
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePartial cation substitution reduces iodide ion transport in lead iodide perovskite solar cells†
Dominic W.
Ferdani‡
a,
Samuel R.
Pering‡
 a,
Dibyajyoti
Ghosh
ab,
Peter
Kubiak
a,
Alison B.
Walker
b,
Simon E.
Lewis
a,
Andrew L.
Johnson
a,
Peter J.
Baker
c,
M. Saiful
Islam
*a and
Petra J.
Cameron
*a
a,
Dibyajyoti
Ghosh
ab,
Peter
Kubiak
a,
Alison B.
Walker
b,
Simon E.
Lewis
a,
Andrew L.
Johnson
a,
Peter J.
Baker
c,
M. Saiful
Islam
*a and
Petra J.
Cameron
*a
aDepartment of Chemistry, University of Bath, Bath BA2 7AY, UK. E-mail: m.s.islam@bath.ac.uk; p.j.cameron@bath.ac.uk
bDepartment of Physics, University of Bath, Bath BA2 7AY, UK
cISIS Neutron & Muon Source, STFC Rutherford Appleton Laboratory, Didcot OX11 0QX, UK
First published on 15th May 2019
Abstract
Halide perovskite solar cells containing a mixture of A-site cations are attracting considerable interest due to their improved stability and high power conversion efficiencies. Ionic transport is known to be an important predictor of perovskite behaviour, but the impact of partial A-site substitution on iodide ion diffusion is poorly understood. Here, we combine ab initio modelling, impedance spectroscopy and muon spin relaxation to investigate the effect on iodide ion transport of incorporating a low concentration of each of seven different sized cations (from small rubidium to large guanidinium) into methylammonium lead iodide. Experimental and simulation results are in good agreement, indicating that these cation substitutions increase the activation energy for iodide ion diffusion. We show for the first time that partial guanidinium substitution into methylammonium lead iodide strongly suppresses iodide ion transport. The insights gained from this multi-technique study are important for the future design of mixed-cation perovskite solar cells with enhanced performance.
Broader contextPerovskite solar cells (PSCs) have been generating headlines over the last five years as their solar conversion efficiency has increased rapidly to rival that of multi-crystalline silicon solar cells. PSCs have several advantages over conventional silicon solar cells, including ease of manufacture and projected low energy pay back times. The cells are named for the ‘perovskite’ crystal structure of the light absorbing layer, which is based on an organic–inorganic iodide material. The perovskite layer is a mixed electronic–ionic conductor, containing mobile iodide ions which can strongly influence the measured properties of the solar cell. In this study we show that by moving from a single organic cation perovskite to a mixed-cation perovskite, iodide ion movement is supressed. We have systematically studied seven different cations and shown that the activation energy for iodide ion motion is linked to the size of the cation that is being substituted into the structure. Our results help to understand the improved stability of mixed cation PSCs. |
Introduction
Hybrid halide perovskites are generating enormous interest for use in next-generation solar cells with their power conversion efficiency rising rapidly to above 23% in less than a decade.1–5 Lead halide perovskites have the general formula APbX3, in which both the ‘A’ site cations and the ‘X’ site anions can be varied to some degree. Recent studies underline the benefits of mixing cations and/or anions in the halide perovskite structure.6–15 In comparison to the archetypal methylammonium lead iodide (MAPbI3), mixed-cation perovskites allow the band gap to be finely tuned, as well as exhibiting enhanced stability.10 Some studies suggest there is lower current–voltage (JV) curve hysteresis in mixed cation devices, even when all the other contact layers remain the same.8,10 Mixed formamidinium (FA) and methylammonium (MA) materials form solid solutions16 with triple-cation perovskite solar cells (PSCs) fabricated by the addition of small amounts of CsI to the MA/FA mixture. Multiple cation cells have the highest efficiencies, with many examples producing over 20%, with the current record of 23.7%.17,18Several larger A-site cations, including azetidinium (Az) and guanidinium (GA), were first identified by structural factors19 and the mixed materials were subsequently prepared.8,11 MA:Az materials, which retain a perovskite structure up to 5% Az substitution, show enhanced stability and better fill factors compared to MAPbI3.8 Guanidinium (GA) substitution improves the PSC performance by increasing the open cell voltage (VOC) and greatly enhancing stability,20 although the effect on ion transport has not been investigated. Jodlowski et al. showed that GA is phase miscible with MAPbI3 at MA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :GA ratios of up to 75:25, and MA0.86GA0.14PbI3 cells can reach efficiencies of over 20%.21
:GA ratios of up to 75:25, and MA0.86GA0.14PbI3 cells can reach efficiencies of over 20%.21
Given the enhanced stability and high efficiencies, what is not fully understood is why mixing A-site cations improves the cell performance. It is widely accepted that iodide ions are mobile in MAPbI3, but the impact of partial A-site cation substitution on iodide ion transport is unclear.22–24
Here, we combine experimental and simulation techniques to investigate, for the first time, the effect on iodide ion diffusion of substituting low concentrations of each of seven different sized cations into MAPbI3 (listed in Table 1). The cation sizes were varied from rubidium and caesium to guanidinium (GA). GA has an ionic radius of 278 pm, indicating significant MA/GA ion size mismatch. All of the cations were included at 5% substitution, giving MA0.95A0.05PbI3. As a result, the compounds retained the same 3D perovskite structure and band gap as MAPbI3, which allowed direct comparison of our results. Ab initio modelling was used to probe the atomic-scale effects of cation substitution on the local structure and ion transport activation energies. Photovoltaic devices were prepared for each mixed-cation composition and investigated by impedance spectroscopy (IS). Finally, powders of cation-substituted perovskites were investigated using muon spin relaxation (μSR) and we show that we can directly detect iodide ion diffusion in MAPbI3 and the doped compounds.
| Cation | Structure | Radius/pm19,25,26 |
|---|---|---|
| Rubidium (Rb) |

|
152 |
| Caesium (Cs) |

|
167 |
| Methylammonium (MA) |

|
217 |
| Azetidinium (Az) |
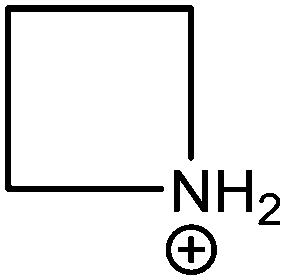
|
250 |
| Formamidinium (FA) |
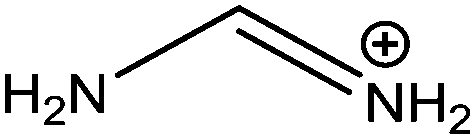
|
253 |
| Dimethylammonium (DM) |
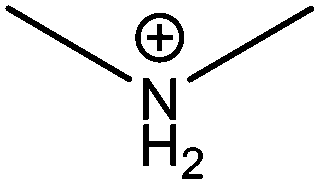
|
272 |
| Acetamidinium (Ac) |
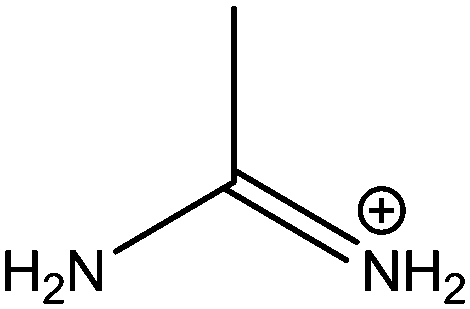
|
277 |
| Guanidinium (GA) |

|
278 |
Our combined studies show strong agreement between the ion transport results obtained from μSR on perovskite powders, ab initio simulations on the atomic scale and IS on full PSC devices. All the cation substitutions in MAPbI3 increase the activation energy for iodide ion transport. In particular, we show that just 5% GA substitution strongly suppresses iodide ion transport in the halide perovskite, which has implications for solar cell performance.
Results & discussion
Partial substitution of a range of A-site cations
The seven A-site cations selected for this study are summarised in Table 1. Although most of the cations are either too large or small to form a 3D perovskite phase on their own, they can be partially substituted into MAPbI3 to create binary mixed-cation systems.19,25,26 As phase separation can occur above certain substitution ratios, A-site doping was limited experimentally to 5% corresponding to MA0.95A0.05PbI3. Although it has been reported that Rb does not act as an A-site cation in FA-based perovskites, it can be incorporated alongside other smaller additives.27,28 Rb has been included here as it may act as an A-site cation in the more compact MA-based perovskite and as it is a very common additive in high efficiency mixed-cation PSCs.X-ray diffraction (XRD) was used to confirm that all seven doped materials had the same perovskite crystal structure and that phase separation had not occurred (Fig. 1). UV/vis measurements confirmed that there was minimal difference in the band gap after substitution, with changes of ±0.05 eV relative to MAPbI3 (Fig. S1a, ESI†). These results are supported by our ab initio simulations which also indicate that at 5% cation substitution there is no major effect on the band gap (Table S1, ESI†).
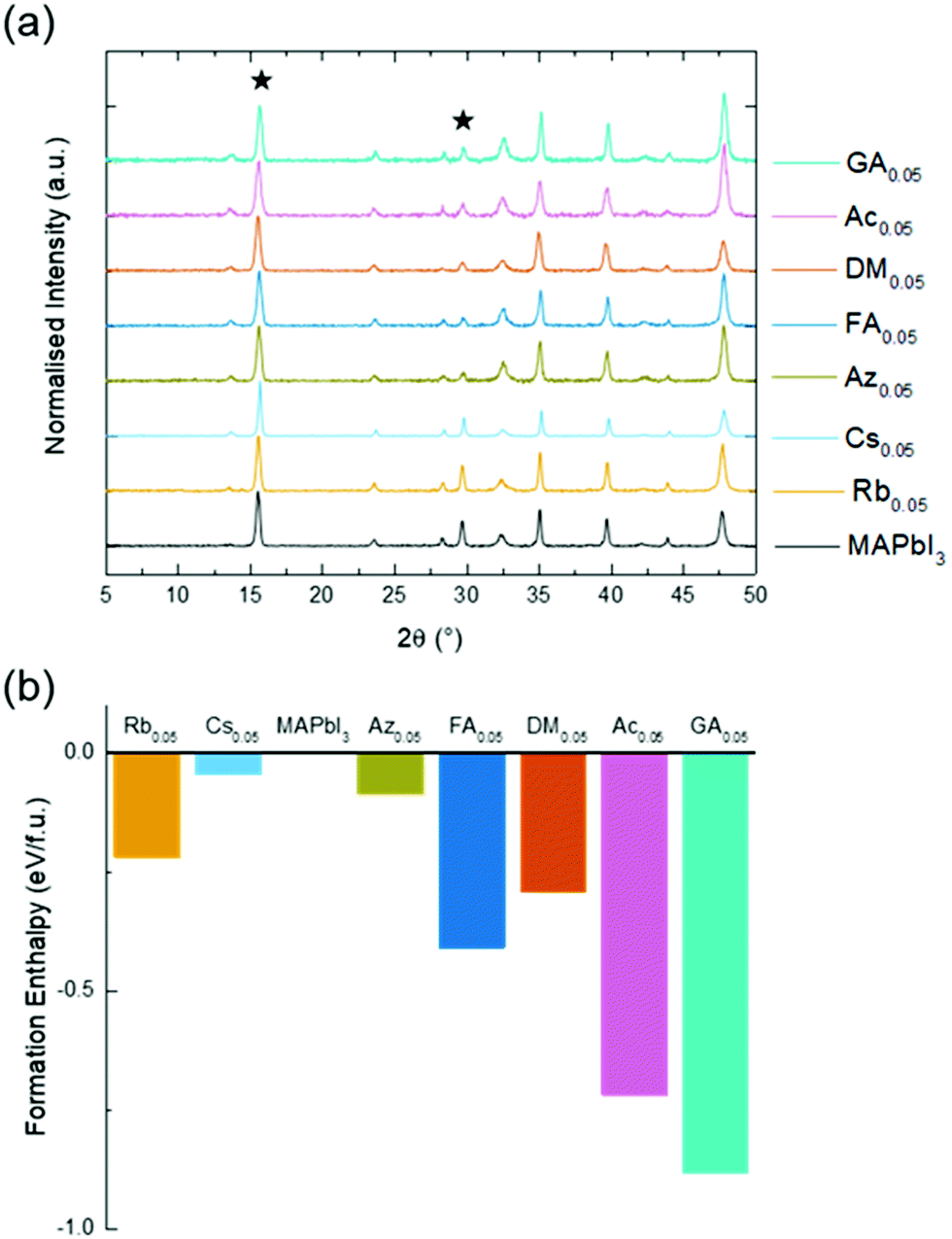 | ||
| Fig. 1 Structural analysis of mixed-cation perovskites MA0.95A0.05PbI3 (a) thin film XRD with the (110) and (220) peaks starred and (b) calculated formation enthalpies of the mixed-cation compound from the component iodide salts and PbI2. | ||
The presence of 5 mol% of the substituted cation was further confirmed by 1H NMR analysis on powders (for Cs, FA and GA substituted MAPbI3; Fig. S2, ESI†). By comparing peak integrals, we show that FA is present at a substitution ratio of 4.35 mol%; GA at 5 mol% and Cs at 4.3 mol%.
The thermodynamic stability of the perovskite phase upon cation substitution was explored using ab initio simulations by calculating the enthalpies of formation of the mixed-cation compound from the component iodide salts and PbI2 (Fig. 1b). As reported previously, pure MAPbI3 shows a very small positive formation enthalpy indicating a degree of metastability.29 More importantly, all the mixed-cation materials exhibit a negative formation enthalpy with GA substitution found to be the most favourable. These results suggest that the thermodynamic stability of MAPbI3 is enhanced by A-site cation substitution, which would suppress the decomposition to the component iodides (for example, MAI, GAI and PbI2). Also, the large cation size of GA (and Ac) promotes hydrogen bonding with iodide anions and these stronger interactions dominate over local strain to help stabilise the structure. Such findings are in good agreement with experimental observations of higher stability against temperature and chemical degradation for mixed-cation perovskites.16
Cation substitution and activation energies for ion transport
There is now a consensus that iodide ion diffusion in lead iodide perovskites has important implications for the behaviour of PSCs.30–38 Our previous work22,39 and other studies24,35,36,40–42 indicate that MAPbI3 is a mixed ionic–electronic conductor and implicate iodide ion transport (mediated by vacancy defects) in the JV curve hysteresis under working conditions, with no significant long-range transport of Pb2+ or MA+ ions. It should be noted that the choice of electron- and hole-selective contacts can also influence hysteresis,43–45 which suggests a complex relationship between the ionic environment and the properties of perovskite–contact interfaces.45 It is also important to note that the scan rate at which the maximum hysteresis is observed varies between devices with a range of contacts, perovskite material compositions and temperatures.46There have been previous ion transport studies on single cation perovskites,22–27 but not the effects of partial substitution of A-site cations as studied here; for example, in terms of comparing single cation perovskites Meloni et al. examined iodide migration in MAPbI3vs. CsPbI3, whereas Oranskaia et al. investigated bromide migration in MAPbBr3vs. FAPbBr3.47 A range of activation energies (Ea) for iodide ion conduction in pure MAPbI3 can be found in the literature (summarised in Table 2);22–24,30–36 most values fall between 0.3 eV and 0.6 eV, with the highest activation energy derived from DFT using the cubic structure.22
| Technique | E a (eV) |
|---|---|
| a Performed on MAPbI3−xClx perovskites. | |
| Ab initio/DFT & chronoamperometry22 | 0.58/0.62 |
| Impedance and IMVS30a |
0.55 |
| Thermally stimulated current31 | 0.5 |
| Temperature dependent capacitance32a |
0.45 |
| Ab initio/DFT33 | 0.44 |
| Impedance24 | 0.43 |
| Temperature dependent JV curves23 | 0.33 |
| Temperature dependent current34a |
0.31 |
| Solid-state NMR35 | 0.17 |
| Classical MD, ab initio/DFT36,37 | 0.08 |
However, the precise effects on ion transport of partial A-site substitutions into MAPbI3 have not been characterised and hence remains poorly understood. Here, we extend our related ab initio simulation work39,48,49 to calculate the relative activation energies for the vacancy-mediated migration of iodide ions in three representative systems: pure tetragonal MAPbI3 and Cs- and GA-substituted compounds. Since the simulations are probing local energy barriers, we investigated the MA0.75A0.25PbI3 composition with a higher substitution level allowing us to better examine A-site cation effects.
Energy profiles were derived for iodide vacancy diffusion, and particular care was taken to ensure full structural convergence to avoid unusually low activation energies. Our results are reported in Fig. 2, revealing three key results. First, the activation energy of 0.44 eV for tetragonal MAPbI3 matches well with previous experimental and computational studies (Table 2). Second, there is a small increase in the activation energy for Cs-substituted MAPbI3. Most significantly, we find a relatively steep increase to 0.78 eV for the GA-substituted material (with no major change with different GA orientations; see ESI,† Section C(iv) and Fig. S9). Hence, for long-range iodide ion diffusion through the perovskite material the rate-limiting barrier will be 0.78 eV, which predicts that iodide ion transport would be suppressed.
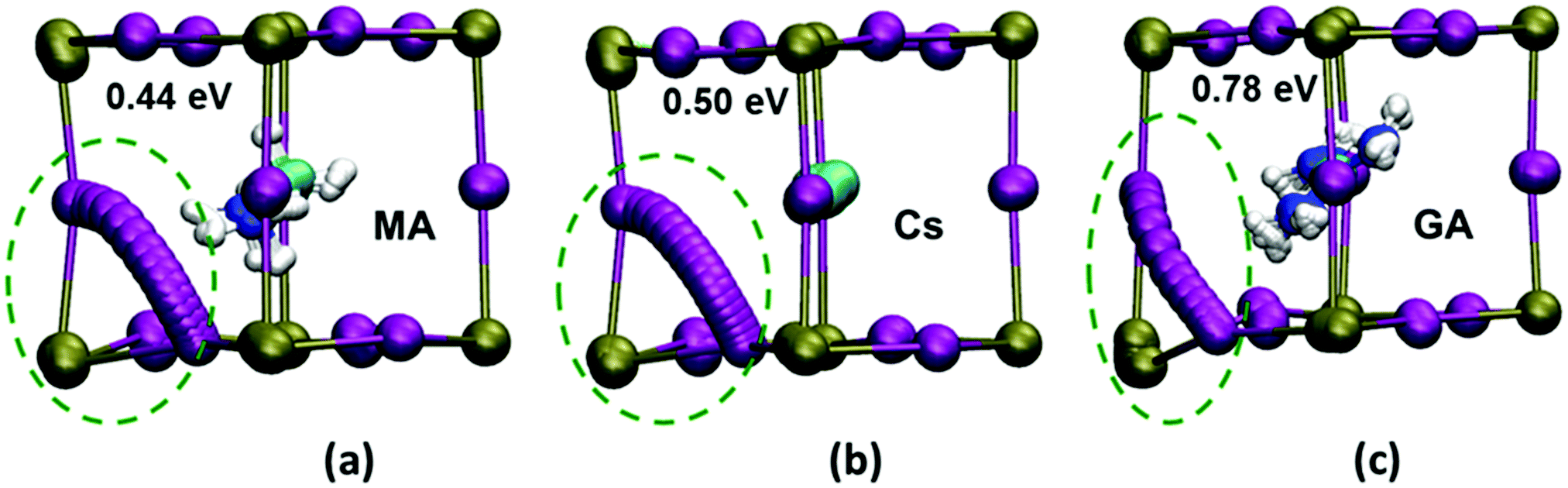 | ||
| Fig. 2 Iodide ion transport paths and energetics. Ab initio simulations of the ion transport paths (using 18 intermediate images), the activation energies and the lattice ion displacements in (a) MAPbI3 (b) MA0.75Cs0.25PbI3 and (c) MA0.75GA0.25PbI3. (Key: Pb, green; I, purple.) Local lattice relaxations near the diffusion path are highlighted by green circles, showing greater structural distortion in the GA-substituted material. Displacement values of the adjacent Pb ion in MAPbI3, MA0.75Cs0.25PbI3 and MA0.75GA0.25PbI3 are 0.2, 0.2 and 0.6 Å respectively. | ||
The simulations also reveal a curved migration path between iodide sites within the Pb/I face for both MAPbI3 and Cs-substituted material, with a small displacement of the neighbouring Pb ion (Fig. 2). In contrast, the presence of the much larger GA cation causes significant steric constraints and distortion of the local inorganic framework leading to a large displacement (0.6 Å) of the neighbouring Pb ion. These structural changes now lead to a curved path that is bowed out of the Pb/I plane (Fig. 2c), resulting in a much higher activation energy in the GA-substituted perovskite. Hence, it is the significant local distortion of the Pb/I cage due to the GA cation size mismatch with MA that leads to the reduction in iodide transport, rather than the exact orientation of the substituted molecular cation. Such atomic-scale mechanistic features are difficult to extract from experiment alone.
To gain insights into relative changes in diffusion rates, we derived iodide ion diffusion coefficients (DI) from the calculated activation energies, assuming a Boltzmann-like barrier-hopping process with a typical attempt frequency of 1012Hz for ionic species at a temperature of 300K. We find that DI would be approximately one order and five orders of magnitude lower for the Cs- and GA-substituted materials respectively; these results predict the suppression of iodide ion transport with GA substitution, which accords well with the muon spin relaxation and IS results described below.
Ion transport in complete solar cells
Planar inverted perovskite solar cells (PSCs) were made with 5 mol% substitution of all the seven cations listed in Table 1. We stress that the inverted FTO/NiO/perovskite/PCBM/bathocuproine/silver cells were prepared with a focus on stability rather than high efficiency to allow effective transport measurements. Indeed, to carry out impedance studies the cells needed to maintain a stable VOC over several hours of measurement. The highest published efficiency for a planar NiOx based MAPbI3 PSC is 16%.50 Our champion cells had efficiencies close to 15% (Fig. S10 and S11; Table S3, ESI†). To allow a direct comparison between cells, devices with 10% (±0.6) efficiency and a VOC at 1 sun of 1 V (±0.05 V) were selected. It should be noted that 5 mol% is not the optimum percentage for each cation in terms of efficiency. Consistency was more important to allow the effect of A-site cations to be compared. AFM images of the mixed-cation films were taken on NiOx coated FTO-glass and showed no differences in crystallite size and coverage between the samples (Fig. S12, ESI†).Temperature-dependent impedance spectroscopy (IS) was carried out on the devices. IS was measured at open circuit under illumination at temperatures between 45 °C and −15 °C. In line with our previous results, three semi-circles were observed in the Nyquist plot for MAPbI3 (Fig. S13, ESI†). We describe these as the high frequency (HF), mid-frequency (MF) and low frequency (LF) responses. The interpretation of impedance spectra for PSCs is still hotly debated in the literature.51–56 Nevertheless, it is generally accepted that the HF response gives the recombination resistance, Rrecomb, and the geometric capacitance, Cgeo. The LF feature is observed between 50 and 0.03 Hz (the time constant, τ, is typically just below 1 s) and the time scale is consistent with slow ionic reorganisation. We have previously attributed the LF response to modulation of Rrecomb as a function of a changing ionic environment. This would occur if, for example, the modulating voltage caused ions to move in such a way to better screen photo-generated charges, which in turn would increase the recombination resistance.
Regardless of the model used to explain the LF response, numerous studies show that it is intrinsically linked to ion diffusion. Bag et al. and Pockett et al. show that an activation energy can be found by plotting the LF time constant as a function of temperature.30,56,57 The activation energy of 0.40 eV for MAPbI3 (Fig. 3b and Table S4, ESI†) is very close to our calculated value and to those measured for iodide ions by a wide range of other techniques (Table 2). It is extremely unlikely that motion of A site cations such as MA+ will be observed on this timescale as their diffusion coefficients are at least three orders of magnitude lower than that for iodide ions.22
 | ||
| Fig. 3 Impedance spectroscopy measurements. (a) Nyquist plots for MA0.95Az0.05PbI3 and MA0.95GA0.05PbI3 at 25 °C and (b) a plot of the activation energy measured by impedance spectroscopy against the A-cation ionic radius relative to methylammonium, MA. There are no activation energies for GA or Ac as ion transport was too slow to be measured in the impedance spectra (the dashed line is a guide to the eye). | ||
Fig. 3 shows representative Nyquist plots for MA0.95Az0.05PbI3 and MA0.95GA0.05PbI3 at 25 °C (see also Fig. S13 and S14, ESI†). Three semi-circles are clearly visible in the Nyquist plot for MA0.95Az0.05PbI3. In contrast, the LF feature disappears completely for MA0.95GA0.05PbI3 (Fig. S14, ESI†) and an activation energy for iodide diffusion can no longer be extracted. These results mirror our ab initio modelling and μSR work and suggest that GA substitution strongly suppresses iodide ion diffusion. Similarly, it was not possible to resolve the LF feature for the second largest cation, Ac. Hence, there are no IS-derived activation energies for either GA or Ac, since ion transport was too slow to be detected.
Activation energies were extracted from the LF impedance response for cells substituted with the other five cations, Rb, Cs, Az, FA, DM (Fig. S15, ESI†), and plotted as a function of ionic radius in Fig. 3(b). The trend with ion size matches well with our calculated activation energies (Fig. 2). The IS results clearly show that partial substitution of MA with smaller or larger cations increases the activation energy for iodide ion transport.
All of the inverted cells prepared for this study showed low hysteresis at the scan rates studied (Fig. S11, ESI†). Despite not being the focus of the present study, iodide ion diffusion in perovskite solar cells has important implications for JV hysteresis, although it is known that hysteresis is also influenced by the nature of the electron- or hole-transport layers. In our study we did not see a reduction in JV-curve hysteresis after cation substitution, but it is important to note that scans were measured at a single scan rate and at room temperature. This is clearly an area that warrants further investigation.
Measuring iodide transport with muon spin relaxation
Muon spin relaxation (μSR) has been widely used to investigate the diffusion of Li+ and Na+ in modern battery materials,58,59 and here we apply the same approach to probe the motion of iodide ions in hybrid perovskites. Muons are non-destructive probes that can detect changes in nuclear dipole fields caused by moving ions inside a material (with full details of the technique given in Section E of the ESI†).μSR was used to probe ion movement in pure MAPbI3 and 5 mol% guanidinium substituted crystallites. To first investigate whether iodide motion could be detected by μSR, experiments were carried out on MAPbI3 and d6-perdeuterated MAPbI3 powders. Measurements on deuterated and protonated cations helped us to separate cation from anion motion. Data were collected between 50 and 410 K with typical raw data shown in Fig. 4a and Fig. S20–S22 (ESI†). The stability of the powders was checked using three separate techniques (ESI,† Section E(ii)). Fig. S19c (ESI†) shows XRD diffractograms of MAPbI3 before and after muon spectroscopy measurements were carried out. Some very small changes in peak intensity were observed, which is unsurprising as the sample has been subjected to a large change in temperature (40 K to 400 K) over 30 hours. Nevertheless, no evidence of PbI2 formation was observed.
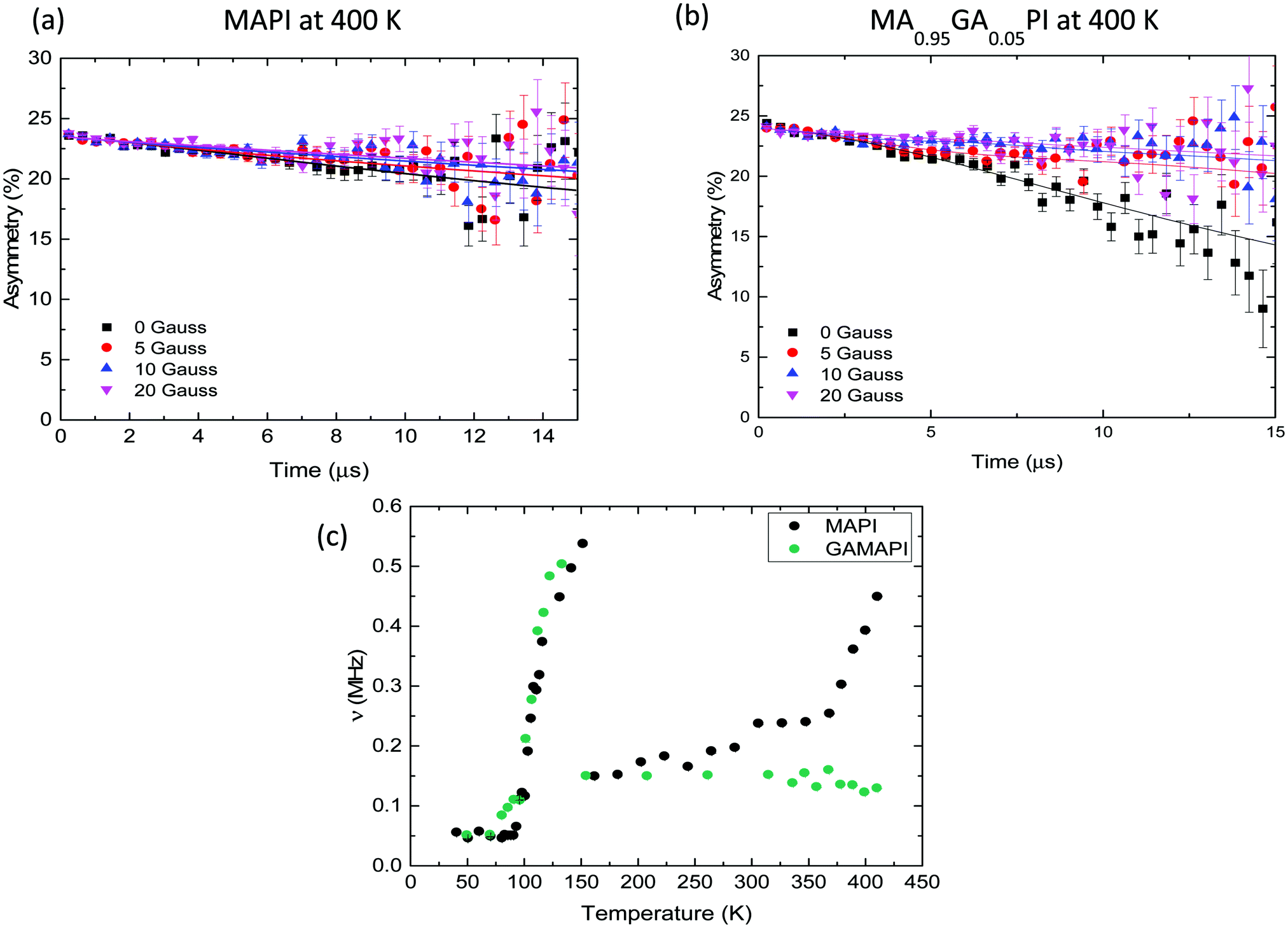 | ||
| Fig. 4 Muon spin relaxation measurements. (a) Raw muon data for MAPbI3 and (b) MA0.95GA0.5PbI3 at 400 K with zero field (squares) and the applied longitudinal fields of 5 G (circles), 10 G (triangles), 20 G (inverted triangle) with fits to a dynamic Kubo–Toyabe function with parameters common to the four fields. (c) Temperature dependence of ν values for MAPbI3 (black) and MA0.95GA0.5PbI3 (green). | ||
In μSR measurements muons are implanted into the sample where they decay into positrons, which are most likely to be emitted in the muon spin direction at the instant of decay. The effect of local fields on the material on the muon spins is detected by asymmetry in the positron counts measured by detectors around the sample. The asymmetry data at each temperature was fitted to a dynamic Gaussian Kubo–Toyabe function multiplied by an exponential relaxation (λ) (details in ESI,† Section E(i)).60 This allowed calculation of the fluctuation rate (ν, due to nuclear magnetic moments flipping the muon spin as they move nearby) and the static width of the disordered local field distribution (Δ, from nuclear magnetic moments at muon implantation sites).
Fig. 4c shows the temperature dependence of ν for MAPbI3. Δ values are shown in Fig. S23 (ESI†). ν increases from 0.05 MHz at 100 K to 0.6 MHz at 150 K, before falling to 0.2 MHz at 160 K. After 160 K, ν slowly increases until above 350 K, where ν increases more quickly in a thermally activated manner. The two large increases in ν are due to the motion of nuclear moments relative to the muon as different processes occur within the range of timescales to which muon spins are sensitive. The activation energies associated with these two processes in MAPbI3 were derived from an Arrhenius fit of the two activated regions (Fig. S24a and b, ESI†), producing values of 0.172 eV and 0.072 eV for the high-temperature and low-temperature processes respectively.
We have assigned the low temperature process to molecular cation reorientation within the Pb/I cage. As shown previously,61,62 MAPbI3 and d6-MAPbI3 are in a less rigid orthorhombic structure at 100 K with the methylammonium cations fully ordered. The {NH3} groups of MA+ align into the square faces of the unit cell and the {PbI6} octahedra are distorted. MAPbI3 undergoes a phase transition from orthorhombic to tetragonal at about 160 K.62–64 At temperatures close to 100 K, ν starts to increase as the muons are affected by increasing molecular cation motion within the Pb/I cages. Above 150 K the fluctuation rate decreases as the perovskite undergoes a phase transition, above the phase transition cation motion is too fast to be detected by μSR. Our results match neutron scattering studies showing that MA cations reorient within the perovskite lattice at temperatures close to 140 K.63
The high-temperature activation energies of 0.174 eV in MAPI and 0.158 eV in d6-MAPI are within the margin of experimental error (Table 3). Although lower than the DFT and IS derived energies, the muon result agrees well with the Ea value for iodide ion diffusion in MAPbI3 obtained from NMR studies (Table 2). NMR and μSR both probe changes in local fields within the bulk of a material so it would be expected that the two methods produce similar results.35
| Perovskite | Low T Ea (eV) | High T Ea (eV) |
|---|---|---|
| MAPbI3 | 0.072 (±0.005) | 0.174 (±0.010) |
| d6-MAPbI3 | 0.082 (±0.003) | 0.158 (±0.019) |
| MA0.95GA0.05PbI3 | 0.051 (±0.005) | Not observed |
The hopping rates in all of the samples are indistinguishable between about 150 K and 320 K as the iodide motion is too slow to be measurable by μSR.64 It is only when the diffusion coefficient rises above 1 × 10−13 cm2 s−1 (at about 360 K) that the ionic motion is on the correct timescale for μSR to measure. It is important to stress that this does not mean that the iodide ion is not moving at room temperature, but simply that it is not being detected by μSR.
Our muon study was then extended to MAPbI3 substituted with 5 mol% GA. Fig. 4b shows the raw data collected at 400 K. The retention of the same perovskite crystal structure was confirmed by powder XRD (Fig. S26, ESI†). Our computational predictions show that GA substitution increases the activation energy for iodide ion migration suggesting that iodide ion transport would be supressed. The μSR results support the computational and IS results as after GA substitution iodide motion can no longer be measured in the material. In MA0.95GA0.05PbI3 the increase in fluctuation rate above room temperature disappears completely and no iodide ion diffusion is detected up to 420 K (Fig. 4c). This result is also good evidence that the high-temperature growth in ν for the other samples is due to iodide ion transport, rather than muon motion, which we discuss fully in the ESI,† Section D. The presence of GA also affects the low-temperature fluctuations of the MA cations; the low-temperature Ea are listed in Table 3. Overall, the μSR results agree with our ab initio simulation and impedance spectroscopy results, clearly showing that adding GA to MAPbI3 reduces iodide ion transport.
Conclusions
Ion transport properties of MAPbI3-based materials and solar cells substituted with low concentrations of seven different sized A-site cations have been investigated using a combination of ab initio simulation, impedance spectroscopy and muon spin relaxation techniques. The study reveals fresh insights into the effect on iodide ion transport of partial substitution of methylammonium by other cations ranging in size from small Rb and Cs to large GA. The largest ion size mismatch for MA is with GA. The experimental and simulation results show strong agreement, indicating that all the cation substitutions increase the activation energy for iodide ion transport relative to pure MAPbI3. We show for the first time that just 5% GA substitution strongly suppresses iodide ion transport, with no observable ion diffusion in the timescale of our measurements. This combined study enhances our fundamental understanding of mixed-cation perovskites, and provides a design strategy for reducing iodide ion transport that has important implications for improving solar cell performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. S. I. and D. G. acknowledge support from the UK Engineering and Physical Sciences Research Council (EPSRC) Archer/HPC facilities through the Materials Chemistry Consortium (EP/L000202). We thank the University of Bath (studentship for S. R. P.), the Centre for Doctoral Training in Sustainable Chemical Technologies (studentship for D. W. F. under grant EP/L016354/1), the Centre for Doctoral Training in New and Sustainable Photovoltaics (studentship for P. S. K. under grant EP/LO1551X/1) and the EPSRC for P. J. C. (grant EP/H026304/1). This work was supported by the Energy Oriented Centre of Excellence (grant number 676629) funded within the European Union Horizon2020 framework. We thank the STFC for the allocated muon beamtime, and Laurie Peter and Alex Aziz for useful discussions.Notes and references
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed.
- J.-P. Correa-Baena, A. Abate, M. Saliba, W. Tress, T. Jesper Jacobsson, M. Gratzel and A. Hagfeldt, Energy Environ. Sci., 2017, 10, 710–727 RSC.
- D. A. Egger, A. Bera, D. Cahen, G. Hodes, T. Kirchartz, L. Kronik, R. Lovrincic, A. M. Rappe, D. R. Reichman and O. Yaffe, Adv. Mater., 2018, 30, 1800691 CrossRef PubMed.
- D. P. McMeekin, G. Sadoughi, W. Rehman, G. E. Eperon, M. Saliba, M. T. Horantner, A. Haghighirad, N. Sakai, L. Korte, B. Rech, M. B. Johnston, L. M. Herz and H. J. Snaith, Science, 2016, 351, 151–155 CrossRef CAS PubMed.
- D. Bi, W. Tress, M. I. Dar, P. Gao, J. Luo, C. Renevier, K. Schenk, A. Abate, F. Giordano, J.-P. Correa Baena, J.-D. Decoppet, S. M. Zakeeruddin, M. K. Nazeeruddin, M. Grätzel and A. Hagfeldt, Sci. Adv., 2016, 2, e1501170 CrossRef PubMed.
- S. R. Pering, W. Deng, J. R. Troughton, P. S. Kubiak, D. Ghosh, R. G. Niemann, F. Brivio, F. E. Jeffrey, A. B. Walker, M. S. Islam, T. M. Watson, P. R. Raithby, A. L. Johnson, S. E. Lewis and P. J. Cameron, J. Mater. Chem. A, 2017, 5, 20658–20665 RSC.
- Y. Sheng, A. Mei, S. Liu, M. Duan, P. Jiang, C. Tian, Y. Xiong, Y. Rong, H. Han and Y. Hu, J. Mater. Chem. A, 2018, 6, 2360–2364 RSC.
- C. Yi, J. Luo, S. Meloni, A. Boziki, N. Ashari-Astani, C. Gratzel, S. M. Zakeeruddin, U. Rothlisberger and M. Gratzel, Energy Environ. Sci., 2016, 9, 656–662 RSC.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, M. Saski, P. Yadav, D. Bi, N. Pellet, J. Lewiński, S. M. Zakeeruddin, M. Grätzel and L. Emsley, J. Am. Chem. Soc., 2018, 140, 3345–3351 CrossRef CAS PubMed.
- J. Lu, L. Jiang, W. Li, F. Li, N. K. Pai, A. D. Scully, C.-M. Tsai, U. Bach, A. N. Simonov, Y.-B. Cheng and L. Spiccia, Adv. Energy Mater., 2017, 7, 1700444 CrossRef.
- J. He, W.-H. Fang, R. Long and O. V. Prezhdo, ACS Energy Lett., 2018, 3, 2070–2076 CrossRef CAS.
- R. J. Stoddard, A. Rajagopal, R. L. Palmer, I. L. Braly, A. K.-Y. Jen and H. W. Hillhouse, ACS Energy Lett., 2018, 3, 1261–1268 CrossRef CAS.
- N. D. Pham, C. Zhang, V. T. Tiong, S. Zhang, G. Will, A. Bou, J. Bisquert, P. E. Shaw, A. Du, G. J. Wilson and H. Wang, Adv. Funct. Mater., 2019, 29, 1806479 CrossRef.
- B. Charles, J. Dillon, O. J. Weber, M. S. Islam and M. T. Weller, J. Mater. Chem. A, 2017, 5, 22495–22499 RSC.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- M. A. Green, Y. Hishikawa, E. D. Dunlop, D. H. Levi, J. Hohl-Ebinger, M. Yoshita and A. W. Y. Ho-Baillie, Prog. Photovoltaics, 2019, 27, 3–12 Search PubMed.
- G. Kieslich, S. Sun and A. K. Cheetham, Chem. Sci., 2014, 5, 4712–4715 RSC.
- N. De Marco, H. Zhou, Q. Chen, P. Sun, Z. Liu, L. Meng, E.-P. Yao, Y. Liu, A. Schiffer and Y. Yang, Nano Lett., 2016, 16, 1009–1016 CrossRef PubMed.
- A. D. Jodlowski, C. Roldán-Carmona, G. Grancini, M. Salado, M. Ralaiarisoa, S. Ahmad, N. Koch, L. Camacho, G. de Miguel and M. K. Nazeeruddin, Nat. Energy, 2017, 2, 972–979 CrossRef CAS.
- C. Eames, J. M. Frost, P. R. F. Barnes, B. C. O’Regan, A. Walsh and M. S. Islam, Nat. Commun., 2015, 6, 7497 CrossRef CAS PubMed.
- S. Meloni, T. Moehl, W. Tress, M. Franckevičius, M. Saliba, Y. H. Lee, P. Gao, M. K. Nazeeruddin, S. M. Zakeeruddin, U. Rothlisberger and M. Graetzel, Nat. Commun., 2016, 7, 10334 CrossRef CAS PubMed.
- T.-Y. Yang, G. Gregori, N. Pellet, M. Grätzel and J. Maier, Angew. Chem., Int. Ed., 2015, 54, 7905–7910 CrossRef CAS PubMed.
- M. Becker, T. Kluner and M. Wark, Dalton Trans., 2017, 46, 3500–3509 RSC.
- W. Travis, E. N. K. Glover, H. Bronstein, D. O. Scanlon and R. G. Palgrave, Chem. Sci., 2016, 7, 4548–4556 RSC.
- M. Saliba, T. Matsui, K. Domanski, J.-Y. Seo, A. Ummadisingu, S. M. Zakeeruddin, J.-P. Correa-Baena, W. R. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Science, 2016, 354, 206–209 CrossRef CAS PubMed.
- M. Zhang, J. S. Yun, Q. Ma, J. Zheng, C. F. J. Lau, X. Deng, J. Kim, D. Kim, J. Seidel, M. A. Green, S. Huang and A. W. Y. Ho-Baillie, ACS Energy Lett., 2017, 2, 438–444 CrossRef CAS.
- A. S. Thind, X. Huang, J. Sun and R. Mishra, Chem. Mater., 2017, 29, 6003–6011 CrossRef CAS.
- A. Pockett, G. E. Eperon, N. Sakai, H. J. Snaith, L. M. Peter and P. J. Cameron, Phys. Chem. Chem. Phys., 2017, 19, 5959–5970 RSC.
- A. Baumann, S. Väth, P. Rieder, M. C. Heiber, K. Tvingstedt and V. Dyakonov, J. Phys. Chem. Lett., 2015, 6, 2350–2354 CrossRef CAS PubMed.
- O. Almora, I. Zarazua, E. Mas-Marza, I. Mora-Sero, J. Bisquert and G. Garcia-Belmonte, J. Phys. Chem. Lett., 2015, 6, 1645–1652 CrossRef CAS PubMed.
- J. Haruyama, K. Sodeyama, L. Han and Y. Tateyama, J. Am. Chem. Soc., 2015, 137, 10048–10051 CrossRef CAS PubMed.
- C. Li, S. Tscheuschner, F. Paulus, P. E. Hopkinson, J. Kießling, A. Köhler, Y. Vaynzof and S. Huettner, Adv. Mater., 2016, 28, 2446–2454 CrossRef CAS PubMed.
- A. Senocrate, I. Moudrakovski, G. Y. Kim, T.-Y. Yang, G. Gregori, M. Grätzel and J. Maier, Angew. Chem., Int. Ed., 2017, 56, 7755–7759 CrossRef CAS PubMed.
- J. M. Azpiroz, E. Mosconi, J. Bisquert and F. De Angelis, Energy Environ. Sci., 2015, 8, 2118–2127 RSC.
- D. Barboni and R. A. De Souza, Energy Environ. Sci., 2018, 11, 3266–3274 RSC.
- J. M. Frost and A. Walsh, Acc. Chem. Res., 2016, 49, 528–535 CrossRef CAS PubMed.
- D. Ghosh, P. Walsh Atkins, M. S. Islam, A. B. Walker and C. Eames, ACS Energy Lett., 2017, 2, 2424–2429 CrossRef CAS.
- E. J. Juarez-Perez, R. S. Sanchez, L. Badia, G. Garcia-Belmonte, Y. S. Kang, I. Mora-Sero and J. Bisquert, J. Phys. Chem. Lett., 2014, 5, 2390–2394 CrossRef CAS PubMed.
- G. Richardson, S. E. J. O’Kane, R. G. Niemann, T. A. Peltola, J. M. Foster, P. J. Cameron and A. B. Walker, Energy Environ. Sci., 2016, 9, 1476–1485 RSC.
- A. Walsh and S. D. Stranks, ACS Energy Lett., 2018, 3, 1983–1990 CrossRef CAS.
- H.-S. Kim, I.-H. Jang, N. Ahn, M. Choi, A. Guerrero, J. Bisquert and N.-G. Park, J. Phys. Chem. Lett., 2015, 6, 4633–4639 CrossRef CAS PubMed.
- P. Calado, A. M. Telford, D. Bryant, X. Li, J. Nelson, B. C. O’Regan and P. R. F. Barnes, Nat. Commun., 2016, 7, 13831 CrossRef CAS PubMed.
- S. A. L. Weber, I. M. Hermes, S.-H. Turren-Cruz, C. Gort, V. W. Bergmann, L. Gilson, A. Hagfeldt, M. Graetzel, W. Tress and R. Berger, Energy Environ. Sci., 2018, 11, 2404–2413 RSC.
- D. Bryant, S. Wheeler, B. C. O’Regan, T. Watson, P. R. F. Barnes, D. Worsley and J. Durrant, J. Phys. Chem. Lett., 2015, 6, 3190–3194 CrossRef CAS.
- A. Oranskaia, J. Yin, O. M. Bakr, J.-L. Brédas and O. F. Mohammed, J. Phys. Chem. Lett., 2018, 9, 5474–5480 CrossRef CAS PubMed.
- N. Aristidou, C. Eames, I. Sanchez-Molina, X. Bu, J. Kosco, M. Saiful Islam and S. A. Haque, Nat. Commun., 2017, 8, 1–10 CrossRef PubMed.
- R. Brenes, C. Eames, V. Bulović, M. S. Islam and S. D. Stranks, Adv. Mater., 2018, 30, 1706208 CrossRef PubMed.
- X. Yin, P. Chen, M. Que, Y. Xing, W. Que, C. Niu and J. Shao, ACS Nano, 2016, 10, 3630–3636 CrossRef CAS PubMed.
- A. Todinova, L. Contreras-Bernal, M. Salado, S. Ahmad, N. Morillo, J. Idígoras and J. J. A. Anta, ChemElectroChem, 2017, 4, 2891–2901 CrossRef CAS.
- A. Guerrero, G. Garcia-Belmonte, I. Mora-Sero, J. Bisquert, Y. S. Kang, T. J. Jacobsson, J. P. Correa-Baena and A. Hagfeldt, J. Phys. Chem. C, 2016, 120, 8023–8032 CrossRef CAS.
- L. Contreras, J. Idigoras, A. Todinova, M. Salado, S. Kazim, S. Ahmad and J. A. Anta, Phys. Chem. Chem. Phys., 2016, 18, 31033–31042 RSC.
- R. S. Sanchez, V. Gonzalez-Pedro, J. W. Lee, N. G. Park, Y. S. Kang, I. Mora-Sero and J. Bisquert, J. Phys. Chem. Lett., 2014, 5, 2357–2363 CrossRef CAS PubMed.
- A. Kovalenko, J. Pospisil, O. Zmeskal, J. Krajcovic and M. Weiter, Phys. Status Solidi RRL, 2017, 11, 1600418 CrossRef.
- A. Pockett, G. E. Eperon, T. Peltola, H. J. Snaith, A. Walker, L. M. Peter and P. J. Cameron, J. Phys. Chem. C, 2015, 119, 3456–3465 CrossRef CAS.
- M. Bag, L. A. Renna, R. Y. Adhikari, S. Karak, F. Liu, P. M. Lahti, T. P. Russell, M. T. Tuominen and D. Venkataraman, J. Am. Chem. Soc., 2015, 137, 13130–13137 CrossRef CAS PubMed.
- J. Sugiyama, K. Mukai, Y. Ikedo, H. Nozaki, M. Månsson and I. Watanabe, Phys. Rev. Lett., 2009, 103, 147601 CrossRef PubMed.
- T. E. Ashton, J. V. Laveda, D. A. MacLaren, P. J. Baker, A. Porch, M. O. Jones and S. A. Corr, J. Mater. Chem. A, 2014, 2, 6238 RSC.
- R. S. Hayano, Y. J. Uemura, J. Imazato, N. Nishida, T. Yamazaki and R. Kubo, Phys. Rev. B: Condens. Matter Mater. Phys., 1979, 20, 850–859 CrossRef CAS.
- M. T. Weller, O. J. Weber, P. F. Henry, A. M. Di Pumpo and T. C. Hansen, Chem. Commun., 2015, 51, 4180–4183 RSC.
- P. S. Whitfield, N. Herron, W. E. Guise, K. Page, Y. Q. Cheng, I. Milas and M. K. Crawford, Sci. Rep., 2016, 6, 35685 CrossRef CAS PubMed.
- A. M. A. Leguy, J. M. Frost, A. P. McMahon, V. G. Sakai, W. Kockelmann, C. Law, X. Li, F. Foglia, A. Walsh, B. C. O’Regan, J. Nelson, J. T. Cabral and P. R. F. Barnes, Nat. Commun., 2015, 6, 7124 CrossRef PubMed.
- J. Sugiyama, J. Phys. Soc. Jpn., 2013, 82, SA023 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ee00476a |
| ‡ These authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2019 |