
Efficient hydrogen peroxide synthesis by metal-free polyterthiophene via photoelectrocatalytic dioxygen reduction†
Wenjun
Fan
ab,
Bingqing
Zhang
ab,
Xiaoyu
Wang
c,
Weiguang
Ma
 a,
Deng
Li
a,
Zhiliang
Wang
a,
Michel
Dupuis
c,
Jingying
Shi
*a,
Shijun
Liao
*b and
Can
Li
*a
a,
Deng
Li
a,
Zhiliang
Wang
a,
Michel
Dupuis
c,
Jingying
Shi
*a,
Shijun
Liao
*b and
Can
Li
*a
aState Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, Liaoning, China. E-mail: jingyingshi@dicp.ac.cn; canli@dicp.ac.cn
bThe Key Laboratory of Fuel Cell Technology of Guangdong Province & The Key Laboratory of New Energy Technology of Guangdong University, School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou, 510641, Guangdong, China. E-mail: chsjliao@scut.edu.cn
cDepartment of Chemical and Biological Engineering, University at Buffalo, State University of New York Buffalo, NY 14260, USA
First published on 26th November 2019
Abstract
Solar hydrogen peroxide (H2O2) produced through the selective two-electron (2e−) oxygen reduction pathway is an ideal alternative to liquid fuel in addition to being a versatile chemical. Up to now, low photocatalytic activity, low selectivity and serious competing reactions have been big hurdles in the production of solar H2O2 in an efficient way. Herein, we report that polyterthiophene (pTTh), a metal-free narrow-bandgap polymeric semiconductor, is an efficient photocathode for H2O2 production in alkaline solution. We found that 2e− selectivity for the ORR is dependent on the pH of electrolytes and approaches 100% at pH ∼ 13. A record-high H2O2 concentration of 110 mmol L−1 is achieved, which is two orders of magnitude higher than the previous photosynthesized H2O2. Furthermore, NiFeOx/BiVO4–pTTh dual-photoelectrodes in photoelectrochemical devices enabled bias-free synthesis of solar H2O2 of concentration ∼90 mmol L−1 for several cycles without any noticeable decay. This extremely high 2e− selectivity is attributed to the intrinsic electrochemical properties of pTTh. Theoretical calculations suggested that the selectivity-determining step in the 2e− process is over ∼200 times faster than that in the 4e− pathway. Our work paves an alternative way of generating liquid solar fuel that is very promising for practical applications.
Broader contextThe importance of hydrogen peroxide (H2O2) has been witnessed by its annual production, approaching 6 million tons in 2016. However, this otherwise highly versatile and green chemical is currently manufactured by the energy-intensive anthraquinone process, which requires complex and large-scale infrastructure. In this regard, H2O2 production from solar energy is an ideal way, in which only water, oxygen and sunlight (H2O + 1/2O2 → H2O2, ΔG = 117 kJ mol−1) are required. Nevertheless, all previously reported particulate photocatalytic systems suffered from poor efficiency in H2O2 generation, due to the serious back reaction by photogenerated holes and the low selectivity of the two-electron (2e−) pathway. In our contribution, we propose a selective metal-free polyterthiophene photocathode combined with a spatially separated photoelectrochemical cell to generate a record high H2O2 concentration of 110 millimole per liter (mM). Our work provides an attractive route for the practical synthesis of liquid solar fuel. |
Introduction
Solar fuel production has attracted increasing attention as the key technology to relieve environmental issues and reduce the reliance on fossil resources.1,2 Substantial efforts have focused on the generation of hydrogen (from overall water splitting) and carbonaceous fuels like methanol (from CO2 reduction) by conversion of solar energy.3–6 In contrast, another type of solar fuel, hydrogen peroxide (H2O2), has received less attention so far.7 Generally, H2O2 could be synthesized via photo(electro)catalytic H2O oxidation or O2 reduction following a selective two-electron (2e−) pathway.8–11 This is the reason why we also call it as one of the solar fuels. Similar to H2 in a H2–O2 fuel cell, H2O2 can also be utilized for electricity generation in a single-compartment H2O2 fuel cell with a theoretical output voltage of above 1.09 V.12 Besides, it is a value-added chemical which has been extensively applied in organic synthesis, pulp bleaching, waste-water treatment and medical disinfection. Meanwhile, it exists in a liquid state at normal temperature and pressure that makes it convenient for storage, transportation, and compatibility with the current energy infrastructure.13 Hence, H2O2 is an ideal solar fuel that deserves increased research.However, the synthesis of solar H2O2 has been extremely challenging up to now. The solar water oxidation reaction (WOR) itself is rather difficult to realize due to the uphill thermodynamics (1.76 V) together with sluggish kinetics.10,14 Besides, the as-synthesized H2O2 can readily decompose at a highly oxidative photoanode because the H2O2 molecule is an excellent hole scavenger.15 Several photoanodes, such as BiVO4 and WO3,10,16,17 combined with highly concentrated hydrogen carbonate solution as an electrolyte have been reported, but the formed H2O2 is not more than 5 millimole per liter (mmol L−1) via the photoelectrocatalytic WOR. Alternatively, solar H2O2 can be obtained from an oxygen reduction reaction (ORR) via a 2e− pathway (eqn (1) under acidic conditions and eqn (2) under alkaline conditions), which is enabled by applying the particulate system or a photoelectrochemical (PEC) cell with a photocathode.
| O2 + 2H+ + 2e− → H2O2 E° = 0.68 vs. RHE | (1) |
| O2 + H2O + 2e− → HO2− + OH− E° = 0.74 vs. RHE | (2) |
For instance, various photocatalysts such as inorganic ZnO, TiO2, and CdS and organic g-C3N4 have been found to be active for the artificial photosynthesis of H2O2 in a particulate system.18–21 Nevertheless, the achieved concentration is limited to a rather low level of a few mmol L−1, which is due to the following facts: (1) the produced H2O2 is susceptible to a back reaction of reoxidation by holes, (2) low activity and selectivity for the 2e− process. Thus, most of the photocatalytic H2O2 generation approaches rely on the use of sacrificial reagents (e.g. alcohol, formic acid and formate) which are intrinsically thermodynamic down-hill processes, and therefore solar energy cannot be stored in the form of chemicals.
In contrast, PEC cells with spatially separated two half-reactions and isolated electrolytes offer solutions to address the loss of H2O2 from the rapid back reaction. The H2O2 generation from PEC cells started with the seminal work of the Eric Daniel Głowacki group using a hydrogen-bonded organic semiconductor as the photocathode, although only 3 mmol L−1 was accumulated.22 Subsequently, a 2e− ORR electrocatalyst was wired with a WO3 photoanode as the driving force and a high concentration of 48 mmol L−1 was achieved in a catholyte after a long illumination duration of 24 h.23 However, to date, very few semiconducting materials have been found to be active for the PEC ORR and thus high-yield synthesis of H2O2via a selective photoelectrocatalytic ORR has not been reported.
Polymeric semiconductors have emerged as a class of promising materials for energy-related conversions. They offer the advantages of low-cost, facile synthesis and broad light absorption.24 As a p-type polymeric semiconductor material, polyterthiophene (pTTh) has been identified to be an active photocathode for the ORR in H2–O2 fuel cells and biofuel cells by our group and other groups.25–27 However, the 2e− selectivity of the pTTh photocathode for the ORR has not been investigated. In the present work, we found that the ORR 2e− selectivity of pTTh is strongly dependent on the pH of the electrolytes and it reaches nearly 100% at pH ∼ 13 without the requirement of any other additives or cocatalysts. We observed that a concentration of solar H2O2 of ∼ 110 mmol L−1 can be achieved, which is by far the most concentrated H2O2 derived via artificial photosynthesis methods.28 Furthermore, by wiring a pTTh photocathode with a NiFeOx/BiVO4 photoanode, the as-fabricated dual-photoelectrode device was capable of self-driven preparation of high-concentration H2O2 for several cycles without any noticeable decay. The high selectivity of the 2e− ORR was investigated by electrochemical experiments and density functional theory calculations. Our work sets an example for artificial photosynthesis of H2O2 with high yield, indicating that H2O2 may be a very promising liquid solar fuel to be developed.
Results and discussion
Characterization of the pTTh photoelectrode
Polyterthiophene (pTTh) films were deposited on three-dimensional carbon paper (CP) or smooth glassy carbon (GC) by electro-polymerization of terthiophene. The corresponding photoelectrodes are denoted as pTTh/CP and pTTh/GC for convenience.29 As shown in Fig. 1a, diffuse reflectance UV-vis spectrum of the pTTh/CP electrode exhibits wide absorption extending to 640 nm. The bandgap of pTTh is estimated to be ca. 2.0 eV from the Tauc plot (Fig. S1, ESI†). The red-brown photoelectrode (Fig. 1a, inset) appears with a flower-like morphology assembled in ultrathin nanosheets (Fig. 1b) as revealed by SEM observation. Notably, the nanoporous structure facilitates the immediate transport of the photogenerated excitons to the polymer–solution interface and their participation in the redox reaction while suppressing the undesirable electron–hole recombination.30 Cyclic voltammetry (CV) measurements were used to evaluate the HOMO and LUMO energy levels – from the ionization potential and the electron affinity, respectively – and determine the bandgap of pTTh (Fig. S2a and b, ESI†). A careful analysis of the CVs of the material (zoom at lower current, Fig. S2b, ESI†) revealed that the onset of oxidation occurred at ca. 0.39 V. The corresponding HOMO energy level was calculated to be −5.13 eV (using the formula given in the ESI†), and the LUMO energy was −3.13 eV by adding the optical bandgap (Eg) to the calculated HOMO energy level. Thus, LUMO and HOMO potentials are calculated to be −1.37 V and 0.63 V, respectively. To obtain more information of the electronic structure, a Mott–Schottky (M–S) analysis was performed on the pTTh/GC electrode (Fig. S2c, ESI†). The M–S plot exhibits a negative slope, which is typical for p-type semiconductors. The flat band potential locates at ca. 0.48 V vs. NHE at pH 13; thus, the conduction band energy level (LUMO level equivalent) is estimated to be ca. −1.42 V, similar to the energy level results of the CVs. Notably, it is thermodynamically favorable for both the oxygen reduction reaction (either 4e− or 2e− pathway) and the hydrogen evolution reaction. Control experiments were conducted on a rotating-ring disk system (Fig. S3, ESI†) to check the PEC activity of pTTh toward the ORR in 0.1 M KOH solution under 100 mW cm−2 light illumination. Remarkably, a pronounced photocurrent was detected in the O2-saturated electrolyte in contrast to a negligible photocurrent in the Ar-saturated electrolyte (Fig. S4 and S5, ESI†), suggesting that the ORR is the dominant photocathodic process.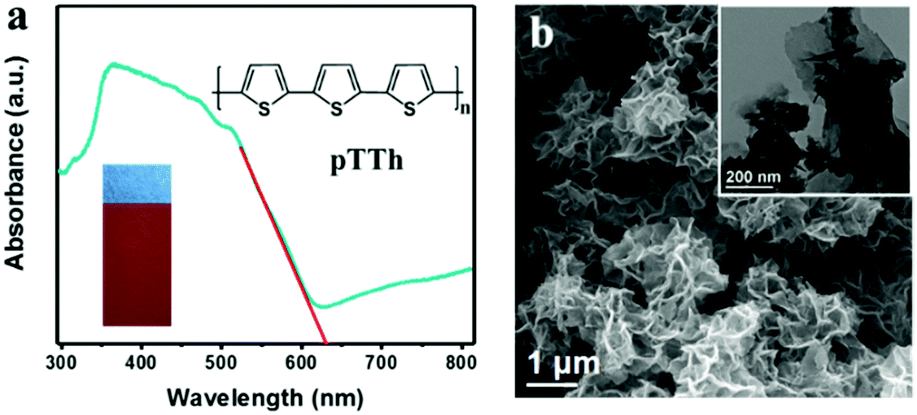 | ||
| Fig. 1 (a) UV-vis diffuse reflectance spectrum of pTTh; the inset shows the molecular structure and the digital photo of the as-prepared pTTh electrode, respectively. (b) SEM image of the pTTh film; the inset shows the TEM image of the pTTh film. | ||
pH-Dependent photocathodic ORR to H2O2
ORR performance is commonly affected by the pH of solutions since it is a proton-coupled electron transfer process.31 To unravel the pH-dependence of the PEC ORR activity of the pTTh photocathode, linear sweep voltammetry (LSV) curves of the pTTh/GC in O2-saturated electrolytes with different pH values were investigated. It can be seen that the photocurrent density is remarkably enhanced with a negative shift of the applied bias for all curves (Fig. 2a). Meanwhile, under the same applied bias, the photocurrent density gradually increases with the increase of pH. In other words, pTTh shows higher PEC ORR activity in alkaline electrolytes than in acidic electrolytes. To demonstrate the pH-dependence more directly, the photocurrent densities at specific applied potentials of 0.6, 0.7 and 0.8 V are plotted together with the onset potentials derived from Fig. 2a with respect to the corresponding pH. As shown in Fig. 2b, it can be seen that both the photocurrent and the onset potential depend greatly on the pH of the solutions. For example, under the bias of 0.6 V, the photocurrent increases from less than −0.2 mA cm−2 at pH 0.1 to about −1.7 mA cm−2 at pH 12.9. And the largest shift of the onset potential is as high as 0.48 V (0.67 V at pH 0.1 and 1.15 V at pH 12.9). The pH of the electrolyte is thus found to be a key parameter to regulate the PEC ORR activity of the pTTh photocathode.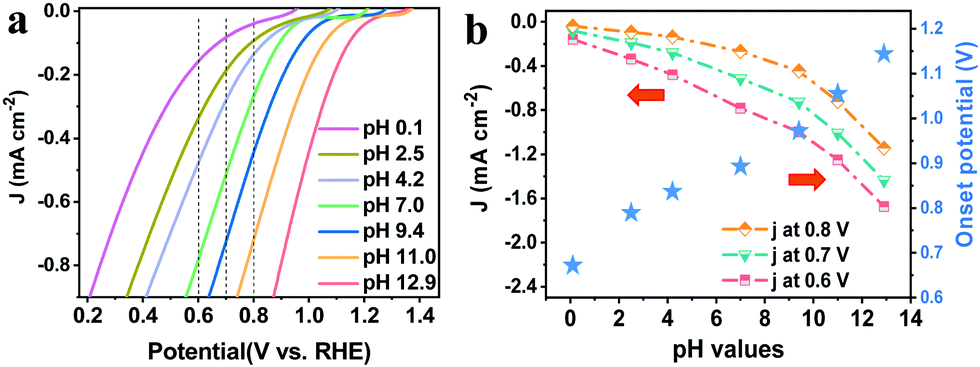 | ||
| Fig. 2 (a) LSVs of the pTTh electrode measured in O2-saturated electrolytes with different pH values under 1 sun illumination. Rotation rate: 1600 rpm. Ionic strength is constant at 0.1 M. (b) Correlation between current densities of the ORR at 0.6, 0.7 and 0.8 V vs. RHE and the pH values (dotted–dashed lines); correlation between onset potentials (at 0.1 mA cm−2) and the pH values (blue stars). | ||
The selectivity for H2O2 generation and its pH-dependence for the pTTh photocathode were investigated by the conventional rotating ring-disk electrode (RRDE) technique (see ESI,† Methods). Fig. 3a exhibits the disk current for the ORR and ring current for the H2O2 oxidation reaction in 0.1 M KOH solution. The onset potentials at the ring and the disc coincide at ∼1.25 V. As the overpotential increases, most of the current in the disc can be accounted for by the amount of H2O2 detected at the ring (dashed–dotted line in Fig. 3a). The H2O2 selectivity with pTTh exceeds 95% in the wide potential range of 0.2 to 1.2 V (Fig. 3b), suggesting that the PEC ORR on pTTh is dominated by a 2e− process with HO2− as the final product in alkaline solution (pH 12.9). By contrast, the H2O2 selectivity gradually decreases with decreasing pH (Fig. 3c) that only maintains ∼50% in strong acidic solution (pH 0.1). Thus, it can be concluded that both the PEC activity and H2O2 selectivity of pTTh for the ORR extremely depend on the pH of electrolytes. In view of the superior activity and selectivity for the 2e− ORR to H2O2 in 0.1 M KOH solution (pH 12.9), the discussion hereafter corresponds to these conditions with saturated O2 unless otherwise stated (HO2− is the deprotonated anion of H2O2 in alkaline media; we will use H2O2 and HO2− interchangeably throughout this work).
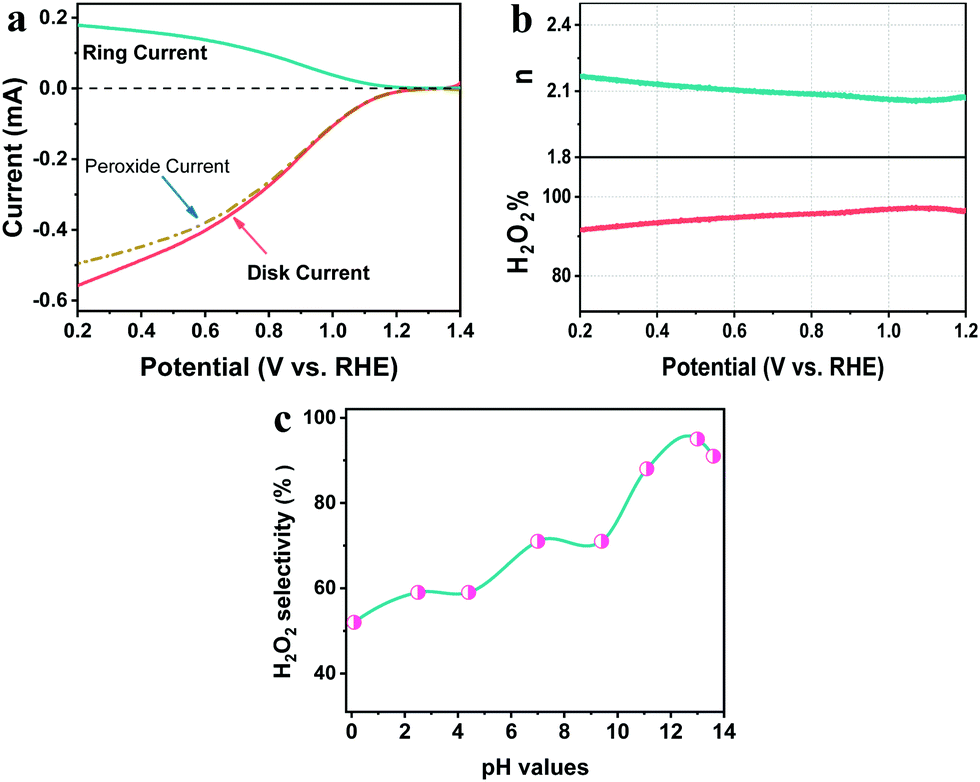 | ||
| Fig. 3 (a) RRDE voltammograms of pTTh in O2 saturated solution at pH 12.9 under 1 sun illumination with the disc current, ring current and current corresponding to hydrogen peroxide obtained from the ring current. The disk potential was scanned at 10 mV s−1 and the ring potential was constant at 1.5 V vs. RHE. (b) Percentage of peroxide and the electron transfer number (n) of pTTh at various potentials, calculated from RRDE data. (c) pH dependent hydrogen peroxide selectivity of pTTh calculated by RRDE results. | ||
To reveal the ORR kinetics of the material, the rotating disk electrode (RDE) measurements were recorded. As shown in Fig. S6 (ESI†), the linearity of the Koutecky–Levich (K–L) plot and near parallelism of the fitting lines over the investigated potential range suggest a first-order reaction kinetics for the ORR with respect to the concentration of dissolved O2 and similar electron transfer numbers at different potentials (Fig. S6b, ESI†).32 Meanwhile, the electron-transfer number (n) is calculated to be approximately 2.0 from the slopes of K–L plots, which agrees well with the above RRDE results. According to the Levich equation (for details see ESI,† Methods), the diffusion-limited current density is calculated to be 2.19 mA cm−2 (2e− pathway) or 4.39 mA cm−2 (4e− pathway) at 1600 rpm.33 At 0.4 V, the polarization curve reaches a current density (ca. 2.0 mA cm−2) close to the limiting value of H2O2 formation, which implies the fast kinetics of the pTTh photocathode toward the 2e− pathway. Therefore, the combined characterization of RRDE and RDE measurements clearly predicted an efficient synthesis of H2O2 by the PEC method on the pTTh photocathode in alkaline solution.
PEC H2O2 production from the pTTh photocathode
To study the synthesis of H2O2via the PEC ORR, the pTTh/CP was used as the photocathode in a three-electrode configuration with 0.1 M KOH as the electrolyte. A Nafion membrane was used to isolate the anolyte from the O2-saturated catholyte in order to prevent the reoxidation of the generated H2O2 by the anode.22 An external bias of 0.65 V was applied on the pTTh under simulated 100 mW cm−2 illumination. Considering that H2O2 is susceptible to temperature and UV irradiation, we conducted control experiments to determine the synthesis conditions (Fig. S7, ESI†), in which 100 mmol L−1 H2O2 in 0.1 M KOH solution was investigated as an example. It can be seen that H2O2 scarcely decomposed in the temperature range of 10–30 °C even after 24 h. Under UV-filtered illumination (λ > 420 nm), more than 90% of H2O2 was retained even when the exposure duration was longer than 20 h. In contrast, the extended wavelength illumination (full spectrum) caused 90% loss within 16 h. During the UV irradiation, the bond between hydrogen and oxygen would cleave and induce a chain reaction according to the Haber–Weiss reaction theory,34 which would lead to the violet decomposition of H2O2. With these combined observations, the reaction temperature was maintained at 20 °C with applied visible light irradiation as the light source.Fig. 4a shows the time courses of H2O2 production at the cathodic side. It can be seen that the concentration of H2O2 increased to 80 mmol L−1 almost linearly within 5 h and an even higher concentration of 110 mmol L−1 was achieved after a sustained PEC reaction for 11 h despite a slight slowing down of the growth rate. The time courses of faradaic efficiency for H2O2 synthesis (Fig. 4a, green) suggest an efficiency of above 90% over the entire time range. In comparison with the previous photosynthesized H2O2, the produced H2O2 in this work achieved a record high concentration which is ca. two orders of magnitude greater than the previous results (Table S1, ESI†). Concerning the solar H2O2 synthesis via direct photocathodic ORR, the generation rate of H2O2 (10 mmol L−1 h−1) is four orders of magnitude higher than that derived on an organic pigment photocathode in acidic solution (3 mmol L−1 in 50 h, i.e. 0.06 mmol L−1 h−1).22 Meanwhile, the generated current stayed almost constant within the whole testing duration as shown in Fig. 4b, which indicates that the parasitic reaction was boosted with the formation of H2O2.
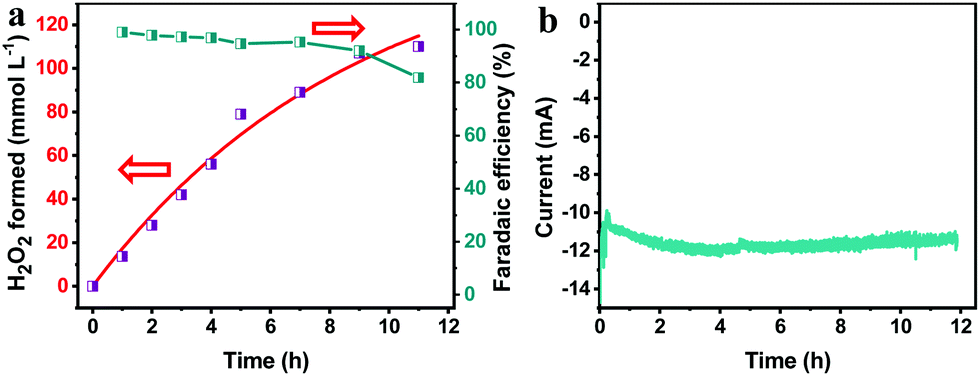 | ||
| Fig. 4 (a) Time course for H2O2 accumulation with the pTTh photocathode (9 cm2) and corresponding faradaic efficiency. The green line shows the faradaic efficiency. The red line is the fitting results using the equation [H2O2] = (Kf/Kd)[1 − exp(−Kdt)]. (b) The corresponding I−t curve measured at 0.65 V (vs. RHE) of pTTh for ORR. Electrolyte: 0.1 M KOH; illumination: 100 mW cm−2 Xe lamp with a solar simulator; counter electrode: Pt; reference electrode: SCE. | ||
To evaluate the competitive H2O2 reduction reaction involved in the production of H2O2, the LSV curves of pTTh/CP in Ar-saturated 0.1 M KOH solution containing various concentrations of H2O2 (0–100 mmol L−1) in the dark and under illumination were compared. Without exposure to light, the reduction of H2O2 is almost negligible (Fig. S8a, ESI†) in spite of the concentration being as high as 100 mmol L−1. Under illumination, the photo-reduction significantly increased with the increased concentration of H2O2. In the presence of 100 mmol L−1 H2O2, the photocurrent reached ca. −0.12 mA cm−2 at a bias of 0.65 V (Fig. S8b, ESI†), accounting for only 15% of the photocurrent generated by the ORR (Fig. S8c, ESI†). This results in the consecutive accumulation of H2O2 with a only slight decrease in faradaic efficiency.
In O2-saturated solution, the actual kinetics can be regarded as zero-order owing to the constant concentration of the dioxygen. For the decomposition of H2O2 by the reduction reaction, it commonly follows the first-order kinetics related to the concentration of H2O2 (denoted as [H2O2]). Therefore, the kinetic equation for PEC synthesis of H2O2 can be expressed as
| [H2O2] = (Kf/Kd)[1 − exp(−Kdt)] |
Unbiased H2O2 production by coupling with the photoanode
The ultimate goal of photosynthetic H2O2 production is to make efficient and stable overall unbiased H2O2 generation devices in a cost-effective way. To demonstrate the remarkable performance of the pTTh photocathode, we constructed a bias-free overall H2O2 production system by wiring it to a BiVO4 photoanode with a parallel illumination configuration (Fig. 5a). BiVO4 is one of the most promising photoanode materials with intriguing properties for the PEC water oxidation reaction.37,38 The energy levels of BiVO4 were estimated by the UV-vis and the Mott–Schottky analysis (Fig. S10, ESI†). The schematic energy diagram and the photogenerated carrier transfer processes are presented in Fig. S11 (ESI†). Owing to the difference between the Fermi levels of the n-type BiVO4 and p-type pTTh, the PEC system produces spontaneously an interior bias between the two photoelectrodes equal to the open-circuit voltage (VOC). To facilitate the interfacial charge transfer, a thin NiFeOx layer was photo-deposited on the BiVO4 surface as a cocatalyst (Fig. S12, ESI†).39 The resulting NiFeOx/BiVO4 photoanode exhibited a much earlier onset potential and remarkably higher photocurrent density (2.5 mA cm−2) at 1.23 V compared with pristine BiVO4 (Fig. S13, ESI†).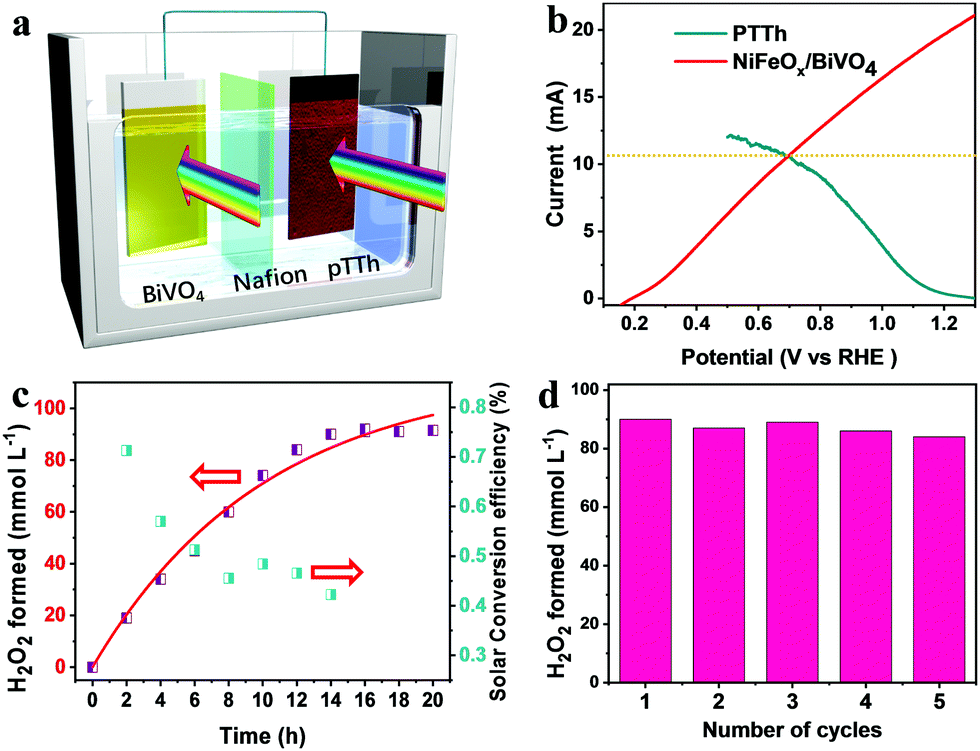 | ||
| Fig. 5 (a) Illustration of the unbiased H2O2 production device, with pTTh as the photocathode and NiFeOx/BiVO4 as the photoanode in 0.1 M KOH and 1 M borate buffer electrolytes, respectively. The setup of the PEC cell is in parallel illumination mode. (b) Intersection of the NiFeOx/BiVO4 photoanode and pTTh photocathode. (c) Time course for H2O2 generation under simulated 100 mW cm−2 Xe illumination and corresponding solar-to-H2O2 efficiency. (d) Concentrations of H2O2 produced with the two-electrode system during repeated operation cycles. | ||
Fig. 5b displays the LSV curves of the pTTh/CP photocathode and the NiFeOx/BiVO4 photoanode in 0.1 M KOH solution and 1M potassium borate buffer, respectively. The working area of both photoelectrodes is 9 cm2 (Fig. S14, ESI†). An intersection point between the two curves corresponds to a current of 10.2 mA. This result predicts that the NiFeOx/BiVO4–pTTh/CP dual-photoelectrodes in a PEC cell are able to self-drive simultaneous H2O oxidation on the photoanode and O2 reduction on the photocathode only with light illumination,40,41 although there is a pH mismatch between the photoanode and photocathode. Meanwhile, prolonged testing of the NiFeOx/BiVO4 photoanode exhibits a stable photocurrent over 20 h under illumination (Fig. S15, ESI†). The time course for the unbiased production of H2O2 is shown in Fig. 5c. As expected, the concentration of H2O2 increases linearly with time and reaches the maximum ∼90 mmol L−1 after 14 h of illumination. The corresponding solar energy conversion efficiency was determined to peak at ca. 0.34% after 2 h of illumination. Recyclability tests revealed that the system can be used for stable H2O2 generation for 5 cycles without any significant decay. Meanwhile, no detectable changes in the morphology and structure for the NiFeOx/BiVO4 electrode (Fig. S16, ESI†), and no changes in the morphology of pTTh (Fig. S17, ESI†) were observed.
The produced alkali HO2− could be directly used in certain applications, for example, pulp and paper bleaching.42 Of note, the produced HO2− is sufficiently concentrated to be applied in an H2O2 fuel cell for electricity generation. With Ni foam as the anode and Prussian blue/carbon paper as the cathode, the H2O2 fuel cell delivered an open circuit potential and a maximum power density of 0.6 V and 2.7 mW cm−2 (Fig. S18, ESI†), respectively. Combining the solar-driven H2O2 production and the H2O2 fuel cell for electricity generation is thus capable of addressing the intermittency of solar irradiation.
Mechanistic understanding for O2 activation
Photoelectrocatalysis is usually regarded as the light-excited electrocatalysis on a semiconducting photoelectrode.43,44 To elucidate the role of light and identify the electrochemical behavior of pTTh, the electrochemical tests were carried out under identical conditions except that they were conducted in the dark. The results showed that the pTTh exhibits a similar 2e− selectivity of the electrocatalytic ORR in 0.1 M KOH to that under illumination (Fig. S19, ESI†). An early onset potential of 0.71 V relative to the 2e− pathway was observed with a low overpotential of 30 mV (Fig. S20, ESI†), although the current density was lower. Furthermore, a similar behavior of pH-dependence was found in which both the electrocatalytic activity and onset potential were enhanced with increasing pH (Fig. S21, ESI†). Meanwhile, the 2e− selectivity for H2O2 production at different pH values remained almost the same between the PEC and electrochemical processes (Fig. S22, ESI†). Based on these results, it can be concluded that pTTh behaves indistinguishably when employed as an electrocatalyst or as a photoelectrode, with the selectivity primarily determined by the pH, independent of whether the electrode is driven by applied potential or light irradiation. Therefore, the electrochemical properties of pTTh are responsible for the high selectivity of H2O2 generation in the basic environment.We carried out density functional theory (DFT) calculations to gain insight into the mechanism and the high selectivity of the 2e− route in the electrochemical production of H2O2 on pTTh in 0.1 M KOH solution. Preliminary calculations indicated that relative energies of the reaction intermediates with a monomer of terthiophene (TTh) were similar to those with a dimer or a trimer of TTh. We used the monomer model (three thiophene rings) for the complete characterization of the reaction steps and the energy profile of the ORR for both thermodynamic and kinetic analysis. Note that the TTh and, for that matter, pTTh prefer a trans configuration in the orientation of the S atoms over a cis configuration. Fig. 6a shows the energy profile of the ORR derived from the Gibbs free energies of the proposed reaction steps (Table S2, ESI†) and Fig. 6c depicts the corresponding reaction steps. It was found that the binding of a dioxygen molecule to the neutral TTh is endothermic both in the gaseous phase and the aqueous phase, while attachment of an electron to TTh is exothermic by ∼2.44 eV in an aqueous phase (ΔG = ∼−2.44 eV). Thus, reduction of pTTh via electron attachment to form the negative ion pTTh− is regarded as the first step (S0 → S1). Addition of an O2 molecule to the pTTh− leads to the formation of a peroxy anion pTTh–O2− (S1 → S2), a step that is exothermic by ∼0.82 eV (ΔG = ∼−3.26 eV). The addition occurs at the α-CH site of a terminal thiophene ring of TTh as the structure favors increased stabilization due to π electron conjugation effects (Fig. S23 and Table S3, ESI†). The peroxy anion abstracts a proton from a water molecule in the alkaline solution while accepting an electron in a proton-coupled step “pTTh–OO− + H2O + e− ⇌ pTTh–OOH− + OH−” (S2 → S3) that is exothermic by ∼3.19 eV and yields a stable intermediate, specifically a pTTh–OOH− species (relative energy ΔG ∼ −6.45 eV). The next step is the cleavage of the CO–OH bond to evolve a hydroxide ion OH− (green lines) or cleavage of the C–OOH bond to evolve a peroxide ion HO2− (red lines), which is the branching point for the 2e− and 4e− pathways (S3 → S0 for the 2e− route or S3 → S4 for the 4e− route). Either product requires one or more steps (protonation to form H2O2 in the 2e− route or a reaction with H2O in the 4e− route). C–OOH bond cleavage leads to the recovery of the starting pTTh. In contrast, CO–OH bond cleavage results in the generation of epoxide pTTh![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O which requires further reduction to evolve the second water molecule.
O which requires further reduction to evolve the second water molecule.
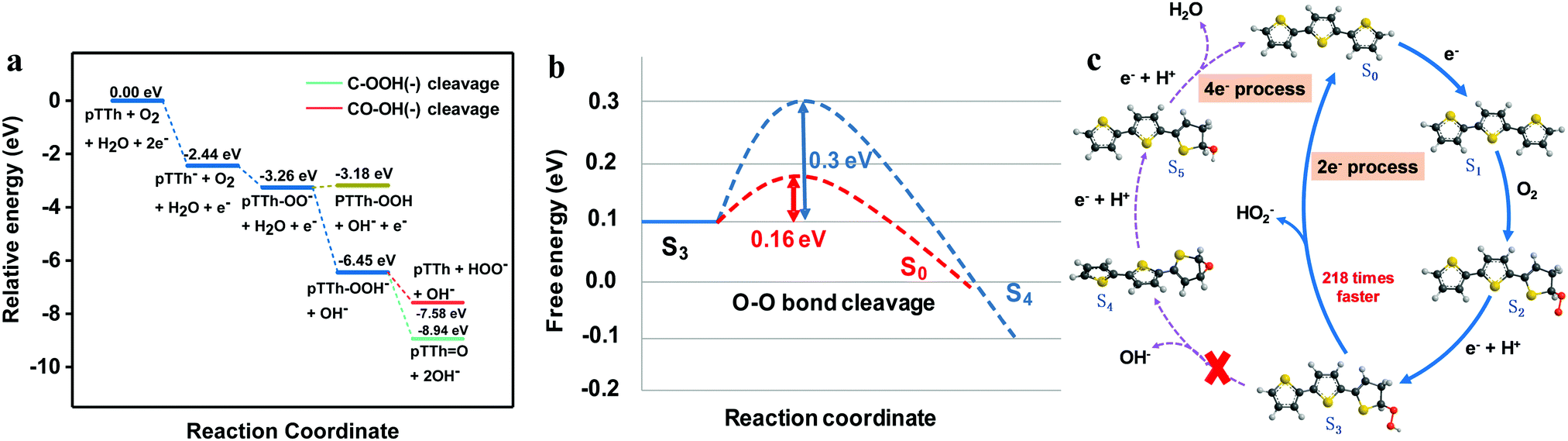 | ||
| Fig. 6 (a) Energy profiles of key possible ORR pathways on pTTh (the energy profile for the overall mechanism is shown in Fig. S24, ESI†); (b) free energy diagram of the branching point for 2e− and 4e− selectivity of the ORR at pH = 13. The blue dashed line represents the O–O bond cleavage pathway, and the red dashed line represents the C–O bond cleavage. (c) Proposed reaction cycles of H2O2 production with the energetically most feasible active sites. S0 is the bare surface, Sx (x = 1, 2, 3, 4, 5) are the structures of the intermediate states involved in the ORR. Red, gray, yellow and black balls represent oxygen, hydrogen, sulphur and carbon, respectively. | ||
Both pathways of peroxide anion HO2− formation and hydroxide anion OH− formation from the same pTTh–OOH− precursor are thermodynamically favorable, exothermic by ∼1.13 eV and ∼2.49 eV, respectively. To establish selectivity required characterizing the kinetics of the two pathways, by determining the transition state structures (Fig. 6b) and applying transition state theory. The transition states (TS) for the C–OOH and CO–OH bond cleavage steps were characterized with the smaller basis set (cc-pVDZ). We then carried out single-point energy evaluations with the larger aug-cc-pVTZ basis set. We verified that the TS's have one and only one vibrational mode with an imaginary frequency (∼330i cm−1 for CO–OH cleavage and ∼233i cm−1 for C–OOH cleavage). At the TS for C–OOH cleavage, the C–OOH distance is ∼1.82 Å while at the TS for CO–OH cleavage, the CO–OH distance is somewhat shorter ∼1.74 Å. After correcting for zero-point vibrational energy, the energy barriers for the cleavage of C–OOH and CO–OH were determined to be ∼+0.16 eV and ∼+0.30 eV (Fig. 6b), respectively. According to transition state theory where the reaction rate k = A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) exp(−ΔG*/kBT) (ΔG* is the energy barrier), the reaction rate for peroxide ion formation (S3 → S0 with ΔG* ∼ +0.16 eV) is ∼218 times faster than the rate for hydroxide ion formation (S3 → S4 with ΔG* ∼ 0.30 eV). Based on the above analysis, we proposed a reaction mechanism in which the 2e− pathway is kinetically favored over the 4e− pathway, leading to the high selectivity for H2O2 production (Fig. 6c).
exp(−ΔG*/kBT) (ΔG* is the energy barrier), the reaction rate for peroxide ion formation (S3 → S0 with ΔG* ∼ +0.16 eV) is ∼218 times faster than the rate for hydroxide ion formation (S3 → S4 with ΔG* ∼ 0.30 eV). Based on the above analysis, we proposed a reaction mechanism in which the 2e− pathway is kinetically favored over the 4e− pathway, leading to the high selectivity for H2O2 production (Fig. 6c).
When considering the effects of pH on the energy profile, the relative energies associated with individual steps in Fig. 6a change only when the number of hydroxide ions changes in a given step. Step S3 → S0 conserves the number of hydroxide ions and pH does not affect its relative energy (exothermicity). In contrast, step S3 → S4 generates one extra hydroxide. The exothermicity of the step increases by δ(ΔE) ∼ 2.3kBT × δ(pH) or δ(ΔE) ∼ 0.059 eV/(unit of pH). From the Bells–Evans–Polanyi rule, an increase in exothermicity leads to a decrease in energy barrier by about half of the increase in exothermicity. To lower the barrier of hydroxide formation (4e− pathway) by ∼0.14 eV requires a change in pH of ∼4.7 pH units. At pH ∼ 9.3 the 4e− pathway would be predicted to be competitive with the 2e− pathway, and is then preferred at lower pH. This finding is broadly in accord with experiment, showing that the proposed mechanism is consistent with the pH dependence observed experimentally.
Conclusions
In summary, a metal-free organic polymer of pTTh enables the synthesis of solar H2O2 in an efficient way by photoelectrochemical reduction of dissolved O2 in alkaline solution. Under a constant bias of 0.65 V (vs. RHE) with visible light irradiation for 11 h, a H2O2 concentration as high as 110 mmol L−1 is achieved, which outperforms previously reported photosynthesis routes by ca. 2 orders of magnitude. Furthermore, a NiFeOx/BiVO4 photoanode wired with the pTTh/CP photocathode enables the bias-free dual-photoelectrode synthesis of solar H2O2 of concentration ∼90 mmol L−1 for several cycles without any noticeable decay. The high selectivity of the pTTh photoelectrode towards 2e− ORR originates from the intrinsic electrochemical properties of pTTh regardless of the incident light. Theoretical calculations suggest that the selectivity-determining step for the 2e− process is over ∼200 times faster than that for the 4e− pathway. This study provides a green, low-cost and efficient route to synthesise H2O2 from water, air and sunlight, which demonstrates promising generation of liquid solar fuel for future applications.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the DICP & QIBEBT (DICP&QIBEBT UN201709), and National Natural Science Foundation of China (No. 21573230, 51971094, 21761142018), and the National Key Research and Development Program of China (No. 2017YFA0204804, No. 2017YFB0102900), the Pioneer Initiative (B) Project of CAS (No. XDB17030200). XW and MD gratefully acknowledge partial start-up support from the University at Buffalo.References
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed.
- K. Dohyung, S. K. Kelsey, H. Dachao and Y. Peidong, Angew. Chem., Int. Ed., 2015, 54, 3259–3266 CrossRef PubMed.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- J. H. Kim, D. Hansora, P. Sharma, J.-W. Jang and J. S. Lee, Chem. Soc. Rev., 2019, 48, 1908–1971 RSC.
- S. Kato, J. Jung, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2013, 6, 3756–3764 RSC.
- Y. Jiang, P. Ni, C. Chen, Y. Lu, P. Yang, B. Kong, A. Fisher and X. Wang, Adv. Energy Mater., 2018, 8, 1801909 CrossRef.
- K. Sayama, ACS Energy Lett., 2018, 3, 1093–1101 CrossRef CAS.
- X. Shi, S. Siahrostami, G.-L. Li, Y. Zhang, P. Chakthranont, F. Studt, T. F. Jaramillo, X. Zheng and J. K. Nørskov, Nat. Commun., 2017, 8, 701 CrossRef PubMed.
- C. Xia, Y. Xia, P. Zhu, L. Fan and H. Wang, Science, 2019, 366, 226–231 CrossRef CAS PubMed.
- S. A. Mousavi Shaegh, N.-T. Nguyen, S. M. Mousavi Ehteshami and S. H. Chan, Energy Environ. Sci., 2012, 5, 8225–8228 RSC.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- J. H. Baek, T. M. Gill, H. Abroshan, S. Park, X. Shi, J. Nørskov, H. S. Jung, S. Siahrostami and X. Zheng, ACS Energy Lett., 2019, 4, 720–728 CrossRef CAS.
- H. Dotan, K. Sivula, M. Grätzel, A. Rothschild and S. C. Warren, Energy Environ. Sci., 2011, 4, 958–964 RSC.
- S. Siahrostami, G.-L. Li, V. Viswanathan and J. K. Nørskov, J. Phys. Chem. Lett., 2017, 8, 1157–1160 CrossRef CAS PubMed.
- K. Fuku and K. Sayama, Chem. Commun., 2016, 52, 5406–5409 RSC.
- M. Teranishi, S.-i. Naya and H. Tada, J. Am. Chem. Soc., 2010, 132, 7850–7851 CrossRef CAS PubMed.
- H.-i. Kim, O. S. Kwon, S. Kim, W. Choi and J.-H. Kim, Energy Environ. Sci., 2016, 9, 1063–1073 RSC.
- Y. Kofuji, Y. Isobe, Y. Shiraishi, H. Sakamoto, S. Tanaka, S. Ichikawa and T. Hirai, J. Am. Chem. Soc., 2016, 138, 10019–10025 CrossRef CAS PubMed.
- A. J. Hoffman, E. R. Carraway and M. R. Hoffmann, Environ. Sci. Technol., 1994, 28, 776–785 CrossRef CAS PubMed.
- M. Jakešová, D. H. Apaydin, M. Sytnyk, K. Oppelt, W. Heiss, N. S. Sariciftci and E. D. Głowacki, Adv. Funct. Mater., 2016, 26, 5248–5254 CrossRef.
- K. Mase, M. Yoneda, Y. Yamada and S. Fukuzumi, Nat. Commun., 2016, 7, 11470 CrossRef CAS PubMed.
- S. Ghosh, N. A. Kouamé, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P.-H. Aubert and H. Remita, Nat. Mater., 2015, 14, 505 CrossRef CAS PubMed.
- B. Zhang, S. Wang, W. Fan, W. Ma, Z. Liang, J. Shi, S. Liao and C. Li, Angew. Chem., Int. Ed., 2016, 55, 14748–14751 CrossRef CAS PubMed.
- B. Zhang, L. He, T. Yao, W. Fan, X. Zhang, S. Wen, J. Shi and C. Li, ChemSusChem, 2019, 12, 1026–1032 CrossRef CAS PubMed.
- B. Kolodziejczyk, O. Winther-Jensen, D. R. MacFarlane and B. Winther-Jensen, J. Mater. Chem., 2012, 22, 10821–10826 RSC.
- S. Fukuzumi, Y.-M. Lee and W. Nam, Chem. – Eur. J., 2018, 24, 5016–5031 CrossRef CAS.
- B. Zhang, W. Fan, T. Yao, S. Liao, A. Li, D. Li, M. Liu, J. Shi, S. Liao and C. Li, ChemSusChem, 2017, 10, 99–105 CrossRef CAS.
- K. Schwinghammer, M. B. Mesch, V. Duppel, C. Ziegler, J. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2014, 136, 1730–1733 CrossRef CAS PubMed.
- Q. Li, B. W. Noffke, Y. Wang, B. Menezes, D. G. Peters, K. Raghavachari and L.-S. Li, J. Am. Chem. Soc., 2014, 136, 3358–3361 CrossRef CAS PubMed.
- K. J. J. Mayrhofer, D. Strmcnik, B. B. Blizanac, V. Stamenkovic, M. Arenz and N. M. Markovic, Electrochim. Acta, 2008, 53, 3181–3188 CrossRef CAS.
- H. Wang, Y. Liang, Y. Li and H. Dai, Angew. Chem., Int. Ed., 2011, 50, 10969–10972 CrossRef CAS PubMed.
- J. Weinstein and B. H. J. Bielski, J. Am. Chem. Soc., 1979, 101, 58–62 CrossRef CAS.
- G.-h. Moon, W. Kim, A. D. Bokare, N.-e. Sung and W. Choi, Energy Environ. Sci., 2014, 7, 4023–4028 RSC.
- S. Zhao, X. Zhao, H. Zhang, J. Li and Y. Zhu, Nano Energy, 2017, 35, 405–414 CrossRef CAS.
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef CAS.
- S. Ye, C. Ding, R. Chen, F. Fan, P. Fu, H. Yin, X. Wang, Z. Wang, P. Du and C. Li, J. Am. Chem. Soc., 2018, 140, 3250–3256 CrossRef CAS PubMed.
- K. Yongbo, J. Qingxin, N. Hiroshi, Y. Taro, K. Akihiko and D. Kazunari, Adv. Energy Mater., 2016, 6, 1501645 CrossRef.
- L. Pan, J. H. Kim, M. T. Mayer, M.-K. Son, A. Ummadisingu, J. S. Lee, A. Hagfeldt, J. Luo and M. Grätzel, Nat. Catal., 2018, 1, 412–420 CrossRef CAS.
- J. H. Kim, J.-W. Jang, Y. H. Jo, F. F. Abdi, Y. H. Lee, R. van de Krol and J. S. Lee, Nat. Commun., 2016, 7, 13380 CrossRef CAS PubMed.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- D. Li, J. Shi and C. Li, Small, 2018, 14, 1704179 CrossRef PubMed.
- E. Pastor, F. L. Formal, M. T. Mayer, S. D. Tilley, L. Francàs, C. A. Mesa, M. Grätzel and J. R. Durrant, Nat. Commun., 2017, 8, 14280 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ee02247c |
| This journal is © The Royal Society of Chemistry 2020 |