 and the NPCNs. It also means that K+ would prefer to combine with the Ti3C2 and NPCNs anchored on the NPCN/Ti3C2 hybrids, which not only greatly decrease the ΔEa of K+, but also facilitate the potassiation process. As presented in Fig. S17 (ESI†), the hybrids modify the electronic structure. In all situations, a net gain of electronic charge between K+ and Ti3C2, the NPCNs and the NPCN/Ti3C2 hybrids can be observed, indicating charge transfer from the adsorbed K to its nearest neighbor atoms. Different from the pure Ti3C2 and NPCNs, the surfaces for the NPCN/Ti3C2 hybrids also show more electron accumulation, indicating stronger charge transfer than the others. It suggests that the NPCNs evidently change the potassiation state of Ti3C2 and provide fast electron transfer pathways, which facilitates the potassiation process. Therefore, the NPCN/Ti3C2 hybrids could not only be more favorable for the K+ adsorption reactions, but also efficiently decrease the ΔEa of K+ and facilitate the potassiation process, theoretically validating the boosted K+ storage kinetics.
Conclusions
In summary, we have employed a facile solution-phase co-feeding and electrostatic attraction self-assembly approach to rationally design novel PDDA-NPCN/Ti3C2 hybrids for high-performance potassium-ion batteries. The hybrids show an obvious synergetic effect of both materials, leading to a high reversible capacity of 358.4 mA h g−1 after 300 cycles at 0.1 A g−1, much larger than ex-Ti3C2 (106.2 mA h g−1) and the NPCNs (280.9 mA h g−1), and long cycling stability of 252.2 mA h g−1 with only 0.03% degradation per cycle within 2000 cycles at 1.0 A g−1. This effect is attributed to the hybrids with stacked structure ensuring intimate contact between Ti3C2 and the NPCNs to efficiently take advantage of both components. The PDDA-NPCN/Ti3C2 hybrids afford enlarged interlayer spacing, a unique 3D porous interconnected conductive network and abundant utilization of active sites, which shortens the electrolyte ion transport pathways and facilitates rapid ionic/electronic transport. The robust hybrids show a durable rate capability and cycling stability through alleviating the volumetric changes caused by phase transformations during the fast charge/discharge process. DFT calculations further reveal that the PDDA-NPCN/Ti3C2 hybrids help with synergistically ameliorating the electrochemical performance by enhancing interfacial K+ adsorption reactions and facilitate the potassiation process. This method opens up the possibility to controllably explore self-assembly of coupled hybrids in energy storage devices.Methods
Methods and any associated references are available in the online version of the paper.Data availability
All data used in this study are available from the corresponding authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the project supported by the Sate Key Program of National Natural Science of China (no. 51532005), National Nature Science Foundation of China (no. 51472148, 51272137, 51602181, 51902188), General Financial Grant from the China Postdoctoral Science Foundation (no. 2015M582088), and the Fundamental Research Fund of Shandong University.Notes and references
- X. Wu, D. P. Leonard and X. Ji, Chem. Mater., 2017, 29, 5031–5042 CrossRef CAS.
- J. C. Pramudita, D. Sehrawat, D. Goonetilleke and N. Sharma, Adv. Energy Mater., 2017, 7, 1602911 CrossRef.
- Y.-S. Xu, S.-Y. Duan, Y.-G. Sun, D.-S. Bin, X.-S. Tao, D. Zhang, Y. Liu, A.-M. Cao and L.-J. Wan, J. Mater. Chem. A, 2019, 7, 4334–4352 RSC.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Adv. Funct. Mater., 2018, 28, 1802938 CrossRef.
- X. Zou, P. Xiong, J. Zhao, J. Hu, Z. Liu and Y. Xu, Phys. Chem. Chem. Phys., 2017, 19, 26495–26506 RSC.
- Q. Zhao, J. Wang, Y. Lu, Y. Li, G. Liang and J. Chen, Angew. Chem., Int. Ed., 2016, 55, 12528–12532 CrossRef CAS PubMed.
- S. Komaba, T. Hasegawa, M. Dahbi and K. Kubota, Electrochem. Commun., 2015, 60, 172–175 CrossRef CAS.
- B. Ji, F. Zhang, X. Song and Y. Tang, Adv. Mater., 2017, 29, 1700519 CrossRef PubMed.
- K. Lei, F. Li, C. Mu, J. Wang, Q. Zhao, C. Chen and J. Chen, Energy Environ. Sci., 2017, 10, 552–557 RSC.
- W. Zhang, J. Mao, S. Li, Z. Chen and Z. Guo, J. Am. Chem. Soc., 2017, 139, 3316–3319 CrossRef CAS PubMed.
- X. Sang, Y. Xie, M. W. Lin, M. Alhabeb, K. L. Van Aken, Y. Gogotsi, P. R. Kent, K. Xiao and R. R. Unocic, ACS Nano, 2016, 10, 9193–9200 CrossRef CAS PubMed.
- Y. Xie, Y. Dall’Agnese, M. Naguib, Y. Gogotsi, M. W. Barsoum, H. L. Zhuang and P. R. Kent, ACS Nano, 2014, 8, 9606–9615 CrossRef CAS PubMed.
- J. Wen, Q. Fu, W. Wu, H. Gao, X. Zhang and B. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 7087–7095 CrossRef CAS PubMed.
- D. Er, J. Li, M. Naguib, Y. Gogotsi and V. B. Shenoy, ACS Appl. Mater. Interfaces, 2014, 6, 11173–11179 CrossRef CAS PubMed.
- R. Zhao, M. Wang, D. Zhao, H. Li, C. Wang and L. Yin, ACS Energy Lett., 2017, 3, 132–140 CrossRef.
- D. Zhao, R. Zhao, S. Dong, X. Miao, Z. Zhang, C. Wang and L. Yin, Energy Environ. Sci., 2019, 12, 2422–2432 RSC.
- J. Li, X. Yuan, C. Lin, Y. Yang, L. Xu, X. Du, J. Xie, J. Lin and J. Sun, Adv. Energy Mater., 2017, 7, 1602725 CrossRef.
- X. Hui, R. Zhao, P. Zhang, C. Li, C. Wang and L. Yin, Adv. Energy Mater., 2019, 9, 1901065 CrossRef.
- P. Lian, Y. Dong, Z.-S. Wu, S. Zheng, X. Wang, W. Sen, C. Sun, J. Qin, X. Shi and X. Bao, Nano Energy, 2017, 40, 1–8 CrossRef CAS.
- M. Naguib, R. A. Adams, Y. Zhao, D. Zemlyanov, A. Varma, J. Nanda and V. G. Pol, Chem. Commun., 2017, 53, 6883–6886 RSC.
- H. Huang, J. Cui, G. Liu, R. Bi and L. Zhang, ACS Nano, 2019, 13, 3448–3456 CrossRef CAS PubMed.
- Y. Tian, Y. An, S. Xiong, J. Feng and Y. Qian, J. Mater. Chem. A, 2019, 7, 9716–9725 RSC.
- R. Ma, X. Liu, J. Liang, Y. Bando and T. Sasaki, Adv. Mater., 2014, 26, 4173–4178 CrossRef CAS PubMed.
- P. Xiong, R. Ma, N. Sakai, L. Nurdiwijayanto and T. Sasaki, ACS Energy Lett., 2018, 3, 997–1005 CrossRef CAS.
- R. Zhao, Z. Qian, Z. Liu, D. Zhao, X. Hui, G. Jiang, C. Wang and L. Yin, Nano Energy, 2019, 65, 104037 CrossRef CAS.
- R. Zhao, L. Zhang, C. Wang and L. Yin, J. Power Sources, 2017, 353, 77–84 CrossRef CAS.
- D. Li, X. Ren, Q. Ai, Q. Sun, L. Zhu, Y. Liu, Z. Liang, R. Peng, P. Si, J. Lou, J. Feng and L. Ci, Adv. Energy Mater., 2018, 8, 1802386 CrossRef.
- L. Fang, J. Xu, S. Sun, B. Lin, Q. Guo, D. Luo and H. Xia, Small, 2019, 15, 1804806 CrossRef PubMed.
- W. Luo, J. Wan, B. Ozdemir, W. Bao, Y. Chen, J. Dai, H. Lin, Y. Xu, F. Gu, V. Barone and L. Hu, Nano Lett., 2015, 15, 7671–7677 CrossRef CAS PubMed.
- Y. Dong, Z. S. Wu, S. Zheng, X. Wang, J. Qin, S. Wang, X. Shi and X. Bao, ACS Nano, 2017, 11, 4792–4800 CrossRef CAS PubMed.
- G. Liao, Y. Gong, L. Zhang, H. Gao, G.-J. Yang and B. Fang, Energy Environ. Sci., 2019, 12, 2080–2147 RSC.
- H. He, D. Huang, Y. Tang, Q. Wang, X. Ji, H. Wang and Z. Guo, Nano Energy, 2019, 57, 728–736 CrossRef CAS.
- K. Share, A. P. Cohn, R. Carter, B. Rogers and C. L. Pint, ACS Nano, 2016, 10, 9738–9744 CrossRef CAS PubMed.
- J. Yan, C. E. Ren, K. Maleski, C. B. Hatter, B. Anasori, P. Urbankowski, A. Sarycheva and Y. Gogotsi, Adv. Funct. Mater., 2017, 27, 1701264 CrossRef.
- X. Ge, C. Gu, Z. Yin, X. Wang, J. Tu and J. Li, Nano Energy, 2016, 20, 185–193 CrossRef CAS.
- M. Boota, B. Anasori, C. Voigt, M. Q. Zhao, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2016, 28, 1517–1522 CrossRef CAS PubMed.
- W. Zhao, J. Peng, W. Wang, B. Jin, T. Chen, S. Liu, Q. Zhao and W. Huang, Small, 2019, 15, 1901351 CrossRef PubMed.
- A. Lipatov, M. Alhabeb, M. R. Lukatskaya, A. Boson, Y. Gogotsi and A. Sinitskii, Adv. Electron. Mater., 2016, 2, 1600255 CrossRef.
- S. Liu, J. Zhou and H. Song, Chem. Commun., 2018, 54, 9825–9828 RSC.
- S. Liu, J. Zhou and H. Song, Adv. Energy Mater., 2018, 8, 1800569 CrossRef.
- C. Li, S. Dong, R. Tang, X. Ge, Z. Zhang, C. Wang, Y. Lu and L. Yin, Energy Environ. Sci., 2018, 11, 3201–3211 RSC.
- Y. Xie, Y. Chen, L. Liu, P. Tao, M. Fan, N. Xu, X. Shen and C. Yan, Adv. Mater., 2017, 29, 1702268 CrossRef PubMed.
- J. Gu, Z. Du, C. Zhang and S. Yang, Adv. Energy Mater., 2016, 6, 1600917 CrossRef.
- Q. Xue, H. Zhang, M. Zhu, Z. Pei, H. Li, Z. Wang, Y. Huang, Y. Huang, Q. Deng, J. Zhou, S. Du, Q. Huang and C. Zhi, Adv. Mater., 2017, 29, 1604847 CrossRef PubMed.
- Y. Huo, Y. Jin, J. Zhu and H. Li, Appl. Catal., B, 2009, 89, 543–550 CrossRef CAS.
- G. Yang, Z. Jiang, H. Shi, T. Xiao and Z. Yan, J. Mater. Chem., 2010, 20, 5301–5309 RSC.
- W. Bao, L. Liu, C. Wang, S. Choi, D. Wang and G. Wang, Adv. Energy Mater., 2018, 8, 1702485 CrossRef.
- Z. Jian, W. Luo and X. Ji, J. Am. Chem. Soc., 2015, 137, 11566–11569 CrossRef CAS PubMed.
- G. Ma, K. Huang, J.-S. Ma, Z. Ju, Z. Xing and Q.-C. Zhuang, J. Mater. Chem. A, 2017, 5, 7854–7861 RSC.
- X. Qi, K. Huang, X. Wu, W. Zhao, H. Wang, Q. Zhuang and Z. Ju, Carbon, 2018, 131, 79–85 CrossRef CAS.
- Z. Ju, P. Li, G. Ma, Z. Xing, Q. Zhuang and Y. Qian, Energy Storage Mater., 2018, 11, 38–46 CrossRef.
- Y. An, H. Fei, G. Zeng, L. Ci, B. Xi, S. Xiong and J. Feng, J. Power Sources, 2018, 378, 66–72 CrossRef CAS.
- J. Yang, Z. Ju, Y. Jiang, Z. Xing, B. Xi, J. Feng and S. Xiong, Adv. Mater., 2018, 30, 1700104 CrossRef PubMed.
- Z. Tai, Q. Zhang, Y. Liu, H. Liu and S. Dou, Carbon, 2017, 123, 54–61 CrossRef CAS.
- Y. Sun, H. Xiao, H. Li, Y. He, Y. Zhang, Y. Hu, Z. Ju, Q. Zhuang and Y. Cui, Chem. – Eur. J., 2019, 25, 7359–7365 CrossRef CAS PubMed.
- Q. Gan, J. Xie, Y. Zhu, F. Zhang, P. Zhang, Z. He and S. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 930–939 CrossRef CAS PubMed.
- A. P. Cohn, N. Muralidharan, R. Carter, K. Share, L. Oakes and C. L. Pint, J. Mater. Chem. A, 2016, 4, 14954–14959 RSC.
- R. A. Adams, J. M. Syu, Y. Zhao, C. T. Lo, A. Varma and V. G. Pol, ACS Appl. Mater. Interfaces, 2017, 9, 17872–17881 CrossRef CAS PubMed.
- L. Kong, J. Zhu, W. Shuang and X.-H. Bu, Adv. Energy Mater., 2018, 8, 1801515 CrossRef.
- L. Wei, Z. Hou and H. Wei, Electrochim. Acta, 2017, 229, 445–451 CrossRef CAS.
- G. SandI, R. E. Winans and K. A. Carrado, J. Electrochem. Soc., 1996, 143, L95–L98 CrossRef CAS.
- H. Gao, L. Song, J. Niu, C. Zhang, T. Kou, Y. Sun, J. Qin, Z. Peng and Z. Zhang, J. Mater. Chem. A, 2019, 7, 13602–13613 RSC.
- Y. Zhang, H. Gao, J. Niu, W. Ma, Y. Shi, M. Song, Z. Peng and Z. Zhang, ACS Nano, 2018, 12, 11678–11688 CrossRef CAS PubMed.
- H. Zhang, K. Xi, K. Jiang, X. Zhang, Z. Liu, S. Guo and H. Zhou, Chem. Commun., 2019, 55, 7910–7913 RSC.
- J. C. Chacón-Torres, L. Wirtz and T. Pichler, Phys. Status Solidi B, 2014, 251, 2337–2355 CrossRef.
- X. Zhao, P. Xiong, J. Meng, Y. Liang, J. Wang and Y. Xu, J. Mater. Chem. A, 2017, 5, 19237–19244 RSC.
- J. Huang, X. Lin, H. Tan and B. Zhang, Adv. Energy Mater., 2018, 8, 1703496 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ee03250a |
| This journal is © The Royal Society of Chemistry 2020 |