
Protonic ceramic electrochemical cells for hydrogen production and electricity generation: exceptional reversibility, stability, and demonstrated faradaic efficiency†
Sihyuk
Choi
 ab,
Timothy C.
Davenport
a and
Sossina M.
Haile
*a
ab,
Timothy C.
Davenport
a and
Sossina M.
Haile
*a
aMaterials Science and Engineering, Northwestern University, Evanston, IL, USA. E-mail: sossina.haile@northwestern.edu
bDepartment of Mechanical Engineering, Kumoh National Institute of Technology, Gumi, Republic of Korea
First published on 13th December 2018
Abstract
We demonstrate exceptional performance for steam electrolysis at intermediate temperatures (500–650 °C) using protonic ceramic electrolyte cells comprised of the proton-permeable, high-activity mixed conductor PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF) as the air electrode, the highly proton-conductive and chemically stable perovskite oxide BaZr0.4Ce0.4Y0.1Yb0.1O3 (BZCYYb4411) as the electrolyte, and a composite of Ni–BZCYYb4411 as the fuel electrode. Cells constructed from this material set have been shown previously to function efficiently in fuel cell mode. We demonstrate here reversible operation, enabling hydrogen production when excess electricity is available and immediate electricity generation from stored hydrogen when power demand is high. The cells are stable under cyclic operation and also under prolonged continuous operation in electrolysis mode, undergoing minimal loss in electrochemical characteristics after 500 h at 550 °C. Microstructurally optimized cells yield a remarkable current density of −1.80 A cm−2 at 600 °C and an operating voltage of 1.3 V, of which, based on an electrochemically deduced faradaic efficiency of 76%, −1.37 A cm−2 contributes to useful hydrogen.
Broader contextProduction of electricity from intermittent solar and wind resources has risen sharply in recent years, creating an urgent need for efficient energy storage. Reversible electrochemical cells enable interconversion between electrical and chemical energy, in particular hydrogen, implying a facile route for meeting this storage challenge. In electrolysis, or water splitting, mode reversible electrochemical cells convert electricity into readily stored, long-lived and transportable chemical fuel, whereas in fuel cell mode they produce electricity on demand with zero emissions. Despite increasing efforts, reversible and efficient operation of such devices has remained elusive. Here, we demonstrate excellent performance and durability upon reversible operation of a cell incorporating a proton-permeable, high-activity mixed conducting oxide as the air electrode, a highly proton-conductive and chemically stable perovskite oxide as the electrolyte, and a composite of Ni and the electrolyte as the fuel electrode. Microstructurally optimized cells yield a remarkable current density of −1.80 A cm−2 at 600 °C and 1.3 V, with an estimated faradaic efficiency of 76%. The operational conditions of protonic ceramic cells, including the absence of liquid water and the absence of steam at the Ni-bearing electrode, provide inherent advantages in electrolysis mode over competing systems. |
Introduction
Production of electricity from solar and wind resources has risen steeply in recent years, accounting, for example, for 7.6% of electricity generation in the US in 2017,1 up from just 0.8% a decade earlier.2 The remarkable inroads of these renewable energy technologies is now driving a push to develop approaches for effectively storing and delivering the inherently intermittent electricity from these natural sources. While many storage strategies are under investigation, reversible electrochemical cells, which interchange chemical energy, in particular hydrogen, with electrical energy, have received only moderate attention.3 Reversible electrochemical cells combine the functions of electrolysis (electrochemical water splitting) and electrochemical power generation from fuel (fuel cell functionality), providing a storage solution that resembles, in some ways, rechargeable batteries. Historically, interest in chemical–electrical interconversion has focused on devices that function in only one direction or the other.4 In particular, electrolysis is a well-established commercial technology and offers the potential for generating high-purity hydrogen, free of carbon emissions.5–9 Similarly, fuel cells have gained traction as devices that can subsequently use this hydrogen in the generation of emissions-free electric power.10–12 Successful integration of these functions in a reversible electrochemical cell would support a long-term energy storage technology deployable at a scale limited only by the size of the fuel storage tanks. Additionally, the hydrogen produced by electrolysis using excess wind and solar power would be attractive for use in the transportation sector.13,14The overall function of a reversible electrochemical cell operating on H2O, O2, H2 and generating either electricity or fuel (as required) is described in generic terms in eqn (1).
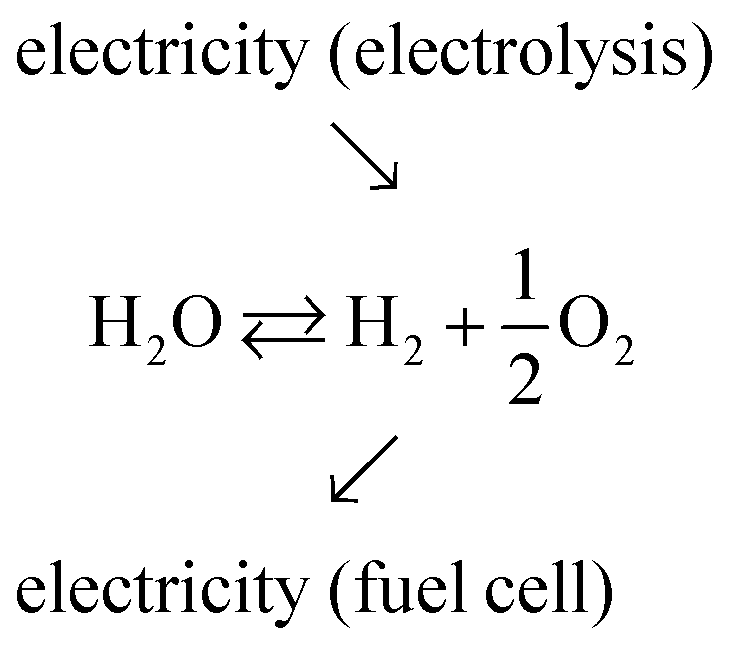 | (1) |
In recognition of their anticipated benefits for energy storage, reversible solid oxide cells have received increasing attention in recent years. As shown in Fig. 1, such cells may be operated using either an oxygen ion conducting electrolyte or a proton conducting electrolyte. When external electricity is applied, the mobile ion is driven across the electrolyte, releasing hydrogen at the fuel electrode and oxygen at the air electrode.20–25 The current density through the cell at a specified voltage (typically 1.3 V, close to the thermal-neutral voltage for steam electrolysis of ∼1.28 V8,9) is generally taken as a surrogate for the hydrogen production rate. In fuel cell mode, each of the steps shown in Fig. 1 occurs in reverse.
 | ||
| Fig. 1 Schematic images of water splitting by electrolysis. (a) Using an O2− conducting electrolyte in an OCEC and (b) using a H+ conducting electrolyte in a PCEC. | ||
Despite the high conductivity of the electrolyte in protonic ceramic electrochemical cells (PCECs),26 the electrolysis performance of these systems lags far behind those of oxygen ion conducting electrolyte cells (OCECs). At the standard operating voltage of 1.3 V and a temperature of 800 °C, the current densities of state-of-the-art OCECs, Table S1 (ESI†), in many instances exceed (in magnitude) 1.0 A cm−2.20–23,27–33 Even at 600 °C Jensen et al.34 obtained an extremely impressive −0.92 A cm−2 from an OCEC. In contrast, the few publications that report on the performance of PCECs indicate, with only three exceptions, current densities of just tens of mA cm−2, irrespective of temperature, Table 1 and Table S2 (ESI†).35–47 In the study by Kim et al.,42 in which an attractive −0.75 A cm−2 was obtained at 1.3 V at 600 °C, the authors propose that the oxide electrolyte used in their work, in fact, transports both oxygen ions and protons, contributing to the high hydrogen production rates. Even recognizing this recent advance, a large disparity exists between the expected and realized performance of PCECs.
| Ref. | Cell configuration (air electrode/electrolyte (thickness)/fuel electrode) | Inlet gas composition | OCV/EN (V) | Current density @ 1.3 V (A cm−2) | |
|---|---|---|---|---|---|
| Air electrode | Fuel electrode | ||||
| For air electrode: SSC = Sm0.5Sr0.5CoO3−δ; LSCF = La0.6Sr0.4Co0.2Fe0.8O3−δ; LNO = La2NiO4+δ; SFM = Sr2Fe1.5Mo0.5O6−δ; SEFC = SrEu2Fe1.8Co0.2O7−δ; NBSCF = NdBa0.5Sr0.5Co1.5Fe0.5O5+δ; BSCF = Ba0.5Sr0.5Co0.8Fe0.2O3−δ; PBSCF = PrBa0.5Sr0.5Co1.5Fe0.5O5+δ; LSM = La0.8Sr0.2MnO3−δ. For electrolyte: BZCY44 = BaZr0.4Ce0.4Y0.2O3; BZCYbCo = BaCe0.48Zr0.40Yb0.10Co0.02O3; BZY10 = BaZr0.9Y0.1O3; BCZD = BaCe0.5Zr0.3Dy0.2O3; BZCY53 = BaZr0.5Ce0.3Y0.2O3; BZY20 = BaZr0.8Y0.2O3; BCZY35 = BaCe0.3Zr0.5Y0.2O3; BZCYYb1711 = BaZr0.1Ce0.7Y0.1Yb0.1O3; BCZY62 = Ba0.98Ce0.6Zr0.2Y0.2O3; BZCYYb4411 = BaZr0.4Ce0.4Y0.1Yb0.1O3. | |||||
| 35 | SSC/BZCY44 (1500 μm)/Pt | Air (20% H2O) | 5% H2/Ar (3% H2O) | 1.02/0.99 | −0.01 |
| 36 | LSC-BZCYbCo/BZCYbCo (45 μm)/Ni–BZCYbCo | Air (30% H2O) | 10% H2/He | 0.98/1.00 | −0.03 |
| 37 | LSCF-BZY10/BZY10 (15 μm)/Ni–BZY10 | Air (3% H2O) | 4% H2/Ar | 0.86/1.02 | −0.05 |
| 38 | LNO-BCZD/BCZD (30 μm)/Ni–BCZD | Air (90% H2O) | 97% H2 (3% H2O) | 0.96/1.00 | −0.16 |
| 39 | SSC-BCZY53/BCZY53 (20 μm)/Ni–BCZY53 | Air (50% H2O) | 100% H2 | 0.97/1.03 | −0.17 |
| 40 | SFM-BZY20/BZY20 (16 μm)/Ni–BZY20 | Air (3% H2O) | 10% H2/N2 | 0.86/1.04 | −0.19 |
| 41 | SEFC-5 wt% BCZY35/BCZY35 (15 μm)/Ni–BCZY35 | Air (10% H2O) | 97% H2 (3% H2O) | 0.99/1.06 | −0.42 |
| 42 | NBSCF-BZCYYb1711/BZCYYb1711 (20 μm)/Ni–BZCYYb1711 | Air (10% H2O) | 90% H2 (10% H2O) | 1.04/1.06 | −0.75 |
| 43 | BSCF-BCZY62/BCZY62 (10–15 μm)/Ni–BCZY62 | Air (2.76% H2O) | 50% H2 (2.76% H2O) | 1.05/1.07 | −1.05 |
| This work | PBSCF/BZCYYb4411 (15 μm)/Ni–BZCYYb4411 (conventional) | Air (3% H2O) | 97% H2 (3% H2O) | 1.05/1.08 | −1.42 |
| This work | PBSCF/PBSCF PLD/BZCYYb4411 (15 μm)/Ni–BZCYYb4411 (PLD-modified) | Air (3% H2O) | 97% H2 (3% H2O) | 1.03/1.08 | −1.92 |
| This work | PBSCF/BZCYYb4411 (15 μm)/Ni–BZCYYb4411 (conventional) | Air (3% H2O) | 5% H2/Ar | 0.92/1.03 | −1.31 |
| This work | LSCF/BZCYYb4411 (15 μm)/Ni–BZCYYb4411 (conventional) | Air (3% H2O) | 97% H2 (3% H2O) | 1.06/1.08 | −0.70 |
| This work | LSM/BZCYYb4411 (15 μm)/Ni–BZCYYb4411 (conventional) | Air (3% H2O) | 97% H2 (3% H2O) | 1.04/1.08 | −0.65 |
A key cause of this performance gap is likely the poor activity of the air electrode for the water oxidation reaction:
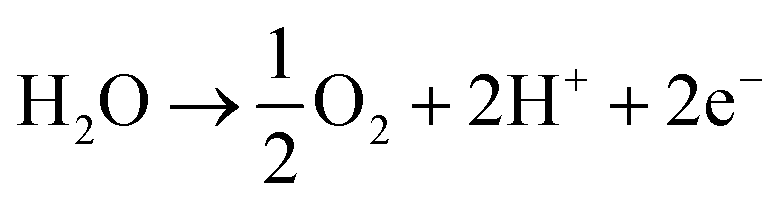 | (2) |
These observations motivated us to explore the behavior of our recently developed material set and fabrication strategy for protonic ceramic fuel cells48 in the context of electrolysis. Specifically, we pursue electrolysis using the electrolyte BaZr0.4Ce0.4Y0.1Yb0.1O3 (BZCYYb4411), which has high conductivity (amongst the highest of known proton conducting oxides), excellent chemical stability under CO2, and satisfactory processability. As the oxygen evolution electrocatalyst we employ PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF), which has high activity for oxygen electroreduction. This material can incorporate large quantities of H2O, supporting electro-reduction reaction via a double-phase boundary pathway such that the entire surface of the mixed conducting oxide is electrochemically active. Using these electrolyte and air electrode materials, along with a composite Ni–BZCYYb4411 as the fuel electrode and a fabrication methodology that addresses ohmic resistance losses due to poor contact between the electrolyte and air electrode, we demonstrate unprecedented hydrogen production rates from a reversible solid oxide electrochemical cell.
Results and discussion
Electrochemical cell preparation
As indicated in Fig. 1b, both the electrolyte and air electrode of a PCEC system operated in electrolysis mode are exposed to high partial pressures of steam. Accordingly, a first evaluation was performed to assess the stability of these components under such conditions, which are not generally encountered in the context of fuel cell operation. Diffraction patterns collected after heat treatment at 500 °C in 20% H2O/air for 24 h, revealed the materials to be stable, with no structural changes within detection limits (Fig. S1, ESI†).After confirmation that this material set, previously demonstrated to have attractive properties for fuel cell application, is compatible with the electrolysis environment, we prepared cells configured in a fuel-electrode supported arrangement. A bilayer structure comprised of a thin, ∼15 μm, layer of BZCYYb4411 supported on a relatively thick, ∼300 μm, NiO–BZCYYb4411 electrode was first prepared by conventional ceramic processing, Fig. 2. A layer of PBSCF was then directly applied onto the electrolyte layer and the entire structure sintered at 950 °C for 4 h in air. The resulting porous air electrode was about 20 μm (Fig. 2a) thick with homogenously distributed and well-connected fine particles of PBSCF (Fig. 2b). At the initiation of the electrochemical measurement, the NiO in the composite fuel electrode was reduced to Ni by exposure to humidified (3% H2O) H2 at 700 °C for 4 h.
 | ||
| Fig. 2 Scanning electron microscopy images of NiO + BZCYYb4411/BZCYYb4411/PBSCF (fuel electrode/electrolyte/air electrode) electrochemical cells. (a) Cross-sectional image showing 15 μm thick, dense electrolyte and porous PBSCF supported on porous fuel electrode. (b) PBSCF air electrode showing retention of porosity after high temperature processing. | ||
Reversible electrochemical performance
The polarization behavior obtained upon supplying 3% H2O humidified hydrogen and 3% H2O humidified air to the fuel and air electrodes, respectively, is shown in Fig. 3 for both electrolysis and fuel cell modes at temperatures between 500 and 650 °C. The cell behavior in fuel cell mode, Fig. 3b, is virtually indistinguishable to what was reported previously using analogous cells operated with dry air supplied to the PBSCF electrode.48 A 3% humidification level was selected for these measurements after establishing that the H2O concentration supplied to the cells, within the range from 3 to 10% H2O, had minimal impact on the electrochemical characteristics (Fig. S2, ESI†). Operation of PCECs in electrolysis mode at low steam concentrations (Table 1), in contrast to the high concentrations required in OCECs (Table S1, ESI†), is possible because the steam is supplied in the former case to the relatively thin air electrode, minimizing mass transport losses resulting from gas-phase diffusion limitations.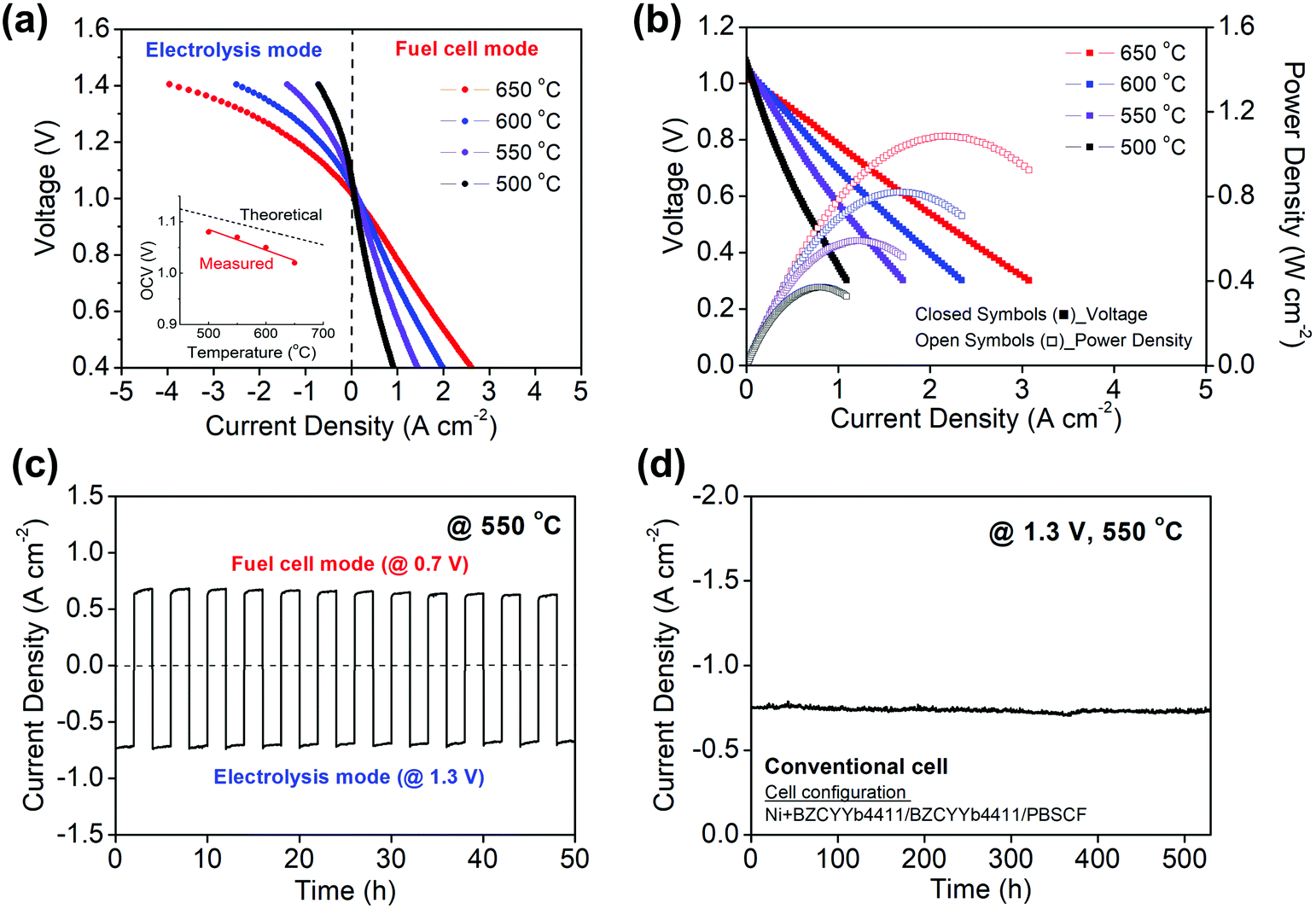 | ||
| Fig. 3 Electrochemical characteristics of a representative Ni + BZCYYb4411/BZCYYb4411/PBSCF conventional cell operated with humidified (3% H2O) H2 and humidified air supplied respectively to the fuel and air electrodes. (a) Polarization curves spanning both electrolysis and fuel cell modes. (b) Polarization and power density curves for fuel cell mode. (c) Continuous cyclic operation between electrolysis (@ 1.3 V) and fuel cell mode (@ 0.7 V) at 550 °C. (d) Temporal evolution at 550 °C of the current density in electrolysis mode at a fixed voltage of 1.3 V showing good stability. | ||
The reversibility of the cell was evaluated by cyclic operation between 1.3 V (electrolysis mode) and 0.7 V (fuel cell mode) at 550 °C (Fig. 3c). The cell current was measured for 2 h at each condition in alternating fashion. No performance degradation was detectable over the course of 12 measurement cycles. Furthermore, at a constant voltage of 1.3 V, the current density was found to be exceptionally stable over the course of 500 h of continuous operation at 550 °C. (Fig. 3d) In light of this electrochemical stability, the absence of microstructural changes in the cell components, as investigated by scanning electron microscopy, is not surprising (Fig. S3, ESI†).
As discussed and quantified below, a fraction of the current that flows through the cell is due to electronic leakage. This is reflected in the difference between the measured and theoretical open circuit voltages (inset, Fig. 3a). Under the reasonable assumption that the electrolyte electronic leakage through protonic ceramic electrolytes does not vary dramatically between various compositions, polarization curves, despite overestimating the protonic current, serve as useful surrogates for comparing cells from different laboratories. In electrolysis mode, the I–V profiles obtained here reflect the highest performance reported to date. At 600 °C, the current density at 1.3 V is −1.42 A cm−2, exceeding by 90% the value reported by Kim et al.42 in their hybrid PCEC, Table 1. At 500 °C, the current density of −0.40 A cm−2 (at 1.3 V) exceeds all prior 600 °C PCEC results. Moreover, if even only ∼50% of the current flowing through the cell is due to protons, the electrical characteristics are attractive in the context of higher temperature OCECs (Table S1, ESI†).
Faradaic efficiency
Beyond high current density at the thermal-neutral voltage, an effective electrolyzer must convert a high fraction of the current into hydrogen, that is, it must have high faradaic efficiency, ηF. Because parasitic reactions are atypical in high temperature solid oxide electrochemical cells, it is common to assume 100% faradaic efficiency in such devices. However, electronic leakage through protonic ceramic electrolytes, the electronic transference number of which increase with increasing temperature and oxygen partial pressure,49–51 can lower this efficiency.24Here, the efficiency characteristics of a Ni–BZCYYb4411/BZCYYb4411/PBSCF electrolysis cell were evaluated by a combination of electrochemical measurements and quantification of the hydrogen production by off-gas analysis using mass spectrometry (MS). To enable detection of the electrochemically produced hydrogen beyond that supplied to the cell, the gas at the fuel electrode was changed to 5% H2 in Ar (the gas supplied to the air electrode remained 3% H2O in air). The current densities measured under these conditions were only marginally lower than with 3% H2O in H2 supplied to the fuel electrode (compare Fig. 3a and 4a).
 | ||
| Fig. 4 Measurement of faradaic efficiency for electrolysis of a representative Ni + BZCYYb4411/BZCYYb4411/PBSCF conventional cell operated with a low concentration of dry H2 (5%, balance Ar) supplied to the fuel and humidified (3% H2O) air supplied to the air electrode. (a) Polarization curves by total current (closed symbol) and only proton current (open symbol). (b) Measured and Nernst OCV as a function of temperature. (c) Ohmic resistance and DC resistance under OCV condition. (d) Measured hydrogen production rates at 500 °C and at the voltages indicated. (e) Measured and calculated faradaic efficiency as a function of applied voltage at several temperatures as indicated and with a change in flow rate, where the default flow rate is 150 ml min−1 and error bars are derived from the time-averaging of the data in (d). (f) Steam-to-hydrogen conversion rate in various temperatures and applied voltages. | ||
Significant with respect to faradaic efficiency is the value of the open circuit voltage (VOC). As is typical of protonic ceramic cells, VOC of the present experiments is detectably lower than the theoretical or Nernst voltage (VN) computed on the basis of the supplied gases and cell temperature, Fig. 4b and Table 2. This loss in voltage, which is more severe when the fuel electrode is supplied with 5% H2 than 97% H2 (consistent with the oxygen partial pressure dependence of the ionic transference number), is a manifestation of the electronic leakage. Thus, the faradaic efficiency measurement carried out here under conditions suitable for detecting the hydrogen generation inherently underestimates the behavior under more realistic conditions, as addressed further below.
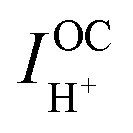 are, respectively, the computed electronic resistance, protonic resistance, polarization resistance, and ionic leakage current. Cells are measured with 5% hydrogen, balance argon, supplied to the fuel electrode and 3% H2O humidified synthetic air supplied to the air electrode
are, respectively, the computed electronic resistance, protonic resistance, polarization resistance, and ionic leakage current. Cells are measured with 5% hydrogen, balance argon, supplied to the fuel electrode and 3% H2O humidified synthetic air supplied to the air electrode
| T, °C | V N, V | V OC, V | R ohmic, Ω cm2 | R DC, Ω cm2 | R e−, Ω cm2 | R H+, Ω cm2 | t H+ | R P, Ω cm2 |
|
|---|---|---|---|---|---|---|---|---|---|
| 650 | 0.99 | 0.87 | 0.21 | 0.41 | 3.24 | 0.22 | 0.94 | 0.25 | 0.27 |
| 600 | 1.02 | 0.92 | 0.27 | 0.56 | 5.71 | 0.28 | 0.95 | 0.34 | 0.16 |
| 550 | 1.05 | 0.96 | 0.34 | 0.88 | 10.2 | 0.35 | 0.97 | 0.61 | 0.09 |
| 500 | 1.08 | 1.01 | 0.42 | 1.83 | 28.3 | 0.43 | 0.99 | 1.53 | 0.04 |
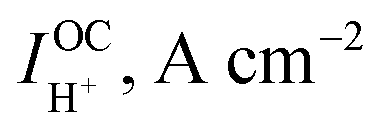
At open circuit the electronic leakage is exactly balanced by the ionic flux such that the net current is zero. Taking the ionic flux to be purely protonic, a reasonable assumption here because oxide ionic conductivity is low in protonic ceramic oxides at these temperatures and because there is no steam supplied to the fuel electrode, the flux balance is expressed as
 | (3) |
The hydrogen leakage rate under open circuit conditions can be estimated from a measurement of the open circuit voltage, Fig. 4b, and the zero-bias electrochemical impedance, Fig. S4 (ESI†). The voltage, Vcell, across an electrochemical cell with a mixed ion and electron conducting electrolyte, is given as
| Vcell = VN − ηC − ηA − ηH+ | (4) |
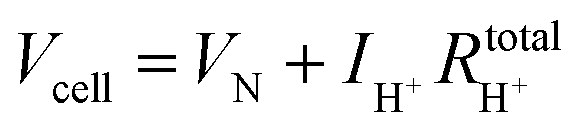 | (5) |
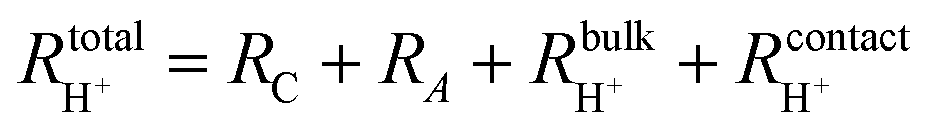 | (6) |
Consideration of the electronic rail implies an open circuit voltage of
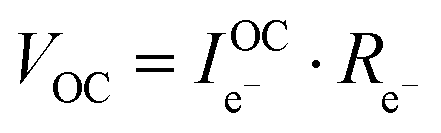 | (7) |
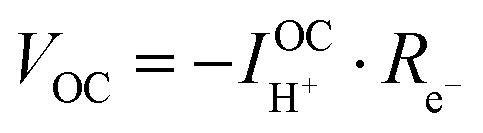 | (8) |
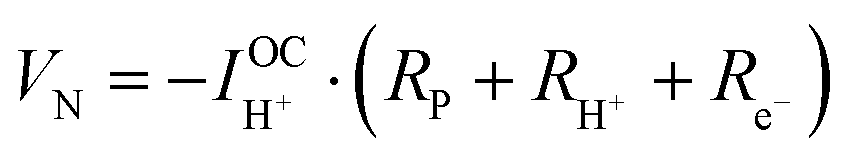 | (9) |
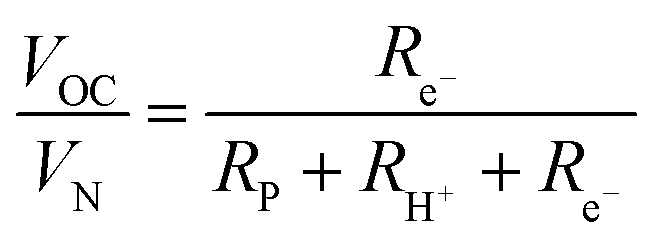 | (10) |
From impedance measurements one obtains two relevant resistance terms. The first is the high frequency ohmic offset
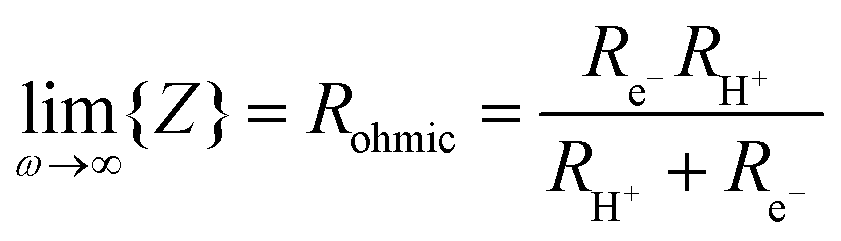 | (11) |
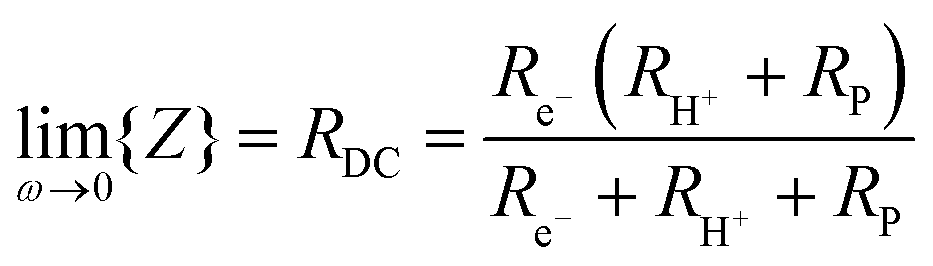 | (12) |
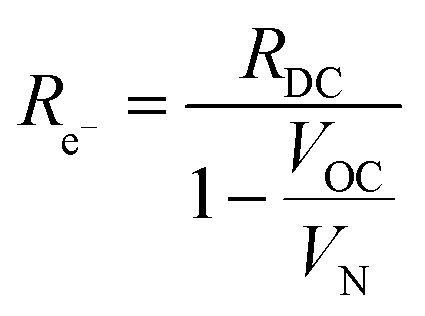 | (13) |
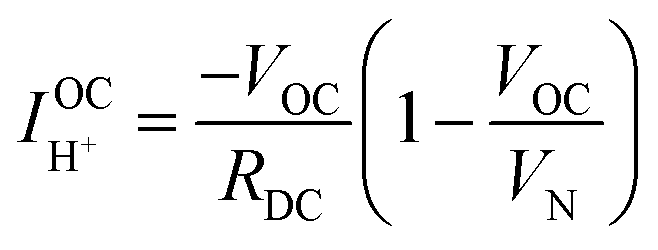 | (14) |
Performing such an analysis for the cell represented in Fig. 4 and Table 2, indicates mean protonic transference numbers (tH+ = σH+/(σH+ + σe−)) ranging from 0.985 at 500 °C to 0.936 at 650 °C. Despite the dominance of the protonic species, the leakage current at 650 °C is 0.268 A cm−2 (or 1.9 ml min−1 cm−2), which falls to 0.036 A cm−2 at 500 °C (or 0.25 ml min−1 cm−2). Although these currents are relatively large, the fraction of hydrogen lost to leakage is rather small, amounting to 0.35% at 650 °C and just 0.05% at 500 °C of the 150 ml min−1 of the hydrogen supplied to the fuel electrode. Moreover, as expected, the ionic transference number is slightly higher under more realistic conditions with high pH2 at the fuel electrode, Table S3 (ESI†).
Away from open circuit, if the cell components behave linearly, the ionic current is given, by rearranging (5):
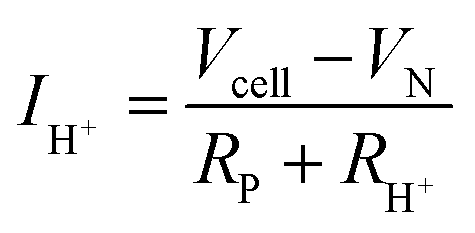 | (15) |
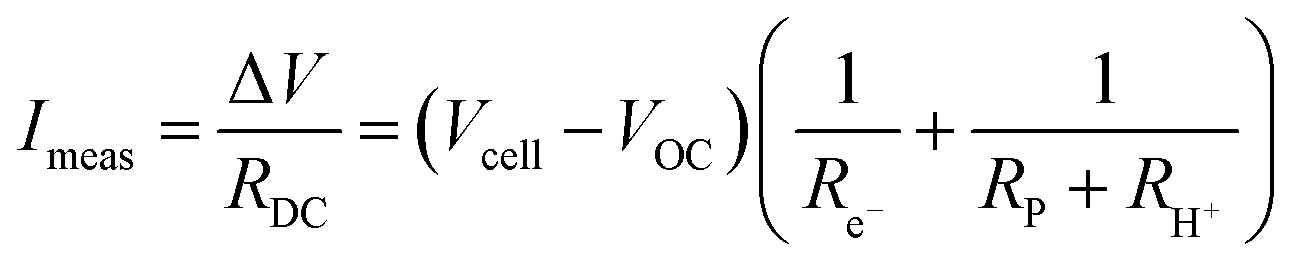 | (16) |
A trace of the hydrogen detected under different voltage conditions, Fig. 4d and Fig. S6 (ESI†), reveals that the protonic current is substantially lower than the total current, Fig. 4a, and accordingly the faradaic efficiency, Fig. 4e, is measurably lower than 100%, ranging from 40 to 75% over the measurement conditions examined here. Moreover, this efficiency is independent of the gas flow rate, as expected for losses due to electronic conductivity in the electrolyte as opposed to gas leaks across the cell. The protonic current in Fig. 4a is taken to be zero at the Nernst potential, as required on thermodynamic grounds. Consistent with eqn (14) and (15), the measured faradaic efficiency generally rises with increasing voltage, reaching a value of almost 75% at 1.3 V at 500 °C. The turn-over at high voltage at 600 °C is attributed to oxidation of the electrolyte, as evidenced by a decrease in Rohmic upon application of bias (not shown). In an absolute sense, the measured values are in reasonable agreement with the predictions based on the open circuit electrical properties. The agreement implies it possible to estimate faradaic efficiency using electrochemical measurements alone and in particular when the fuel electrode is exposed to 97% H2 and hydrogen production is difficult to directly observe. We estimate that the faradaic efficiencies are ∼10% higher under the more reducing conditions, Table S3 (ESI†), rising from 77% at 650 °C to 87% at 500 °C for an operating voltage of 1.3 V. Moreover, deviation from the theoretical curves as a consequence of electrolyte oxidation under bias is also likely to be less detrimental to ηF under high pH2 rather than under low pH2 conditions.
Literature values quoted for faradaic efficiency of PCECs operated at 600 °C and 1.3 V generally range from 65 to 95%.35,36,40 However, these values may be overestimated due to the challenges of detecting a small increase in gas hydrogen concentration (during electrolysis) over a large background (as already present in the gas supplied to and flowing past the fuel electrode). In a recent study by Huan et al., instead of hydrogen production, the oxygen generated at the air electrode was measured.41 Such an approach has the benefit of eliminating the large background signal inherent to measurements of hydrogen production, however, faradaic efficiencies are expected to be somewhat overestimated relative to operational conditions as a consequence of the relatively reducing conditions at the air electrode. Despite the latter effect, values of ηF in the range of 20–40% were recorded at 700 °C, reflecting high electronic leakages encountered in PCECs during high temperature operation in electrolysis mode. The result underscores the critical advance of developing air electrodes that have high activity at 600 °C and even lower.
Related to faradaic efficiency is the steam-to-hydrogen conversion rate. Significantly, despite increases in electronic leakage, the conversion, Fig. 4f, monotonically increases with temperature (and voltage) at fixed steam flow-rate, up to the maximum temperature at which hydrogen production was measured (600 °C). The behavior is a result of the enhanced ion transport and electrocatalysis rates, which overwhelm the enhanced electron hole transport as temperature is increased. In large-scale OCEC stack testing, steam-to-hydrogen conversion rates are typically fixed at ∼50%,54 which presumably balances the energy cost of heating large quantities of steam against the electrochemical performance losses at low reactant concentrations. While the highest conversion here was ∼19%, one can readily anticipate higher conversion simply by increasing cell voltage and/or decreasing steam flow-rate.
Ultimately, overall energy storage efficiency is likely to be a decisive factor for electrolysis technology adoption. In an electrochemical system comprising a stack of multiple reversible cells, this efficiency will depend not only on material properties and operating conditions, but also on device configuration. In particular, thermal management will surely play a key role. A recent analysis suggests that state-of-the-art oxide based systems, with cells of much poorer electrochemical characteristics than those demonstrated here, but negligible electronic leakage, can deliver round-trip efficiencies of 55–60%, with theoretical efficiencies, assuming tight thermal integration, approaching 100%.55 These efficiency metrics, in combination with the flexibility of chemical energy storage, render reversible oxide electrochemical cells with the performance metrics obtained here intriguing technologies for integration with intermittent, carbon-free electricity sources.
Cell design and characteristics of the air electrode
While the intrinsic chemical properties of the electrode and electrolyte materials are expected to be the key factors in the high performance of our reversible PCECs, differences in cell fabrication procedures can have profound influence on polarization behavior. Thus, it is critical to both assess reproducibility and exploit fabrication techniques known to provide performance advantages. In particular, we have previously shown that application of a thin dense layer of the air electrode material between the electrode and the porous air electrode can significantly improve contact between these two components, decreasing cell ohmic resistance. Here, four cells without, Fig. S7 (ESI†), and three cells with, Fig. S8 (ESI†), a ∼100 nm thin PLD layer were compared, and results from a representative PLD-modified cell are presented in Fig. 5. The variation in current density at any given voltage between cells of the same type was less than 20%, whereas the current density was higher, on average, for the PLD-modified cells by a remarkable 30%. The beneficial impact of the PLD layer on the ohmic resistance and its negligible impact on the electrochemical reaction resistance were moreover confirmed (Fig. S9, ESI†). The current density at 600 °C and 1.3 V, of almost 2 A cm−2, far exceeds, by more than a factor of two, any prior result obtained using a protonic ceramic electrolyte (Table 1). Accounting for an estimated faradaic efficiency of 76%, Table S3 (ESI†), the protonic current density at these conditions is 1.36 A cm−2, far exceeding the performance of electrolysis cells operated at this temperature using either proton conducting or oxygen ion conducting electrolytes (Table S1, ESI†). Significantly, long-term cell stability was maintained in the PLD-modified cells over the course of a 300 h measurement as evidenced both by the unchanging current density, Fig. 5b, and absence of microstructural evolution (Fig. S10, ESI†). | ||
| Fig. 5 Electrochemical characteristics of a representative Ni + BZCYYb4411/BZCYYb4411/PBSCF PLD/PBSCF PLD-modified cell operated with humidified (3% H2O) H2 and humidified air supplied respectively to the fuel and air electrodes. (a) Polarization curves spanning both electrolysis and fuel cell modes. (b) Temporal evolution at 550 °C of the current density in electrolysis mode at a fixed voltage of 1.3 V showing good stability. | ||
To provide a direct comparison to other air electrode materials, cells in which the PBSCF air electrode was replaced with other known solid oxide fuel cell (SOFC) cathode materials were prepared. Candidates examined were Sm0.5Sr0.5CoO3−δ (SSC), La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF), and La0.8Sr0.2MnO3−δ (LSM). Based on an initial screening for chemical compatibility, SSC was eliminated due to its reactivity with BZCYYb4411, Fig. S11 (ESI†). The polarization characteristics of cells fabricated with LSCF or LSM (Fig. S12, ESI†) reveal that the PBSCF electrode, rather than cell-to-cell variations or differences in fabrication procedures between different laboratories, is indeed the major factor in the high performance of the cells of the present work, in both fuel cell and electrolysis modes (Fig. 6). The electrolysis current densities, for example, are almost an order of magnitude lower using these alternative electrodes than they are for cells incorporating PBSCF, Table 1. It is noteworthy that PBSCF has been recently found to display excellent activity for oxygen evolution even in room-temperature aqueous cells, comparable to that of IrO2.56
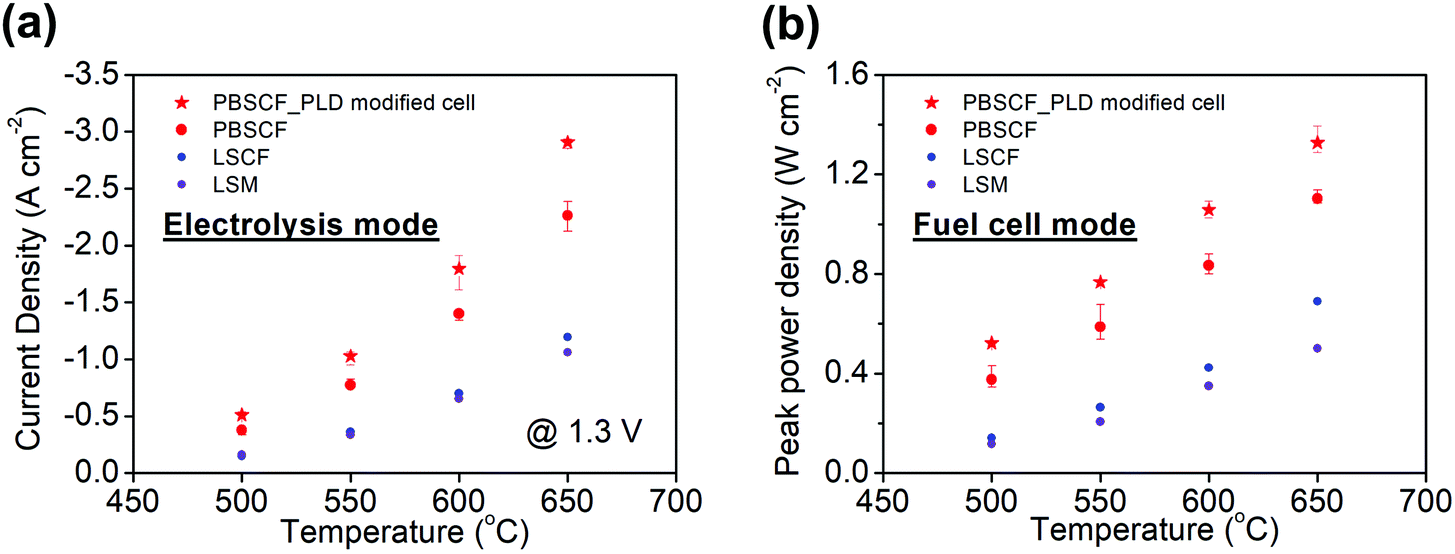 | ||
| Fig. 6 Summary of electrochemical performance of various air electrode materials in both (a) electrolysis mode and (b) fuel cell mode. The error bars for the PBSCF air electrode data are based on the averaging of all measurements in Fig. S7 and S8 (ESI†), and reflect the high level of reproducibility. | ||
Concluding remarks
We demonstrate record performance for steam electrolysis using protonic ceramic electrolyte cells. Introduction of a dense layer of the air electrode material between the electrolyte and the porous air electrode by PLD dramatically decreased the cell ohmic resistance, resulting in an increase in current density in electrolysis mode of 30%. The faradaic efficiency as measured under low hydrogen supply ranged from 40 to 75% at 1.3 V, while the electrochemical characteristics of the cells under high hydrogen supply imply ηF values in the range of 74 to 87% for realistic electrolysis operational conditions. Even accounting for the current loss due to electronic leakage, estimated hydrogen production rates at 1.3 V of 15.0 and 9.5 ml min−1 cm−2 were obtained at 650 and 600 °C, respectively.The excellent performance of the Ni–BZCYYb4411/BZCYYb4411/PBSCF protonic ceramic electrochemical cell of this work is attributed to the activity of the PBSCF air electrode towards the oxygen evolution and reduction reactions, whereas the depressed faradaic efficiency is attributed to electronic leakage through the BZCYYb4411 electrolyte. The excellent stability in electrolysis mode is attributed to the absence of steam, an otherwise corrosive agent, at the fuel cell electrode, and the absence of oxygen bubble formation in the vicinity of the air electrode, as can occur for an oxygen ion conducting electrolyte. Operability at low steam concentrations is attributed to the minimal H2O gas-phase diffusion resistance through the thin air electrode suggests that high steam-to-hydrogen conversion ratios will be possible upon optimization of system parameters.
In addition to enabling electrolysis at more moderate temperatures (400–650 °C)26 than a cell relying on an oxygen ion conductor (typically 700–900 °C),20–23,27–34 the protonic ceramic electrochemical cell (PCEC) offers distinct operational advantages. In particular, the H2O concentration at the fuel electrode, whether the device is operated in fuel cell mode or in electrolysis mode, can be negligible. In fuel cell mode this implies that the hydrogen fuel does not become diluted during operation, and thus, unspent fuel is easily recycled, a benefit that is well-recognized.9,26,57 In electrolysis mode, absence of steam at the fuel electrode avoids mass transport limitations through the structure-supporting fuel electrode, while also generating tremendous benefit in terms of material stability. By far the most commonly employed electrocatalyst in the fuel electrode of both oxide ion conducting and protonic ceramic electrolytes is Ni. However, Ni suffers from oxidation and irreversible agglomeration under high partial pressures of steam, causing serious performance degradation in continuous electrolysis operation.58,59 These degradation modes can be expected to be absent in PCEC systems. Another benefit may arise from the manner in which the mobile species are evolved from the electrolyte. In the case of the OCEC, oxygen evolution at the interface between the electrolyte and the air electrode causes delamination and eventual cell failure.60,61 Such a mechanism is not expected to be operative when hydrogen is evolved, suggesting a stability benefit to the air electrode from PCEC implementation in addition to the stability benefit accrued at the fuel electrode. Consistent with these anticipated PCEC benefits, no degradation in cell current density was observed in our PBSCF/BZCYYb4411/BZCYYb4411 + Ni cells after 500 h of electrolysis at 1.3 V and 550 °C.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded primarily by the U.S. Department of Energy, through ARPA-e Contract DE-AR0000498, via subcontract from United Technologies Research Center. This work made use of the Cohen X-ray Diffraction Laboratory at Northwestern University, which has received support from MRSEC program (NSF DMR-1121262) at the Materials Research Center, the Pulsed Laser Deposition facility of Northwestern University, and the EPIC Facility of Northwestern University's NUANCE Center. These facilities receive support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205); the MRSEC program (NSF DMR-1121262) at the Materials Research Center; the International Institute for Nanotechnology (IIN); the Keck Foundation; and the State of Illinois, through the IIN.References
-
R. Hankey, Independent Statistics & Analysis, U.S. Department of Energy, Washington, DC 20585, 2018 Search PubMed
.
- J. Luna-Camara, U.S. Department of Energy, Washington, DC, 20585, 2008.
- H. Chen, T. N. Cong, W. Yang, C. Tan, Y. Li and Y. Ding, Prog. Nat. Sci., 2009, 19, 291–312 CrossRef CAS
- Z. Abdin, C. J. Webb and E. M. Gray, Prog. Energy Combust. Sci., 2009, 35, 231–244 CrossRef
- J. T. S. Irvine, D. Neagu, M. C. Verbraeken, C. Chatzichristodoulou, C. Graves and M. B. Mogensen, Nat. Energy, 2016, 1, 15014 CrossRef CAS
- C. Graves, S. D. Ebbesen, S. H. Jensen, S. B. Simonsen and M. B. Mogensen, Nat. Mater., 2015, 14, 239–244 CrossRef CAS PubMed
- S. Y. Gómez and D. Hotza, Renewable Sustainable Energy Rev., 2016, 61, 155–174 CrossRef
- S. D. Ebbesen, S. H. Jensen, A. Hauch and M. B. Mogensen, Chem. Rev., 2014, 114, 10697–10734 CrossRef CAS PubMed
- L. Bi, S. Boulfrad and E. Traversa, Chem. Soc. Rev., 2014, 43, 8255–8270 RSC
- A. Hauch, S. D. Ebbesen, S. H. Jensen and M. Mogensen, J. Mater. Chem., 2008, 18, 2331–2340 RSC
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed
- N. Q. Minh, J. Am. Ceram. Soc., 1993, 76, 563–588 CrossRef CAS
- K. Christopher and R. Dimitrios, Energy Environ. Sci., 2012, 5, 6640–6651 RSC
- J. D. Holladay, J. Hu, D. L. King and Y. Wang, Catal. Today, 2009, 139, 244–260 CrossRef CAS
- H. Ito, N. Miyazaki, M. Ishida and A. Nakano, Int. J. Hydrogen Energy, 2016, 41, 5803–5815 CrossRef CAS
- B. Paul and J. Andrews, Renewable Sustainable Energy Rev., 2017, 79, 585–599 CrossRef
- S. D. Song, H. M. Zhang, X. P. Ma, Y. N. Zhang and B. L. Yi, Prog. Chem., 2006, 18, 1375–1380 CAS
- Y. F. Wang, D. Y. C. Leung, J. Xuan and H. Z. Wang, Renewable Sustainable Energy Rev., 2016, 65, 961–977 CrossRef CAS
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS
- A. Jun, J. Kim, J. Shin and G. Kim, Angew. Chem., Int. Ed., 2016, 55, 12512–12515 CrossRef CAS PubMed
- K. J. Yoon, M. Biswas, H.-J. Kim, M. Park, J. Hong, H. Kim, J.-W. Son, J.-H. Lee, B.-K. Kim and H.-W. Lee, Nano Energy, 2017, 36, 9–20 CrossRef CAS
- K. Hosoi, T. Sakai, S. Ida and T. Ishihara, Electrochim. Acta, 2016, 194, 473–479 CrossRef CAS
- M. A. Laguna-Bercero, R. D. Bayliss and S. J. Skinner, Solid State Ionics, 2014, 262, 298–302 CrossRef CAS
- H. Iwahara, T. Esaka, H. Uchida and N. Maeda, Solid State Ionics, 1981, 3-4, 359–363 CrossRef CAS
- H. Iwahara, H. Uchida and N. Maeda, J. Power Sources, 1982, 7, 293–301 CrossRef CAS
- E. Fabbri, D. Pergolesi and E. Traversa, Chem. Soc. Rev., 2010, 39, 4355–4369 RSC
- M. A. Laguna-Bercero, H. Monzon, A. Larrea and V. M. Orera, J. Mater. Chem. A, 2016, 4, 1446–1453 RSC
- T. Chen, M. Liu, C. Yuan, Y. Zhou, X. Ye, Z. Zhan, C. Xia and S. Wang, J. Power Sources, 2015, 276, 1–6 CrossRef CAS
- M. J. López-Robledo, M. A. Laguna-Bercero, A. Larrea and V. M. Orera, J. Power Sources, 2018, 378, 184–189 CrossRef
- J. Lin, L. Chen, T. Liu, C. Xia, C. Chen and Z. Zhan, J. Power Sources, 2018, 374, 175–180 CrossRef CAS
- Y. Wang, T. Liu, M. Li, C. Xia, B. Zhou and F. Chen, J. Mater. Chem. A, 2016, 4, 14163–14169 RSC
- T. Liu, Y. Wang, Y. Zhang, S. Fang, L. Lei, C. Ren and F. Chen, Electrochem. Commun., 2015, 61, 106–109 CrossRef CAS
- J. Yan, Z. Zhao, L. Shang, D. Ou and M. Cheng, J. Power Sources, 2016, 319, 124–130 CrossRef CAS
- S. H. Jensen, C. Graves, M. Mogensen, C. Wendel, R. Braun, G. Hughes, Z. Gao and S. A. Barnett, Energy Environ. Sci., 2015, 8, 2471–2479 RSC
- T. Kobayashi, K. Kuroda, S. W. Jeong, H. Kwon, C. Zhu, H. Habazaki and Y. Aoki, J. Electrochem. Soc., 2018, 165(5), F342–F349 CrossRef CAS
- M. A. Azimova and S. McIntosh, Solid State Ionics, 2011, 203, 57–61 CrossRef CAS
- L. Bi, S. P. Shafi and E. Traversa, J. Mater. Chem. A, 2015, 3, 5815–5819 RSC
- J. Lyagaeva, N. Danilov, G. Vdovin, J. Bu, D. Medvedev, A. Demina and P. Tsiakaras, J. Mater. Chem. A, 2016, 4, 15390–15399 RSC
- F. He, D. Song, R. Peng, G. Meng and S. Yang, J. Power Sources, 2010, 195, 3359–3364 CrossRef CAS
- L. Lei, Z. Tao, X. Wang, J. P. Lemmon and F. Chen, J. Mater. Chem. A, 2017, 5, 22945–22951 RSC
- D. Huan, N. Shi, L. Zhang, W. Tan, Y. Xie, W. Wang, C. Xia, R. Peng and Y. Lu, ACS Appl. Mater. Interfaces, 2018, 10, 1761–1770 CrossRef CAS PubMed
- J. Kim, A. Jun, O. Gwon, S. Yoo, M. Liu, J. Shin, T.-H. Lim and G. Kim, Nano Energy, 2018, 44, 121–126 CrossRef CAS
- Y. Yoo and N. Lim, J. Power Sources, 2013, 229, 48–57 CrossRef CAS
- Y. Gan, J. Zhang, Y. Li, S. Li, K. Xie and J. T. S. Irvine, J. Electrochem. Soc., 2012, 159, F763–F767 CrossRef CAS
- S. Li and K. Xie, J. Electrochem. Soc., 2013, 160, F224–F233 CrossRef CAS
- L. Gan, L. Ye, M. Liu, S. Tao and K. Xie, RSC Adv., 2016, 6, 641–647 RSC
- S. Li, R. Yan, G. Wu, K. Xie and J. Cheng, Int. J. Hydrogen Energy, 2013, 38, 14943–14951 CrossRef CAS
- S. Choi, C. J. Kucharczyk, Y. Liang, X. Zhang, I. Takeuchi, H.-I. Ji and S. M. Haile, Nat. Energy, 2018, 3, 202–210 CrossRef CAS
- D. Han, Y. Noda, T. Onishi, N. Hatada, M. Majima and T. Uda, Int. J. Hydrogen Energy, 2016, 41, 14897–14908 CrossRef CAS
- J. Guan, S. Dorris, U. Balachandran and M. Liu, Solid State Ionics, 1997, 100, 45–52 CrossRef CAS
- T. Schober and H. Bohn, Solid State Ionics, 2000, 127, 351–360 CrossRef CAS
- W. Lai and S. M. Haile, Phys. Chem. Chem. Phys., 2008, 10, 865–883 RSC
- M. Liu and H. Hu, J. Electrochem. Soc., 1996, 143(6), L109–L112 CrossRef CAS
- Q. Fang, L. Blum and N. H. Menzler, J. Electrochem. Soc., 2015, 162(8), F907–F912 CrossRef CAS
- S. Santhanam, M. P. Heddrich, M. Riedel and K. A. Friedrich, Energy, 2017, 141, 202–214 CrossRef
- B. Zhao, L. Zhang, D. Zhen, S. Yoo, Y. Ding, D. Chen, Y. Chen, Q. Zhang, B. Doyle, X. Xiong and M. Liu, Nat. Commun., 2017, 8, 14586 CrossRef CAS PubMed
- M. Ni, M. K. H. Leung and D. Y. C. Leung, Int. J. Hydrogen Energy, 2008, 33, 4040–4047 CrossRef CAS
- T. Matsui, R. Kishida, J.-Y. Kim, H. Muroyama and K. Eguchi, J. Electrochem. Soc., 2010, 157, B776–B781 CrossRef CAS
- R. Knibbe, M. L. Traulsen, A. Hauch, S. D. Ebbesen and M. Mogensen, J. Electrochem. Soc., 2010, 157, B1209–B1217 CrossRef CAS
- F. M. Sapountzi, J. M. Gracia, C. J. Weststrate, H. O. A. Fredriksson and J. W. Niemantsverdriet, Prog. Energy Combust. Sci., 2017, 58, 1–35 CrossRef
- K. Chen and S. P. Jiang, J. Electrochem. Soc., 2016, 163, F3070–F3083 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ee02865f |
| This journal is © The Royal Society of Chemistry 2019 |