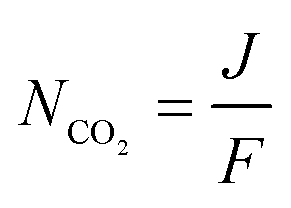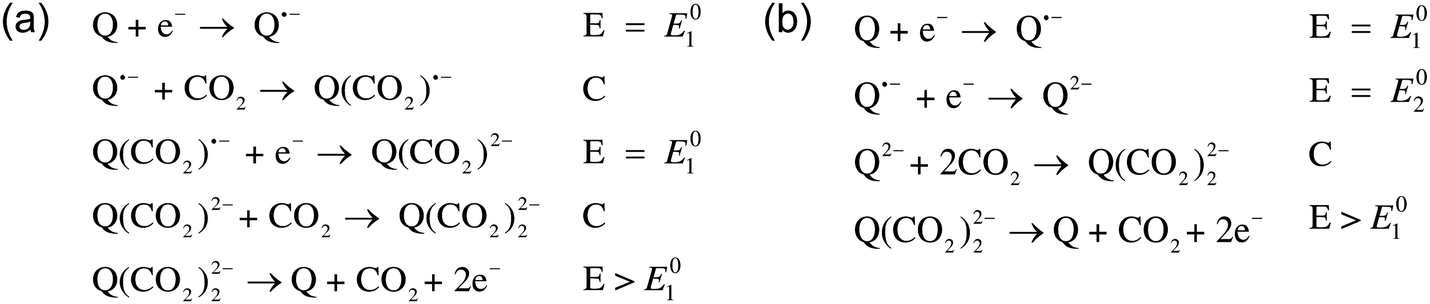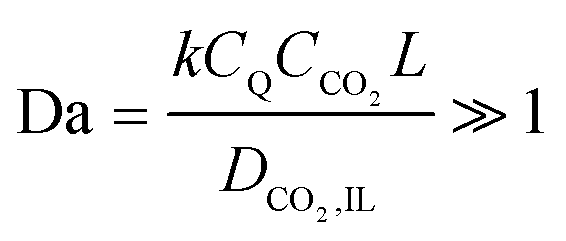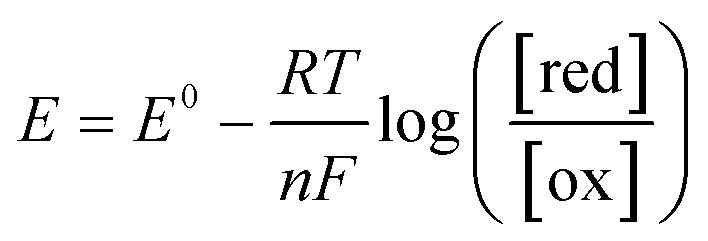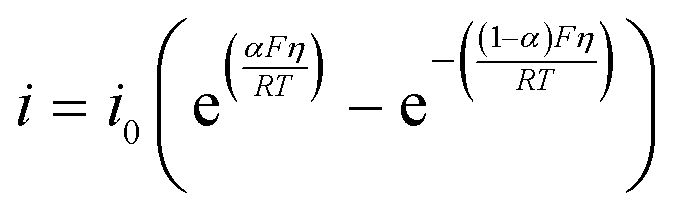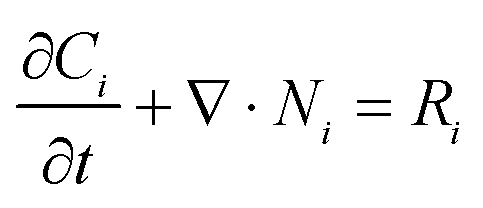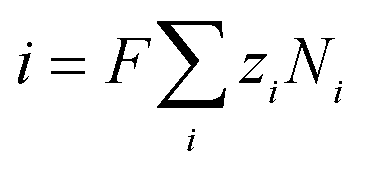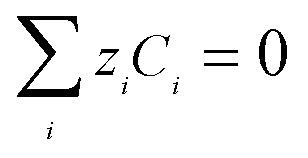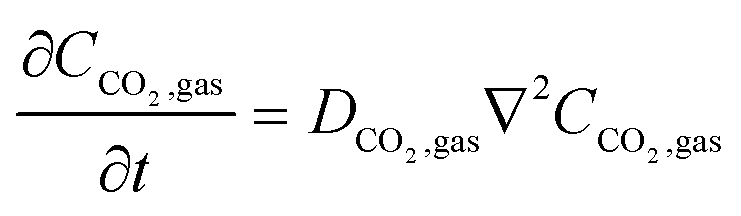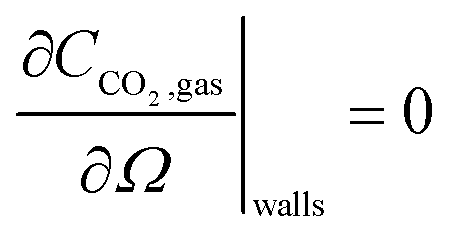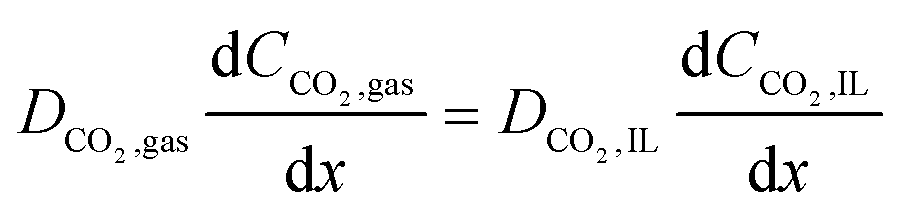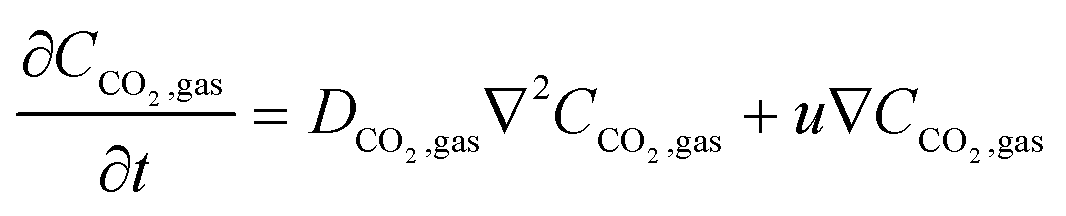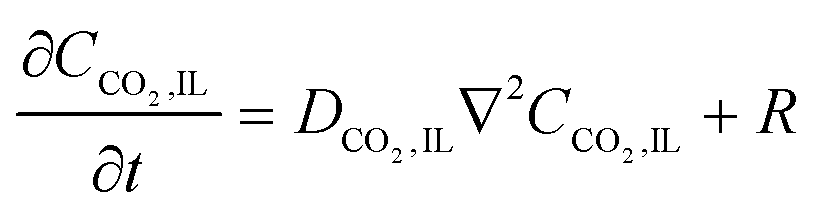Faradaic electro-swing reactive adsorption for CO2 capture†
Received
28th July 2019
, Accepted 30th September 2019
First published on 1st October 2019
Abstract
Carbon capture is one of the foremost methods for curtailing greenhouse gas emissions. Incumbent technologies are inherently inefficient due to thermal energy losses, large footprint, or degradation of sorbent material. We report a solid-state faradaic electro-swing reactive adsorption system comprising an electrochemical cell that exploits the reductive addition of CO2 to quinones for carbon capture. The reported device is compact and flexible, obviates the need for ancillary equipment, and eliminates the parasitic energy losses by using electrochemically activated redox carriers. An electrochemical cell with a polyanthraquinone–carbon nanotube composite negative electrode captures CO2 upon charging via the carboxylation of reduced quinones, and releases CO2 upon discharge. The cell architecture maximizes the surface area exposed to gas, allowing for ease of stacking of the cells in a parallel passage contactor bed. We demonstrate the capture of CO2 both in a sealed chamber and in an adsorption bed from inlet streams of CO2 concentrations as low as 0.6% (6000 ppm) and up to 10%, at a constant CO2 capacity with a faradaic efficiency of >90%, and a work of 40–90 kJ per mole of CO2 captured, with great durability of electrochemical cells showing <30% loss of capacity after 7000 cylces.
Broader context
There is a clear scientific consensus on the role of anthropogenic greenhouse gas (GHG) emissions in the unusually wild fluctuations of global climate patterns over the last century, to which carbon dioxide is the largest contributor. This has generated a scientific tide in the study of carbon removal techniques over the last three decades, which has slowly gathered momentum, culminating in the 2015 Paris Agreement, which heavily emphasized the role of carbon removal by tackling emissions or via negative emissions technologies. While a path forward for carbon capture is being shaped by a mix of incumbent actors and newcomers, the great majority of the proposed processes rely on conventional concepts. Moreover, most of these processes have a large balance of plant which hinders their potential to flexibly adapt to the multi-scale nature of carbon capture. Herein, we demonstrate a new approach which presents a paradigm shift in adsorption type processes and carbon capture. The electro-swing basis of our process allows its linear scaling with desired capacity and lends it geometric flexibility. It also allows carbon capture from streams of any concentration, and its release into any carrier streams, including pure CO2, with a low balance of plant.
|
Introduction
With the alarming increase in the atmospheric concentration of carbon dioxide (CO2) and its implications for global climate pattern developments,1,2 mitigation of climate change through curtailment of anthropogenic CO2 emissions has been one of the most urgent socioeconomic and scientific problems in the global arena over the last decade.3 To this end, a number of technologies have been developed for the large-scale capture of CO2 from combustion and other industrial processes to produce high-purity CO2 streams for storage or valorization.4 The most mature of these technologies are solvent scrubbing, mainly amine scrubbing,5 and oxyfuel combustion,6 which target high CO2 concentration streams (>10%). These approaches have a large footprint and, when retrofitted to a process, can require major modifications to the plant. Consequently, there has been a major effort to develop new materials and processes for high efficiency CO2-capture, including sorbents for pressure and temperature swing adsorption systems,7 and membranes for selective transport of the CO2.8 Furthermore, many potential applications of carbon capture require compact devices due to space limitations, such as in the direct capture of CO2 from tailpipe exhausts on board mobile sources, in which there is growing interest given the large contribution of transportation exhaust to greenhouse gas emissions (33.5% of U.S. CO2 emissions in 2016).9
In addition to the capture of CO2 from direct combustion processes, there is a need to remove CO2 from enclosed spaces for ventilation purposes in buildings and car cabins, or for cabin environmental control systems on board spacecraft and submarines, where the maximum allowed CO2 concentration in habitable spaces is 5000 ppm (or 0.5%).10 The first of such systems was developed by Winnick et al., for the electrochemical capture of CO2 in spacecraft cabins using molten carbonates.11 However, the low concentration of CO2 in such applications poses a challenge, mainly due to the low driving forces for mass transfer and the large quantities of other species present in air in addition to CO2.12 Thus, carbon capture is a multi-scale problem, where the CO2-rich streams to be treated vary greatly in volume, concentration and composition, and different criteria need to be fulfilled to ensure optimal processing depending on whether sources are industrial or small-scale (e.g., power plants or oil and gas heaters), concentrated or dilute (exhausts from combustion or air in confined spaces), and clean or contaminated with other pollutants.
Many of the CO2-capture chemical processes that involve a capture agent such as amines or solid sorbents require temperature and/or pressure swings to release the captured CO2 and regenerate the agents for further capture. These swings result in inefficiencies due to energy wasted in heating solvents and sorbents, pressurizing feed gas, or drawing a vacuum for desorption. Electrochemical systems can minimize such parasitic energy losses as they can be operated at near isothermal conditions, with significantly higher efficiencies than their thermal-swing (TSA) and pressure-swing (PSA) adsorption counterparts.13 One mode of electrochemical capture of CO2 is through the use of a redox-active carrier.
Electrochemically mediated selective transport of chemical species was first reported by Ward et al.,14 where a redox-active carrier (ferrous ion) was used to transport nitric oxide across a membrane. Since then, a number of systems have been developed for transporting chemical species by redox-active carriers that are activated at one electrode, to bind with the target species, and deactivated at the opposite electrode, to release the target and regenerate the carrier.15,16 Systems that have been proposed for the concentration of CO2 through this approach have been based on a number of different carrier molecules, such as quinones,17–20 4,4′-bipyridine,21 and thiolates.22,23 Quinones are of particular interest to this work for their superior electrochemical performance, serving as redox-active carriers for CO2 in electrochemically mediated separation processes. DuBois et al. demonstrated this possibility, and studied the thermodynamics of an electrochemical CO2 pumping system that utilizes quinones.18 More work followed, where Scovazzo et al. demonstrated the electrochemical separation of CO2 from <1% concentration gas mixtures using 2,6-di-tert-butyl-1,4-benzoquinone as a carrier in ionic liquid (IL) and organic solvent electrolytes media,19 while Gurkan et al. screened a number of ILs to serve as suitable electrolytes for quinone carriers in an electrochemically mediated selective transport system for CO2.20 All of these systems, however, require the transport of the electrolyte and the dissolved carrier molecules between two electrodes in an electrochemical cell for capture and release of CO2. This limits their implementation in a number of applications where the requirement for flow systems and pumping, and the large footprint, are problematic.
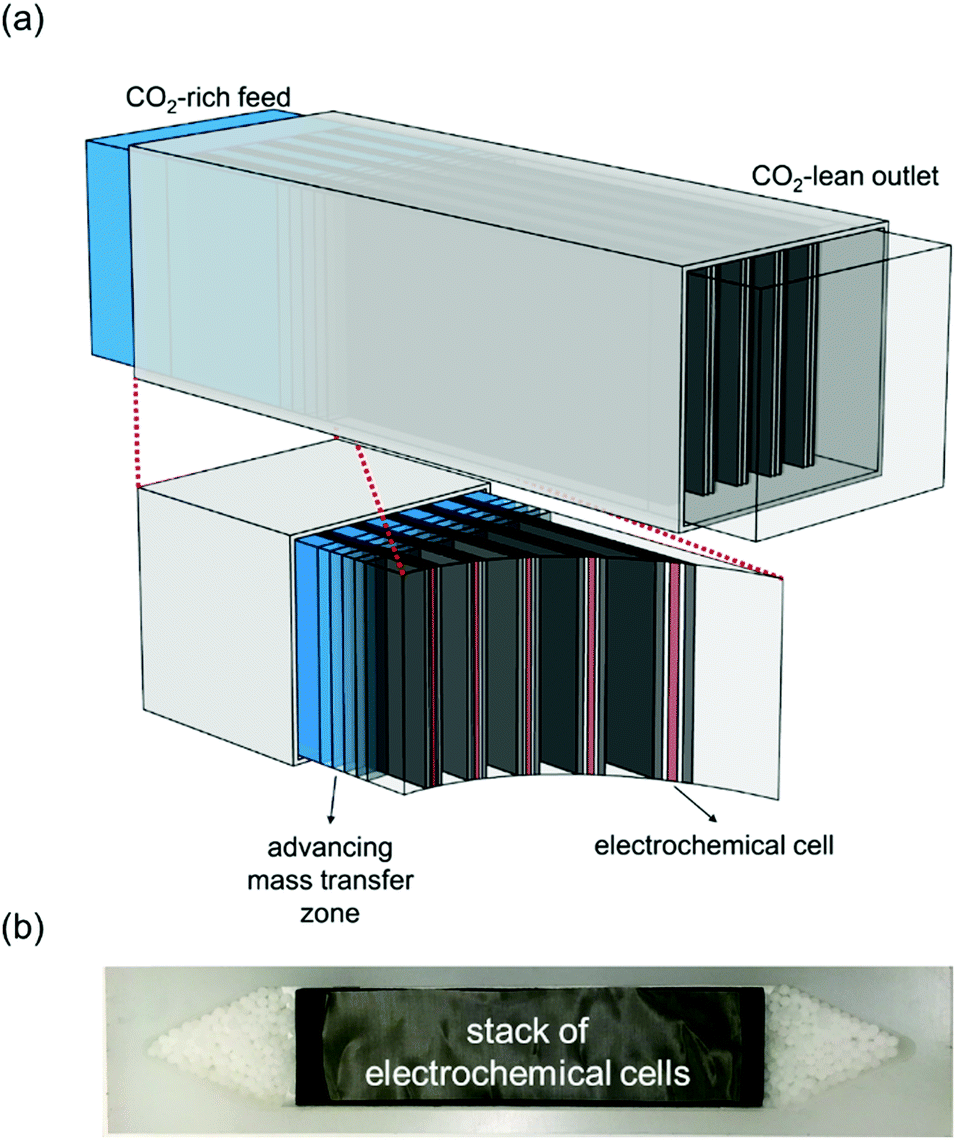
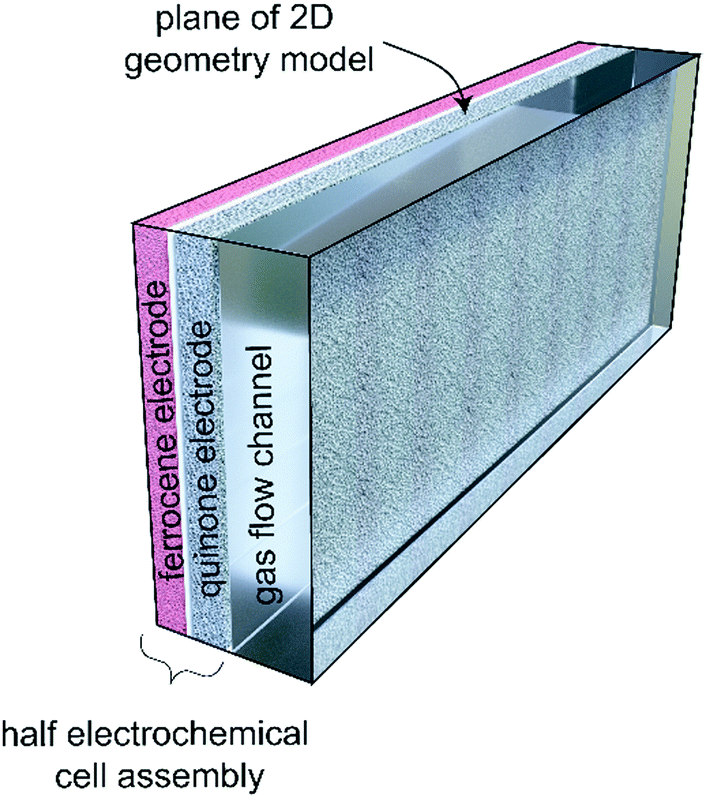
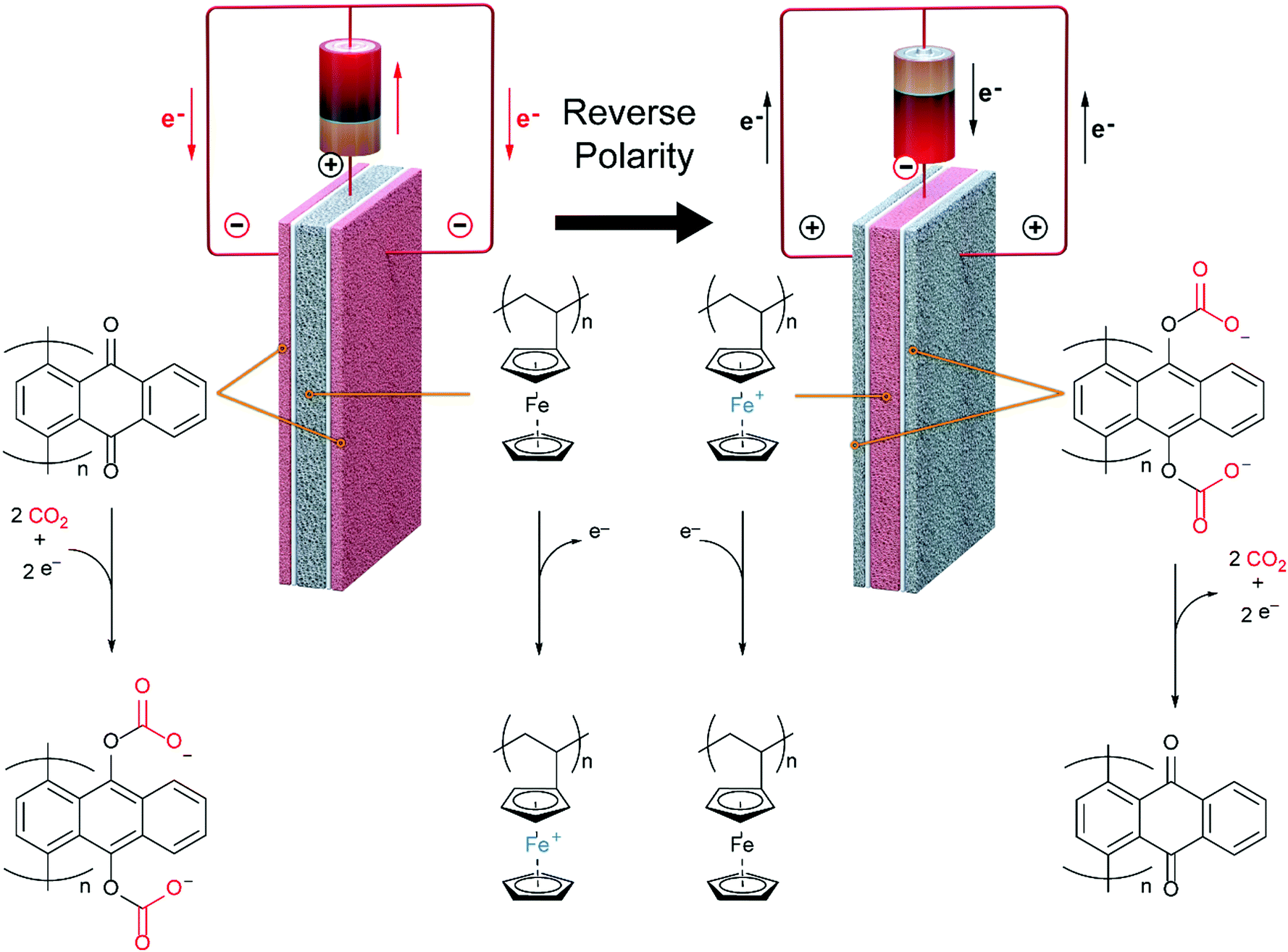
In this work, we develop a compact electrochemical device for CO2 capture that eliminates the requirement for complex flow systems, and that can be effective in the treatment of both high and low concentration feed streams. A quinone-utilizing faradaic electrochemical cell that fulfils these requirements and operates as an Electro-Swing Adsorption (ESA) process is illustrated schematically in Fig. 1. The cell is comprised of two cathode electrode substrates coated with a CO2-binding quinone–carbon nanotube (Q–CNT) composite sandwiching an anode electrode substrate coated with a ferrocene–CNT (Fc–CNT) composite, with separator membranes between the electrodes. This cell architecture is employed to maximize the CO2-binding surface area of the cell exposed to gas flow in a parallel passage adsorbent contactor design where stacks of these cells form parallel gas channels. The Fc–CNT electrode serves as an electron source and sink for the reduction and oxidation, respectively, of the Q–CNT electrodes to regulate the uptake and release of the CO2. Wetting of porous non-woven carbon fiber mats used as the electrode substrates by a room temperature ionic liquid (RTIL) electrolyte enables effective ionic currents to pass through the electrolyte on activation and deactivation of the electrodes, and permits the diffusion of CO2 into the electrolyte-wetted cathodes during capture.
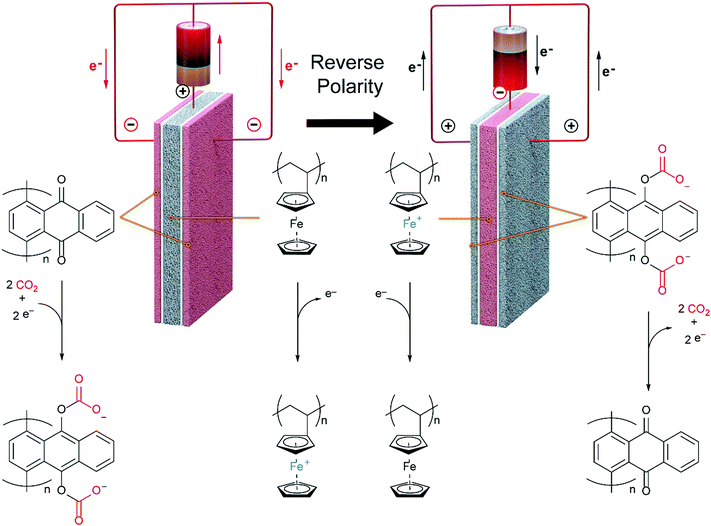 |
| | Fig. 1 Schematic of a single electro-swing adsorption electrochemical cell with porous electrodes and electrolyte separators. The outer electrodes, coated with poly-1,4-anthraquinone composite, can capture CO2 on application of a reducing potential via carboxylation of quinone, and release the CO2 on reversal of the polarity. The inner polyvinylferrocene-containing electrode serves as an electron source and sink for the quinone reduction and oxidation, respectively. | |
The effectiveness of the electro-swing concept for CO2 capture and release was demonstrated in a sealed system in which cell polarity was cycled to regulate the activation and deactivation of the carrier, i.e. reduction and oxidation of the redox active quinone. This was in lieu of the advection of the carrier electrolyte solution between two electrodes with a difference in potential.19 The performance of flow-through modules constructed with stacks of the assembled electrode cells was also assessed. The CO2 capture processes were modelled to provide greater insight into the operational principles for this technology.
Ultimately, the goal of this work was to demonstrate an electrochemically activated packed parallel passage contactor adsorption system for the removal of CO2 from flue gas and other CO2-rich streams. The regeneration of the bed takes place on reversal of the polarity of the stack in an ESA, with analogies to PSA systems, where adsorption takes place at high pressure and desorption at a lower pressure,24 or TSA systems, where adsorption takes place at low temperatures and the fixed bed is regenerated at higher temperatures.25
Results and discussion
Design of the electrochemical cell
Thin films of crystallized organic compounds have been studied as a means of immobilizing CO2 redox-active carriers onto electrode substrates.26 Redox-active polymers have long been the preferred electrode material when using organic molecules in electrochemical cells, however, because polymers are less soluble in the electrolyte than their corresponding redox-active repeating units, and thus the self-discharge of the cell can be avoided, and its cycling performance improved.27 For example, quinone polymers have been investigated as suitable cathode materials in lithium-ion batteries,28 and pseudocapacitive energy storage devices.29 These polymers are usually made into composites with conductive high-surface area carbon materials, such as carbon black, graphite or carbon nanotubes (CNT), to enhance the overall conductivity of the electrode material and increase the area of the polymer exposed to the electrolyte.30 Here, we used a polyanthraquinone–CNT (PAQ–CNT) composite for the cathode and polyvinylferrocene–CNT (PVFc–CNT) composite for the anode.31 The electrolyte used was the RTIL 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([Bmim][TF2N]), selected for its low volatility, which allows for flow of gas over the wetted electrodes without loss of electrolyte to evaporation or entrainment.
A sealed chamber was constructed in which CO2 capture and release on electrochemical activation and deactivation of the quinones, respectively, was indicated by the concomitant pressure changes in the chamber. A flow cell was also fabricated to house stacks of the electrochemical cells to allow for capture of CO2 from flowing gas streams of compositions similar to those of flue gases, air in aircraft cabins and buildings, and in the ambient environment. The adsorption effectiveness was assessed through analysis of the breakthrough profiles.
The chemical specificity of quinones can provide near 100% purity of released CO2, which, when combined with a well-designed process in which 100% CO2 recovery is achieved, puts the technology much closer to the upper-right corner on the recovery-purity Pareto plot than those based on current CO2 sorbents.32,33
Polyanthraquinone electrodes.
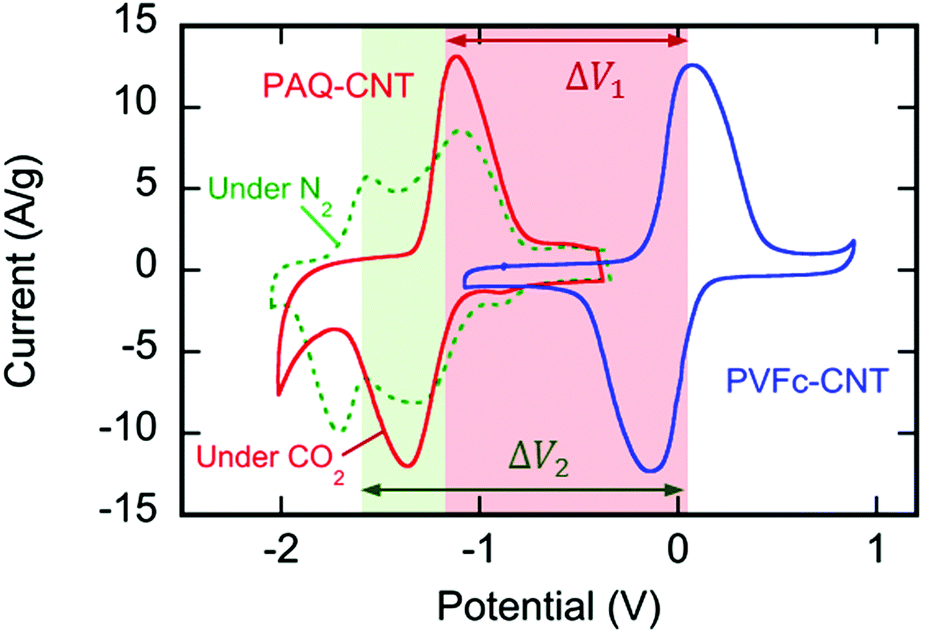
Poly(1,4-anthraquinone) (P14AQ), shown in Fig. 1, was synthesized and incorporated into an ink with multi-walled carbon nanotubes (MWCNT) in N-methyl-2-pyrrolidone (NMP). Non-woven carbon fiber mat electrode substrates of thickness 150 μm (35 g m−2) were dip-coated in the ink and dried very slowly to obtain a composite that uniformly coated the substrate at ∼1.2 mg cm−2, shown in Fig. 2(a). The composite structure allows for a greater utilization of the anthraquinone repeating units in the polymers, and thus larger currents under applied potentials. The CNT mesh, shown in Fig. 2(f), provides a backbone to improve the conductivity of the conjugated PAQ via π–π interactions with the anthraquinone units (Fig. S3, ESI†); it also provides a porous matrix decorated by the polymer chains for better electrolyte penetration. This results in a larger surface area of the polymer being exposed to electrolyte, and an increased conductivity in the composite.34 The π–π interaction of the polymer chains and CNTs, and the subsequent decoration of the nanotubes with the polymer chains, shown in Fig. 2(e), also improved the suspension of the CNTs in the polymer solution, which resulted in very well dispersed inks. The ratio of PAQ to CNT was optimized, with the highest current per mass of active material (polymer) obtained at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, PAQ:CNT ratio by weight (Fig. S4, ESI†). The cyclic voltammograms in Fig. 3 shown in green (under N2) and red (under CO2) indicate the strong redox activity of these electrode surfaces; the differences in the current response following potential scans are discussed later.
:3, PAQ:CNT ratio by weight (Fig. S4, ESI†). The cyclic voltammograms in Fig. 3 shown in green (under N2) and red (under CO2) indicate the strong redox activity of these electrode surfaces; the differences in the current response following potential scans are discussed later.
 |
| | Fig. 2 (a) SEM micrograph of the cathode non-woven carbon mat coated with P14AQ–CNT, with details of coated and uncoated areas. (b, c and f) SEM micrographs of increasing magnification of carbon fibers coated with P14AQ–CNT. (d) SEM micrograph of the uncoated carbon fibers. (e) TEM of PAQ–CNT showing the amorphous polyanthraquinone decorating the MWCNT, a result of the π–π interaction. | |
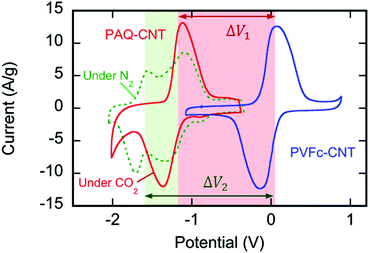 |
| | Fig. 3 Superimposed CVs of PVFc–CNT ( ) and P14AQ–CNT ( ) and P14AQ–CNT ( ) under N2 and ( ) under N2 and ( ) under CO2 in [Bmim][TF2N], at 20 mV s−1, vs. Fc, at T ∼ 21 °C. The two potential windows are shown; ΔV1 under CO2 and ΔV2 under N2. ) under CO2 in [Bmim][TF2N], at 20 mV s−1, vs. Fc, at T ∼ 21 °C. The two potential windows are shown; ΔV1 under CO2 and ΔV2 under N2. | |
Polyvinylferrocene electrode.
A counter electrode with a reduction potential more positive than the oxidative CO2 release potential of PAQ–CNT is required to make an electrochemical cell that can effectively capture and release CO2. Therefore, the anode electrode material for the electrochemical cell with PAQ–CNT cathode was a composite of polyvinylferrocene and CNT (PVFc–CNT), shown in Fig. 1. This was prepared by dispersing MWCNTs in a solution of PVFc in NMP. The optimum ratio of the composite was found to be 1:1, PVFc:CNT, by weight. The electrode substrate was dip-coated in the ink and dried slowly to yield a composite that uniformly coated the substrate at ∼1.4 mg cm−2, as shown in Fig. 4. The CVs of the PVFc–CNT electrode in N2 and CO2 saturated [Bmim][TF2N] are identical, and are indicated by the blue line in Fig. 3. PVFc–CNT has a reduction potential of E0 = −0.03 V, against ferrocene (Fc).
 |
| | Fig. 4 (a) SEM micrograph of the anode non-woven carbon mat coated with PVFc–CNT with details of coated and uncoated areas. (b and c) SEM micrographs of carbon fibers coated with PVFc–CNT, the squares indicates the region which is magnified in the next micrograph. (d) SEM micrograph of the magnified polymer-coated CNTs from a different area on the electrode. | |
The electrochemical cell.
The PAQ–CNT electrodes were assembled into an electrochemical cell in which CO2 was able to be captured and released upon potential swings. This was accomplished by layering a PVFc–CNT counter electrode between the two PAQ–CNT electrodes to balance the charge of these electrodes, as shown in Fig. 5(d). In a CO2-rich environment, this afforded a polymer-based battery with an electrochemical window of ∼1.2 V, where all the charge from the ferrocene oxidation, at its reduction potential, was balanced by the charge transferred to the two-electron reduction of quinone, at its first reduction potential. This window is defined by the reduction potential of PVFc–CNT and the first reduction potential of PAQ–CNT, shown in Fig. 3 as ΔV1. However, in the absence of CO2 or other such electrophiles, mainly under N2, PAQ retains its two one-electron reductions, with half of the charge of the ferrocene oxidation being balanced by the reduction of quinone at the first potential and the other half by its reduction at the second potential. This results in a wider electrochemical window, shown in Fig. 3 as ΔV2, and the behavior of the cell resembles that of an asymmetric pseudocapacitor.35
 |
| | Fig. 5 (a) Custom-made sealed chamber for closed system experiments with pressure transducer to monitor the changes in pressure as CO2 is adsorbed and desorbed upon cycling of the cell potential. The internal of the sealed chamber (b) with and (c) without the insulating cup. (d) Layers of the electrochemical cell assembled in the sealed chamber. | |
The capacities of the anode and cathodes of the cell, evaluated by gravimetric charge calibration of the two electrode composite materials (Fig. S5, ESI†), were balanced by requiring that mP14AQ–CNT = 1.12mPFVc–CNT, which allowed for some excess PVFc–CNT.
Carbon capture
Two different types of units were fabricated to demonstrate the effectiveness of the electro-swing process. The first was a sealed chamber into which the electrodes were inserted. Changes in pressure were measured as CO2 was adsorbed and then released on cycling of the potential applied to the electrodes. The second approach used a flow adsorption in which the CO2 exiting the device was monitored, with the breakthrough point reflecting the overall capacity of the electrodes. All carbon capture experiments were conducted at T ∼ 21 °C.
CO2 capture and release in a batch system.
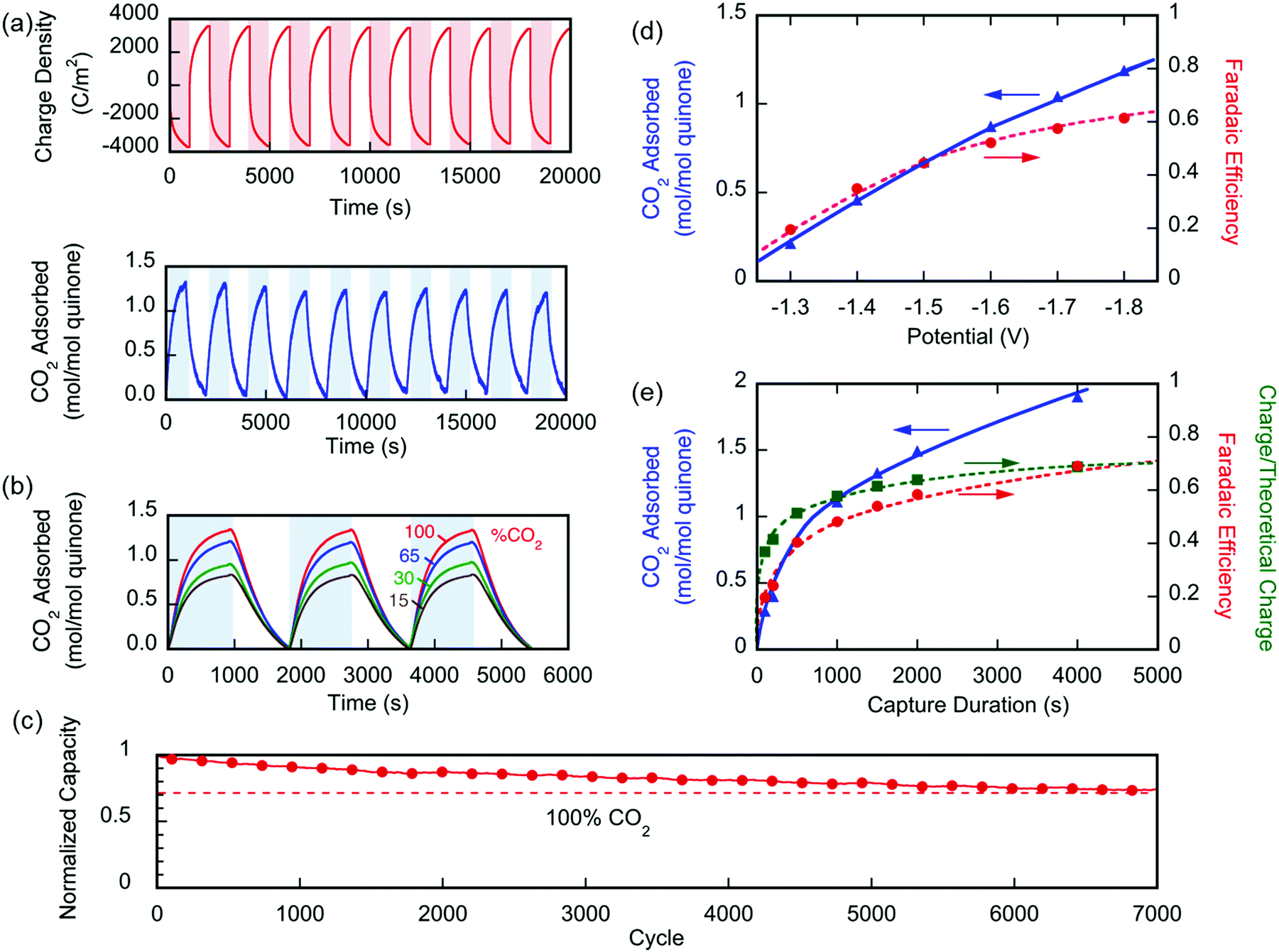
An electrochemical cell (4 cm2 of total exposed area) with the three layers, such as that illustrated in Fig. 1, was tested for the capture and release of CO2 in a sealed chamber, shown in Fig. 5. This cell had ∼4.8 mg of PAQ–CNT deposited, i.e. 5.8 μmol of quinone, or a capacity for 11.6 μmol of CO2. From the monitored pressure changes in the chamber of known internal void volume, the number of moles of CO2 captured and released upon charge and discharge of the cell at constant potentials, were calculated. The chamber was purged with CO2 at the desired concentrations and sealed at atmospheric pressure. The pressure inside was allowed to equilibrate at constant temperature before the electrochemical experiments were started.
The performance of the electrochemical cell was evaluated in chronoamperometric (potentiostatic) experiments, with capture at 1.3 V and release at 0.5 V, i.e. ΔV = 0.8 V. Fig. 6(a) shows the change in the number of moles of CO2 captured from pure gas over 10 cycles (blue), along with the charge transferred to and from the cell (red). It shows that ∼65% of the quinone units were utilized, i.e. 7.4 μmol of CO2 captured. This may be due to incomplete charging of the quinones that are not exposed to electrolyte. From the charge transferred during charging and discharge (∼800 mC), and the moles of CO2 captured and released, a faradaic efficiency
| | 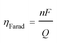 | (1) |
of up to 90% was estimated, with some of the charge lost to double layer charging of the porous electrodes. Here,
F is Faraday's constant,
Q is the charge transferred, and
n is the number of moles of useful electrons transferred in the reaction,
i.e. used for the capture of CO
2, which in this reaction, is equivalent to the number of moles of CO
2. This efficiency results in ∼86 kJ per mole of CO
2 captured and released, as determined by the relation
| | 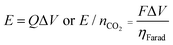 | (2) |
where Δ
V is the difference between the applied charge and discharge potentials.
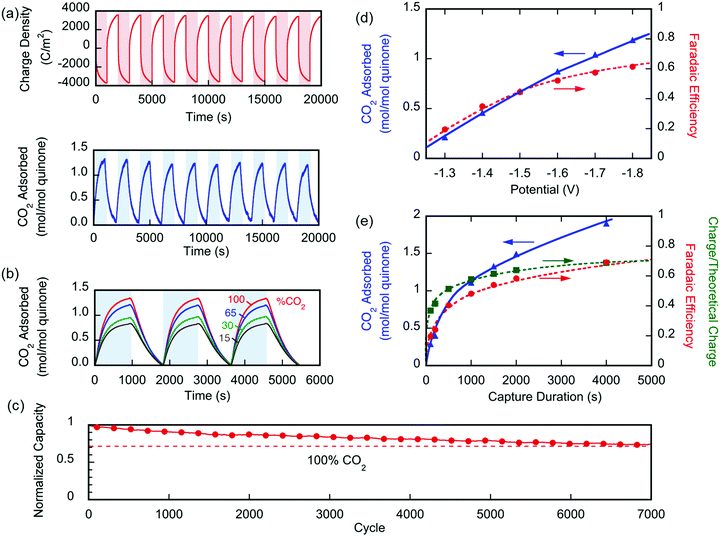 |
| | Fig. 6 (a) Changes in the number of moles of CO2 captured upon charging and discharge of the electrochemical cell over 10 cycles, normalized by the moles of quinone on the electrode ( ). The CO2 captured from and released to the chamber tracks the charge applied to the electrochemical cell, normalized by the area of the cell ( ). The CO2 captured from and released to the chamber tracks the charge applied to the electrochemical cell, normalized by the area of the cell ( ). (b) The CO2 captured under different feed concentrations. (c) Capacity of cell over 7000 cycles. In a different set of experiments using a larger cell and cavity, (d) shows the effect of varying charging potential for a 1000 s capture and (e) shows the effect of varying the capture duration at −1.8 V capture potential. These experiments were conducted at T ∼ 21 °C. ). (b) The CO2 captured under different feed concentrations. (c) Capacity of cell over 7000 cycles. In a different set of experiments using a larger cell and cavity, (d) shows the effect of varying charging potential for a 1000 s capture and (e) shows the effect of varying the capture duration at −1.8 V capture potential. These experiments were conducted at T ∼ 21 °C. | |
Fig. 6(b) shows similar results for the capture of CO2 from gases of different concentrations ranging between 15 and 100%. Again, quinone utilization was high, but dependent on the initial gas concentration, owing not to capacity limitations, but to a reduction on sorption dynamics due to mass transfer limitations with lower concentration driving forces. The electrochemical cell showed great stability with <30% loss in capacity over 7000 cycles of capture and release, Fig. 6(c). The gradual loss in capacity is mainly due to the electromigration of the shorter polymer chains, Fig. S1 (ESI†), from the electrode and into the electrolyte, towards the opposite electrode.
In a different set of experiments where a larger cell (∼450 cm2 of total exposed area) was used, CO2 transport limitations became more prominent due to the larger internal volume of the cavity used. In this set, the effect of applying an overpotential was studied, where capture potentials of greater than ΔV1 (in Fig. 3) were applied during a 1000 s capture experiment. Fig. 6(d) shows that upon increasing the applied capture potential more CO2 is captured for the duration of the experiment, which indicates the activation of more quinone units on the electrodes during the charging half-cycle. Charging the system at −1.8 V for varying durations, Fig. 6(e), shows that the system is fully charged after 2000 s, but more CO2 is captured, i.e. more of the quinones are utilized, at increasing durations of capture. This demonstrates the transport limitations of the electrodes, as explained in later sections.
CO2 capture and release in flow systems.
The adsorption bed assembled consisted of a few electrochemical cells stacked in the flow device to allow the flow of gas in between, as illustrated in Fig. 7(a). The blue regions indicate the presence of CO2, and the decrease in blue intensity within the channel indicates the adsorption front, ahead of which the bed is still empty, and behind which the bed, i.e. the electrode assembly, is saturated with CO2 and cannot adsorb more. A number of different cells were constructed in the flow bed configuration shown in Fig. 7(b). The exposed area of a typical cell was 18 cm × 10 cm, with ∼0.2 g of P14AQ–CNT composite; at ∼70% useful quinones, this amounted to the adsorption of ∼9–14 cm3 of CO2. The channels between the cells were typically ∼500 μm thick, and bed volumes were about 35 cm3 with a total bed weight of ∼1.5 g bringing the productivity of the bed to 0.01–0.02 kg CO2 per kgbedtb, where tb is the breakthrough time, i.e. the time to reach saturation of the electrodes or for the outlet CO2 concentration to begin to increase. At feed flow rates of ∼10 mL min−1 and 15% CO2, this productivity is on par with the TSA systems reported by Ntiamoah et al.36
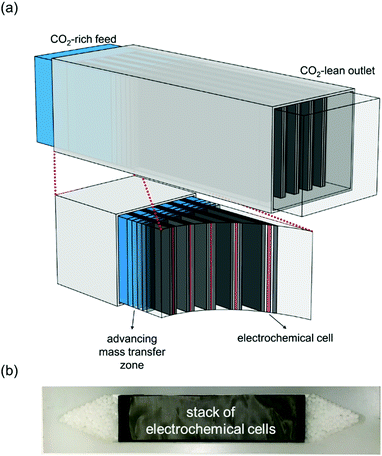 |
| | Fig. 7 (a) Schematic illustration of the parallel passage electrochemical cell contactor. The blue region indicates the saturated zone and the development of the mass transfer zone. (b) Photograph of a flow bed with a stack of the electrochemical cells. | |
It can be shown that axial flow dispersion within individual channels was minimal at most flow rates, and that the transport resistances resided primarily within the electrodes. Imperfect packing of the cells in the flow device by hand did, in some cases, lead to significant channeling and large-scale device axial dispersion, however, with broad breakthrough curves, but it is anticipated that under optimal manufacturing conditions these effects can be reduced significantly.
The chronoamperometric experiments performed in the sealed system were repeated in a flow system. Mixtures of N2 and CO2 at different compositions were flown past the stack of assembled electrochemical cells. Upon reduction of the PAQ–CNT electrodes, CO2 was removed (captured) from the inlet stream, and the decrease in the concentration of CO2 was detected at the outlet stream. In these experiments the bed was fully activated by charging at a constant potential greater than ΔV22 to guarantee the complete reduction of the all accessible quinone units prior to adsorption. Experiments with an initial half activation, i.e., at ΔV1, followed by adsorption at constant potential, did not result in different breakthrough curves, mainly because the kinetics of the reduction and the adsorption reactions are much faster than the rates of mass transfer of CO2 into the film, as discussed later.
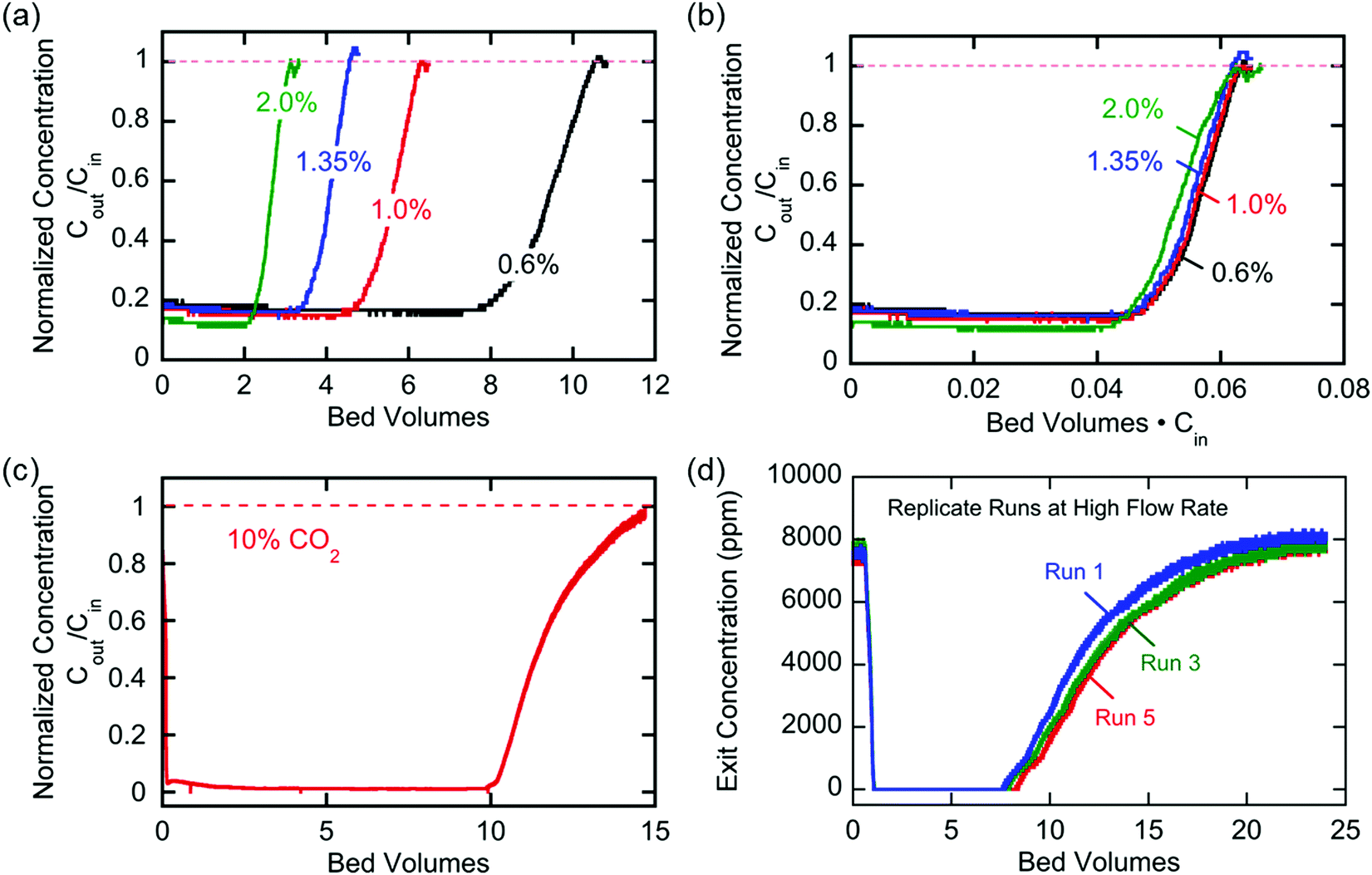
The breakthrough profiles obtained for one flow system are shown in Fig. 8(a). Breakthrough was achieved at smaller bed volumes of gas treated for higher feed CO2 concentrations, where the bed volume is a time measure which represents the total volume of gas treated at any given time relative to the volume of the bed itself. The breakthrough curves superimpose when normalized by the inlet CO2 concentration, shown in Fig. 8(b), which indicates a consistent behavior of the bed, i.e. equal utilization of the overall adsorption capacity regardless of the inlet concentration, an indication of a rectangular equilibrium isotherm. Note in this particular module that, although the breakthrough curves themselves were sharp, prior to breakthrough there was a consistent effluent concentration of 20% of the feed concentration, the result of a bypassing of the active cells by 20% of the total gas flow. The overall capacity of the bed can be calculated from the area between the line at Cout/Cin = 1 and the normalized breakthrough curve. A contactor bed of a desired overall capacity can be designed by linear scaling from this overall capacity. With a larger bed, CO2 was fully captured from a stream at 10% inlet concentration, which is on the higher end for carbon capture applications as it mimics combustion exhaust. Up to 10 bed volumes of feed were treated before breakthrough was achieved, indicating capacity of greater than one bed volume of pure CO2. It has similarly been shown that complete removal of CO2 with a smaller stack of cells (a scaled down contactor), five replicate capture experiments were conducted on an inlet stream of ∼0.8% CO2, Fig. 8(d), which mimics conditions for ventilation of confined living spaces, such as space modules, submarines and buildings. The breakthrough curves were unaffected by the prior history of the bed, indicating that complete regeneration and restoration of the bed capacity was attained during the desorption following each adsorption run.
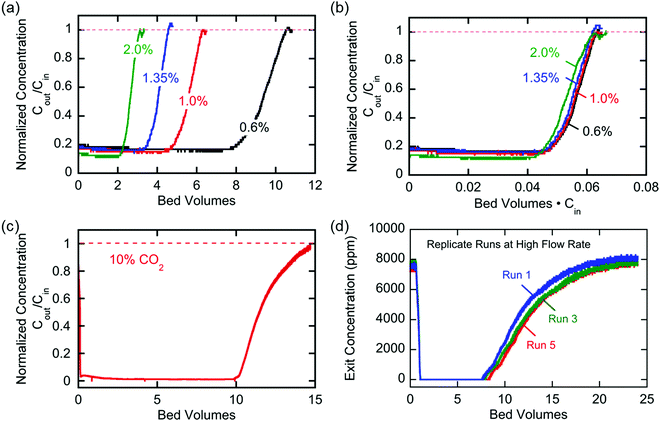 |
| | Fig. 8 (a) Breakthrough profiles obtained at four inlet concentrations. (b) Same breakthrough profiles in (a) normalized by the inlet concentrations. (c) Breakthrough profile obtained from a large system operating at ∼10% inlet concentration. (d) Breakthrough profiles obtained from five replicate runs of a smaller system operating at ∼0.8% inlet concentration. These experiments were conducted at T ∼ 21 °C. | |
The broadness of the breakthrough profiles in Fig. 8(c) and (d) can be attributed to the variation in channel width. Since the electrochemical cell stacks are hand-assembled and fitted into the bed, some variation in channel width along the same channel and the parallel channels can cause an uneven distribution of flow resulting in overlapping of multiple breakthrough profiles, which culminate in a broad overall profile. Also, mass-transfer dispersion plays a significant role in the broadness of the profiles. As is evident from the long durations for an effective capture in Fig. 6(e), and discussed later, mass transfer of CO2 into the electrodes is the rate controlling step, and this causes axial dispersion that widens the mass transfer zone in the channel, illustrated in Fig. 7(a), resulting in broad breakthrough profiles in this case.
Mechanism of CO2 capture
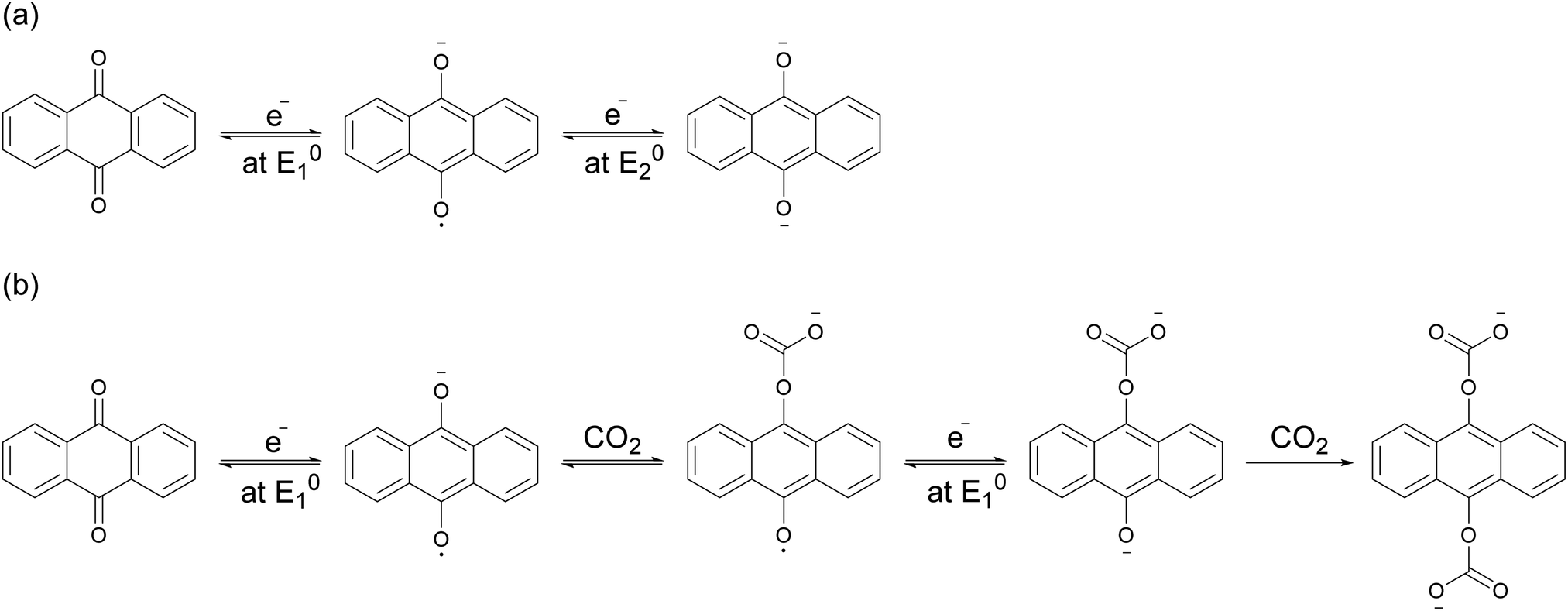
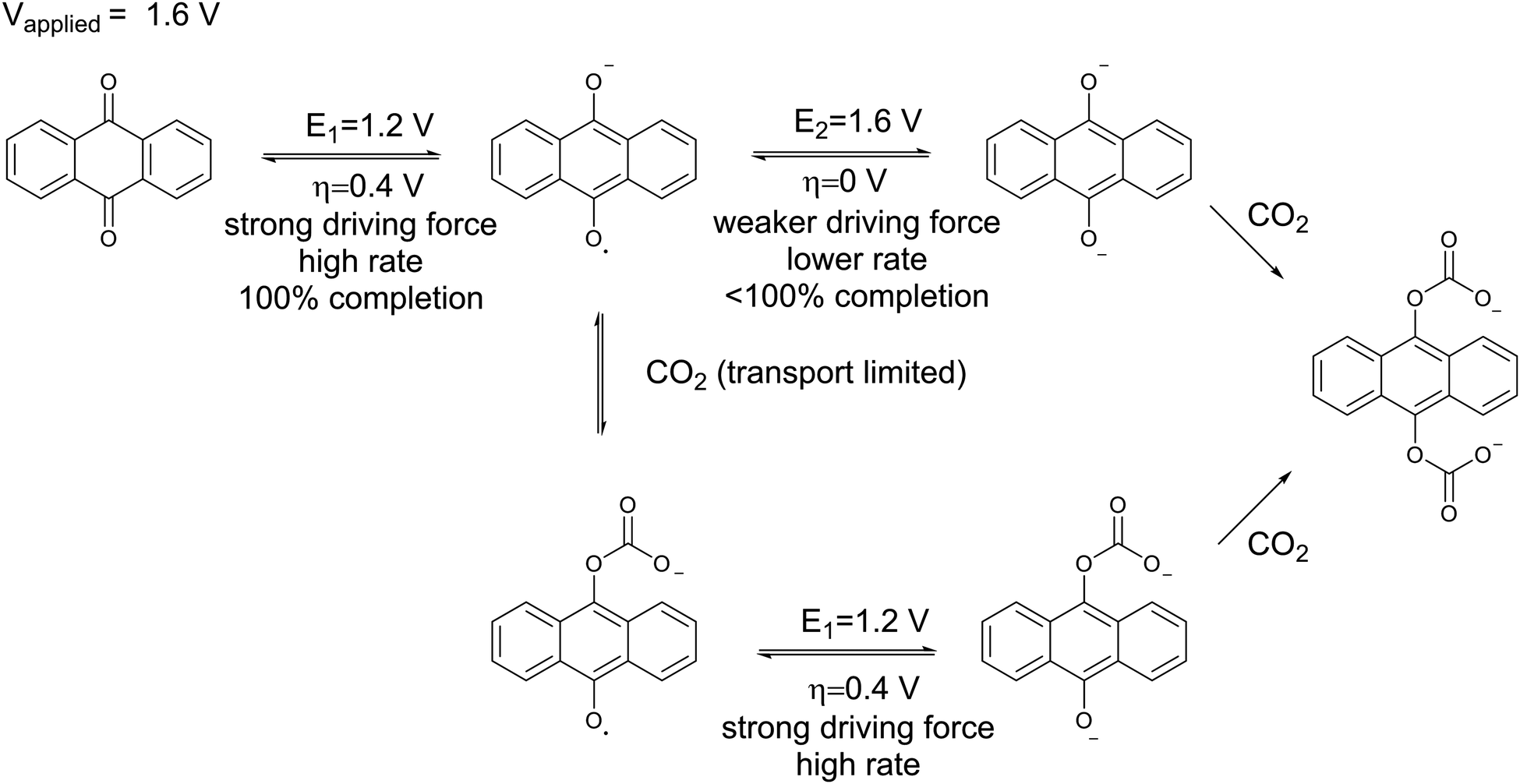
Mizen and Wrighton first demonstrated the reductive addition of CO2 to 9,10-phenanthrenequinone in aprotic electrolytes. Quinones undergo two reversible one-electron reductions in aprotic electrolytes, at two reduction potentials, as shown in Scheme 1(a) for anthraquinone. But in the presence of CO2, a single two-electron quasi-reversible reduction takes place at the first reduction potential, as shown in Scheme 1(b), where the CO2 molecule carboxylates the quinone resulting in a carbonate salt.37 Upon the first reduction of a quinone molecule, a semiquinone radical anion is produced. Some semiquinones, with sufficient electron density, i.e. sufficient Lewis base strength, can react with CO2 in a nucleophilic addition to form a carbonate salt. Semiquinones with lower electron density – due to attached electron withdrawing groups – do not have the nucleophilic strength to react with CO2.38 A semiquinone carboxylated by the reductive addition of CO2 has its negative charge isolated from the rest of the molecule by a σ-bond, rendering the carboxylated quinone aromatic ring(s) relatively neutral. Hence, the two-electron reduction of the parent quinone under CO2 occurs at the first reduction potential. The second reduction generates another nucleophilic center, i.e. the second oxygen of the quinone, which reacts with a second CO2 molecule to form a bis(carbonate) dianion salt – the quinol dianion diadduct. A similar behavior is observed when quinones are reduced in protic electrolyte media and the reduced quinones react with protons to form two hydroxyl groups via a two-electron reduction.39
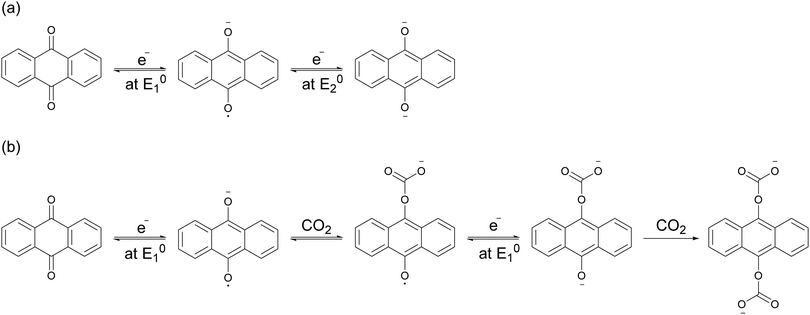 |
| | Scheme 1 (a) Two single-electron reduction waves of anthraquinone in the absence of electrophiles. (b) One two-electron reduction wave of anthraquinone in the presence of CO2. | |
The nucleophilic addition reaction of reduced quinones occurs only with electrophiles with sufficient Lewis acidity, such as CO2, and not N2, hence the great selectivity in adsorption. Furthermore, water, in the treated gas stream, has minimal effect on carbon capture, mainly because the hydrophobicity of the IL used presents a considerable mass transfer resistance for water with very little retention,40 and any residual water in the IL phase merely stabilizes the reduced quinones,41 with no significant change to the electrochemical or chemical steps of the carbon capture process.20 While the presence of oxygen can cause the oxidation of some reduced quinones with a sufficiently negative reduction potential, through the formation of the superoxide radical anion O2˙−, it is possible to avoid any reactions with oxygen in the feed stream by carefully tuning the electron density of the quinone polymer.42
The cyclic voltammogram (CV) of the PAQ–CNT composite on the electrode substrate in [Bmim][TF2N] saturated with N2, the green line in Fig. 3, shows two redox couples corresponding to the two reduction potentials of anthraquinone. The cathodic and anodic peaks, however, are broader than those reported for solutions of quinone molecules in electrolytes,20 which can be ascribed to the slower electron exchange between differently-reduced redox units, i.e. neutral quinone, semiquinone (radical anion) and quinol dianion, along the π-conjugated backbone of the polymer,43 and also to the diffusion and electromigration of electrolytic species into and out of the porous PAQ–CNT film.44 The expected disappearance of the second redox couple of the CV under CO2 was observed, as shown by the red line in Fig. 3, and attributed to the carboxylation of the reduced quinone units along the polymer chain facilitating the second electron reduction of anthraquinone at the first reduction potential, with the consequent increase in the current at that potential. The PAQ–CNT electrode can therefore be used to remove CO2 selectively from a gas mixture via nucleophilic addition, and regenerated via an electrochemical oxidation, with release of the CO2, as shown earlier. The two reduction potentials under N2 are E01 = −1.21 V and E02 = −1.63 V, vs. Fc. Under CO2 these two potentials coalesce to yield a single quasi-reversible redox couple at the first reduction potential observed under N2, i.e. E01.
The separation of the oxidation and reduction peaks in the CVs reported is large, indicating sluggish electron transfer to the quinone repeat units on the polymer, and a thick diffusion layer in the porous electrode.44 While the ratio of polymer to CNT was optimized for the greatest gravimetric capacity, it is not the ideal ratio for conductivity. Some of the conductivity takes place through the backbone of the polymer, which contributes to the slow electron transfer. This, however, does not present a problem in the design of the contactor separation unit, since the rate limiting step in the CO2 capture process is the transport of CO2 and not electron transfer.
The double carboxylation (CO2 addition) of anthraquinone to form the bis(carbonate) dianion follows an ECEC mechanism, as shown in Scheme 2(a). During a constant-current charging of the cell in the presence of a flux of CO2 towards the quinone units equal to, or higher than, the rate of the electrochemical reduction of the quinones, both of the reduction electrochemical reactions take place at the first reduction potential, E01. The molar flux of CO2, NCO2, towards the electrode (moles per unit area per second) should match the current density, J, at the electrode (charge per unit area per second), as in,
| | 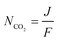 | (3) |
Conversely, if the flux of CO
2 is lower than the rate of reduction, some of the semiquinones produced in the first reduction are not carboxylated but are instead reduced to quinol dianions at the more negative second potential,
E02, as shown in
Scheme 2(b). In this case, for the same number of moles of semiquinone being reduced, and hence electrons,
i.e. charge, transferred, more energy is required in accordance with
eqn (2).
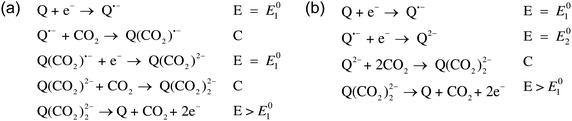 |
| | Scheme 2 Reaction steps of the double carboxylation of quinones (a) in high and (b) low CO2 fluxes towards the anthraquinone electrode. E represents an electrochemical reaction step. C represents a chemical reaction step. | |
CO2 deprivation was not a problem in the highly saturated electrolyte in the CV electrochemical characterizations, where the concentration of CO2 in the electrolyte was about 20 times higher than that of the quinones on the electrode. However, in an adsorption system, where the electrolyte is not saturated with CO2 there needs to be a careful balance between the advective and diffusive fluxes of CO2 towards the anthraquinone cathodes for reductive addition, and the rate of the reduction of anthraquinone. This can be better controlled by charging the cell, i.e. reducing the anthraquinone, at a constant potential that is sufficient to drive the first reduction of anthraquinone but not the second, i.e. charging the cell at a potential difference that is between ΔV1 and ΔV2. With operation at this constant potential, the flux of CO2 does not affect the energy of binding, since the second electron is acquired by a semiquinone at the first reduction potential, but only after the first CO2 addition occurs; following this second reduction, a second CO2 molecule can bind to the dianion monoadduct. When the first reduction is allowed in a CO2-lean environment at a constant potential, the current observed after the initial surge, which results from the first reduction, is proportional to the flux of CO2 towards the anthraquinone electrode. Nevertheless, this current ultimately diminishes due to the decrease in the number of semiquinones available to react with CO2. In a CO2-rich environment, at constant potential charging, the observed current after the initial surge is proportional to the transport of CO2 into the electrode and the rate of the addition reaction which follows the nucleophilic addition mechanism between semiquinones and dissolved CO2,
where
R is the rate of the reaction and
k is the rate constant. The electrochemical steps proceed at a faster rate than the chemical reactions, but both types of reactions have rates that are faster than the rate of diffusion of CO
2 in the IL, leading to a large Damköhler number,
| | 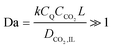 | (5) |
where
CQ is the initial concentration of anthraquinone repeat units on the electrode,
CCO2 is the concentration of CO
2 in the IL,
L is the thickness of the IL-wetted porous electrode, which is about 150 μm, and
DCO2,IL is the diffusion coefficient of CO
2 in the IL and is about 4 × 10
−10 m
2 s
−1. While it is the semiquinone that participates in the reaction here, the initial concentration
CQ is a good approximation for estimating the order of magnitude of Da. The rate constant can be estimated to be 10
2 M
−1 s
−1,
37 resulting in Da ∼10
4. Large values of Da indicate that mass transfer will control the process and thus the charging and subsequent carboxylation of the quinones can be assumed to be in equilibrium with the prevailing CO
2 concentration at any position and at any time. Therefore, with a reduced thickness of the electrodes, the CO
2 mass transfer rates will approach that of the nucleophilic addition reaction.
Modeling of CO2 capture
To further understand the behavior of the cell during constant-current and constant-potential charging at various CO2 concentrations, a two-dimensional model of the cell and the adjacent gap was set up in COMSOL®, with the geometry shown in Fig. 9. The model accounted for all the chemical and electrochemical reactions described in Scheme 2, in addition to the electrochemical oxidation of the ferrocene. The equilibrium potentials of all electrochemical reactions were predicted by the Nernst equation| | 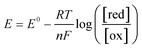 | (6) |
where E0 is the standard reduction potential, n is the number of moles of electrons involved, R is the gas constant, T is the temperature, [red] is the concentration of the reduced species and [ox] is the concentration of the reactant with high oxidation number. In the first two electrochemical reactions shown in Scheme 2(a), the reduced species are Q˙− and Q(CO2)2−, while the species of higher oxidation number in these two reactions are Q and Q(CO2)˙− respectively. The standard reduction potential for both reactions is E01. While the first of the two electrochemical reactions in Scheme 2(b) is the same as that of Scheme 2(a), the second reaction has Q2− as the reduced species and Q˙− as the species with higher oxidation number, with a standard reduction potential of E02. Likewise, in the anode oxidation reaction, Fc+ (ferrocenium) is the oxidized species and Fc (ferrocene) is the reduced species at E0 = −0.03 V. The current densities of these reactions were described by the Butler–Volmer equation,| | 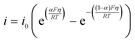 | (7) |
where η is the overpotential and α, the symmetry factor, was approximated to be 0.5 for all the electrochemical reactions. The exchange current, i0, is given by| | | i0 = k0[red]α[ox](1−α) | (8) |
The governing mass transfer equations for all the species involved in the electrochemical reaction, including the supporting electrolyte cations and anions, is| | 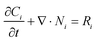 | (9) |
where Ci is the concentration of the species, Ri is the rate of reaction in which the species is involved and Ni is the flux, which is zero for all immobilized faradaic species: anthraquinone in its different oxidation and binding states, and ferrocene. For mobile species, mainly the supporting electrolyte ions, the flux is given by| | | Ni = −Di∇Ci − ziuiFCi∇Φ | (10) |
where zi is the valence, ui is the molar mobility, F is Faraday's constant and Φ is the electric potential. The current density can be obtained from the flux of charged mobile species in the electrolyte| | 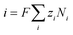 | (11) |
and the overall electroneutrality is maintained by ensuring that| | 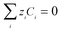 | (12) |
The boundary conditions for these equations are ground (Φ = 0 V) for the anthraquinone electrode (colored grey in Fig. 9), and either constant current or constant potential at the ferrocene electrode (pink). The electrochemical model is the same for both sealed and flow systems, although the transport of CO2 in the gas phase (shown as a transparent block) is different in the two scenarios.
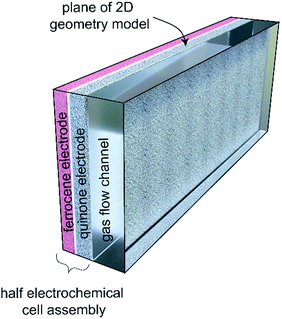 |
| | Fig. 9 Cross-section of the electrochemical cell used in the simulations. | |
In the sealed system, CO2 transport occurs via diffusion toward the electrode only, while in flow CO2 is also advected down the channel. In the sealed system, the partial pressure of the gas decreases during capture, while in flow the CO2 concentration changes along the gas channel with time, and the concentration of the gas leaving the device is a measure of the total effectiveness of the adsorption bed capacity and transport and kinetic limitations within the system. The total transport of CO2 is modelled by transport equations in each of the two phases, gas and IL. In the gas phase, the mass transfer governing equation in the sealed system is given by
| | 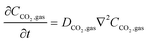 | (13) |
where
CCO2,gas is the concentration of CO
2 in the gas phase, obtained from its partial pressure and the ideal gas law. This governing equation has a no-flux boundary condition at the three walls
| | 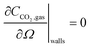 | (14) |
and at the gas–IL interface, the fluxes normal to the interface are set to be equal
| | 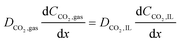 | (15) |
where
CCO2,IL is the concentration of dissolved CO
2 in the IL phase. In the flow system, the flux of CO
2 has an advective component as well and the governing equation becomes
| | 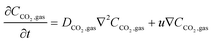 | (16) |
where
u is the velocity of the gas flowing in the channel. The inlet is set at a constant concentration inflow and the outlet at zero gauge pressure. The mass transfer governing equation for dissolved CO
2 in the IL phase, for both sealed and flow systems, is given by
| | 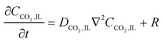 | (17) |
where
R is a bulk reaction term describing the rate of the chemical reactions shown in
Scheme 2(a). Simple rate equations, first order in all reactants, are used. A no-flux boundary condition is set at the walls. At the gas–electrode interface, the concentration of dissolved CO
2 is set to
where
β is the adjusted Henry's constant which accounts for the equilibrium partition of CO
2 between the gas and IL phases.
The purpose of this simulation is to obtain a semi-quantitative analysis of the behavior of the cell under different conditions, and, therefore, order of magnitude estimates are made for the electrochemical parameters of the model.
Model results for batch system.
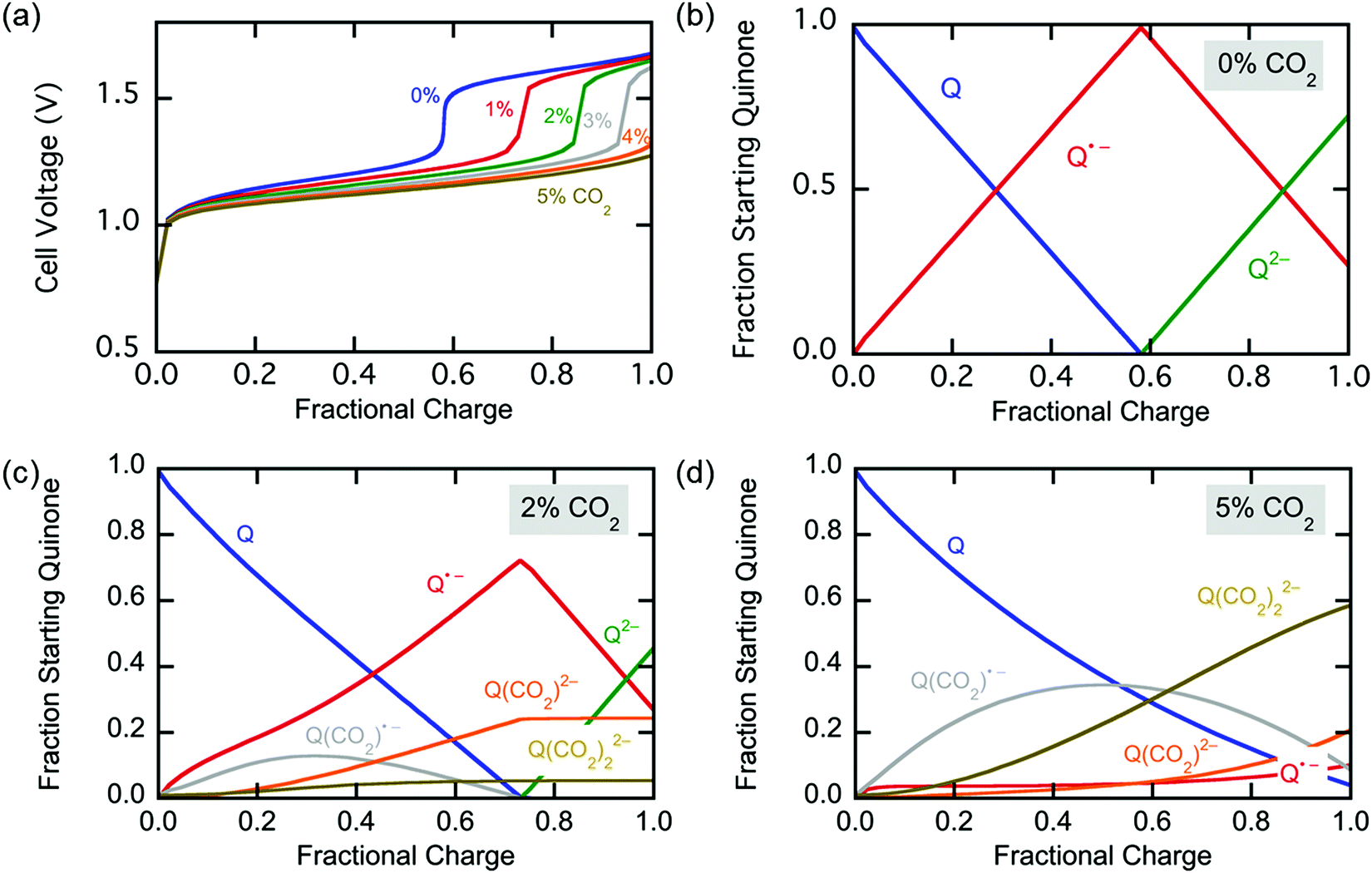
The simulation of constant-current charging at 0–5% CO2 concentrations in the sealed system in which the gas channel shown in Fig. 9 served as a gas headspace, shows results that are consistent with the aforementioned mechanism of CO2 capture. In addition to the cell voltage, the concentrations of all the cathode quinonic species were studied during the charging process. Fig. 10 shows the cell voltages at 0–5% CO2 and the concentration profiles of cathode species at 0, 2, and 5% CO2. The cell voltage profiles in Fig. 10(a) show that as the concentration of CO2 increases, less of the cell charging occurs at the higher potential. At 0% CO2 the cell simply traverses both reduction potentials of the quinone, and the species profiles in Fig. 10(b) show two consecutive conversions; the first is the reduction of quinone to semiquinone, and the second is the reduction of semiquinone to quinol dianion. The second reaction starts only after all the quinone is converted to semiquinone, and to further accommodate the incoming electrons at the constant current, the second, more energetic reduction takes place until all faradaic reactions are exhausted. Here, the simulation was stopped just short of full conversion to avoid numerical complications that arise from the exhaustion of faradaic material, i.e. complete charging. At 2% CO2, upon generation of the semiquinone, shown in Fig. 10(c), the modest diffusive flux of CO2 allows for the formation of the semiquinone monoadduct which soon accepts a second electron at the first reduction potential of quinone, hence extending the cell charging period spent at the lower potential. This dianion monoadduct can then react with a second CO2 molecule, to form the dianion diadduct. But, upon the depletion of CO2 in the vicinity of the cathode, due to a charging rate that is faster than the diffusive flux of CO2 into the electrolyte at 2% headspace concentration, the semiquinone is converted to the dianion at the higher second potential, while the concentrations of the dianion monoadduct and the dianion diadduct remain constant. This is reflected in the potential profile at 2% CO2 in Fig. 10(a). However, at 5% CO2, the diffusive flux of CO2 is adequate, and barely any semiquinone can be detected since it is converted to semiquinone monoadduct, and other subsequent species, soon after generation, shown in Fig. 10(d). Here, all the electrochemical reactions take place at the first potential and none of the semiquinone is converted to quinol dianion.
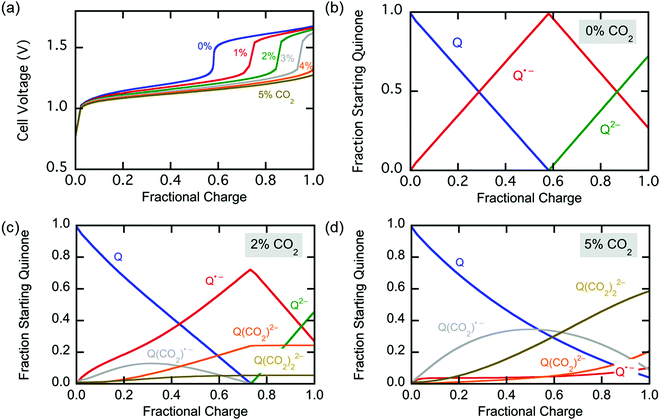 |
| | Fig. 10 Simulation of charging the electrochemical cell at different CO2 concentrations at constant current. (a) Potential difference of the cell. The change in concentration of quinone with charge is shown at (b) 0%, (c) 2% and (d) 5% CO2. | |
Hence, the optimum charging method of the cell, i.e. CO2 capture, is to operate at a constant potential just larger than the first potential window ΔV1. The greater the charging voltage, the faster the reduction reaction due to the larger overpotential. For practical reasons, and to avoid large current surges, the cell can be charged at a constant current until the desired charging potential difference is reached. This will be between ΔV1 and ΔV2 based on the rate of charging desired. At this point the bed is activated for capture by making half the total capture capacity available. Then the cell can be set at that constant potential until saturation of the cathodes is achieved. The second half of the capture capacity is made available as more CO2 flows in.
It is also possible to fully activate the bed by charging at a potential difference ΔVHigh > ΔV1, at which point the cells are disconnected and allowed to saturate with no further supply of current, until breakthrough is achieved, and the bed is regenerated by discharging the cell and releasing pure CO2. The energy recovered however, cannot exceed QΔV1, resulting in an energy loss of greater than Q(ΔVHigh − ΔV1).
Model results for flow system.
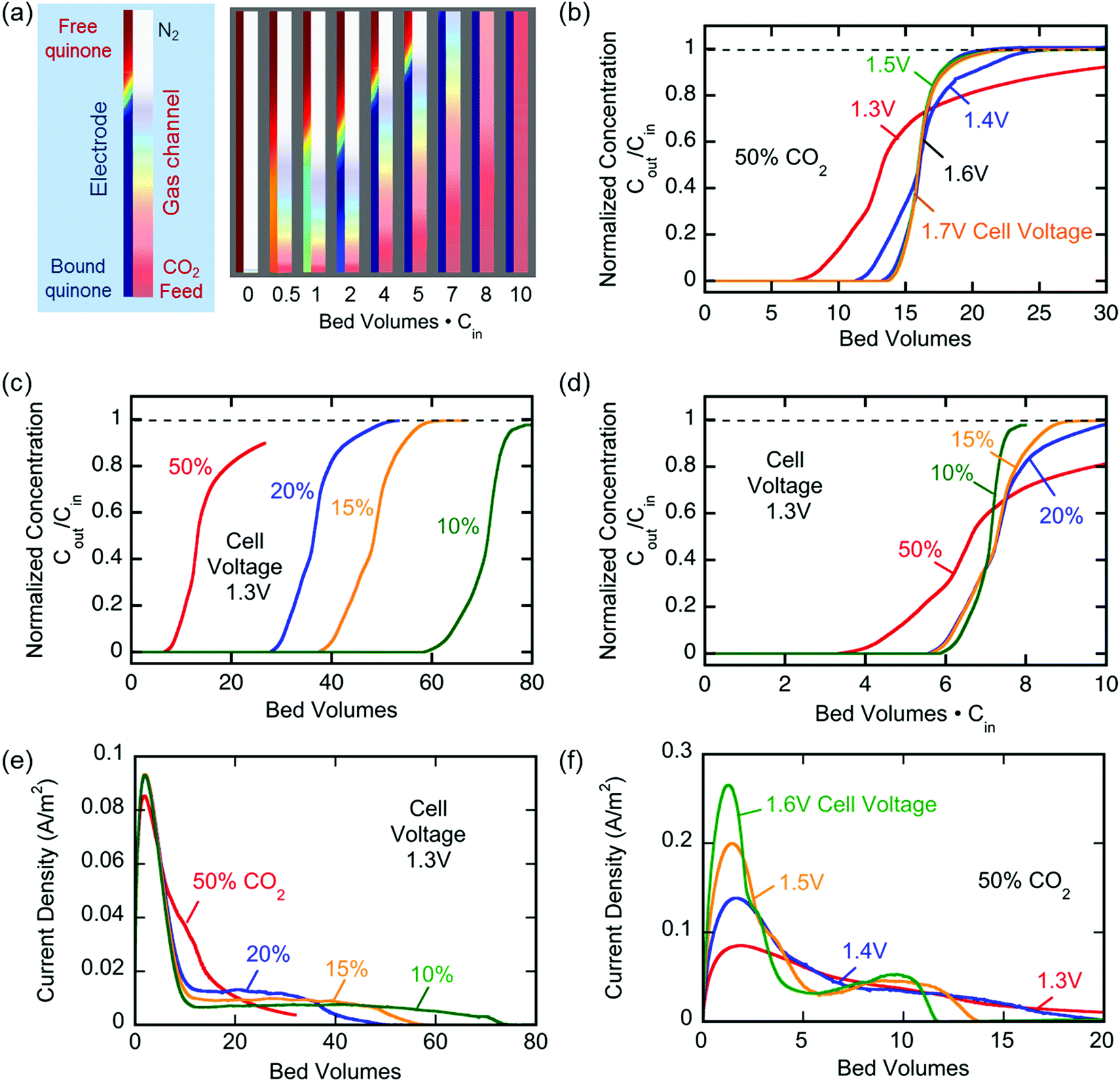
The model described above was also used to simulate the capture of CO2 from a channel under flow conditions. The gas flowed parallel to the electrodes in the cell, a cross-section of which is shown in Fig. 9. The simulated concentration profiles in the channel and the quinone electrode are shown as a function of the number of bed volumes in Fig. 11(a). The gas phase profiles are consistent with a dispersed plug flow in the channels, with a reasonably sharp adsorption front, while the linear CO2 profiles in the electrode are a result of the significant diffusional limitations in this region. Of particular interest is the depletion of unbound quinonic species in the cathode as removal of CO2 from the channel proceeds; as the cathode becomes saturated with CO2, the concentration of CO2 increases along the length of the channel until breakthrough is achieved at around 8 bed volumes, after which further saturation is allowed until the outlet concentration is equal to the inlet concentration at around 10 bed volumes. Fig. 11(b) and (c) show the breakthrough profiles as a function of cell voltage for a given feed concentration, and of feed concentration for a given cell voltage, respectively. The number of bed volumes required increases with decreasing feed concentrations, a result of the fixed overall bed capacity and the rectangular nature of the adsorption isotherm of this system. In the absence of transport and reaction limitations, all breakthrough curves should collapse when the bed volume is multiplied by the feed fraction of the adsorbate; the fact that this is not exactly so in Fig. 11(d) indicates that mass transfer limitations do play a role in the uptake of CO2 by the activated electrodes. Nevertheless, the overall capacity, indicated by the trapezoidal area under Cout/Cin = 1 and the breakthrough curves, remained constant. However, the effect of a stronger driving force on improved mass transfer is evident in Fig. 11(b), where at charging potentials that are closer to ΔV2, more of the dianion is generated in a Nernstian distribution, which provides two reaction sites for CO2. This results in a stronger driving force in the adsorption of CO2 and hence improved mass transfer, which yields a sharper breakthrough profile.
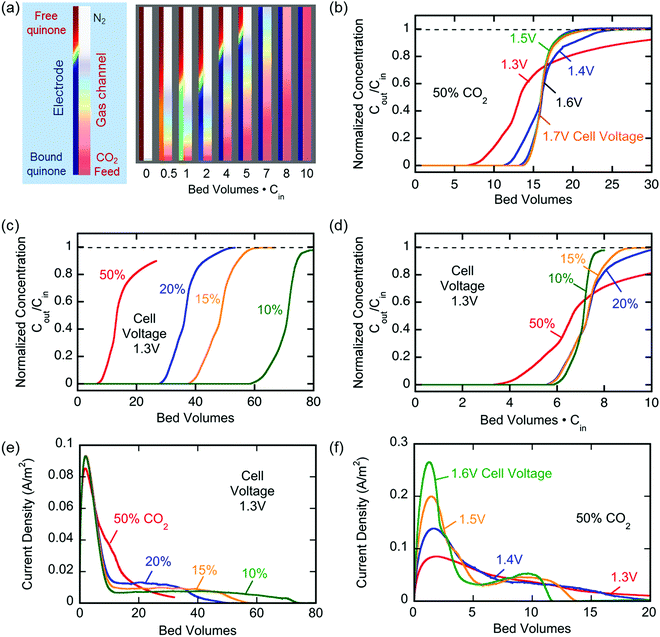 |
| | Fig. 11 (a) Breakthrough profiles from simulation at 50% CO2 and charging potential of 1.7 V; inset: the concentration of CO2 in the channel and the concentration of unbound quinonic species in the cathode with bed volume. (b) Breakthrough profiles at multiple capture potentials at 50% inlet concentration. (c) Breakthrough profiles from simulation at multiple inlet concentrations at a charging potential of 1.3 V. (d) Normalized breakthrough profiles of the capture experiments in (c). (e) The current across the electrochemical cells during the capture experiments in (b). (f) The current across the electrochemical cells during the capture experiments in (c). | |
Fig. 11(e) and (f) show the current across the electrochemical cell during the capture experiments in (c) and (b) respectively. Fig. 11(e) shows that at low potential charging, i.e. 1.3 V, an initial spike in current is observed which results from the reduction of the quinones on the electrodes to semiquinone, as explained earlier. This is followed by a region of constant current which is proportional to the flux of CO2 into the electrodes, since it reacts with the semiquinones and allows them to accept a second electron at the low potential. As the electrodes become saturated and all the semiquinone is depleted, the current decreases. This region of constant current followed by the sharp decrease is very broad for the 10% case, but much shorter for the 50% case, where there is a brief transition from the initial spike to the decreasing current region manifested in a shoulder. However, Fig. 11(f) shows that as the applied capture potential increases, the current profile for the same inlet concentration changes dramatically. This is elucidated in Scheme 3; as the applied potential approaches ΔV2, all of the quinone is reduced to semiquinone and a Nernstian distribution of semiquinone and quinol dianion is generated, evident by the increasing current in the first peak.
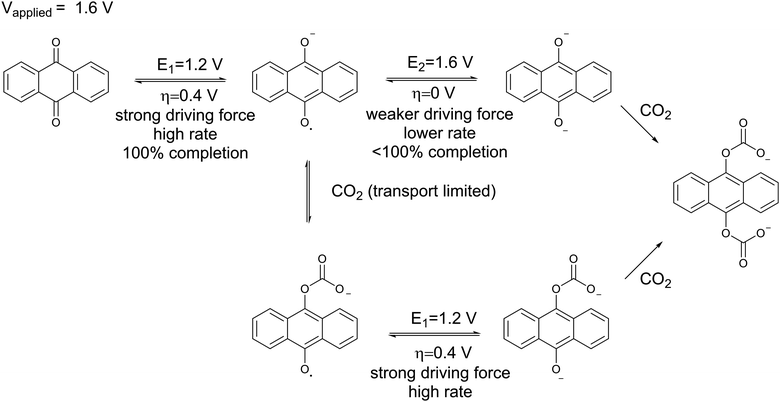 |
| | Scheme 3 The reduction of anthraquinone at a potential higher than its second reduction potential with a limited flux of CO2. | |
Thus, CO2 reacts with the Nernstian mixture of semiquinone and quinol dianion, where in the case of the latter no further reaction, chemical or electrochemical, occurs, but in the case of the former a semiquinone monoadduct species is formed, which can be reduced to the dianion monoadduct at ΔV1. Therefore, the applied potential presents a large overpotential for this reaction, hence the second increase in the current which corresponds to the electron transfer for this reduction. Following these steps, all the quinone moieties in the electrodes are in their doubly charged states and no further electron transfer occurs. This double peak behavior of the current is the result of the balance between the rate of electron transfer and the flux of CO2, which relates to the earlier discussions. At charging potentials higher than ΔV2e.g. ΔVHigh, not shown here, a larger first peak is expected where all the quinone is converted to semiquinone then dianion, i.e. two consecutive single-electron reductions, after which the current drops to zero and only chemical addition reactions occur, Scheme 2(b).
Energetic analysis of CO2 release
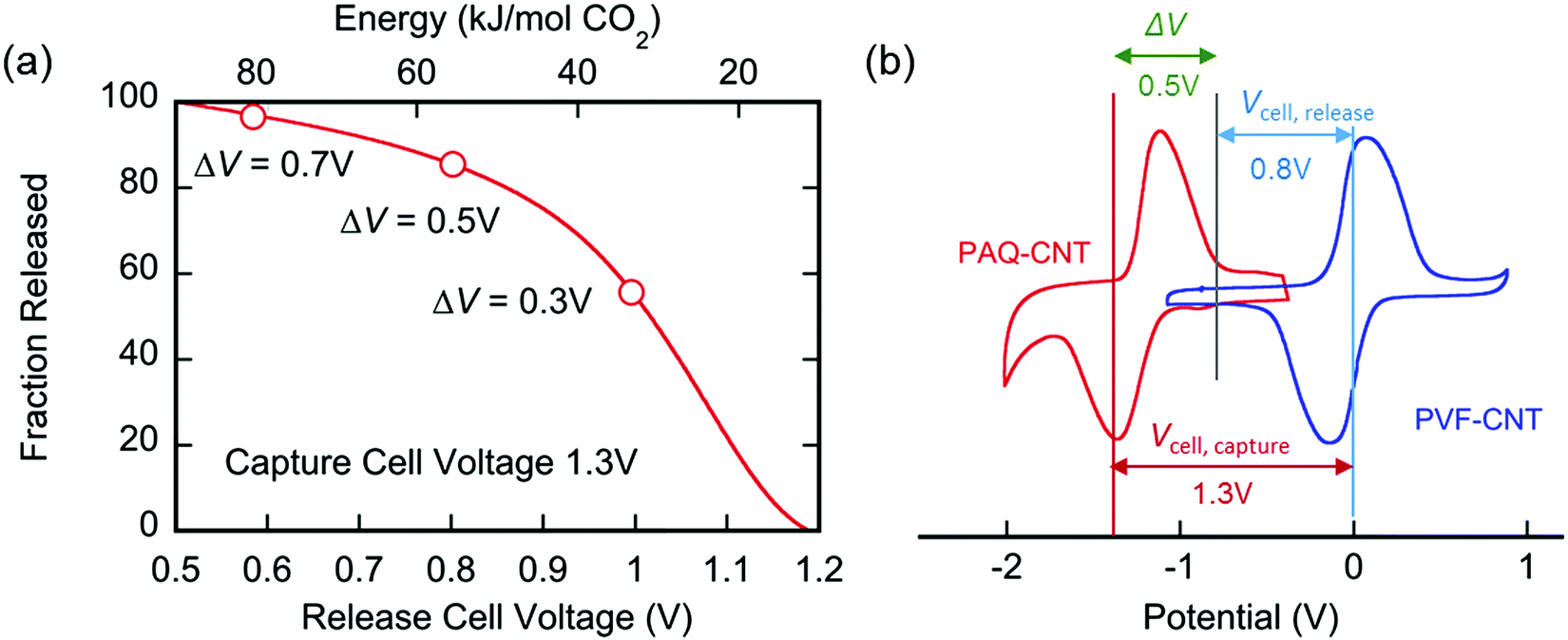
The applied release potential determines the energy recovered from the cell upon the release of CO2, and must, therefore, be selected carefully. While quinones under N2 are expected to be reduced and oxidized reversibly without any significant loss of energy, in an electrophile-rich environment, such as CO2, the reduction is only quasi-reversible due to the energetic penalty of breaking a bond formed with CO2. Therefore, some of the energy is lost in the process, and it is imperative to minimize this loss, since it is directly related to energy per mole of CO2 concentrated (captured and released) in a CO2 capture process. The last step in the CO2 capture, Scheme 1(b), i.e. the addition of a second molecule of CO2 to the semiquinone monoadduct to form a dianion diadduct, is chemically irreversible for the duration of the cycle,37 and the release reaction takes place via an electrochemical oxidation of the diadduct. This oxidation occurs at a more positive potential than the first reduction potential E01. Thus, the more positive the release potential, the larger the overpotential and the faster and more complete is the release of the bound CO2. However, a more positive release voltage corresponds to less energy recovered during discharge, and thus more work done per cycle of capture and release, which is given by eqn (2).
Consequently, to reduce energy losses per cycle, a less positive potential can be used in a constant potential discharge. While the capture cell voltage can be maintained at fairly minimal levels by setting it close to ΔV1, with some diminishment in rate, release at high cell voltages can result in a partial release of CO2, due to the partial oxidation of the bis(carbonate) quinone units (dianion diadduct units), and a subsequent underutilization of anthraquinone. After a few cycles of capture and release at a higher potential, a constant equilibrium rate of utilization of anthraquinone is achieved. This is akin to the working capacity of a pressure-swing adsorption system.45 Thus partial release can be achieved at higher cell discharge potentials as shown in Fig. 12. This allows for a much smaller window of operation, which at the ∼90% faradaic efficiency reported earlier, an energy use of ∼43 kJ mol−1, with 60% utilization of accessible quinone is possible. This is on par with other carbon capture systems in efficiency and energy consumption,46,47 including thermal swing systems.48
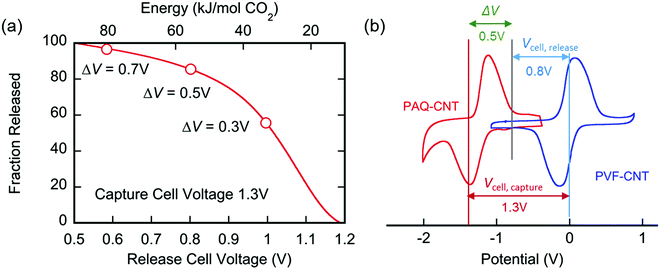 |
| | Fig. 12 Fraction of CO2 released from the PAQ–CNT electrode with release voltage. At a constant capture cell voltage of 1.3 V, less of the bed is recovered with increasing release cell voltage, but the energy per mole of CO2 captured and released also decreases. | |
Solid sorbent materials used in carbon capture applications have enthalpies of CO2 adsorption of the same order, e.g., Metal–Organic Frameworks (MOFs) with 30–50 kJ mol−1 for some types of MOF-74,49 and ∼100 kJ mol−1 for some amine functionalized MOFs,50 or zeolites, such as 13X at 40 kJ mol−1.51 However, these are not process energetics but rather enthalpies of adsorption. Therefore, processes which are designed to utilize these materials incur greater energetic penalties associated with the techniques used for delivering the energy for desorption, i.e. heat and pressure change, whereas the energetics reported herein correspond to that of the process, which unlike those utilizing solid sorbents, has a binary affinity for CO2.
Moreover, if a precise rate of release is desired, the cell can be discharged at a constant current. In this case, the flux of the released CO2 is proportional to the current density, as given by eqn (3). The discharge current and time, control the charge transferred, which is the extensive variable in the work of the process. This method of release is seldom utilized in swing adsorption processes, because it is the intensive, rather than the extensive variable, that is modulated, e.g., temperature in thermal swing, pressure in pressure swing, and, here, potential. Nevertheless, this can be useful for fast regeneration of the bed at high currents, where long periods of capture and short periods of release are desired. But due to the incomplete discharge of the electrochemical cells, in analogy with high current discharge of batteries,30 the bed will only be partially regenerated.
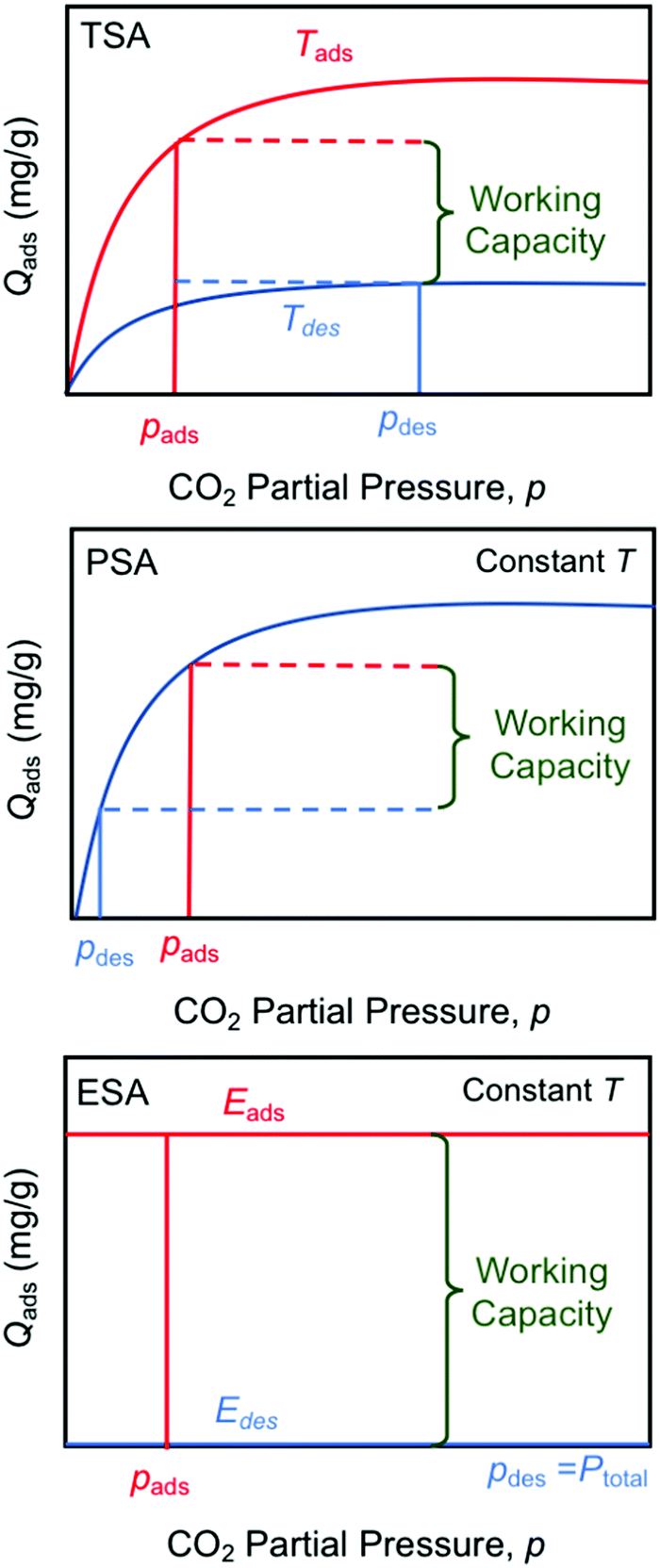
Swing operations
The electro-swing adsorption (ESA) technology developed in this work has a number of perceived advantages over other swing technologies such as temperature- (TSA) and pressure-swing (PSA) adsorption in terms of both its mode of operation and its thermodynamic adsorption characteristics, as illustrated in Fig. 13. Temperature-swing operations require heating and cooling of the entire adsorption matrix to release the adsorbed compounds, rather than just targeting the adsorption sites themselves, which introduces many thermal inefficiencies to the operation. The heating is typically accomplished with high-temperature steam as the sweep gas, with subsequent condensation of the steam to recover the pure CO2 product stream, whereas cooling is achieved during the blowdown phase in preparation for the next sorption cycle. With pressure-swing operations, the release of the captured material occurs on reduction of the adsorbate partial pressure in the sweep to reverse the mass transfer driving force, which can be achieved either by drawing a vacuum on the bed, or by using low pressure steam as the sweep gas. The product leaving the bed is dilute, and requires a second separation step, such as the condensation of steam, and external compression of the resulting CO2 to bring it to the desired pressures for subsequent utilization or for transportation and subsurface sequestration in geologic formations. Moreover, in both TSA and PSA operations, the working capacity is strongly dependent on the shape of the Langmuir- (or Freundlich-) type isotherm, with significantly lower capacities at lower concentrations, which limits the degree of regeneration of the adsorbent, or the partial pressure of the recovered adsorbate in the sweep stream. The electro-swing adsorbents, on the other hand, are characterized by a rectangular adsorption isotherm in which the saturation loading capacity in the activated state is independent of the gas phase concentration, so the adsorbent is equally effective over a wide range of feed concentrations from ambient conditions through to those characteristic of large scale power and industrial facilities. When deactivated, the adsorbent has negligible capacity for the CO2 even at high CO2 gas phase concentrations such that the working capacity of the adsorbent is independent of operating conditions. Thus, the sweep stream during the desorption process can be pure CO2 at the desired pressure, subject primarily to equipment design constraints, which greatly simplifies the adsorption/desorption cycle relative to those of the TSA and PSA processes. The results in Fig. 6(a) show that release into a pure CO2 gas environment is facile and fast, and complete if sufficient time is allowed to account for the kinetics of the process.
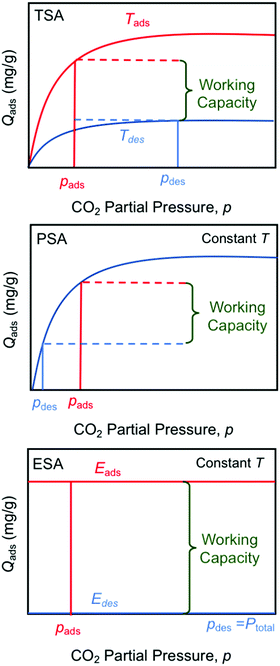 |
| | Fig. 13 Comparison of temperature- (TSA), pressure- (PSA) and electro- (ESA) swing operations showing the impact of sorption isotherms on total working capacity. | |
Conclusions
The electro-swing operations outlined in this paper offer a new approach to the exploitation of electrochemically mediated complexation of CO2 with redox-active species for the mitigation of CO2 emissions, and for control of CO2 levels in confined environments. Electro-swing adsorption has been shown to be equally effective, at a constant capacity, for the capture of CO2 at concentrations typical of fossil fuel combustion flue gases (∼10%) and in confined ambient environments (0.6–0.8%). These advances have been made possible by the development of new electrode materials in which redox-active moieties, such as quinones, that can bind CO2 in one oxidation state but not in another are immobilized onto the electrode substrates. These functionalized electrodes were incorporated into an electrochemical cell assembly to facilitate their cyclical electrochemical reduction and oxidation. The cells demonstrated exceptional capture and release of CO2 over more than 7000 cycles at 60–70% quinone utilization, 90% faradaic efficiency, and 90 kJ mol−1 at 100% bed utilization and 43 kJ mol−1 at 60%. A significant advantage of the quinone-functionalized electrodes is that they have a CO2 capacity that is essentially independent of the inlet feed concentration, in contrast to the capacities of sorbents typically used in PSA or TSA operations, where Langmuir-type equilibria limit capacity at lower concentrations.
The strong performance of the electro-swing process allows for numerous applications where CO2 removal and/or storage is of interest. The ESA device can be easily integrated into processes in a plug-and-play fashion due to its simple design, and minimum requirement for ancillary equipment, primarily a regulated power source and sink (load). An initial techno-economic analysis (not reported here) shows that such carbon capture systems can be economically feasible with costs ranging from $50–$100 per tonne CO2 depending on the feed concentrations and applications under consideration.
It is projected that further optimization of the ESA process can be obtained through refinement of the electrode chemistries and their assembly into compact adsorption devices to address a wide range of CO2 mitigation strategies.
Experimental
Materials
Dichloro-1,4-anthraquinone (>98%) and 2,2′-bipyridyl (>99%) were purchased from TCI America. Bis(1,5-cyclooctadiene)nickel(0), 1,5-cyclooctadiene (>99%), MWCNT (>98%, O.D. × L 6–13 nm × 2.5–20 μm), anhydrous N,N-dimethylformamide (DMF, >99.5%) and N-methyl-2-pyrrolidinone (NMP, >98%) were purchased from Sigma-Aldrich. Poly(vinylferrocene) (PVFc, MW ∼ 50000 g mol−1) was purchased from Polysciences, Inc. 1-Butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide (>99%) was purchased from IOLITEC, Ionic Liquids Technologies GmbH. Bone dry 3.0 grade carbon dioxide and industrial grade nitrogen gases were purchased from Airgas. Non-woven carbon mat (34 g m−2) was purchased from Marktek, Inc. Whatman-2 filter paper was purchased from General Electric – Health. Celgard battery separator was purchased from Celgard. Stainless steel (SS) mesh (316, 500 × 500) was purchased from Gerard Daniel International. DMF was dried over 3 Å molecular sieves over three stages and purged with N2 for 12 h. All other reagents were used without further modification.
Methods
Synthesis of poly(1,4-anthraquinone) (P14AQ).
The synthetic procedure followed the work of Yamamoto et al.43 A solution of 1.510 g (5.45 mmol) 1,4-dichloroanthraquinone in 20 mL DMF (dry and O2-free), was added dropwise to a solution of 2.000 g (7.27 mmol) of bis(1,5-cyclooctadiene)nickel(0), 1.134 g (7.27 mmol) of 2,2′-bipyridyl and 0.7 mL (5.45 mmol) of 1,5-cyclooctadiene in 50 mL DMF (dry and O2-free), stirring at 65 °C under Nitrogen. The mixture reacted at 65 °C for 72 h. After cooling to room temperature, 100 mL of 1 M hydrochloric acid (HCl) was added to reaction mixture and allowed to stir at room temperature for 1 h. The mixture was then filtered and washed with 200 mL of 1 M HCl, twice with 200 mL of de-ionized water, twice with 100 mL DMF followed by 100 mL water and 100 mL methanol washes. The residue was dried under vacuum for 24 h. The procedure yielded 1.084 g polymer (96% yield). The resulting solid was extracted for low molecular weight chains via Soxhlet extraction with DMF for 5 h, to obtain 0.890 g of long chain polymers.
Polymer–multi-walled carbon nanotube (MWCNT) composites P14AQ–CNT.
The composite was made by ultra-sonicating using Cole-Parmer 750 Watt Ultrasonic Processor a suspension of 40 mg of P14AQ (as made) in 40 mL of NMP at 5 °C for 20 minutes, followed by adding 120 mg MWCNT into the polymer suspension to be sonicated for 20 minutes, resulting in a well-dispersed 4 mg mL−1 ink.
PVFc–CNT.
The composite was made by ultra-sonicating using a suspension of 160 mg of PVFc in 40 mL of NMP at 5 °C for 20 minutes, followed by adding 160 mg MWCNT into the polymer suspension to be sonicated for 20 minutes, resulting in a well-dispersed 8 mg mL−1 ink.
Preparation of electrodes.
Non-woven carbon mat were dip-coated into the polymer–CNT inks multiple time to deposit a certain mass of the composite. These were dried for 40 min in an oven at 120 °C and ambient pressure between dips, and then dried at the same conditions for 12 h after the final dip. These were used for electrochemical characterizations and for cell assembly.
Electrochemical cell assembly.
Each electrochemical cell was assembled by carefully laminating 9 layers and the ILs in the following order: SS current collector; P14AQ–CNT coated carbon mat; IL; electrolyte separator; IL; PVFc–CNT coated carbon mat electrolyte separator; SS current collector; PVFc–CNT coated carbon mat; IL; electrolyte separator; IL; P14AQ–CNT coated carbon mat; SS current collector. The electrolyte separators were either Whatman-2 cellulose filter paper, and the IL was [Bmim][Tf2N].
Electrochemistry.
Electrochemical measurements were carried out using a Parstat 3000-DX potentiostat with VersaStudio software from Princeton Applied Research. Cyclic voltammetry was conducted in a standard three-electrode glass cell with a polymer–CNT coated substrate as working electrode, platinum wire as the counter electrode and Leakless Ag|AgCl Reference Electrode (purchased from eDAQ). The IL electrolyte was stored at ∼20 mbar and purged with N2 or CO2 for 30 min before recording the CV. All CV measurements were compensated for internal resistance of the cell. Ferrocene was added at the end of CV experiments for reference adjustment. Potentiostatic and galvanostatic experiments were conducted in a two-electrode mode. All electrochemical and experiments were conducted at T ∼ 21 °C.
Microscopy.
Non-woven carbon mats were imaged using high-resolution scanning electron microscope (HR-SEM), Zeiss Merlin. The electron high tension was between 3–6 kV, and the high efficiency secondary electron (HE-SE2) detector was used in an analytic column mode. Diluted suspensions of P14AQ–CNT (0.1 mg mL−1) were dropcast onto holey-carbon transmission electron microscopy (TEM) grid for imaging using HR-TEM, JEOL 2010 Advanced High Performance TEM.
Gas mixes.
Mixtures of N2 and CO2 were made for the sealed chamber batch and the open flow capture and release experiments. These were made by controlling the flow rates using Aalborg Mass Flow Controller GFC-17 (0–50 mL min−1) for the CO2 and (0–50 mL min−1) for the N2 for experiments with >5% CO2 and (0–1000 mL min−1) for experiments with <5% CO2.
Sealed chamber capture and release.
The electrochemical cell(s) were placed in an insulating sealed chamber that was charged with different CO2 concentrations (with nitrogen as the balance) at ∼1 atm and ∼21 °C by purging for 2 h. Potential difference was applied across the power feed-throughs that were connected to the electrochemical cells, and the decrease and increase in pressure in the chamber, upon capture and release, respectively, was monitored using a Omega® PXM409-USBH pressure transducer with a range of 0–15 psia and an accuracy of 0.08% BSL.
Flow capture and release experiments.
The assembled electrochemical cells were stacked in a flow cell with the P14AQ–CNT electrode sides tracing parallel passages. The stack of these cells, separated by ∼500 μm, was placed in a gas flow chamber (the experimental setup) and gas was flown past them. Mixtures of nitrogen and carbon dioxide at ∼10 mL min−1 and various inlet concentrations of CO2 at T ∼ 21 °C were studied. The bed volume of the system was 70 cm3. The concentration of CO2 in the mixture at the outlet was measured using CO2-Meter SprintIR® with an accuracy of 100 ppm.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by an MIT Energy Initiative Seed Fund grant, and by Eni Eni S.p.A. We thank Jonathan Thomas and Ziad Mansour for their assistance in coating and assembly of electrodes, and Michael J. Tarkanian and Mark Belanger for their guidance and help with the fabrication of the adsorption beds. This work made use of the DMSE microscopy facility and machine shop, and the Edgerton Student Shop at MIT.
References
- F. Giorgi, P. H. Whetton, R. G. Jones, J. H. Christensen, L. O. Mearns, B. Hewitson, H. vonStorch, R. Francisco and C. Jack, Geophys. Res. Lett., 2001, 28, 3317–3320 CrossRef.
- W. Cai, A. Santoso, G. Wang, S.-W. Yeh, S.-I. An, K. M. Cobb, M. Collins, E. Guilyardi, F.-F. Jin, J.-S. Kug, M. Lengaigne, M. J. McPhaden, K. Takahashi, A. Timmermann, G. Vecchi, M. Watanabe and L. Wu, Nat. Clim. Change, 2015, 5, 849–859 CrossRef.
- J. Tollefson and K. R. Weiss, Nature, 2015, 528, 315–316 CrossRef CAS PubMed.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M. C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130–189 RSC.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- F. L. Horn and M. Steinberg, Fuel, 1982, 61, 415–422 CrossRef CAS.
- J. Wang, L. Huang, R. Yang, Z. Zhang, J. Wu, Y. Gao, Q. Wang, D. O’Hare and Z. Zhong, Energy Environ. Sci., 2014, 7, 3478–3518 RSC.
- A. Brunetti, F. Scura, G. Barbieri and E. Drioli, J. Membr. Sci., 2010, 359, 115–125 CrossRef CAS.
- United States Environmental Protection Agency, Inventory of U.S. Greenhouse Gas Emissions and Sinks: 1990–2016, 2018.
- ASHRAE, ASHRAE 62.1-2016, Ventilation for Acceptable Indoor Air Quality, 2016.
- J. Winnick, H. Toghiani and P. D. Quattrone, AIChE J., 1982, 28, 103–111 CrossRef CAS.
- S. Choi, M. L. Gray and C. W. Jones, ChemSusChem, 2011, 4, 628–635 CrossRef CAS PubMed.
- M. C. Stern, F. Simeon, H. Herzog and T. A. Hatton, Energy Environ. Sci., 2013, 6, 2505 RSC.
- W. J. Ward, Nature, 1970, 227, 162–163 CrossRef CAS PubMed.
- N. Jemaa, H. J. Walls, R. D. Noble, D. E. Wedman and C. A. Koval, AIChE J., 1993, 39, 867–875 CrossRef CAS.
- P. A. Terry, R. D. Noble, D. Swanson and C. A. Koval, AIChE J., 1997, 43, 1709–1716 CrossRef CAS.
- W. L. Bell, A. Miedaner, J. C. Smart, D. L. DuBois and C. E. Verostko, J. Aerosp., 1988, 97, 544–552 Search PubMed.
-
D. L. DuBois, A. Miedaner, W. Bell and J. C. Smart, Electrochemical and Electrocatalytic Reactions of Carbon Dioxide, Elsevier, 1993 Search PubMed.
- P. R. Scovazzo, J. Electrochem. Soc., 2003, 150, D91–D98 CrossRef CAS.
- B. Gurkan, F. Simeon and T. A. Hatton, ACS Sustainable Chem. Eng., 2015, 3, 1394–1405 CrossRef CAS.
- R. Ranjan, J. Olson, P. Singh, E. D. Lorance, D.
A. Buttry and I. R. Gould, J. Phys. Chem. Lett., 2015, 6, 4943–4946 CrossRef CAS PubMed.
- P. Singh, J. H. Rheinhardt, J. Z. Olson, P. Tarakeshwar, V. Mujica and D. A. Buttry, J. Am. Chem. Soc., 2017, 139, 1033–1036 CrossRef CAS PubMed.
- J. H. Rheinhardt, P. Singh, P. Tarakeshwar and D. A. Buttry, ACS Energy Lett., 2017, 2, 454–461 CrossRef CAS.
- P. Xiao, J. Zhang, P. Webley, G. Li, R. Singh and R. Todd, Adsorption, 2008, 14, 575–582 CrossRef CAS.
- T. Harada, F. Simeon, E. Z. Hamad and T. A. Hatton, Chem. Mater., 2015, 27, 1943–1949 CrossRef CAS.
-
D. H. Apaydin, E. D. Głowacki, E. Portenkirchner and N. S. Sariciftci, Angew. Chem., Int. Ed., 2014, 53, 6819–6822 Search PubMed.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280 RSC.
- Z. Song, Y. Qian, M. L. Gordin, D. Tang, T. Xu, M. Otani, H. Zhan, H. Zhou and D. Wang, Angew. Chem., 2015, 127, 14153–14157 CrossRef.
- Y. Zhou, B. Wang, C. Liu, N. Han, X. Xu, F. Zhao, J. Fan and Y. Li, Nano Energy, 2015, 15, 654–661 CrossRef CAS.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS PubMed.
- D. S. Achilleos and T. A. Hatton, ACS Appl. Mater. Interfaces, 2016, 8, 32743–32753 CrossRef CAS PubMed.
- M. Hefti, L. Joss, Z. Bjelobrk and M. Mazzotti, Faraday Discuss., 2016, 192, 153–179 RSC.
- L. Riboldi and O. Bolland, Int. J. Greenhouse Gas Control, 2015, 39, 1–16 CrossRef CAS.
- X. Mao, F. Simeon, D. S. Achilleos, G. C. Rutledge and T. A. Hatton, J. Mater. Chem. A, 2013, 1, 13120 RSC.
- T. Brousse, D. Belanger and J. W. Long, J. Electrochem. Soc., 2015, 162, A5185–A5189 CrossRef CAS.
- A. Ntiamoah, J. Ling, P. Xiao, P. A. Webley and Y. Zhai, Ind. Eng. Chem. Res., 2016, 55, 703–713 CrossRef CAS.
- M. B. Mizen and M. S. Wrighton, J. Electrochem. Soc., 1989, 136, 941 CrossRef CAS.
- T. Nagaoka, N. Nishii, K. Fujii and K. Ogura, J. Electroanal. Chem., 1992, 322, 383–389 CrossRef CAS.
- P. S. Guin, S. Das and P. C. Mandal, Int. J. Electrochem., 2011, 2011, 1–22 CrossRef.
- P. Bonĥte, A. P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef PubMed.
- P. A. Staley, E. M. Lopez, L. A. Clare and D. K. Smith, J. Phys. Chem. C, 2015, 119, 20319–20327 CrossRef CAS.
- Q. Li, C. Batchelor-McAuley, N. S. Lawrence, R. S. Hartshorne and R. G. Compton, ChemPhysChem, 2011, 12, 1255–1257 CrossRef CAS PubMed.
- T. Yamamoto and H. Etori, Macromolecules, 1995, 28, 3371–3379 CrossRef CAS.
- R. G. C. Ian Streeter, G. G. Wildgoose and L. Shao, Sens. Actuators, B, 2008, 133, 462–466 CrossRef.
-
D. M. Douglas, M. Ruthven, S. Farooq and K. S. Knaebel, Pressure swing adsorption, VCH Publishers, 1994 Search PubMed.
- M. C. Stern, F. Simeon, H. Herzog and T. A. Hatton, Energy Procedia, 2013, 37, 1172–1179 CrossRef CAS.
- M. C. Stern and T. A. Hatton, RSC Adv., 2014, 4, 5906 RSC.
- A. Ntiamoah, J. Ling, P. Xiao, P. A. Webley and Y. Zhai, Ind. Eng. Chem. Res., 2016, 55, 703–713 CrossRef CAS.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocellà, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- J. S. Lee, J. H. Kim, J. T. Kim, J. K. Suh, J. M. Lee and C. H. Lee, J. Chem. Eng. Data, 2002, 47, 1237–1242 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ee02412c |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
T. Alan
Hatton
and
T. Alan
Hatton

 ) and P14AQ–CNT (
) and P14AQ–CNT ( ) under N2 and (
) under N2 and ( ) under CO2 in [Bmim][TF2N], at 20 mV s−1, vs. Fc, at T ∼ 21 °C. The two potential windows are shown; ΔV1 under CO2 and ΔV2 under N2.
) under CO2 in [Bmim][TF2N], at 20 mV s−1, vs. Fc, at T ∼ 21 °C. The two potential windows are shown; ΔV1 under CO2 and ΔV2 under N2.

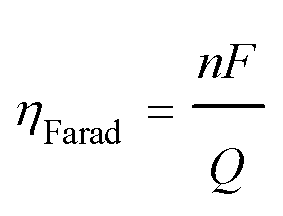
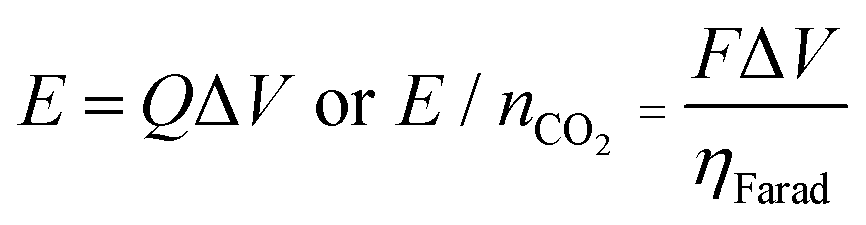
 ). The CO2 captured from and released to the chamber tracks the charge applied to the electrochemical cell, normalized by the area of the cell (
). The CO2 captured from and released to the chamber tracks the charge applied to the electrochemical cell, normalized by the area of the cell ( ). (b) The CO2 captured under different feed concentrations. (c) Capacity of cell over 7000 cycles. In a different set of experiments using a larger cell and cavity, (d) shows the effect of varying charging potential for a 1000 s capture and (e) shows the effect of varying the capture duration at −1.8 V capture potential. These experiments were conducted at T ∼ 21 °C.
). (b) The CO2 captured under different feed concentrations. (c) Capacity of cell over 7000 cycles. In a different set of experiments using a larger cell and cavity, (d) shows the effect of varying charging potential for a 1000 s capture and (e) shows the effect of varying the capture duration at −1.8 V capture potential. These experiments were conducted at T ∼ 21 °C.