
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSinglet oxygen from cation driven superoxide disproportionation and consequences for aprotic metal–O2 batteries†
Eléonore
Mourad
a,
Yann K.
Petit
 a,
Riccardo
Spezia
b,
Aleksej
Samojlov
a,
Francesco F.
Summa
c,
Christian
Prehal
a,
Christian
Leypold
a,
Nika
Mahne
a,
Christian
Slugovc
a,
Olivier
Fontaine
de,
Sergio
Brutti
*f and
Stefan A.
Freunberger
*a
a,
Riccardo
Spezia
b,
Aleksej
Samojlov
a,
Francesco F.
Summa
c,
Christian
Prehal
a,
Christian
Leypold
a,
Nika
Mahne
a,
Christian
Slugovc
a,
Olivier
Fontaine
de,
Sergio
Brutti
*f and
Stefan A.
Freunberger
*a
aInstitute for Chemistry and Technology of Materials, Graz University of Technology, Stremayrgasse 9, Graz 8010, Austria. E-mail: freunberger@tugraz.at
bLaboratoire de Chimie Théorique, UMR 7616 CNRS, Sorbonne Université, CC 137, 4, Place Jussieu, 75252 Paris Cedex 05, France
cDipartimento di Scienze, Università della Basilicata, V.le Ateneo Lucano 10, 85100 Potenza, Italy
dInstitut Charles Gerhardt Montpellier, UMR 5253, CC 1701, Université Montpellier, Place Eugène Bataillon, 34095 Montpellier Cedex 5, France
eRéseau sur le Stockage Electrochimique de l’Energie (RS2E), CNRS FR3459, 33 rue Saint Leu, 80039 Amiens, France
fDipartimento di Chimica, Università di Roma La Sapienza, P.le A. Moro 5, 00185 Roma, Italy. E-mail: sergio.brutti@unibas.it
First published on 16th July 2019
Abstract
Aprotic alkali metal–oxygen batteries require reversible formation of metal superoxide or peroxide on cycling. Severe parasitic reactions cause poor rechargeability, efficiency, and cycle life and have been shown to be caused by singlet oxygen (1O2) that forms at all stages of cycling. However, its formation mechanism remains unclear. We show that disproportionation of superoxide, the product or intermediate on discharge and charge, to peroxide and oxygen is responsible for 1O2 formation. While the overall reaction is driven by the stability of peroxide and thus favored by stronger Lewis acidic cations such as Li+, the 1O2 fraction is enhanced by weak Lewis acids such as organic cations. Concurrently, the metal peroxide yield drops with increasing 1O2. The results explain a major parasitic pathway during cell cycling and the growing severity in K–, Na–, and Li–O2 cells based on the growing propensity for disproportionation. High capacities and rates with peroxides are now realized to require solution processes, which form peroxide or release O2via disproportionation. The results therefore establish the central dilemma that disproportionation is required for high capacity but also responsible for irreversible reactions. Highly reversible cell operation requires hence finding reaction routes that avoid disproportionation.
Broader contextDecarbonizing the energy system requires energy storage with large capacity but equally low economic and ecological footprint. Alkali metal–O2 batteries are considered outstanding candidates in this respect. However, they suffer from poor cycle life as a result of cathode degradation. Formation of the highly reactive singlet oxygen has been proposed to cause this degradation, but formation mechanisms have remained unclear. Here, we show that the singlet oxygen source is the disproportionation of thermodynamically unstable superoxide intermediates to the peroxides. The revealed mechanism conclusively explains the strongly growing degree of degradation when going from K–O2 to Na–O2 and Li–O2 cells. A major consequence is that highly reversible cell operation of Li–O2 and Na–O2 cells requires them to form and decompose the peroxides without disproportionation. Achieving this requires finding new reaction routes. The work lays the mechanistic foundation to fight singlet oxygen as the predominant source of degradation in metal–O2 cells. |
Introduction
Advancing electrochemical storage beyond the limits of current batteries has become the focus of much cutting-edge research and hence has caused immense interest in rechargeable non-aqueous alkali metal–O2 batteries. They operate by reversibly forming/decomposing superoxides or peroxides of Li, Na, or K at a porous cathode according to| O2 + xe− + xM+ ↔ MxO2 (M = Li, Na, K) | (1) |
The parasitic reactions cause deviations from the ideal cell reaction in eqn (1). Key measures for parasitic chemistry are the ratio of O2 consumed/evolved and peroxide or superoxide formed/decomposed per electron passed on discharge/charge. Parasitic reactions form significant amounts of side products such as alkali carbonate, carboxylates, or CO2.2,3,13,15,17,20–22,26,27 The severity of parasitic chemistry increases in the order K–O2, Na–O2, and Li–O2 with typical (su)peroxide yields of ∼98–100%, ∼90–95%, and 50–90%, respectively, and similar O2 yields on recharge.3,5,10,13,15,17,20–22,26–29 Peroxide rather than superoxide as the product increases the severity, particularly on charge, where the voltage climbs inexorably due, in large parts, newly formed parasitic products.15,17,20–22,25,30,31
The parasitic reactions have long been predominantly ascribed to the direct reactivity of electrolyte or carbon with superoxides and peroxides owing to their basicitiy, nucleophilicity, or radical nature.2,3,15,17,19–22,27,28 Nevertheless, these reactivities fail to conclusively explain the mentioned pattern of parasitic reactions. Specifically, the extent of side reactions would suggest the reactivity to seemingly severely grow in the order KO2 < NaO2 < LiO2, and superoxides to be less reactive than peroxides, which opposes chemical intuition suggesting KO2 to be the most reactive. KO2 can, however, cycle highly reversible as recently shown by Lu et al.,5 which forcefully demonstrates that other degradation pathways than superoxide attack must prevail. Only recently, the highly reactive singlet oxygen (1O2 or 1Δg), the first excited state of ground state triplet oxygen (3O2 or 3Σg−), has been revealed to form upon cycling in Li–O2 and Na–O2 cells and to predominantly cause the side reactions.32–341O2 forms during discharge, rest, and from the onset of charge at rates that match the rates of parasitic chemistry occurring in cells. How 1O2 forms is unclear but must be deeply rooted in the way (su)peroxides form or decompose.
Discharge commences with O2 reduction to superoxide (MO2). Whether it further reacts to the peroxide via a second electrochemical 1 e− transfer
| MO2 + e− + M+ → M2O2 | (2) |
| 2MO2 → M2O2 + O2 | (3) |
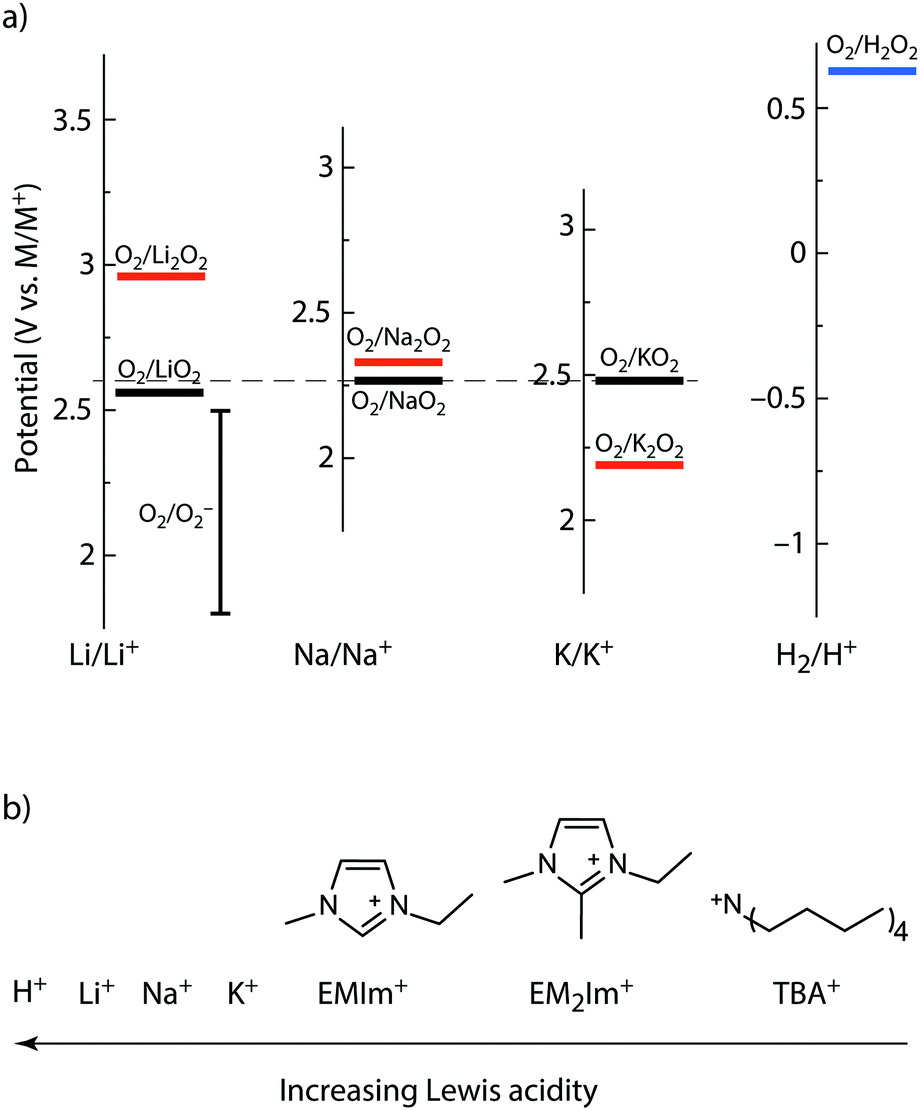 | ||
| Fig. 1 Thermodynamics of alkali peroxides and superoxides and the Lewis acidity of the here used cations. (a) Standard potentials of the O2/MO2 and O2/M2O2 redox couples on the M/M+ scales with M = Li, Na, K as well as for the O2/H2O2 couple. The scales are brought to a common scale based on their M/M+ standard potentials. The dashed horizontal line indicates the O2/KO2 couple. The O2/LiO2 potential is adopted from ref. 45, but also values between 2.29 and 2.46 V vs. Li/Li+ have been reported.28,43 O2/O2− denotes the potential range reported for O2 reduction in TBA+ electrolytes.9,37,41,45,46 (b) Schematic Lewis acidity order of the used cations. | ||
Here, we show that superoxide disproportionation in aprotic media releases significant fractions of 1O2 and we derive mechanistic descriptors for 1O2vs.3O2 release backed by simulations. While the strong Lewis acids Li+ and Na+ stabilize peroxide versus superoxide and drive the overall reaction, the 1O2 fraction is higher with Na+, the weaker Lewis acid. Also present even weaker Lewis acids enhance the 1O2 fraction massively. Larger 1O2 fractions go along with smaller peroxide yield. The results explain a major degradation pathway, explain the growing parasitic chemistry in K–, Na–, and Li–O2 cells based on the growing propensity for disproportionation, and show that counteracting 1O2 formation requires finding reaction routes that avoid superoxide disproportionation. Given that large capacities and rates require solution processes that rely on disproportionation steps, the results establish a central dilemma for high capacity metal–O2 cells.
Experimental
Materials
Salts contained either the bis(trifluoromethane)sulfonimide (TFSI−) or ClO4− anion. These anions have similar donor numbers and exert therefore minor changes on the considered solution equilibria47,48 and an analogous effect of TBA+ addition as confirmed in Fig. S1 (ESI†). LiTFSI, NaClO4, KClO4, TBATFSI were dried under vacuum for 24 h at 80 °C. Dimethoxyethane (DME) and tetraethylene glycol dimethyl ether (TEGDME) were dried over lithium, distilled and further dried and stored over activated molecular sieves. The water content as measured by Karl–Fischer titration was below 5 ppm. 9,10-Dimethylanthracene (DMA) was recrystallized from ethanol. Lithium peroxide (Li2O2) was synthesized as described previously.49 Its purity was confirmed by XRD, FTIR spectroscopy, and carbonate/carboxylate analysis.50Electrochemical methods
Metal–O2 cells with integrated pressure transducer were of the type PAT-Cell-Press from EL-Cell GmbH (Hamburg, Germany) with custom modified cathode plunger as described earlier.51 Electrochemical tests were run on a potentiostat/galvanostat (SP-300 or MPG-2, Bio-Logic). Free standing carbon/PTFE electrodes were made from a slurry of Super P carbon/PTFE binder (9/1, w/w) using isopropanol. Li1−xFePO4/C black/PTFE (8/1/1, m/m) counter electrodes were prepared analogously. For the Li2O2/C/PTFE (1/8/1, m/m) electrodes, Li2O2 was first ground with Super P (1/9, m/m) for 1.5 h in a planetary ball mill (Pulverisette 7, Fritsch) at 200 rpm with ZrO2 grinding balls under Ar. ATR-FTIR and XRD confirmed purity thereafter. A Super P/PTFE mixture (1/1, m/m) was made with isoproanol and dried under vacuum at 120 °C. Then, Li2O2/C and C/PTFE powders were mixed and pressed onto steel grids. Celgard separators and the electrodes were first washed with isoproanol and water (1/1, v/v) and subsequently with acetone. Electrodes and separators were dried under vacuum at 120 °C for 24 h. The counter electrode had three-fold the expected capacity of the working electrode. Typical working electrodes had a carbon mass loading of 1 mg and the cells were assembled with 100 μL electrolyte. Before discharge, cells were purged with high-purity O2 (N5.0).Spectroscopic methods
The mass spectrometry (MS) setup was built in-house and described previously.33,50 The sample setup consisted of a glass vial with a volume of 7 mL equipped with a stirring bar. A PEEK plug with glued-in PEEK tubes and an exchangeable septum is sealed against the glass vial with a flat rubber seal. Reagents were added through a septum using a gas-tight syringe (Hamilton). All solutions were degassed with N2 to remove dissolved CO2 and O2. The headspace was purged to the MS using 5 mL min−1 high purity Ar 6.0. To measure the rate of O2 evolution during the disproportionation reaction, a high-precision pressure transducer (Omega, PAA35X) was connected to the closed vessel instead of the MS. Reagents were added with a gas-tight syringe through glued-in tubing.High-performance liquid chromatography (HPLC) was used to determine the degree of the DMA-to-DMA-O2 conversion as described earlier.33 From chemical experiments, the filtered electrolyte was diluted with DME to ∼1 mgDMA mL−1. From cells, the electrolyte was extracted from all cell components using 400 μL DME, sonicated for 10 min under exclusion of light and heat, centrifuged and the supernatant was transferred and DME removed under a N2 stream at room temperature. The residue was dissolved in 500 μL DME and a volume of 2 μL was injected into the HPLC.
The amount of peroxide in a sample was measured by UV-vis spectroscopy of the Ti(IV)-peroxo complex in combination with mass spectrometry as described previously.50 The acidic environment also evolves CO2 from carbonates which was measured by MS. The samples in Fig. 2 and 3 from which Li2O2 and CO2 yield were obtained were prepared separately to the ones for 1O2 and 3O2 yield since DMA is incompatible with the Ti(IV) peroxo complex. Measurements given in bar graphs are from typically three or more repetitions. Repeatability is illustrated in Fig. S4 (ESI†).
Computational methods
Energies were calculated for solvated species with a solvent dielectric constant of 7.28 (1,1,2-trichloroethane, a value close to short chain glymes, like DME) by density functional theory (DFT) calculations by adopting a computational approach validated previously and benchmarked on post-Hartree–Fock calculations.16 The M06-2X functional and the 6-31++G(d,p) basis set (unrestricted)52 was used and solvation effects incorporated using a self-consistent reaction field (SCRF) in continuum solvation model C-PCM.53 The final computational accuracy for the reaction energies that do not involve the 1O2 species is estimated to be 0.05 eV. The pure O2 (1Δg) molecule computed at the unrestricted M062X level shows unsatisfactory geometry and frequencies, similarly to the B3LYP functional, due to the well-known spin-contamination problem.54 This unavoidable computational limitation at DFT level leads to an underestimate of the 3Σg− → 1Δg energy difference and thus to a worse computational accuracy for all 1O2 release reactions, estimated to 0.1–0.15 eV. All structures were relaxed to their energy ground state and vibrational stability checked for all the reported reagents, intermediates, and products. The Gibbs energy of each molecular/ionic species was calculated at 298 K by considering zero-point energies and thermal effects. All calculations were done using Gaussian16.55 Superoxide dimers we checked for all symmetric and asymmetric cases for all four conformers suggested by Bryantsev et al.56 and reported values are for the most stable ground state structures. The structures are shown in Fig. S2 and S3 (ESI†). The reaction energy for the precipitation to solid peroxides was calculated with thermochemical cycles starting from DFT calculations, the assessed thermodynamic properties of solid phases and for neutral atoms in the gas phase.57 The thermodynamics of the TBA+O2− ion couple was calculated at the same level of theory by relaxing the solid ionic couple in the simulated solvent to a ground state minimum.58Results and discussion
Probing singlet oxygen from superoxide disproportionation
We studied the disproportionation reaction| 2O2− → O22− + x3O2 + (1 − x)1O2 | (4) |
As a bimolecular reaction, superoxide disproportionation passes via M(O2)2M dimers (with M being any of the cation in Fig. 1b).35,41,56 We hypothesize that the energetics of pathways to 3O2 and 1O2 will sensitively depend on the cations involved. Therefore, to learn about the reaction mechanism, we intentionally influence the intermediates by using, next to pure Li+ or Na+ electrolytes, also their mixtures with TBA+ that itself would not drive disproportionation; the overall driving force to Li2O2 or Na2O2 remains unchanged while an asymmetric M(O2)2TBA intermediate dimer can be expected to be destabilized due to weaker O2−–TBA+ than O2−–M+ interactions9,37,45 and hence to change the energetics and relative yields of 3O2 and 1O2 evolution.
To probe for 1O2, we used 9,10-dimethylanthracene (DMA) as a chemical trap that fulfils the requirements for the non-aqueous (electro)chemical environment: it selectively forms the endoperoxide (DMA-O2) in contact with 1O2. DMA and DMA-O2 can be quantified by HPLC as detailed in the Methods, are electrochemically stable in the required potential range, and are stable towards superoxide and peroxide.33,34
Disproportionation of chemically produced superoxide
To probe for 3O2 and 1O2 yields from superoxide disproportionation, we first brought solid KO2 in contact with Li+, Na+, K+, and TBA+ electrolytes based on tetraethyleneglycol dimethylether (TEGDME) that also contained DMA, Fig. 2a. TEGDME was used since it is a common solvent for metal–O2 cells.1,2,9,13 The reaction was done in a closed vessel with the head space continuously purged to a mass spectrometer (MS) for gas analysis and the DMA-to-DMA-O2 conversion measured at the end of the experiment. When KO2 was brought in contact with the Li+ electrolyte, the O2 concentration rose sharply and ceased within 2 h, which indicates disproportionation as reported before.9,23,41 Quantifying the total O2 reveals that the KO2 has nearly quantitatively reacted and has resulted in ∼93% 3O2 and ∼2% 1O2 of the total O2 amount expected from eqn (4), i.e., 1 mol O2 per 2 mol KO2. KO2 in Na+ electrolyte equally resulted in disproportionation as reported recently.36 We found a continuous reaction, which does not come to completion within 2 h. The lower rate is in accord with the lower driving force (Fig. 1a) and the total O2 after this time shows that ∼8% of the KO2 have reacted of which 12% resulted in 1O2. KO2 in contact with K+ and TBA+ electrolyte did not evolve an appreciable amount of O2 as expected. These results show that superoxide disproportionation in presence of alkali cations yields significant fractions of 1O2 with its fraction increasing as Lewis acidity of the cations decreases.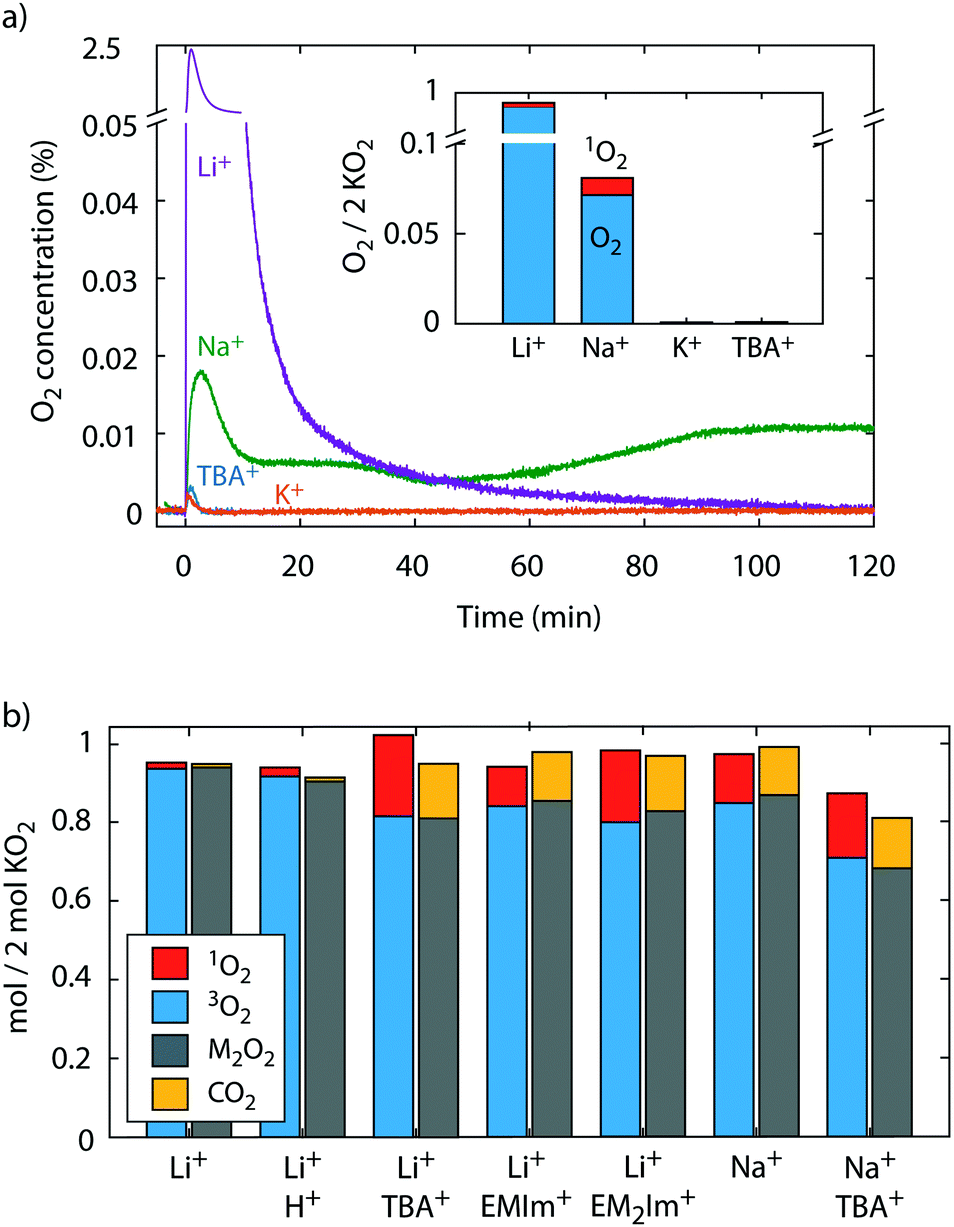 | ||
| Fig. 2 1O2 from superoxide disproportionation in presence of various cations. (a) O2 evolution versus time upon mixing KO2 with TEGDME electrolytes containing 0.1 M of the indicated cations and 30 mM 9,10-dimethylanthracene (DMA). The inset shows the evolved 3O2 (as measured by MS) and 1O2 (as measured as DMA-O2 by HPLC) after 2 h reaction time. (b) Obtained O2, 1O2, and Li2O2 (or Na2O2) upon reacting KO2 in TEGDME that contained equimolar 18-crown-6, 30 mM DMA, 0.5 M Li+ (or Na+), and either no additive, 0.1 M TBA+, EMIm+, or EM2Im+, or F3CCOOH. The scale means mol of O2, 1O2, Li2O2, or CO2 per 2 mol of KO2. I.e., ideally 1 mol O2 and 1 mol M2O2 would form according to 2KO2 + 2M+ → M2O2 + O2 + 2K+. Error bars are given in Fig. S4 (ESI†). | ||
To avoid the complexities of reactions at a solid, we further investigated the disproportionation of KO2 solvated by the crown ether 18-crown-6 (1,4,7,10,13,16-hexaoxacyclooctadecane). Additionally to 3O2 and 1O2, we also measured the Li2O2 or Na2O2 yield, respectively, after O2 evolution ceased using established procedures with photometry of the [Ti(O2)OH]+ complex after adding acidic TiOSO4 solution, which also evolves CO2 from formed carbonate.4,50 The CO2 amount serves as a proxy for the amount of side products. We added either pure Li+ electrolyte or combination of Li+ with H+, TBA+, EMIm+, or EM2Im+. We further added pure Na+ or Na+/TBA+ electrolytes. The results are shown in Fig. 2b. The result with pure Li+ resembles the one with solid KO2 in Fig. 2a; the 3O2 and Li2O2 yields were ∼93% and the 1O2 ∼2%. With F3CCOOH as H+ source we found ∼91% for 3O2 and peroxide yield and 3% for 1O2 yield and thus vanishingly more 1O2 than without acid. This is in accord with reported minor 1O2 yields from proton assisted superoxide disproportionation in Li-free media.59,60
Mixtures of Li+ and the weakly Lewis acidic organic cations, however, increase 1O2 very strongly; the 3O2 and Li2O2 yields dropped to ∼80–85% and the 1O2 rose to ∼10–20%. Carbonaceous side products as indicated by CO2 evolution also rose similarly. Adding weak Lewis acids into the disproportionation reaction not only raised the 1O2 yield massively, but concurrently boosted the reaction rates. We measured the 3O2 evolution kinetics from superoxide disproportionation by means of the pressure rise in a closed reaction vessel (Fig. S5, ESI†). Values compared to the kinetics with Li+ alone are 5-fold with EMIm+ and TBA+ and 8-fold with EM2IM+. The mechanistic implications of this finding are discussed later together with the theoretical results.
Given that organic cations provoke high 1O2 amounts, we assessed their stability in the system. Tetraalkylammoniums have been shown previously to be stable with superoxide.41 We probed whether imidazoliums would be reactive with superoxide or 1O2 and whether they would quench the latter and thus reduce the DMA-O2 yield, which then would underestimate the 1O2 yield. When EMIm+ and EM2Im+ were exposed to KO2 in TEGDME for 1 h, 1H-NMR spectra show a large number of new peaks (Fig. S6, ESI†), indicating decomposition in accord with previous reports.61 Exposing imidazoliums for 1 h to 1O2, generated photochemically as detailed in the Supplementary Methods (ESI†), left the 1H-NMR spectra largely unchanged (Fig. S7, ESI†). We do, however, not exclude a certain reactivity. Imidazoliums in high concentrations show a noticeable ability to quench 1O2 to 3O2, which suggests that measured 1O2 yields with imidazolium are likely underestimated (Fig. S8 and Supplementary Note 1, ESI†). Overall, enhanced 1O2 formation and instability with superoxide both make imidazoliums unsuitable for metal–O2 cells.
Turning to superoxide disproportionation in Na+ and mixed Na+/TBA+ electrolytes, we find for pure Na+ an analogous result to Fig. 2a: ∼13% 1O2, 85% 3O2 and 87% Na2O2. For the mixed Na+/TBA+ electrolyte the 3O2 and Na2O2 yields further dropped to around 70% and the 1O2 rose to ∼16%. Together with the results for Li+/TBA+ mixtures, the higher levels of 1O2 with the less Lewis acidic Na+ suggest that weaker Lewis acidic cations favour 1O2 evolving pathways. Another common result for all conditions in Fig. 2b is that the amounts of 3O2 and alkali peroxide closely match each other and that a larger fraction of missing peroxide is related to a larger amount of 1O2 formed.
Disproportionation during Li–O2 cell cycling
To probe whether the above observed disproportionation phenomena that yield 1O2 also explain 1O2 formation in cells, we performed analogous electrochemical experiments in Li–O2 cells. Li–O2 was chosen since disproportionation is most significantly driven by thermodynamics (Fig. 1a). We focus on TBA+ as the weak Lewis acid since it avoids the further complications of imidazolium instability with O2−. Considering first discharge, we constructed cells as detailed in the Experimental section with carbon black cathodes and TEGDME electrolytes containing 30 mM DMA and either only 0.1 M Li+ or a total of 1 M salt with a Li+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TBA+ ratio of 1:9 or 1:99. The cells were discharged at constant current and the O2 consumption followed using a pressure transducer as shown in Fig. 3a and Fig. S9 (ESI†). At the end of discharge, electrolyte and cathodes were extracted and analysed for the amount of 1O2, Li2O2, and carbonate. The results are shown in Fig. 3b with all values expressed as mol per 2 mol e− passed. Hence, ideally 2 mol e− would give 1 mol Li2O2 according to eqn (1).
:TBA+ ratio of 1:9 or 1:99. The cells were discharged at constant current and the O2 consumption followed using a pressure transducer as shown in Fig. 3a and Fig. S9 (ESI†). At the end of discharge, electrolyte and cathodes were extracted and analysed for the amount of 1O2, Li2O2, and carbonate. The results are shown in Fig. 3b with all values expressed as mol per 2 mol e− passed. Hence, ideally 2 mol e− would give 1 mol Li2O2 according to eqn (1).
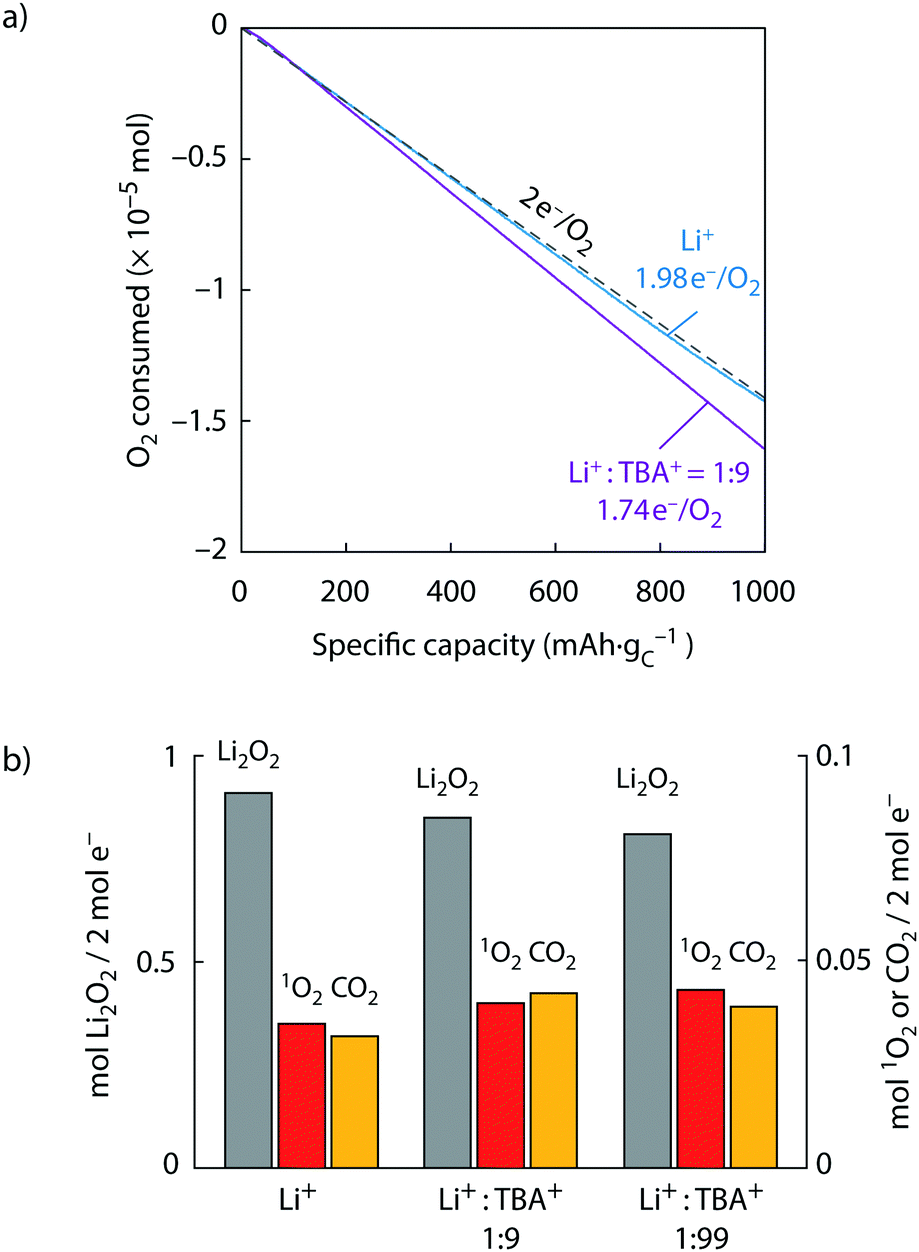 | ||
| Fig. 3 Superoxide disproportionation and 1O2 formation during Li–O2 cell discharge. (a) O2 consumption vs. capacity upon discharge of carbon black electrodes at a rate of 100 mA gC−1 in O2 saturated TEGDME electrolytes that contained 30 mM DMA and either 0.1 M Li+ or a total of 1 M salt with a Li+:TBA+ ratio of 1:9 or 1:99. The 1:99 ratio is given in Fig. S9 (ESI†) and voltage profiles in Fig. S10 (ESI†). (b) Obtained Li2O2, 1O2, and Li2CO3 (expressed as CO2) per 2 e− passed in the cells shown in (a). | ||
Discharge in pure Li+ electrolyte resulted in a ratio of 1.98 e−/O2, close to the ideal ratio of 2, and a Li2O2 yield of 94%, which both is in accord with previous reports for similar cells.11,15,20–22,33,50 The 1O2 yield was ∼3% and hence similar to that found in Fig. 2 for O2− disproportionation in Li+ electrolyte. With mixed Li+/TBA+ electrolytes with a Li+:TBA+ ratio of 1:9 (1:99), the e−/O2 ratio and Li2O2 yield dropped to 1.74 (1.70) e−/O2 and 85% (81%), respectively. Concurrently, the amount of 1O2 and carbonate increased as the Li2O2 yield decreased. Increasing 1O2 yield together with decreasing Li2O2 yield as the electrolyte is changed from Li+ to Li+/TBA+ mix mirrors the results in Fig. 2 for the chemical experiments. Considering further the e−/O2 ratios, the ideal value of 2 results from the sinks for the initially formed O2−: a second 1 e− reduction to peroxide or disproportionation to 3O2, which both give an overall 2 e−/O2 process. e−/O2 ratios lower than 2 imply more efficient sinks to exist for the 1 e− product O2− than a second reduction or disproportionation to 3O2. Given the known stability of TBA+ with O2−,36,41 their reaction can be excluded as the sink to cause the decrease to 1.74 (1.70) e−/O2. Instead, the lower ratio is in accord with TBA+ enhancing the 1O2 fraction from O2− disproportionation. Discharge with imidazoliums instead of TBA+ further corroborates their unsuitability as seen in even lower e−/O2 ratios of 1.42 and 1.2 for EM2Im+ and EMIm+, respectively (Supplementary Note 2 and Fig. S11, ESI†). The results on discharge are in accord with the chemical experiments shown in Fig. 2, which have shown that O2− disproportionation partly releases 1O2 and that the 1O2 fraction increases with the presence of TBA+. Overall, the results show that O2− disproportionation is the source of 1O2 on discharge, which further implies that discharge in the investigated TEGDME electrolyte passes at least significantly via disproportionation next to a possible second 1 e− reduction of the LiO2 intermediate viaeqn (2).
Turning to cell charge, we probed whether TBA+ analogously reveals 1O2 formation by O2− disproportionation. Li2−xO2 or soluble superoxide species were reported as intermediates on charge that disproportionate to form Li2O2 and O2.1,40,42,43 This reaction may hence equally be the source of 1O2 and sensitive to cations. We constructed Li2O2-packed working electrodes as detailed in the Experimental section. Li2O2 was ball milled with carbon black to ensure intimate contact between the two and the resulting powder was used to form composite electrodes using PTFE binder. We charged them in electrolytes that contained either only Li+ or a Li+/TBA+ mix and measured the amount of 3O2 and 1O2 by means of the pressure in the cell head space and DMA conversion, Fig. 4. The charge voltage was limited to 3.95 V since this voltage was reported to be the upper limit for quasi-equilibrium decomposition in TEGDME.40 Pressure evolution with pure Li+ electrolyte (Fig. 4a) shows similarly to previous reports11,20,21,40 an elevated value of 2.40 e−/O2 and thus ∼83% of the expected O2 evolved based on charge passed. 1O2 formation shows that the 3O2 loss is connected with 1O2 formation. When Li2O2 was charged in Li+/TBA+ electrolyte (Fig. 4b), the e−/O2 ratio rose to 2.95 and hence only ∼68% of the expected 3O2 evolved. Roughly doubled missing 3O2 evolution goes along with the 1O2 amount being more than doubled. To exclude the suggested 1O2 evolution from a direct 2 e− oxidation of Li2O2 above 3.5 V,32 we also restricted the charging voltage to 3.45 V, which shows similar results as with charge limited to 3.95 V (Supplementary Note 3, ESI†). Analogously to the experiments on discharge (Fig. 3), presence of TBA+ increased the fraction of 1O2 from O2− disproportionation with concurrently dropping 3O2 fraction. Proportional correlation between missing 3O2 evolution and 1O2 yield suggest in either case superoxide disproportionation to be a major O2 evolution and 1O2 generation pathway.
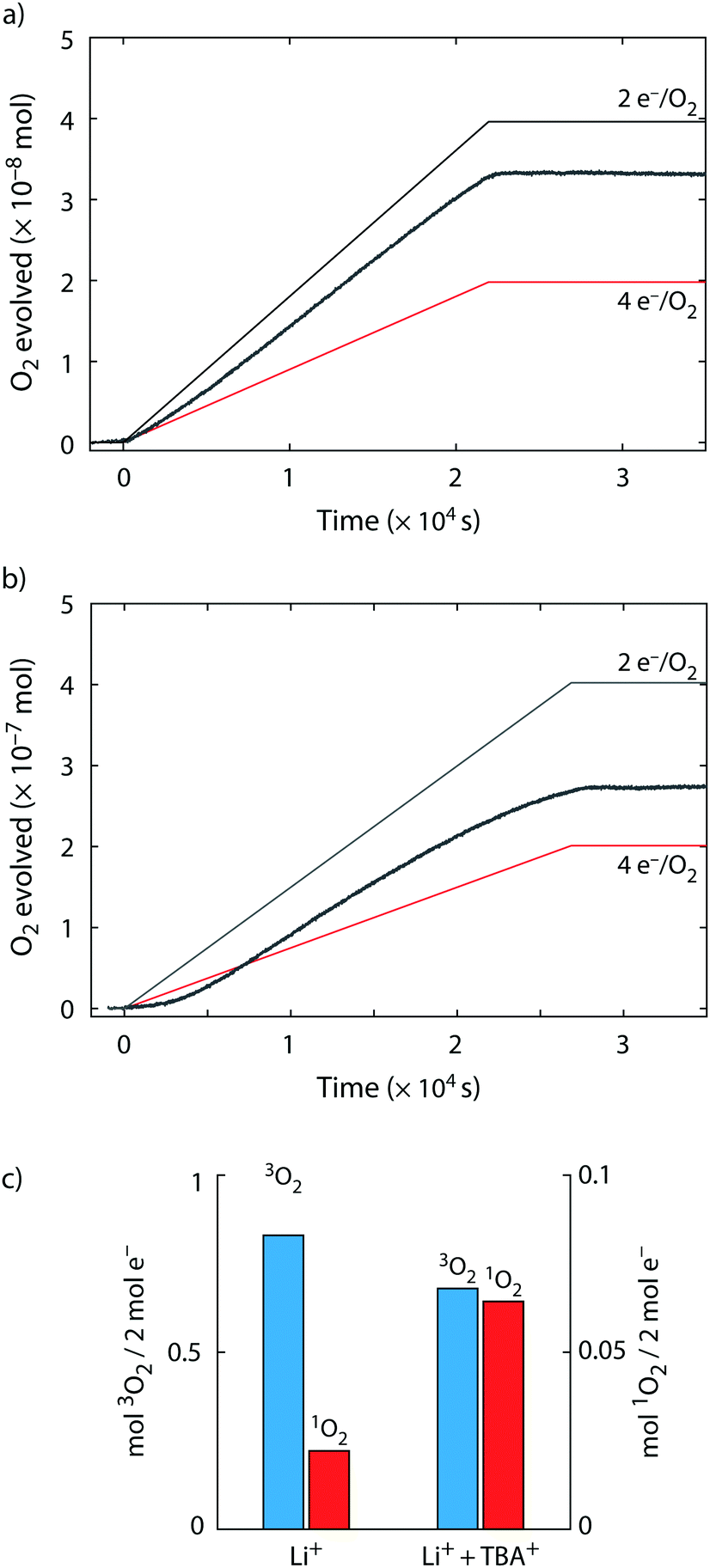 | ||
| Fig. 4 Superoxide disproportionation and 1O2 formation during Li–O2 cell charge. (a and b) O2 evolution vs. time upon charge of carbon black/Li2O2/PTFE (9/1/1, m/m) composite electrodes in TEGDME electrolyte containing 30 mM DMA and 0.1 M Li+ (a) or 0.1 M Li+ and 0.9 M TBA+ (b). Electrodes were charged at a rate of 10 mA gC−1 until 3.95 V and then kept at open circuit until the pressure was stable. (c) 3O2 and 1O2 obtained per 2 e− passed for the cells shown in (a) and (b). | ||
Taken together, the results from the chemical and electrochemical experiments show that superoxide disproportionation, driven by the higher stability of the peroxide with strong Lewis acids, generates in part 1O2. Simultaneous presence of weakly Lewis acidic organic cations increases the 1O2 yield markedly in the chemical and electrochemical experiments. These results (a) corroborate that superoxide disproportionation is a main pathway for the second electron transfer from superoxide to peroxide during discharge and O2 evolution during charge and (b) show that superoxide disproportionation is the 1O2 source during cell cycling.
A direct consequence of this finding is that the extent to which 1O2 can form on discharge and charge is governed by the extent to which disproportionation is responsible for the second electron transfer. The latter has been subject to many important studies recently and current understanding is that dominance of one or the other is governed by the LiO2 solvation vs. surface adsorption.2,62 Except for very poorly LiO2 solvating electrolytes such as MeCN, disproportionation appears to dominate even in only slightly more solvating glymes and certainly in any solvent with higher donor number, which is further enhanced by Li salt anions that dissociate weakly.6,63 Partition between second reduction/disproportionation has been, for example, investigated in glyme and DMSO by Shao-Horn who found at least significant disproportionation in glyme at low overpotentials.64 Peng calculated potentials where O2−* could be directly reduced to Li2O2* in DMSO and found disproportionation to dominate above 2 V vs. Li/Li+ on Au(111). Considering the latter, catalysts could potentially favour a second electron transfer already at higher voltages. We are, however, not aware of any study showing this possibility on discharge, but recent work by Lu suggests that catalysts could favour direct oxidation on charge.40,65,66 Another potential way could be redox mediators as suggested for quinones on discharge.7,8 However, proof that this fully suppresses superoxide disproportionation is still missing.
Energetics of singlet oxygen generation
To better understand the energetics of disproportionation and particularly why weak Lewis acids boost 1O2 formation, we performed density functional theory (DFT) calculations for pathways leading to 3O2 and 1O2. We considered LiO2 and NaO2 disproportionation as well as the asymmetric pairings of LiO2 and NaO2 with HO2 and TBAO2. Energies were calculated for solvated species using the continuous C-PCM solvation model with a mean dielectric constant of ε = 7.28 (resembling glyme) and using the hybrid GGA DFT M06-2X functional and the most favourable pathways are shown in Fig. 5. All energies are relative to the free superoxide monomers (2LiO2 or 2NaO2) to help understanding how cations other than Li+ or Na+ change the energetics relative to pure Li+ or Na+ electrolytes due to ion association/dissociation equilibria. A reaction energy beyond 1 eV implies an at least as high activation barrier and hence slow kinetics at room temperature.18 Starting from the doublet superoxide monomers, reactions may follow singlet or triplet pathways through the formation of a superoxide dimer M(O2)2M′ (M being Li+ or Na+, and M′ being M, H+ or TBA+). The dimers release singlet or triplet O2 and singlet MO2M′ peroxide that may further ion exchange to M2O2 and precipitate as solid M2O2(s).
beyond 1 eV implies an at least as high activation barrier and hence slow kinetics at room temperature.18 Starting from the doublet superoxide monomers, reactions may follow singlet or triplet pathways through the formation of a superoxide dimer M(O2)2M′ (M being Li+ or Na+, and M′ being M, H+ or TBA+). The dimers release singlet or triplet O2 and singlet MO2M′ peroxide that may further ion exchange to M2O2 and precipitate as solid M2O2(s).
 | ||
| Fig. 5 Reaction free energy profiles for superoxide disproportionation. (a) LiO2 disproportionation with itself, O2− or HO2 to Li2O2 and molecular oxygen. (b) NaO2 disproportionation with itself or O2− to Na2O2 and molecular oxygen. Pathways to release 3O2 and 1O2 are indicated by full and dashed lines, respectively. All species are computed in the solvated state except for the final solid peroxide. The computational method is M06-2X6-31**G++C-PMD (ε = 7.28). Numerical values are given in Tables S2–S4 (ESI†). Further asymmetric alkali superoxide pairings are considered in Supplementary Note 5 (ESI†). | ||
We consider first the symmetric LiO2 and NaO2 cases (Fig. 5a and b, red traces). For LiO2, the triplet 3Li(O2)2Li dimer is slightly stabilized compared to two monomers and releases Li2O2 + 3O2 weakly endergonic, followed by strongly stabilizing Li2O2 precipitation to solid Li2O2(s), which is the main overall driving force (Fig. 5a). Our results are in accord with previous works that analysed the route from LiO2 to 3O2 in the gas35,56 and solution phase41 and which are summarized in Fig. S13 (ESI†) for comparison. The path that we find for 1O2 release appears possible but slower with a thermodynamic barrier of ∼1 eV to the singlet 1Li(O2)2Li dimer followed by downhill 1O2 release and Li2O2(s) precipitation. The symmetric NaO2 case (Fig. 5b) is in either case uphill to the dimers but with their order being reversed (relative energies of singlet/triplet M(O2)2M dimers are analysed in detail in Supplementary Note 4, ESI†); the singlet 1Na(O2)2Na dimer forms with an energy increase of 0.83 eV less endergonic than the triplet 3Na(O2)2Na with ∼1.2 eV barrier. However, ongoing 1O2 release is further endergonic by 0.5 eV while 3O2 release is exergonic by −0.5 eV. The single step thermodynamic barrier towards 1O2 release from NaO2 is hence ∼0.1 eV higher than the barrier towards 3O2. The following precipitation of solid Na2O2(s) makes both singlet and triplet path overall exergonic, but less than for LiO2 disproportionation. Together, the relative single step barriers and overall driving forces rationalize our experimental findings: LiO2 disproportionates fast and the strongly differing barriers between singlet and triplet path cause relatively small 1O2 fractions. NaO2 disproportionates slowly and the more similar barriers cause larger 1O2 fractions.
Turning to proton mediated O2− disproportionation, our thermodynamic calculations for the asymmetric LiO2 + HO2 pairing suggest much easier 3O2 than 1O2 formation (Fig. 5a, blue traces). 3O2 and the mixed HLiO2 peroxide form in an exergonic single step reaction 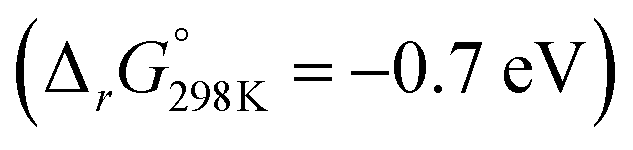 without a stable intermediate dimer. In contrast, the singlet path faces a barrier of 0.46 eV to the singlet 1Li(O2)2H dimer, which releases HLiO2 and 1O2 in a by −0.52 eV exergonic step. Analogous results were obtained for the NaO2 + HO2 pairing (Fig. S14, ESI†). The singlet path is in either case much more demanding and will result in minor 1O2 yields. This is in accord with our experimental finding in Fig. 2, which shows insignificant additional 1O2 with protons compared to pure Li+ electrolyte. It is also in accord with reported negligible 1O2 yields from proton mediated O2− disproportionation in Li+ and Na+ free media.59,60 We conclude from the calculations, in accord with the experiments, that proton sources cause minor additional 1O2 compared to disproportionation in Li+ electrolyte.
without a stable intermediate dimer. In contrast, the singlet path faces a barrier of 0.46 eV to the singlet 1Li(O2)2H dimer, which releases HLiO2 and 1O2 in a by −0.52 eV exergonic step. Analogous results were obtained for the NaO2 + HO2 pairing (Fig. S14, ESI†). The singlet path is in either case much more demanding and will result in minor 1O2 yields. This is in accord with our experimental finding in Fig. 2, which shows insignificant additional 1O2 with protons compared to pure Li+ electrolyte. It is also in accord with reported negligible 1O2 yields from proton mediated O2− disproportionation in Li+ and Na+ free media.59,60 We conclude from the calculations, in accord with the experiments, that proton sources cause minor additional 1O2 compared to disproportionation in Li+ electrolyte.
Turning to the case of the asymmetric pairing of superoxide with Li+ and the weakly Lewis acidic TBA+, our initial hypothesis was that weaker O2−–TBA+ than O2−–M+ interactions9,37,45 would destabilize intermediates, reduce barriers, and hence make 1O2 more favourable. In support of that, the experiments have shown higher kinetics and 1O2 yields with TBA+ (Fig. 2–4 and Fig. S5, ESI†) and the calculations in Fig. 5 confirm the suggested reasons. Considering the weak association of the TBA+O2− ion pair even in low dielectric constant solvents like DME  , TBAO2 may be approximated by the free solvated O2− anion. Solvent dependent O2/LiO2 and O2/TBAO2 standard potentials have been measured and computed by Shao-Horn et al.45 and found to differ by 1.24 V in DME, which agrees well with our estimate of 1.21 eV for the dissociation energy of LiO2 to free O2− anions (Fig. 5a, black traces). Note that O2− does not have to form via dissociation of LiO2, but may form as a transient species upon O2− generation. Ongoing triplet and singlet paths initially form 3Li(O2)2− and 1Li(O2)2− dimers that are stabilized versus LiO2 + O2− by −0.52 eV and −0.49 eV, respectively. Ongoing pathways to the charged LiO2− peroxide species plus 1O2 or 3O2 would face prohibitively high barriers because of the large dissociation energy of Li2O2 → LiO2− + Li+. Instead, our calculations reveal other facile pathways: the Li(O2)2− dimers can easily exchange TBA+ for Li+ and hence feed into the symmetric Li(O2)2Li pathways discussed above and shown in the red traces in Fig. 5a. Crucially, the presence of TBA+ decreases the barrier towards 1O2, the endergonicity of the most unfavourable step to the 1Li(O2)2Li dimer, from ∼1 eV to a mere 0.27 eV. Analogously, the asymmetric NaO2 + O2− pairing passes via Na(O2)2− and Na(O2)2Na dimers and the barrier towards 1O2 decreases from 1.2 eV to 0.4 eV. Overall, the weak Lewis acid TBA+ opens paths to bypass the most unfavourable reaction steps und hence strongly facilitates 1O2 evolution.
, TBAO2 may be approximated by the free solvated O2− anion. Solvent dependent O2/LiO2 and O2/TBAO2 standard potentials have been measured and computed by Shao-Horn et al.45 and found to differ by 1.24 V in DME, which agrees well with our estimate of 1.21 eV for the dissociation energy of LiO2 to free O2− anions (Fig. 5a, black traces). Note that O2− does not have to form via dissociation of LiO2, but may form as a transient species upon O2− generation. Ongoing triplet and singlet paths initially form 3Li(O2)2− and 1Li(O2)2− dimers that are stabilized versus LiO2 + O2− by −0.52 eV and −0.49 eV, respectively. Ongoing pathways to the charged LiO2− peroxide species plus 1O2 or 3O2 would face prohibitively high barriers because of the large dissociation energy of Li2O2 → LiO2− + Li+. Instead, our calculations reveal other facile pathways: the Li(O2)2− dimers can easily exchange TBA+ for Li+ and hence feed into the symmetric Li(O2)2Li pathways discussed above and shown in the red traces in Fig. 5a. Crucially, the presence of TBA+ decreases the barrier towards 1O2, the endergonicity of the most unfavourable step to the 1Li(O2)2Li dimer, from ∼1 eV to a mere 0.27 eV. Analogously, the asymmetric NaO2 + O2− pairing passes via Na(O2)2− and Na(O2)2Na dimers and the barrier towards 1O2 decreases from 1.2 eV to 0.4 eV. Overall, the weak Lewis acid TBA+ opens paths to bypass the most unfavourable reaction steps und hence strongly facilitates 1O2 evolution.
Consequences for metal–O2 batteries
Recognizing that 1O2 formation is deeply rooted in the way current metal–O2 cells operate has serious consequences on aspects to avoid and on directions that should be gone. First, caution must be exercised with weak Lewis acids as electrolytes or additives. This is supported by the selected quaternary ammonium and imidazolium cations, which are prototypical motifs for the cations used so far in ionic liquid electrolytes for metal–O2 cells. Imidazoliums readily decompose with superoxide. Significantly, we could show that the organic cation's weak Lewis acidity rather than its chemical nature massively boosts 1O2 formation. Given that ionic liquid cations suitable for electrolytes are most typically weak Lewis acids, the effect can likely be generalized. Favoured 1O2 formation explains why quantitative studies of metal–O2 chemistry with a broad variety of ionic liquids have shown worse parasitic chemistry on discharge and charge than molecular electrolytes.19,20,24Second, protic additives drive 1O2 formation insignificantly but may drive parasitic chemistry in other ways. This is in accord with reports that found increased side reactions when water or other Brønsted acids were added.11,29,67 The previous suggestion that proton sources could cause 1O2 in Na–O2 cells34 can now be revised to NaO2 disproportionation being the 1O2 source. Protons may be a remaining source of instability in K–O2 cells despite thermodynamic stability of KO2 in K+ electrolytes.5,10,26,38 Meticulously excluding impurities has hence allowed for impressive cyclability of K–O2 batteries.5
Finally, the most prominent consequence is that situations bound for superoxide disproportionation must be avoided. Cells based on metastable LiO2 or NaO2 as target products likely lack the practically required tolerance to slow discharge and rest periods; the superoxides gradually convert to peroxide and side products.3,14,25,27,30,34,35 Peroxide products are preferred as they are much higher in energy density and the thermodynamically stable products.28,31,68 Cycling them highly reversible requires finding routes to form and decompose them without superoxide disproportionation steps. Potential ways to do so are catalysts40,65,66 or redox mediators.7,8
Conclusions
In conclusion, we describe the mechanism for 1O2 formation and hence a main driver parasitic chemistry across alkali metal–O2 cells. We show that superoxide disproportionation forms the 1O2, and we clarify the reaction mechanism and governing factors in detail. The mechanism explains the growing parasitic chemistry in K–, Na–, and Li–O2 cells as well as between superoxide and peroxide products based on the growing propensity for disproportionation. The strong Lewis acids H+, Li+ and Na+ stabilize peroxide versus superoxide and hence drive disproportionation. 1O2 yields grow in this order with H+ causing insignificant 1O2 and strongly growing 1O2 fractions with Li+ and Na+. Importantly, weak Lewis acids such as TBA+ alone do not drive disproportionation, but, when combined with strong Lewis acids, strongly reduce the reaction barriers towards 1O2 and cause substantially larger fractions of 1O2. This calls for caution with ionic liquid electrolytes that comprise such weak Lewis acidic cations. The results explain major degradation routes of metal–O2 cells. Given that achieving high capacities and rates requires solution routes on discharge and charge, which in turn favour disproportionation reactions, the results establish the central dilemma that disproportionation is both important for high capacity/high rate and responsible for degradation. Future work should hence focus on finding routes for peroxide discharge and charge that avoid superoxide disproportionation.Authors contributions
E. M., Y. K. P., A. S., C. L., N. M., S. A. F. developed the methods and/or did the experiments. R. S., F. F. S., S. B. did calculations. C. P., C. S., O. F., S. B., S. A. F. discussed the results. S. A. F. and S. B. conceived the work and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. A. F. is indebted to the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 636069) and the Austrian Federal Ministry of Science, Research and Economy and the Austrian Research Promotion Agency (grant no. 845364). We thank EL-Cell GmbH (Hamburg, Germany) for the test cells. Likewise, we thank M. Winkler of Acib GmbH and G. Strohmeier for HPLC measurements, S. Borisov, W. Yin, and A. Grimaud for discussions, and R. Saf for help with the MS.Notes and references
- Y.-C. Lu, B. M. Gallant, D. G. Kwabi, J. R. Harding, R. R. Mitchell, M. S. Whittingham and Y. Shao-Horn, Energy Environ. Sci., 2013, 6, 750–768 RSC.
- D. Aurbach, B. D. McCloskey, L. F. Nazar and P. G. Bruce, Nat. Energy, 2016, 1, 16128 CrossRef CAS.
- T. Liu, G. Kim, M. T. L. Casford and C. P. Grey, J. Phys. Chem. Lett., 2016, 7, 4841–4846 CrossRef CAS PubMed.
- P. Hartmann, C. L. Bender, M. Vračar, A. K. Dürr, A. Garsuch, J. Janek and P. Adelhelm, Nat. Mater., 2012, 12, 228–232 CrossRef PubMed.
- G. Cong, W. Wang, N.-C. Lai, Z. Liang and Y.-C. Lu, Nat. Mater., 2019, 18, 390–396 CrossRef CAS PubMed.
- C. M. Burke, V. Pande, A. Khetan, V. Viswanathan and B. D. McCloskey, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9293–9298 CrossRef CAS PubMed.
- X. Gao, Y. Chen, L. Johnson and P. G. Bruce, Nat. Mater., 2016, 15, 882–888 CrossRef CAS PubMed.
- T. Liu, J. T. Frith, G. Kim, R. N. Kerber, N. Dubouis, Y. Shao, Z. Liu, P. C. M. M. Magusin, M. T. L. Casford, N. Garcia-Araez and C. P. Grey, J. Am. Chem. Soc., 2018, 140, 1428–1437 CrossRef CAS PubMed.
- L. Johnson, C. Li, Z. Liu, Y. Chen, S. A. Freunberger, J.-M. Tarascon, P. C. Ashok, B. B. Praveen, K. Dholakia and P. G. Bruce, Nat. Chem., 2014, 6, 1091–1099 CrossRef CAS PubMed.
- W. Wang, N.-C. Lai, Z. Liang, Y. Wang and Y.-C. Lu, Angew. Chem., Int. Ed., 2018, 57, 5042–5046 CrossRef CAS PubMed.
- N. B. Aetukuri, B. D. McCloskey, J. M. García, L. E. Krupp, V. Viswanathan and A. C. Luntz, Nat. Chem., 2014, 7, 50–56 CrossRef PubMed.
- C. Xia, R. Black, R. Fernandes, B. Adams and L. F. Nazar, Nat. Chem., 2015, 7, 496–501 CrossRef CAS PubMed.
- L. Lutz, W. Yin, A. Grimaud, D. Alves Dalla Corte, M. Tang, L. Johnson, E. Azaceta, V. Sarou-Kanian, A. J. Naylor, S. Hamad, J. A. Anta, E. Salager, R. Tena-Zaera, P. G. Bruce and J.-M. Tarascon, J. Phys. Chem. C, 2016, 120, 20068–20076 CrossRef CAS.
- I. Landa-Medrano, R. Pinedo, X. Bi, I. Ruiz de Larramendi, L. Lezama, J. Janek, K. Amine, J. Lu and T. Rojo, ACS Appl. Mater. Interfaces, 2016, 8, 20120–20127 CrossRef CAS PubMed.
- B. D. Adams, R. Black, Z. Williams, R. Fernandes, M. Cuisinier, E. J. Berg, P. Novak, G. K. Murphy and L. F. Nazar, Adv. Energy Mater., 2015, 5, 1400867 CrossRef.
- M. Carboni, A. G. Marrani, R. Spezia and S. Brutti, Chem. – Eur. J., 2016, 22, 17188–17203 CrossRef CAS PubMed.
- R. Black, A. Shyamsunder, P. Adeli, D. Kundu, G. K. Murphy and L. F. Nazar, ChemSusChem, 2016, 9, 1795–1803 CrossRef CAS PubMed.
- V. S. Bryantsev, V. Giordani, W. Walker, M. Blanco, S. Zecevic, K. Sasaki, J. Uddin, D. Addison and G. V. Chase, J. Phys. Chem. A, 2011, 115, 12399–12409 CrossRef CAS PubMed.
- S. Das, J. Højberg, K. B. Knudsen, R. Younesi, P. Johansson, P. Norby and T. Vegge, J. Phys. Chem. C, 2015, 119, 18084–18090 CrossRef CAS.
- B. D. McCloskey, D. S. Bethune, R. M. Shelby, T. Mori, R. Scheffler, A. Speidel, M. Sherwood and A. C. Luntz, J. Phys. Chem. Lett., 2012, 3, 3043–3047 CrossRef CAS PubMed.
- B. D. McCloskey, J. M. Garcia and A. C. Luntz, J. Phys. Chem. Lett., 2014, 5, 1230–1235 CrossRef CAS PubMed.
- B. D. McCloskey, A. Valery, A. C. Luntz, S. R. Gowda, G. M. Wallraff, J. M. Garcia, T. Mori and L. E. Krupp, J. Phys. Chem. Lett., 2013, 4, 2989–2993 CrossRef CAS PubMed.
- R. Black, S. H. Oh, J.-H. Lee, T. Yim, B. Adams and L. F. Nazar, J. Am. Chem. Soc., 2012, 134, 2902–2905 CrossRef CAS PubMed.
- M. Piana, J. Wandt, S. Meini, I. Buchberger, N. Tsiouvaras and H. A. Gasteiger, J. Electrochem. Soc., 2014, 161, A1992–A2001 CrossRef CAS.
- J. Kim, H. Park, B. Lee, W. M. Seong, H.-D. Lim, Y. Bae, H. Kim, W. K. Kim, K. H. Ryu and K. Kang, Nat. Commun., 2016, 7, 10670 CrossRef CAS PubMed.
- N. Xiao, R. T. Rooney, A. A. Gewirth and Y. Wu, Angew. Chem., Int. Ed., 2018, 57, 1227–1231 CrossRef CAS PubMed.
- S. Y. Sayed, K. P. C. Yao, D. G. Kwabi, T. P. Batcho, C. V. Amanchukwu, S. Feng, C. V. Thompson and Y. Shao-Horn, Chem. Commun., 2016, 52, 9691–9694 RSC.
- C. L. Bender, P. Hartmann, M. Vračar, P. Adelhelm and J. Janek, Adv. Energy Mater., 2014, 4, 1–10 Search PubMed.
- K. U. Schwenke, M. Metzger, T. Restle, M. Piana and H. A. Gasteiger, J. Electrochem. Soc., 2015, 162, A573–A584 CrossRef CAS.
- D. Zhai, H.-H. Wang, J. Yang, K. C. Lau, K. Li, L. A. Curtiss and K. Amine, J. Am. Chem. Soc., 2013, 135, 15364–15372 CrossRef CAS PubMed.
- C. L. Bender, D. Schröder, R. Pinedo, P. Adelhelm and J. Janek, Angew. Chem., Int. Ed., 2016, 55, 4640–4649 CrossRef CAS PubMed.
- J. Wandt, P. Jakes, J. Granwehr, H. A. Gasteiger and R.-A. Eichel, Angew. Chem., Int. Ed., 2016, 55, 6892–6895 CrossRef CAS PubMed.
- N. Mahne, B. Schafzahl, C. Leypold, M. Leypold, S. Grumm, A. Leitgeb, G. A. Strohmeier, M. Wilkening, O. Fontaine, D. Kramer, C. Slugovc, S. M. Borisov and S. A. Freunberger, Nat. Energy, 2017, 2, 17036 CrossRef CAS.
- L. Schafzahl, N. Mahne, B. Schafzahl, M. Wilkening, C. Slugovc, S. M. Borisov and S. A. Freunberger, Angew. Chem., Int. Ed., 2017, 56, 15728–15732 CrossRef CAS PubMed.
- U. Das, K. C. Lau, P. C. Redfern and L. A. Curtiss, J. Phys. Chem. Lett., 2014, 5, 813–819 CrossRef CAS PubMed.
- C. Sheng, F. Yu, Y. Wu, Z. Peng and Y. Chen, Angew. Chem., Int. Ed., 2018, 57, 9906–9910 CrossRef CAS PubMed.
- C. J. Allen, J. Hwang, R. Kautz, S. Mukerjee, E. J. Plichta, M. A. Hendrickson and K. M. Abraham, J. Phys. Chem. C, 2012, 116, 20755–20764 CrossRef CAS.
- Y. Chen, Z. P. Jovanov, X. Gao, J. Liu, C. Holc, L. R. Johnson and P. G. Bruce, J. Electroanal. Chem., 2018, 819, 542–546 CrossRef CAS.
- C. V. Amanchukwu, H.-H. Chang, M. Gauthier, S. Feng, T. P. Batcho and P. T. Hammond, Chem. Mater., 2016, 28, 7167–7177 CrossRef CAS.
- Y. Wang, N.-C. Lai, Y.-R. Lu, Y. Zhou, C.-L. Dong and Y.-C. Lu, Joule, 2018, 2, 2364–2380 CrossRef CAS.
- Y. Zhang, X. Zhang, J. Wang, W. C. McKee, Y. Xu and Z. Peng, J. Phys. Chem. C, 2016, 120, 3690–3698 CrossRef CAS.
- S. Ganapathy, B. D. Adams, G. Stenou, M. S. Anastasaki, K. Goubitz, X.-F. Miao, L. F. Nazar and M. Wagemaker, J. Am. Chem. Soc., 2014, 136, 16335–16344 CrossRef CAS PubMed.
- S. Kang, Y. Mo, S. P. Ong and G. Ceder, Chem. Mater., 2013, 25, 3328–3336 CrossRef CAS.
- Y.-C. Lu and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 4, 93–99 CrossRef PubMed.
- D. G. Kwabi, V. S. Bryantsev, T. P. Batcho, D. M. Itkis, C. V. Thompson and Y. Shao-Horn, Angew. Chem., Int. Ed., 2016, 55, 3129–3134 CrossRef CAS PubMed.
- Z. Peng, S. A. Freunberger, L. J. Hardwick, Y. Chen, V. Giordani, F. Bardé, P. Novák, D. Graham, J.-M. Tarascon and P. G. Bruce, Angew. Chem., Int. Ed., 2011, 50, 6351–6355 CrossRef CAS PubMed.
- M. Schmeisser, P. Illner, R. Puchta, A. Zahl and R. van Eldik, Chem. – Eur. J., 2012, 18, 10969–10982 CrossRef CAS PubMed.
- W. Linert, A. Camard, M. Armand and C. Michot, Coord. Chem. Rev., 2002, 226, 137–141 CrossRef CAS.
- M. M. Ottakam Thotiyl, S. A. Freunberger, Z. Peng and P. G. Bruce, J. Am. Chem. Soc., 2013, 135, 494–500 CrossRef CAS PubMed.
- B. Schafzahl, E. Mourad, L. Schafzahl, Y. K. Petit, A. R. Raju, M. O. Thotiyl, M. Wilkening, C. Slugovc and S. A. Freunberger, ACS Energy Lett., 2017, 3, 170–176 CrossRef.
- Y. K. Petit, C. Leypold, N. Mahne, E. Mourad, L. Schafzahl, C. Slugovc, S. M. Borisov and S. A. Freunberger, Angew. Chem., Int. Ed., 2019, 58, 6535–6539 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS.
- M. González, R. Valero, J. M. Anglada and R. Sayós, J. Chem. Phys., 2001, 115, 7015–7031 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- V. S. Bryantsev, M. Blanco and F. Faglioni, J. Phys. Chem. A, 2010, 114, 8165–8169 CrossRef CAS PubMed.
- M. W. Chase, Jr., NIST-JANAF thermochemical tables, 4th edn, Washington, DC: American Chemical Society; New York: American Institute of Physics for the National Institute of Standards and Technology, 1998 Search PubMed.
- P. D. C. Dietzel, R. K. Kremer and M. Jansen, J. Am. Chem. Soc., 2004, 126, 4689–4696 CrossRef CAS PubMed.
- J. M. Aubry, J. Rigaudy, C. Ferradini and J. Pucheault, J. Am. Chem. Soc., 1981, 103, 4965–4966 CrossRef CAS.
- E. J. Nanni, R. R. Birge, L. M. Hubbard, M. M. Morrison and D. T. Sawyer, Inorg. Chem., 1981, 20, 737–741 CrossRef CAS.
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed.
- H.-D. Lim, B. Lee, Y. Bae, H. Park, Y. Ko, H. Kim, J. Kim and K. Kang, Chem. Soc. Rev., 2017, 46, 2873–2888 RSC.
- D. Sharon, D. Hirsberg, M. Salama, M. Afri, A. A. Frimer, M. Noked, W. Kwak, Y.-K. Sun and D. Aurbach, ACS Appl. Mater. Interfaces, 2016, 8, 5300–5307 CrossRef CAS PubMed.
- D. G. Kwabi, M. Tułodziecki, N. Pour, D. M. Itkis, C. V. Thompson and Y. Shao-Horn, J. Phys. Chem. Lett., 2016, 7, 1204–1212 CrossRef CAS PubMed.
- Y. Wang and Y.-C. Lu, Angew. Chem., Int. Ed., 2019, 58, 6962–6966 CrossRef CAS.
- Y. Wang, Z. Liang, Q. Zou, G. Cong and Y.-C. Lu, J. Phys. Chem. C, 2016, 120, 6459–6466 CrossRef CAS.
- D. G. Kwabi, T. P. Batcho, S. Feng, L. Giordano, C. V. Thompson and Y. Shao-Horn, Phys. Chem. Chem. Phys., 2016, 18, 24944–24953 RSC.
- S. A. Freunberger, Nat. Energy, 2017, 2, 17091 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting methods, figures, tables and notes. See DOI: 10.1039/c9ee01453e |
| This journal is © The Royal Society of Chemistry 2019 |