
Dyadic promotion of photocatalytic aerobic oxidation via the Mott–Schottky effect enabled by nitrogen-doped carbon from imidazolium-based ionic polymers†
Hong
Zhong
a,
Can
Yang
b,
Lizhou
Fan
a,
Zhihua
Fu
a,
Xue
Yang
a,
Xinchen
Wang
 b and
Ruihu
Wang
*a
b and
Ruihu
Wang
*a
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China
bState Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, Fuzhou, 350002, China
First published on 11th December 2018
Abstract
Metal/semiconductor systems are one type of promising heterogeneous photocatalyst for solar conversion. The injection of hot electrons from photoactivated metals to semiconductors is the rate-determining step owing to the Schottky barrier created at the interface. It is highly desirable to develop new approaches for promoting hot electron transfer. Herein, we present one type of new Mott–Schottky-type photocatalytic material consisting of TiO2 nanosheets and Ru nanoparticles (NPs) coated with nitrogen-doped carbon (TiO2@NC-Ru-T). They are readily available through the conformal coating of TiO2 with a main-chain imidazolium-based ionic polymer (ImIP), followed by anion exchange with perruthenate and subsequent pyrolysis; the sintering of Ru NPs is effectively inhibited by ImIP, generating small-sized and well-dispersed Ru NPs. The nitrogen-doped carbon in TiO2@NC-Ru-T both strengthens the performance of Ru NPs and facilitates photoelectron transfer from photoactivated Ru NPs to TiO2 through a Mott–Schottky contact. The dyadic effects greatly promote selective aerobic oxidation of alcohols with air as an oxidant under visible light irradiation. This work provides a feasible protocol for improving visible light absorption and charge/electron transfer in photocatalytic reactions, and holds great promise for developing a new type of solar-to-chemical energy conversion reaction.
Broader contextThe development and application of photocatalytic technology have captured considerable interest for solving the issues of global energy shortage and environmental pollution by converting solar energy into chemical fuels. Great efforts have been directed towards the improvement of the photocatalytic efficiency in the visible-light region. Metal/semiconductor systems represent a class of promising heterogeneous photocatalysts with broad absorption extending to the visible light region. The injection of hot electrons from photoactivated metals to semiconductors is a rate-determining step because a certain amount of energy is required to overcome the Schottky barrier at the metal/semiconductor interface. Herein, we develop a facile and general approach for promoting hot electron transfer by coating TiO2 nanosheets and Ru nanoparticles with nitrogen-doped carbon. The presence of nitrogen-doped carbon not only results in the formation and stabilization of small-sized Ru nanoparticles, but also improves visible light absorption, which enhances photocatalytic efficiency in the visible light region. This study shows good generality for the synthesis of metal/semiconductor photocatalytic materials with a low Schottky barrier and ultrastable small-sized metal nanoparticles for various organic photosyntheses. |
Introduction
Energy consumption by the chemical industry is one of the major sources of pollution owing to the generation of greenhouse gases.1–3 Photochemical reactions are among the promising processes for the synthesis of various chemicals from the viewpoints of green and sustainable chemistry.4–7 Photocatalytic aerobic oxidation is of considerable importance in practical application. Several types of substrates, such as alcohols,8 amines,9 hydrocarbons10 and sulfides,11 are successfully oxidized. Current interest has been directed toward the improvement of the photocatalytic efficiency in the visible-light region.12–14 Metal nanoparticle/semiconductor heterojunctions are one of powerful systems for the creation of visible-light-driven photocatalysts owing to strong absorption of metal nanoparticles (NPs) in the visible region and concomitant hot electron injection into the semiconductor.12 It is generally recognized that metal–support interactions in the heterojunctions have imposed tremendous influence on the size of metal NPs, optical properties of semiconductors and metal–semiconductor charge transfer.15–18 However, the rate-determining step for these systems is still the electron transfer from photoactivated metal NPs to semiconductors.19–21 In addition, the metal–support interactions are not strong enough to resist sintering/aggregation of metal NPs, which makes the dispersion and stabilization of NPs on the supports a daunting challenge. Therefore, the construction of effective Mott–Schottky contacts between the metal NPs and semiconductive supports is required, and the ability is inevitably related to the structure of semiconducting materials.17With the development of new methods for the modification of semiconductors, such as TiO2, various types of metal–support interactions have been proposed in metal NPs/TiO2 heterojunction systems.22,23 Nitrogen-doped carbons (NCs) are a category of conjugated materials capable of creating favorable Mott–Schottky heterojunctions at the interface of TiO2 and metal NPs for high electron mobility, thus substantially enhancing the activity and stability of photocatalysts.24,25 It could be expected that the conformal surface coverage of TiO2 and metal NPs using NCs could facilitate hot electron transfer from metal NPs to TiO2 during photocatalysis. Meanwhile, NCs possess some unique advantages, such as suppressing phase transition of TiO2 from anatase to rutile, promoting mass transfer of substrates and accelerating deprotonation of substrates in aerobic oxidation reactions. Despite these attractive merits, the application of NC-coated metal NPs and TiO2 in photocatalytic aerobic oxidation has not been reported hitherto.
The main-chain imidazolium-based ionic polymers (ImIPs) are promising precursors to synthesize NCs with high nitrogen contents and good electronic conductivity.26 The imidazolium-based cationic groups are located homogeneously in the host backbones to form highly crosslinked networks. The relatively weak interaction between host networks and counter halide anions provides the possibility of exchanging with various metal-containing anion precursors,27 thus resulting in controllable loading and homogeneous distribution of metal precursors within ImIPs. ImIPs could serve as robust physical barriers to effectively resist the sintering of metal NPs, which is beneficial for the formation of small-sized metal NPs and the improvement of metal–support interactions. As a proof-of-concept study, herein, we present a simple yet effective strategy for the fabrication of TiO2@NC-Ru-T with well-dispersed Ru NPs. The electron transfer between Ru NPs and TiO2 is greatly promoted by the Mott–Schottky effect enabled by NC. TiO2@NC-Ru-T shows significantly enhanced photocatalytic activity and excellent stability in aerobic oxidation of alcohols under visible light irradiation.
Results and discussion
Synthesis of TiO2@NC-Ru-T
The synthetic route of TiO2@NC-Ru-T is shown in Fig. 1 TiO2 nanosheets were prepared through hydrothermal treatment of tetrabutyl titanate in hydrofluoric acid solution.28 ImIP was readily coated on the TiO2 surface through the quaternization reaction of 1,2,4,5-tetrakis(bromomethyl)benzene and two equivalents of bis(1H-imidazol-1-yl)methane to form TiO2@ImIP. The anion exchange between bromide in ImIP and perruthenate gave rise to TiO2@ImIP-Ru, and the subsequent pyrolysis at relatively high temperatures under a nitrogen atmosphere generated TiO2@NC-Ru-T, in which T indicates the pyrolysis temperatures of 500, 600, 700, 800 and 900 °C. | ||
| Fig. 1 Schematic illustration for the synthesis of TiO2@NC-Ru-T. | ||
Microstructural characterization of TiO2@ImIP and TiO2@ImIP-Ru
Transmission electron microscopy (TEM) images reveal that TiO2 nanosheets have a side length of 20–50 nm and a thickness of 2–5 nm (Fig. 2a); they are stacked one above the other to form a plate morphology (Fig. S1, ESI†). After being coated with ImIP, the marginal thickness of ImIP in TiO2@ImIP is in the range of 3–6 nm (Fig. 2d–f). High-annular dark-field scanning TEM (HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDS) elemental mapping further indicate that TiO2 nanosheets are effectively covered with ImIP (Fig. S2, ESI†). The top-view and cross-sectional high-resolution TEM (HRTEM) images show that the intervals between two lattice fringes in TiO2 are 0.350 and 0.235 nm (Fig. 2e and f), corresponding to the (101) and (001) facets,29,30 respectively, which are consistent with that of TiO2 (Fig. 2b and c). Notably, the morphology and structure of TiO2@ImIP are intact after anion exchange (Fig. 2g–i). Perruthenate is homogeneously distributed within ImIP to serve as a charge-balanced anion of the host backbone (Fig. S3, ESI†). | ||
| Fig. 2 (a, d and g) TEM image, (b, e and h) cross-sectional and (c, f and i) top-view HRTEM images for TiO2, TiO2@ImIP and TiO2@ImIP-Ru, respectively. Blue arrows in d and g are indicative of ImIP. | ||
The powder X-ray diffraction (XRD) pattern shows that the as-synthesized TiO2 nanosheets exhibit the typical reflection peaks of the anatase phase (Fig. S4, ESI†).31,32 The anatase phase is well maintained after the coating with ImIP and subsequent anion exchange, and no additional diffraction peaks from ImIP and perruthenate are detected. The simultaneous appearance of the characteristic peaks of TiO2 and ImIP in the Fourier-transform infrared spectroscopy (FTIR) spectrum of TiO2@ImIP indicates successful combination of ImIP and TiO2 (Fig. S5, ESI†). In the solid-state 13C NMR spectrum of TiO2@ImIP (Fig. S6, ESI†), the resonance peaks at 61 and 52 ppm correspond to methylene carbon atoms bonded between two imidazolium and between imidazolium and phenyl rings,27,33 respectively. The peaks at 149–106 ppm are assigned to the aromatic carbon atoms. Notably, the FTIR and solid-state 13C NMR spectra of TiO2@ImIP-Ru are almost identical to those of TiO2@ImIP.
Microstructural characterization of TiO2@NC-Ru-T
The nitrogen adsorption/desorption isotherms and the corresponding pore size distribution show that TiO2, TiO2@ImIP and TiO2@ImIP-Ru possess predominantly mesopores (Fig. S7, ESI†), while TiO2@NC-Ru-T shows hierarchical porosity (Fig. S8 and Table S1, ESI†); the micropores in TiO2@NC-Ru-T are mainly derived from NC. XRD patterns show that the anatase phase of TiO2 is well sustained in TiO2@NC-Ru-500, TiO2@NC-Ru-600 and TiO2@NC-Ru-700, while TiO2@NC-Ru-800 shows a typical diffraction pattern of the rutile phase with co-existence of a small amount of the anatase phase (Fig. 4a). It has been reported that a phase transition from pure anatase to rutile occurs above 550 °C,34,35 and the enhanced phase transition temperature of TiO2@NC-Ru-T reveals that ImIP on the TiO2 surface enhances its thermal stability.36 However, further increasing the pyrolysis temperature to 900 °C generates amorphous TiO2 with a minor amount of the anatase phase, which suggests that the phase evolution of TiO2 from anatase to rutile proceeds from the bulk to the surface. Notably, the characteristic peak of Ru(101) at 43.9° could be clearly identified for all samples.37The effects of pyrolysis temperature on the morphology and microstructure of TiO2@NC-Ru-T were investigated by TEM analyses. As shown in Fig. 3 and Fig. S9–S12 (ESI†), well-dispersed Ru NPs could be observed in all samples. With the increase in temperature, the average size of Ru NPs slightly increases owing to the low Tammann temperature and high surface energies.38 The average sizes of Ru NPs in TiO2@NC-Ru-500, TiO2@NC-Ru-600 and TiO2@NC-Ru-700 are 1.90 ± 0.20, 2.35 ± 0.20 and 2.85 ± 0.40 nm, respectively (Table S1, ESI†), which are much smaller than those in noble metal NPs obtained at the same pyrolysis temperatures.39 Notably, the remarkable sintering resistance of Ru NPs has been also demonstrated even at 800 and 900 °C. The average size of Ru NPs in TiO2@NC-Ru-800 and TiO2@NC-Ru-900 are as low as 3.20 ± 0.50 and 3.90 ± 0.50 nm, respectively. In the HRTEM images of TiO2@NC-Ru-800, the typical (110) facet of rutile could be clearly identified with a lattice fringe of 0.325 nm (Fig. S11d, ESI†), suggesting the phase transformation from anatase to rutile at 800 °C.40 Some lattice fringes of anatase could still be found in the HRTEM images of TiO2@NC-Ru-800 and TiO2@NC-Ru-900 (Fig. S11e and S12e, ESI†), which are in agreement with the results of their XRD patterns. The favorable effects of ImIP on Ru NPs were further validated by direct pyrolysis of ImIP-Ru at 700 °C; the resultant sample is denoted as NC-Ru-700. The average size of Ru NPs in NC-Ru-700 is 2.25 ± 0.20 nm (Fig. S13, ESI†). In sharp contrast, obvious agglomeration and uneven distribution of Ru NPs are observed in TiO2-Ru-500 (Fig. S14, ESI†), which further highlights the importance of ImIP in the stabilization and dispersion of Ru NPs.
 | ||
| Fig. 3 (a and b) TEM, (c) HRTEM, (d) HAADF-STEM and (e) EDS elementary mapping images for TiO2@NC-Ru-700. The inset in (a) shows the size distribution of Ru NPs. Blue arrows in c are indicative of NC. | ||
To better understand the structures and properties of TiO2@NC-Ru-T, TEM images were further investigated through choosing TiO2@NC-Ru-700 as a model sample. HAADF-STEM and EDS elementary mapping images show that Ti (green), C (red) and Ru (orange) atoms are homogeneously distributed in the samples (Fig. 3d and e), and the peripheral domains are mainly composed of C and Ru, which further reveals that TiO2 is conformally coated with NC. The pyrolysis temperature has exerted a crucial effect on the properties of the surface carbon materials. The lattice fringe of the graphitic carbon becomes clearer upon increasing the pyrolysis temperature of TiO2@ImIP-Ru, suggesting the increment of graphite degree in NC,41 which is favorable for electron transfer in photocatalytic reactions.
Spectra and conductivity characterization of TiO2@NC-Ru-T
The roles of NC are further disclosed by Raman spectroscopy (Fig. 4b). NC in TiO2@NC-Ru-T shows the characteristic D-band and G-band at 1346 and 1588 cm−1, respectively.26 The intensity ratios of D-band/G-band gradually decrease from 1.16 in TiO2@NC-Ru-500 to 0.87 in TiO2@NC-Ru-900 with increasing pyrolysis temperature (Table S1, ESI†). The Raman spectra of TiO2@NC-Ru-500, TiO2@NC-Ru-600 and TiO2@NC-Ru-700 show the characteristic peaks of anatase in the range of 50–1000 cm−1 (Fig. S15, ESI†).36 When compared with TiO2 and TiO2-Ru-500, an obvious red shift indicates a strong interaction between NC and TiO2,36 which is helpful for electron transfer from Ru NPs to TiO2. The surface compositions in TiO2@NC-Ru-T were investigated by X-ray photoelectron spectroscopy (XPS). The high-resolution N 1s XPS spectra show the presence of pyridinic N (398.5 eV), pyrrolic N (399.7 eV) and graphitic N (401.1 eV) (Fig. S16, ESI†).42 In the Ru 3d XPS spectra of TiO2@NC-Ru-T, the binding energy peaks of Ru 3d5/2 negatively shift by 0.42 eV when compared with those in TiO2-Ru-500 (Fig. 4c), which is attributed to the coordination interaction of nitrogen atoms in NC with Ru NPs.43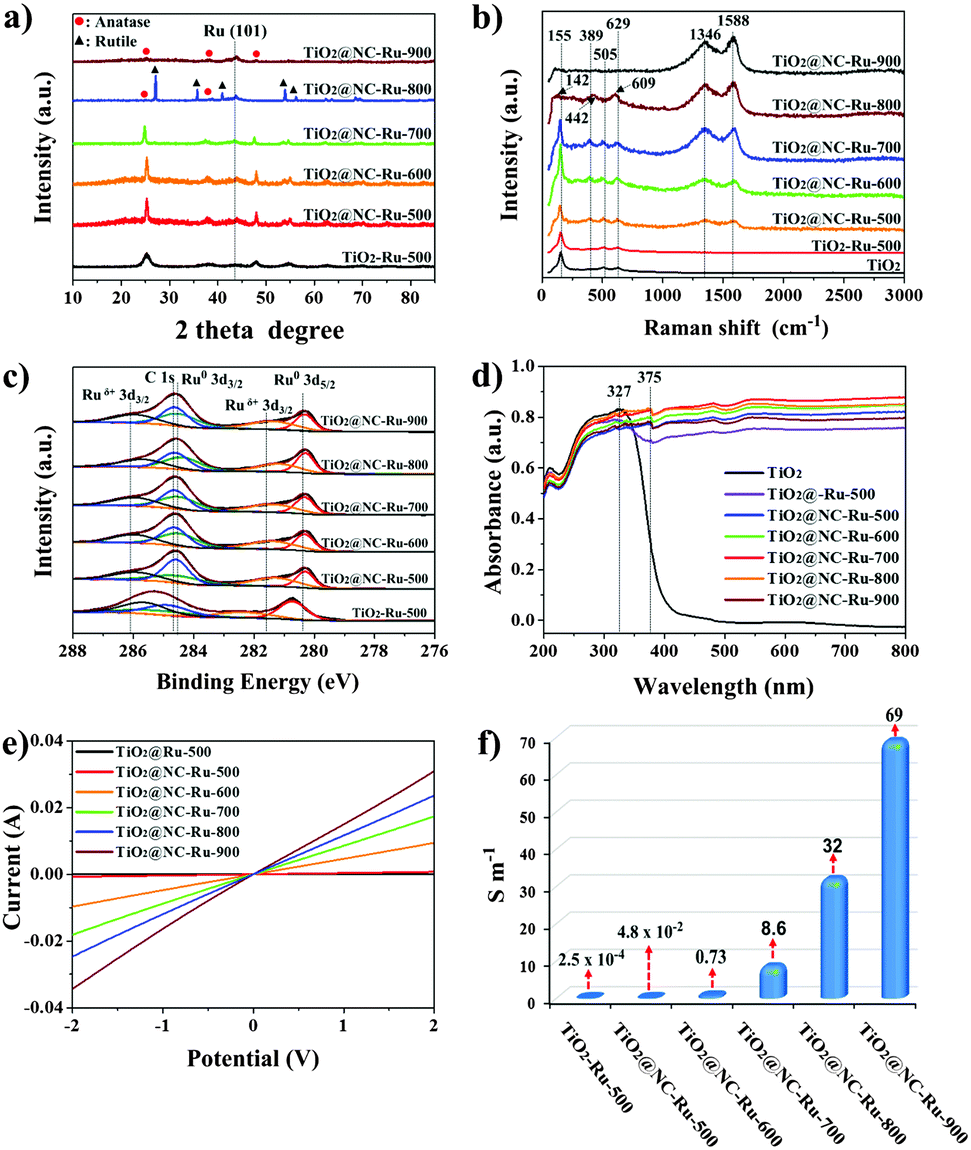 | ||
| Fig. 4 (a) XRD patterns, (b) Raman spectra, (c) Ru 3d XPS spectra, (d) UV-vis diffuse reflection spectra, (e) I–V plots and (f) electronic conductivities for TiO2-Ru-500 and TiO2@NC-Ru-T. | ||
The optical properties of TiO2, TiO2-Ru-500 and TiO2@NC-Ru-T were investigated by UV-vis diffuse reflection spectroscopy (Fig. 4d). All samples exhibit strong absorption at <400 nm, which is assigned to the intrinsic absorption of TiO2.44,45 TiO2-Ru-500 and TiO2@NC-Ru-T show significantly enhanced absorption in the whole visible light region, which indicates that photocatalysis could cover the UV and visible light range in these samples. The energy band structures of NC and TiO2 were also investigated. Cyclic voltammetry (CV) measurements show that the bandgap values (Egap) of TiO2 and NC are 2.56 and 2.82 eV, respectively (Fig. S17, ESI†).46 The positive slopes in the Mott–Schottky plots show that both TiO2 and NC are n-type semiconductors (Fig. S18 and S19, ESI†),47 and the corresponding flatband potentials are −0.86 and −0.57 eV vs. Ag/AgCl, respectively. Thus, the valence band edge potentials of TiO2 and NC are calculated to be 1.70 and 2.25 eV, respectively (Fig. S20, ESI†).
Since TiO2 and Ru NPs are conformally coated with NC, a favorable electron contact between Ru NPs and TiO2 could be achieved. The electronic conductivities of TiO2-Ru-500 and TiO2@NC-Ru-T were recorded by the I–V curves under ambient conditions (Fig. 4e), and the values calculated from the slopes of the I–V curves are provided in Fig. 4f.48 As expected, TiO2-Ru-500 exhibits a nearly insulating behavior with an electronic conductivity of 2.5 × 10−4 S m−1, while the conductivity increases drastically to 4.8 × 10−2 S m−1 in TiO2@NC-Ru-500. The curve slopes and electronic conductivities in TiO2@NC-Ru-T further increase with increasing pyrolysis temperature. The conductivities in TiO2@NC-Ru-700, TiO2@NC-Ru-800 and TiO2@NC-Ru-900 are as high as 8.6, 32 and 69 S m−1, respectively. Obviously, the formation of the Mott–Schottky heterojunction in NC greatly facilitates the electron transfer in the whole material. Moreover, the conductivities mainly rely on the graphite degree of NC, and are independent of the phase state of TiO2. The high conductivity in anatase-based materials could effectively compensate for the relatively low electron transfer rate from Ru NPs to the anatase surface, thus greatly enhancing their photocatalytic activities.
Photocatalytic properties of TiO2@NC-Ru-T
Aerobic oxidation is a typical probe reaction to identify the influences of NC on photocatalytic performance. The photocatalytic activity of TiO2@NC-Ru-T was initially evaluated by the aerobic oxidation of benzyl alcohol using air as an oxidant in H2O without any additives. As shown in Fig. 5a, when the reaction was performed in the dark at 25 °C for 6 h, TiO2@NC-Ru-500, TiO2@NC-Ru-600 and TiO2@NC-Ru-700 gave 9–11% conversion of benzyl alcohol. The similar conversion is probably ascribed to their analogous Ru NP sizes. This conclusion has been further validated by NC-Ru-700 with similar Ru NP size, for which an 11% conversion was obtained under identical conditions. However, the conversions of benzyl alcohol in TiO2@NC-Ru-800 and TiO2@NC-Ru-900 were decreased to 7% and 5%, respectively, owing to the gradual increment of the average size of Ru NPs. In sharp contrast, the use of TiO2-Ru-500 with larger particle size and wider size distribution gave a conversion as low as 3%. These results show that the catalytic activity in the dark is closely related to the size of the Ru NPs, and is independent of the electronic conductivity of the catalysts. Strikingly, the conversion of benzyl alcohol in TiO2-Ru-500 was increased to 9% when the reaction was initiated by visible light irradiation from a Xe lamp (λ > 420 nm visible filter). The conversions of benzyl alcohol in TiO2@NC-Ru-500, TiO2@NC-Ru-600 and TiO2@NC-Ru-700 were gradually increased to 33%, 39% and 43%, respectively. However, TiO2@NC-Ru-800 and TiO2@NC-Ru-900 showed much inferior catalytic activity although they possessed superior electronic conductivity, which probably originates from the phase transformation of TiO2 from anatase to rutile and the amorphous phase, respectively. It should be mentioned that the oxidation reaction both in the dark and under visible light irradiation generated benzaldehyde in >99% selectivity. TiO2@NC-Ru-700 exhibits the highest catalytic activity which is more than four times higher than those in the dark and of TiO2-Ru-500, clearly highlighting the importance of the conformal coating of Ru NPs and anatase with NC in facilitating aerobic oxidation. The apparent quantum yield (AQE) is estimated to be 10.7% under monochromatic irradiation at 420 nm. The action spectrum analysis reveals that the absorption bands of TiO2@NC-Ru-700 correlate well with the AQE in the photocatalytic oxidation of benzyl alcohol (Fig. S21, ESI†), which suggests that enhanced catalytic cativity is triggered by the photoabsorption of the catalyst.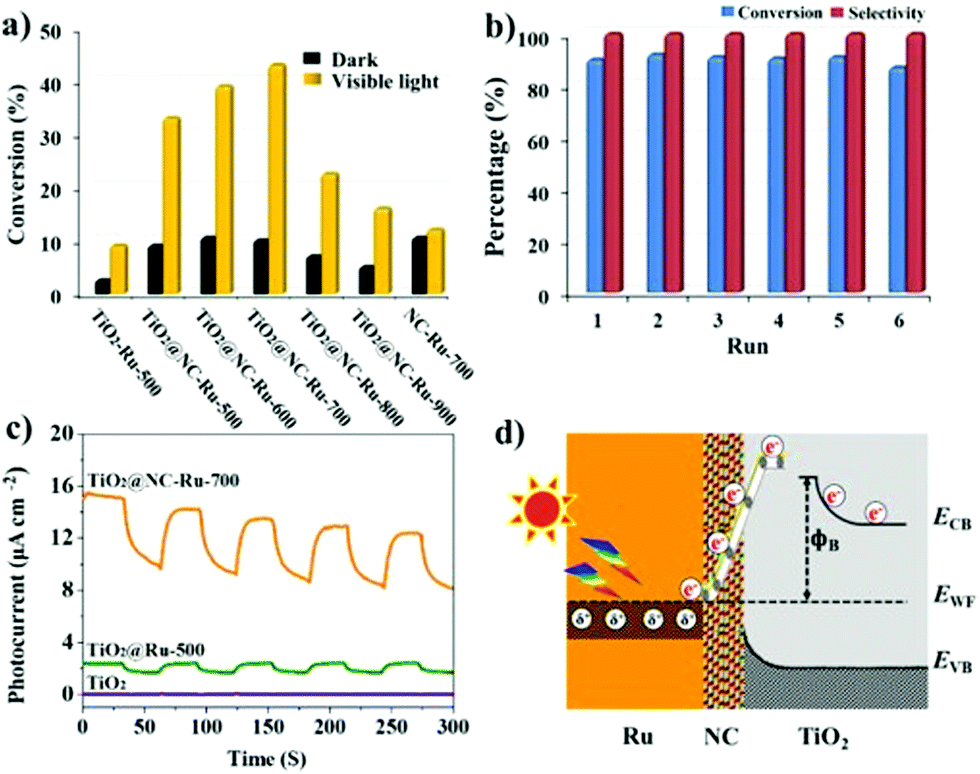 | ||
| Fig. 5 (a) The conversion of benzyl alcohol for respective catalysts in the dark and under visible light irradiation; (b) recyclability of TiO2@NC-Ru-700 in the aerobic oxidation of benzyl alcohol upon direct visible light exposure. Reaction conditions: benzyl alcohol (0.5 mmol), Ru (1.0 mol%), H2O (1 mL), air (1 atm), 6 h; (c) time-dependent photocurrent response of TiO2, TiO2@-Ru-500 and TiO2@NC-Ru-700; (d) proposed mechanism for the electron transfer from photoactivated Ru NPs to TiO2. | ||
The kinetic curve for photocatalytic oxidation of benzyl alcohol was also investigated. As shown in Fig. S22 (ESI†), the conversion of benzyl alcohol increased rapidly in the first 3 h and then went up slowly, but the selectivity remained above 99% in the entire catalytic process. Notably, direct visible light irradiation in TiO2@NC-Ru-700 gave the target product in 90% conversion (entry 1, Table 1). Control experiments using as-prepared TiO2 nanosheets or TiO2@NC-700 as catalysts were also performed. Negligible conversion of benzyl alcohol was observed when the reactions were conducted in the dark and under visible light irradiation (entries 2 and 3, Table 1). The photocatalytic activities of TiO2@NC-Ru-700 were also investigated under acidic and alkaline conditions. The conversion of benzyl alcohol greatly decreased in more than 0.1 M of HCl aqueous solution, while the alkaline environment is beneficial for the catalytic reaction (Fig. S23, ESI†). Impressively, the conversion of benzyl alcohol was increased to 61% at 25 °C when pure oxygen was used as an oxidant (entry 4). However, when the reaction was carried out under a nitrogen atmosphere, no conversion of benzyl alcohol was detected, which provides the evidence that O2 actually functions as the oxidant in the catalytic system. To our best knowledge, most of the reported photocatalytic aerobic oxidation reactions chose O2 as an oxidant owing to competitive absorption between the nitrogen molecule and oxygen molecule in photocatalysis;49,50 the use of cheap and readily available air as an oxidant holds great promise for practical application.
| Entry | Substrate | Product | Tem. | Con.b (%) | Sel.b (%) |
|---|---|---|---|---|---|
| a Reaction conditions: alcohol (0.5 mmol), TiO2@NC-Ru-700 (Ru 1 mol%), H2O (1 mL), air (1 atm) at room temperature (RT, 25 °C) or ambient temperature (AT, direct visible light irradiation, ∼56 °C) for 6 h. b Yields and selectivity were determined by GC. c TiO2 nanosheet or TiO2@NC-700 was used as the catalyst in the dark. d TiO2 nanosheet or TiO2@NC-700 was used as the catalyst under visible light irradiation. e Oxygen was used as an oxidant. f Reaction time was elongated to 12 h. g Hot filtration experiment. The reaction time was 2 h, the conversions in parentheses are from the reaction of the filtrate for additional 4 h. | |||||
| 1 |

|

|
RT | 43 | >99 |
| AT | 90 | >99 | |||
| 2c |

|

|
RT | <1 | >99 |
| 3d |

|

|
RT | <1 | >99 |
| 4e |

|

|
RT | 61 | >99 |
| 5 |
|

|
RT | 46 | >99 |
| AT | 91 | >99 | |||
| 6 |

|

|
RT | 43 | >99 |
| AT | 90 | >99 | |||
| 7 |

|

|
RT | 45 | >99 |
| AT | 93 | >99 | |||
| 8 |
|

|
RT | 26 | >99 |
| AT | 78 | >99 | |||
| 9 |
|

|
RT | 23 | >99 |
| AT | 72 | >99 | |||
| 10 |

|

|
RT | 19 | >99 |
| AT | 56 | >99 | |||
| 11 |

|

|
RT | 24 | >99 |
| AT | 77 | >99 | |||
| 12 |

|

|
RT | 39 | >99 |
| AT | 86 | >99 | |||
| 13f |

|

|
RT | 67 | >99 |
| AT | 100 | >99 | |||
| 14g |

|
|
RT | 26 | >99 |
| RT | (26) | >99 | |||
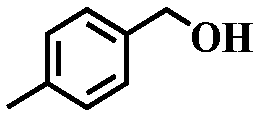
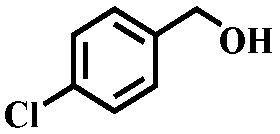
To further explore the generality of this catalytic system, a variety of alcohols were employed as substrates in the aerobic oxidation reaction using 1.0% TiO2@NC-Ru-700 as a catalyst and air as an oxidant both at room temperature (25 °C) and under direct visible light exposure (56 °C).51 As shown in Table 1, the benzyl alcohols bearing electron-donating groups, such as –Me and –OMe, gave rise to the corresponding products in 43–46% conversions at 25 °C (entries 5–7), which are higher than those (19–26%) from the substrates bearing electron-withdrawing groups, such –Cl and –F (entries 8 and 9). Notably, their conversions were increased to 90–93% and 72–78%, respectively, upon direct visible light exposure (entries 1 and 5–9). The steric hindrance of the substituents at para- and meta-positions in the benzyl alcohols has no appreciable effect on the catalytic activity in both cases. The catalytic system is also effective for the oxidation of aliphatic alcohols and cinnamyl alcohol, and the target products were obtained in moderate conversion and 100% selectivity upon direct visible light exposure (entries 10 and 11). Interestingly, when secondary benzyl alcohol was used as a substrate without any base or additives, the corresponding ketone was obtained in 39 and 86% conversions at room temperature and under direct visible light exposure, respectively (entry 12). The conversions were increased to 67 and 100%, respectively, when the reaction time was elongated to 12 h (entry 13).
The recyclability of TiO2@NC-Ru-700 was examined in the aerobic oxidation of benzyl alcohol under direct visible light exposure for 6 h. After the reaction, the recovered catalyst was separated by centrifugation, and directly used for the next run. Strikingly, TiO2@NC-Ru-700 could be used for at least six runs without obvious loss of catalytic activity and selectivity (Fig. 5b). The heterogeneous behavior of this catalytic system was verified by hot filtration experiment. After the reaction was run for 2 h, the Ru-containing catalytic species was quickly removed by filtration, and the photocatalytic reaction of the filtrate was continued for an additional 4 h; negligible change in conversion and selectivity was observed (entry 14, Table 1). Inductively coupled plasma (ICP) analyses showed tha Ru content in the recovered TiO2@NC-Ru-700 was identical to that in the as-prepared sample. In sharp contrast, an apparent degradation of catalytic activity in TiO2-Ru-500 was observed after the second run (Fig. S24, ESI†). The recovered TiO2@NC-Ru-700 after consecutive reactions for six runs (TiO2@NC-Ru-700-6run) was also investigated. As shown in Fig. S25 (ESI†), the XRD pattern of TiO2@NC-Ru-700-6run has no appreciable change in comparison with that of TiO2@NC-Ru-700. In TEM images of TiO2@NC-Ru-700-6run (Fig. 6), no obvious agglomeration of Ru NPs is observed. The Ru NPs are still uniformly dispersed with the retention of the average size of 2.90 ± 0.4 nm. Therefore, besides sintering resistance of Ru NPs, NC also plays an important role in preventing Ru NPs from aggregating during photocatalytic cycles.
 | ||
| Fig. 6 (a and b) TEM, (c) HRTEM images and (d) the size distribution of Ru NPs for TiO2@NC-Ru-700-6run after consecutive reactions for six runs. Blue arrows in f are indicative of NC. | ||
Photoelectric properties and mechanistic studies
To better understand the roles of NC in the photocatalytic reaction, the charge transfer behaviors in TiO2@Ru-500 and TiO2@NC-Ru-700 were investigated by Mott–Schottky plots, Hall effect measurement and time-dependent photocurrent response. The Mott–Schottky curves show that both TiO2@Ru-500 and TiO2@NC-Ru-700 are n-type semiconductors (Fig. S26, ESI†); the smaller slope in TiO2@NC-Ru-700 suggests that its charge transfer ability is much higher than that of TiO2@Ru-500,47 which is consistent with the results of their I–V curves (Fig. 4e and f). Hall effect measurement at room temperature provides a carrier concentration of 2.036 × 1018 cm−3 and a mobility of 0.0947 cm2 V−1 s−1 in TiO2@NC-Ru-700, while the carrier concentration in TiO2@-Ru-500 falls to a degree lower than the Hall effect measurement limit. The significant difference indicates the importance of NC in charge carrier separation and transfer. The photoinduced charge-transfer properties were also investigated by photocurrent–time (I–t) curves with five on–off cycles under intermittent visible light irradiation (k > 420 nm). As shown in Fig. 5c. TiO2@-Ru-500 provides a weak photocurrent of 2.4 μA cm−2, which is increased to 12.6 and 14.2 μA cm−2 in NC-Ru-700 and TiO2@NC-Ru-700, respectively, while the photocurrent of TiO2 is negligible due to its weak absorption. These results are consistent with their UV-visible absorption spectra and photocatalytic activity analyses. The higher photocurrent in TiO2@NC-Ru-700 means that more photogenerated electrons transfer from the composite to the counter electrode via the external circuit,52–54 which is mainly attributed to the formation of the Mott–Schottky heterojunction between TiO2 and metal NPs enabled by NC. The highly conductive NC promotes the injection of hot electrons from photoactivated Ru NPs to the TiO2 conduction band and simultaneously inhibits photo-produced electron/hole (e−/h+) recombination. The proposed electron transfer process is shown in Fig. 5d and Fig. S27 (ESI†). The metal/semiconductor heterojunction usually creates a Schottky barrier (ϕB) at the interface.44,55 Upon visible light irradiation, a collective oscillation of sp or d band electrons on Ru NPs occurs, and the intraband or interband excitation to the sp-conduction band is promoted.12,44 The height of ϕB strongly affects the efficiency of the electron transfer. High ϕB in TiO2-Ru-500 is unfavorable for the electron transfer from photoactivated Ru NPs to TiO2. However, the decreased ϕB in NC is preferable for the electron injection from Ru NPs to NC, and the electron accumulation in the NC conduction band results in a negative shift of its conduction band potential.12 This promotes consecutive electron transfer from Ru NPs to NC and then to the TiO2 surface.56 Thus, photocurrent and photocatalytic activities are significantly enhanced in TiO2@NC-Ru-700.The mechanism for the aerobic oxidation of alcohols over TiO2@NC-Ru-T has been proposed.15,45 As depicted in Fig. 7, the reaction in the dark is initiated by O2 activation on the anionic sites of Ru NPs; the alcohols are simultaneously adsorbed on the surface of the catalytic sites. High content of nitrogen atoms on the surface of TiO2@NC-Ru-T could both promote adsorption of the alcohols and increase the electron density of Ru NPs. The activated O2 species remove the H atom of the alcohols and produce hydroperoxide and alcoholate species on the surface of Ru NPs; subsequent α-hydride elimination of the –CH– group from the alcoholate species affords the aldehyde or ketone as the target products. In the photocatalytic oxidation reaction, incident photons are absorbed by Ru NPs through interband transitions and/or the localized surface plasmon resonance, and the photoexcited electrons are injected into the TiO2 conduction band. The electron transfer rate in traditional TiO2-supported metal NPs is usually low probably due to the weak binding and small contact area between TiO2 and metal NPs. Encouragingly, the electron injection in TiO2@NC-Ru-T may resort to NC owing to their Mott–Schottky contact and high conductivity of NC, and the electron injection from photoactivated Ru NPs to anatase is greatly accelerated. After O2 is reduced into peroxide anionic species, subsequent removal of a H atom in alcohols produces the hydroperoxide and alcoholate species. Further abstraction of a H atom from the –CH– group of the alcoholate species gives rise to the target product. These mechanisms suggest that the catalytic reaction both in the dark and under visible light irradiation is initiated by the activation of O2, followed by the formation of hydroperoxide and alcoholate species. It is generally acceptable that the aerobic oxidation reaction synchronically proceeds through both of the cases.
 | ||
| Fig. 7 Proposed mechanism for aerobic oxidation of alcohols over TiO2@NC-Ru-T (a) in the dark and (b) under visible light irradiation. | ||
On the basis of the above-mentioned observations, ImIP and ImIP-derived NC play pivotal roles in the formation and stabilization of small-sized Ru NPs as well as the improvement of photocatalytic performance: (1) ImIP sustains homogeneous distribution of perruthenate in TiO2@ImIP-Ru. The sintering of Ru NPs is effectively resisted by ImIP-derived NC during pyrolysis, which is crucial for the formation of small-sized Ru NPs, even at a pyrolysis temperature as high as 900 °C. (2) The phase transformation temperature of TiO2 from anatase to rutile is enhanced, which is favorable for the formation of highly conductive carbon materials with the retention of the anatase phase at high pyrolysis temperature. (3) The good conjugation of carbon materials improves visible light absorption, which enhances the photocatalytic efficiency in the visible light region. (4) The high content of nitrogen atoms in NC not only accelerates the adsorption of alcohols on the catalytic sites through hydrogen bonding interactions, but also is beneficial for the deprotonation of alcohols and the formation of Ru-alcoholate species. (5) The presence of conductive NC lowers the Schottky barrier and accelerates hot electron transfer from photoactivated Ru NPs to NC and then to the TiO2 surface, thus leading to efficient O2 reduction on the TiO2 surface. (6) The aggregation of Ru NPs is effectively inhibited by NC. The size and distribution of Ru NPs are almost intact after consecutive catalytic cycles. These merits synergistically improve the photocatalytic performance in air under visible light irradiation.
Conclusions
We have developed a facile and general strategy to boost photocatalytic performance of metal NPs/semiconductor systems via the Mott–Schottky effect enabled by NC. ImIP plays crucial roles in inhibiting the Ostwald ripening of metal NPs during pyrolysis. NC significantly increases the electron density of Ru NPs and the absorption of visible light. Importantly, the electron transfer from photoactivated Ru NPs to TiO2 is greatly accelerated by the formation of Mott–Schottky heterojunctions in conductive NC, which facilitates the generation of the active oxygen species and Ru-alcoholate species. The dyadic effects of NC significantly improve the photocatalytic performance in aerobic oxidation of alcohols under visible light irradiation. In summary, this study provides a type of new metal/semiconductor heterojunction material with a low Schottky barrier and ultrastable small-sized metal NPs. The synthetic strategy could be extended to a broad range of photocatalysts consisting of other noble metals and semiconductors for various organic photosyntheses.Experimental
Materials
The TiO2 nanosheets28 and bis(1H-imidazol-1-yl)methane33 were prepared according to the modified literature methods. Other chemicals were commercially available and used without further purification.Synthesis of TiO2@ImIP
A mixture of TiO2 (800 mg) and 1,2,4,5-tetrakis(bromomethyl)benzene (448 mg, 1.0 mmol) in MeCN (40 mL) and DMF (10 mL) was ultrasonically dispersed for 0.5 h, and then a solution of bis(1H-imidazol-1-yl)methane (296 mg, 2.0 mmol) in MeCN (40 mL) and DMF (10 mL) was added and stirred at 85 °C for 6 h. The resutant white precipitate was collected by filtration, washed with MeCN (3 × 30 mL) and dried in vacuo. Yield: 1498 mg (97%). FTIR (KBr cm−1): 3415 (s), 3081 (w), 1627 (m), 1550 (m), 1439 (w), 1342 (w), 1161 (s), 757 (m), 465 (s).Synthesis of TiO2@ImIP-Ru
TiO2@ImIP (500 mg) was added to an aqueous solution (50 mL) of KRuO4 (271 mg, 1.33 mmol). After the mixture was stirred vigorously at room temperature for 24 h, the resultant powders were washed thoroughly with copious H2O to remove excess KRuO4, and then dried in vacuo at 80 °C for 12 h to afford TiO2@ImIP-Ru. Yield: 527 mg (86%). FTIR (KBr cm−1): 3415 (s), 3150 (w), 1641 (s), 1558 (m), 1383 (w), 1161 (m), 965 (m), 465 (s).General procedures for the synthesis of TiO2@NC-Ru-T
TiO2@ImIP-Ru (500 mg) in a ceramic boat was heated to an appropriate pyrolysis temperature at a heating rate of 5 °C min−1 under a N2 atmosphere, and then kept at the pyrolysis temperature for 2 h. After being cooled to room temperature, the resultant black powders were treated with 10 M aqueous KOH solution at 100 °C for 24 h, followed by washing with water and methanol, and dried at 80 °C in vacuo overnight to afford TiO2@NC-Ru-T, in which T denotes the pyrolysis temperature of TiO2@ImIP-Ru at 500, 600, 700, 800 and 900 °C.Synthesis of TiO2-Ru-500
The TiO2 nanosheets (500 mg) were added to an aqueous KRuO4 (271 mg, 1.33 mol) solution (50 mL), and the mixture was stirred at room temperature for 24 h. The resultant solid was washed with copious H2O to remove excess KRuO4, and then dried in vacuo at 80 °C for 12 h. The obtained powders were reduced under H2/Ar stream (10%) at 500 °C for 2 h giving rise to TiO2-Ru-500. The Ru content in TiO2-Ru-500 was 0.280 mmol g−1 as determined by ICP.Synthesis of NC-Ru-700
ImIP (300 mg) was added to an aqueous KRuO4 (325 mg, 1.60 mol) solution (50 mL), and the mixture was stirred at room temperature for 24 h. The resultant solid was washed with copious H2O to remove excess KRuO4, and then dried in vacuo at 80 °C for 12 h. The obtained powders were put into a ceramic boat and were heated to 700 °C at a heating rate of 5 °C min-1 under a N2 atmosphere. After being kept at 700 °C for 2 h, the resultant powders were treated with 10 M aqueous KOH solution at 100 °C for 24 h, followed by washing with water and methanol, and dried at 80 °C in vacuo overnight to afford NC-Ru-700. The Ru content in TiO2-Ru-500 was 0.537 mmol g−1 as determined by ICP.General procedures for photocatalytic aerobic oxidation of alcohols
The photocatalytic aerobic oxidation was carried out in a quartzose reactor equipped with a magnetic stirrer and an air ball. The mixture of substrate (0.5 mmol), catalysts (1% Ru) and H2O (1 mL) was stirred in the dark or under visible light irradiation for 6 h. After the reaction was completed, the product was extracted with diethyl ether, and the conversion and selectivity were determined by GC.General procedures for recyclability test
After the photocatalytic oxidation of benzyl alcohol, the product was extracted with diethyl ether. The organic layer was analyzed by GC. The black powder in the aqueous phase was separated by filtration and washed with acetone. The resultant solid was used for the next run with the addition of fresh benzyl alcohol and H2O.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21603228, 21673241 and 21471151), and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB20000000).References
- H. M. Jeon, J. Edmonds, N. Bauer, L. Clarke, B. Fisher, B. P. Flannery, J. Hilaire, V. Krey, G. Marangoni, R. Mi, K. Riahi, H. Rogner and M. Tavoni, Nature, 2014, 514, 482–485 CrossRef PubMed.
- A. Otto, T. Grube, S. Schiebahn and D. Stolten, Energy Environ. Sci., 2015, 8, 3283–3297 RSC.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473–486 RSC.
- Y. Zhang, J. Guo, L. Shi, Y. Zhu, K. Hou, Y. Zheng and Z. Tang, Sci. Adv., 2017, 3, e1701162 CrossRef PubMed.
- H. Kim, O. S. Kwon, S. Kim, W. Choi and J. H. Kim, Energy Environ. Sci., 2016, 9, 1063–1073 RSC.
- D. Kim, K. K. Sakimoto, D. Hong and P. A. Yang, Angew. Chem., Int. Ed., 2015, 54, 3259–3266 CrossRef CAS PubMed.
- Y. Sugano, Y. Shiraishi, D. Tsukamoto, S. Ichikawa, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2013, 125, 5403–5407 CrossRef.
- D. S. Ovoshchnikov, B. G. Donoeva and V. B. Golovko, ACS Catal., 2015, 5, 34–38 CrossRef CAS.
- J. Lin, Z. Li, J. Kan, S. Huang, W. Su and Y. Li, Nat. Commun., 2017, 8, 14353 CrossRef PubMed.
- X. Lang, W. R. Leow, J. Zhao and X. Chen, Chem. Sci., 2015, 6, 1075–1082 RSC.
- D. Tsukamoto, Y. Shiraishi, Y. Sugano, S. Ichikawa, S. Tanaka and T. Hirai, J. Am. Chem. Soc., 2012, 134, 6309–6315 CrossRef CAS PubMed.
- A. Li, P. Zhang, X. Chang, W. Cai, T. Wang and J. Gong, Small, 2015, 11, 1892–1899 CrossRef CAS PubMed.
- D. Ding, K. Liu, S. He, C. Gao and Y. Yin, Nano Lett., 2014, 14, 6731–6736 CrossRef CAS PubMed.
- Y. Shiraishi, H. Sakamoto, Y. Sugano, S. Ichikawa and T. Hirai, ACS Nano, 2013, 7, 9287–9297 CrossRef CAS PubMed.
- S. T. Zhang, C. M. Li, H. Yan, M. Wei, D. Evans and G. X. Duan, J. Phys. Chem. C, 2014, 118, 3514–3522 CrossRef CAS.
- H. Su, K. X. Zhang, B. Zhang, H. H. Wang, Q. Y. Yu, X. H. Li, M. Antonietti and J. S. Chen, J. Am. Chem. Soc., 2017, 139, 811–818 CrossRef CAS PubMed.
- S. Wang, Y. Gao, S. Miao, T. Liu, L. Mu, R. Li, F. Fan and C. Li, J. Am. Chem. Soc., 2017, 139, 11771–11778 CrossRef CAS PubMed.
- E. Grabowsk, M. Marchelek, T. Klimczuk, W. Lisowski and A. Zaleska-Medynsk, J. Catal., 2017, 350, 159–173 CrossRef.
- H. Kobayashi and S. Higashimoto, Appl. Catal., B, 2015, 170, 135–143 CrossRef.
- J. Guo, Y. Zhang, L. Shi, Y. Zhu, M. F. Mideksa, K. Hou, W. Zhao, D. Wang, M. Zhao, X. Zhang, J. Lv, J. Zhang, X. Wang and Z. Tang, J. Am. Chem. Soc., 2017, 139, 17964–17972 CrossRef CAS PubMed.
- R. Jiang, B. Li, C. Fang and J. Wang, Adv. Mater., 2014, 26, 5274–5309 CrossRef CAS PubMed.
- W. Zhan, Y. Shu, Y. Sheng, H. Zhu, Y. Guo, L. Wang, Y. Guo, J. Zhang, G. Lu and S. Dai, Angew. Chem., Int. Ed., 2017, 129, 4565–4569 CrossRef.
- Z. Jiang, C. Zhu, W. Wan, K. Qian and J. Xie, J. Mater. Chem. A, 2016, 4, 1806–1818 RSC.
- C. Genovese, M. E. Schuster, E. K. Gibson, D. Gianolio, V. Posligua, R. Grau-Crespo, G. Cibin, P. P. Wells, D. Garai, V. Solokha, S. K. Calderon, J. J. Velasco-Velez, C. Ampelli, S. Perathoner, G. Held, G. Centi and R. Arrigo, Nat. Commun., 2018, 9, 935 CrossRef PubMed.
- H. Pan, Z. Cheng, Z. Xiao, X. Li and R. Wang, Adv. Funct. Mater., 2017, 1703936 CrossRef.
- Y. Gong, H. Zhong, W. Liu, B. Zhang, S. Hu and R. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 776–786 CrossRef CAS PubMed.
- X. Han, Q. Kuang, M. Jin, Z. Xie and L. Zheng, J. Am. Chem. Soc., 2009, 131, 3152–3153 CrossRef CAS PubMed.
- S. SelcuK and A. Selloni, Nat. Mater., 2016, 15, 1107–1112 CrossRef CAS PubMed.
- D. N. Pei, L. Gong, A. Y. Zhang, X. Zhang, J. J. Chen, Y. Mu and H. Q. Yu, Nat. Commun., 2015, 6, 8696 CrossRef PubMed.
- M. Li, Y. Chen, W. Li, X. Li, H. Tian, X. Wei, Z. Ren and G. Han, Small, 2017, 13, 1604115 CrossRef PubMed.
- P. l. Yan, X. Wang, X. Zheng, R. Li, J. Han, J. Shi, A. Li, Y. Gan and C. Li, Nano Energy, 2015, 15, 406–412 CrossRef CAS.
- H. Zhao, L. Li, Y. Wang and R. Wang, Sci. Rep., 2014, 4, 5478 CrossRef PubMed.
- J. Zhang, Q. Xu, Z. Feng, M. Li and C. Li, Angew. Chem., Int. Ed., 2008, 47, 1766–1769 CrossRef CAS PubMed.
- E. Baldini, /. L. Chiodo, A. Dominguez, M. Palummo, S. Moser, M. Yazdi-Rizi, G. Auböck, B. P. P. Mallett, H. Berger, A. Magrez, C. Bernhard, M. Grioni, A. Rubio and M. Chergui, Nat. Commun., 2017, 8, 13 CrossRef CAS.
- L. W. Zhang, H. B. Fu and Y. F. Zhu, Adv. Funct. Mater., 2008, 18, 2180–2189 CrossRef CAS.
- L. Lin, K. Wang, K. Yang, X. Chen, X. Fu and W. T. Dai, Appl. Catal., B, 2017, 204, 440–4555 CrossRef CAS.
- W. Zhan, Q. He, X. Liu, Y. Guo, Y. Wang, L. Wang, Y. Guo, A. Y. Borisevich, J. Zhang, G. Lu and S. Dai, J. Am. Chem. Soc., 2016, 138, 16130–16139 CrossRef CAS PubMed.
- G. Zhou, R. Dou, H. Bi, S. Xie, Y. Pei, K. Fan, M. Qiao, B. Sun and B. Zong, J. Catal., 2015, 332, 119–126 CrossRef CAS.
- J. J. M. Vequizo, H. Matsunaga, T. Ishiku, S. Kamimura, T. Ohno and A. Yamakata, ACS Catal., 2017, 7, 2644–2651 CrossRef CAS.
- Y. Zhang, X. Liu, Y. Fan, X. Guo, L. Zhou, Y. Lv and J. Lin, Nanoscale, 2016, 8, 15281–15287 RSC.
- L. Li, C. Cui, H. Fan and R. Wang, ChemSusChem, 2017, 10, 4921–4926 CrossRef CAS PubMed.
- L. He, F. Weniger, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 12582–12594 CrossRef CAS PubMed.
- Y. Shiraishi, D. Tsukamoto, Y. Sugano, A. Shiro, S. Ichikawa, S. Tanaka and T. Hirai, ACS Catal., 2012, 2, 1984–1992 CrossRef CAS.
- Z. Zheng, F. Zhuge, Y. Wang, J. Zhang, L. Gan, X. Zhou, H. Li and T. Zhai, Adv. Funct. Mater., 2017, 27, 1703115 CrossRef.
- Y. Li, H. Zhong, R. Li, Y. Zhou, C. Yang and Y. Li, Adv. Funct. Mater., 2006, 16, 1705–1716 CrossRef CAS.
- W. Zhou, W. Li, J. Q. Wang, Y. Qu, Y. Yang, Y. Xie, K. Zhang, L. Wang, H. Fu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 9280–9283 CrossRef CAS PubMed.
- G. Liu, L. C. Yin, J. Pan, F. Li, L. Wen, C. Zhen and H. M. Cheng, Adv. Mater., 2015, 27, 3507–3512 CrossRef CAS.
- X. Lang, W. R. Leow, J. Zhao and X. Chen, Chem. Sci., 2015, 6, 1075–1082 RSC.
- C. Su, R. Tandiana, B. Tian, A. Sengupta, W. Tang, J. Su and K. P. Loh, ACS Catal., 2016, 6, 3594–3599 CrossRef CAS.
- S. Sarina, H. Zhu, E. Jaatinen, Q. Xiao, H. Liu, J. Jia, C. Chen and J. Zhao, J. Am. Chem. Soc., 2013, 135, 5793–5801 CrossRef CAS.
- P. Zhang, P. Wu, S. Bao, Z. Wang, B. Tian and J. Zhang, Chem. Eng. J., 2016, 306, 1151–1161 CrossRef CAS.
- W. Zhou, Y. Guan, D. Wang, X. Zhang, D. Liu, H. Jiang, J. Wang, X. Liu, H. Liu and S. Chen, Chem. – Asian J., 2014, 9, 1648–1654 CrossRef CAS.
- X. Yang, H. Zhao, J. Feng, Y. Chen, S. Gao and R. Cao, J. Catal., 2017, 351, 59–66 CrossRef CAS.
- W. Schottky, Z. Phys., 1939, 113, 367–414 CrossRef CAS.
- A. Furube, L. Du, K. Hara, R. Katoh and M. Tachiya, J. Am. Chem. Soc., 2007, 129, 14852–14853 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ee02727g |
| This journal is © The Royal Society of Chemistry 2019 |