Highly active atomically dispersed CoN4 fuel cell cathode catalysts derived from surfactant-assisted MOFs: carbon-shell confinement strategy†
Received
14th September 2018
, Accepted 23rd October 2018
First published on 23rd October 2018
Abstract
Development of platinum group metal (PGM)-free catalysts for oxygen reduction reaction (ORR) is essential for affordable proton exchange membrane fuel cells. Herein, a new type of atomically dispersed Co doped carbon catalyst with a core–shell structure has been developed via a surfactant-assisted metal–organic framework approach. The cohesive interactions between the selected surfactant and the Co-doped zeolitic imidazolate framework (ZIF-8) nanocrystals lead to a unique confinement effect. During the thermal activation, this confinement effect suppressed the agglomeration of Co atomic sites and mitigated the collapse of internal microporous structures of ZIF-8. Among the studied surfactants, Pluronic F127 block copolymer led to the greatest performance gains with a doubling of the active site density relative to that of the surfactant-free catalyst. According to density functional theory calculations, unlike other Co catalysts, this new atomically dispersed Co–N–C@F127 catalyst is believed to contain substantial CoN2+2 sites, which are active and thermodynamically favorable for the four-electron ORR pathway. The Co–N–C@F127 catalyst exhibits an unprecedented ORR activity with a half-wave potential (E1/2) of 0.84 V (vs. RHE) as well as enhanced stability in the corrosive acidic media. It also demonstrated high initial performance with a power density of 0.87 W cm−2 along with encouraging durability in H2–O2 fuel cells. The atomically dispersed Co site catalyst approaches that of the Fe–N–C catalyst and represents the highest reported PGM-free and Fe-free catalyst performance.
Broader context
To enable large-scale commercialization of proton exchange membrane fuel cells (PEMFCs), low-cost yet high-performance cathode catalysts for the sluggish oxygen reduction reaction (ORR) are urgently needed. Among the platinum group metal (PGM)-free catalysts explored, Fe–N–C catalysts have demonstrated excellent activity. However, improved durability is desperately needed. One of likely reasons causing the catalyst stability issue is due to the attack by H2O2 (and/or derived free radicals). To minimize the formation of hydroperoxyl radicals from the Fenton reactions (Fe2+ + H2O2), it is desirable to develop ORR cathode catalysts which are both platinum group metals (PGM)-free and Fe-free, such as Co-based catalysts. This work reports an effective strategy to significantly enhance the ORR activity of atomically dispersed Co–N–C catalysts by using surfactant capping onto metal–organic framework (MOF) precursors, which can extend to other catalyst systems for electrochemical energy conversion.
|
Introduction
Proton exchange membrane fuel cells (PEMFCs) hold great promise for energy conversion due to their high power density, low operating temperature, and carbon-free emissions.1,2 However, large-scale application of PEMFCs has been greatly hampered by high cost and insufficient durability of platinum group metal (PGM) cathode catalysts for the oxygen reduction reaction (ORR). Therefore, the replacement of PGM catalysts with highly active, inexpensive, and durable PGM-free catalysts made from earth-abundant materials is desirable to promote the widespread application of PEMFCs.3–6 Although tremendous progress has been made in PGM-free catalysts, practical implementation still faces great challenges in acidic media due to the insufficient kinetics and material instability.7,8 The most promising class of catalysts investigated so far is transition metal and nitrogen co-doped carbon catalysts (M–N–C, M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Fe, Co, Ni, Mn, etc.) synthesized through a high temperature approach by pyrolyzing metals, nitrogen, and carbon precursors. Such-synthesized M–N–C catalysts have exhibited promising activity and selectivity towards ORR in acid media,9–11 following the order of Fe > Co > Cu > Mn > Ni.12–14 Recently, Fe–N–C catalysts have demonstrated significantly improved activity approaching that of Pt/C in challenging acidic media.3,5,11,15–18 Unfortunately, Fe-based catalysts suffer from poor stability, challenged by insufficient understanding of their degradation mechanisms during fuel cell operation.19–22 The possible degradation may be attributed to (1) leaching of the nonprecious metal sites from catalysts,19,21,23,24 (2) the attack by H2O2 (and/or free radicals),20 and (3) protonation of the active site or adjacent N dopants followed by anion adsorption.25 Recently, Dodelet et al., proposed a new mechanism concerning micropore flooding as an explanation for the rapid initial performance loss.22 In addition, a deactivation mechanism has been verified by Jaouen et al., providing new insights that durable Fe–N–C catalysts can be retained in PEMFCs if rational strategies to minimize the amount of H2O2 or reactive oxygen species produced during the ORR are developed.26 Thus, the possible Fenton's reactions between Fe and H2O2, which generate hydroxyl and hydroperoxyl radical species, are likely one of reasons causing the degradation of current Fe–N–C catalysts along with the degradation of organic ionomers within the electrodes and the membranes in PEMFCs.26,27 Thus, it is essential to develop Fe-free catalysts to completely address the Fenton reaction issue and to acquire the understanding of the degradation mechanisms. Alternatively, Co-based catalysts would have far less deleterious effects in this regard and appear to be the ideal candidate for Pt-free and Fe-free catalysts.12,13,28–30 When compared to the Fe–N–C catalysts,15 current Co–N–C ones have far less activity and higher yield of H2O2 during the ORR in acids,28,29,31–39 which still requires significant effort to improve their performance.
Fe, Co, Ni, Mn, etc.) synthesized through a high temperature approach by pyrolyzing metals, nitrogen, and carbon precursors. Such-synthesized M–N–C catalysts have exhibited promising activity and selectivity towards ORR in acid media,9–11 following the order of Fe > Co > Cu > Mn > Ni.12–14 Recently, Fe–N–C catalysts have demonstrated significantly improved activity approaching that of Pt/C in challenging acidic media.3,5,11,15–18 Unfortunately, Fe-based catalysts suffer from poor stability, challenged by insufficient understanding of their degradation mechanisms during fuel cell operation.19–22 The possible degradation may be attributed to (1) leaching of the nonprecious metal sites from catalysts,19,21,23,24 (2) the attack by H2O2 (and/or free radicals),20 and (3) protonation of the active site or adjacent N dopants followed by anion adsorption.25 Recently, Dodelet et al., proposed a new mechanism concerning micropore flooding as an explanation for the rapid initial performance loss.22 In addition, a deactivation mechanism has been verified by Jaouen et al., providing new insights that durable Fe–N–C catalysts can be retained in PEMFCs if rational strategies to minimize the amount of H2O2 or reactive oxygen species produced during the ORR are developed.26 Thus, the possible Fenton's reactions between Fe and H2O2, which generate hydroxyl and hydroperoxyl radical species, are likely one of reasons causing the degradation of current Fe–N–C catalysts along with the degradation of organic ionomers within the electrodes and the membranes in PEMFCs.26,27 Thus, it is essential to develop Fe-free catalysts to completely address the Fenton reaction issue and to acquire the understanding of the degradation mechanisms. Alternatively, Co-based catalysts would have far less deleterious effects in this regard and appear to be the ideal candidate for Pt-free and Fe-free catalysts.12,13,28–30 When compared to the Fe–N–C catalysts,15 current Co–N–C ones have far less activity and higher yield of H2O2 during the ORR in acids,28,29,31–39 which still requires significant effort to improve their performance.
Metal–organic frameworks (MOF), especially zeolitic-imidazole frameworks (ZIFs), have been used as precursors to synthesize atomically dispersed Co–N–C catalysts, due to their unique capability to form a large number of CoNx active sites and yield porous structures through a thermal activation.37,40 The resultant catalysts have exhibited well-dispersed atomic CoNx active sites, which correlated with good catalytic activity. However, there is still a significant activity gap between the Co–N–C and Pt/C catalysts, such as at least 60 mV of half-wave potential in acidic aqueous electrolytes.37 Simply increasing Co metal content in the precursors is found to be ineffective, because it gives rise to severe aggregation of Co metal during high-temperature treatments.41 Thus, new strategies to effectively control the synthesis of CoNx active sites with high density are extremely desirable, but very challenging. Here, we develop an innovative surfactant-assisted MOF approach to preparing core–shell structured Co–N–C catalysts, which was inspired by the strong interactions between surfactants and nanocrystal particles in solution phases.42–44 Due to the confinement role of surfactants covering onto the ZIF-8 nanocrystals, core–shell structured and atomically dispersed Co–N–C@surfactant catalysts with significantly increased active site density were obtained versus other synthetic routes. Among the surfactants studied, F127 block copolymer (PEO100-PPO65-PEO100) was explored to identify as the optimal surfactant. Advanced electron microscopy and X-ray absorption fine structure (XAFS) measurements show that the CoN4 are atomically dispersed and are more abundant in the catalysts synthesized using surfactant F127 versus those synthesized without surfactant or other surfactants. The Co–N–C@F127 catalyst exhibits exceptionally enhanced ORR activity with a half-wave potential (E1/2) of 0.84 V (vs. RHE) in acidic electrolyte. To the best of our knowledge, this ORR activity exceeds those values of any previously-known PGM-free and Fe-free catalysts (see Table S1, ESI†) and is comparable to that of the state-of-the-art Fe–N–C catalysts.15 Density functional theory (DFT) calculations were used to elucidate the chemical nature of the active sites capable of catalyzing the ORR via four electrons (4e−) pathway. Fuel cell tests further confirm that the Co–N–C@F127 catalyst can perform as an efficient cathode in PEMFCs. Thus, exploration of alternative high-performance Co–N–C catalysts would provide more insightful understanding on degradation mechanisms and open a new avenue to design advanced PGM-free catalysts for vital applications in PEMFCs.
Results and discussion
Catalyst synthesis and morphology
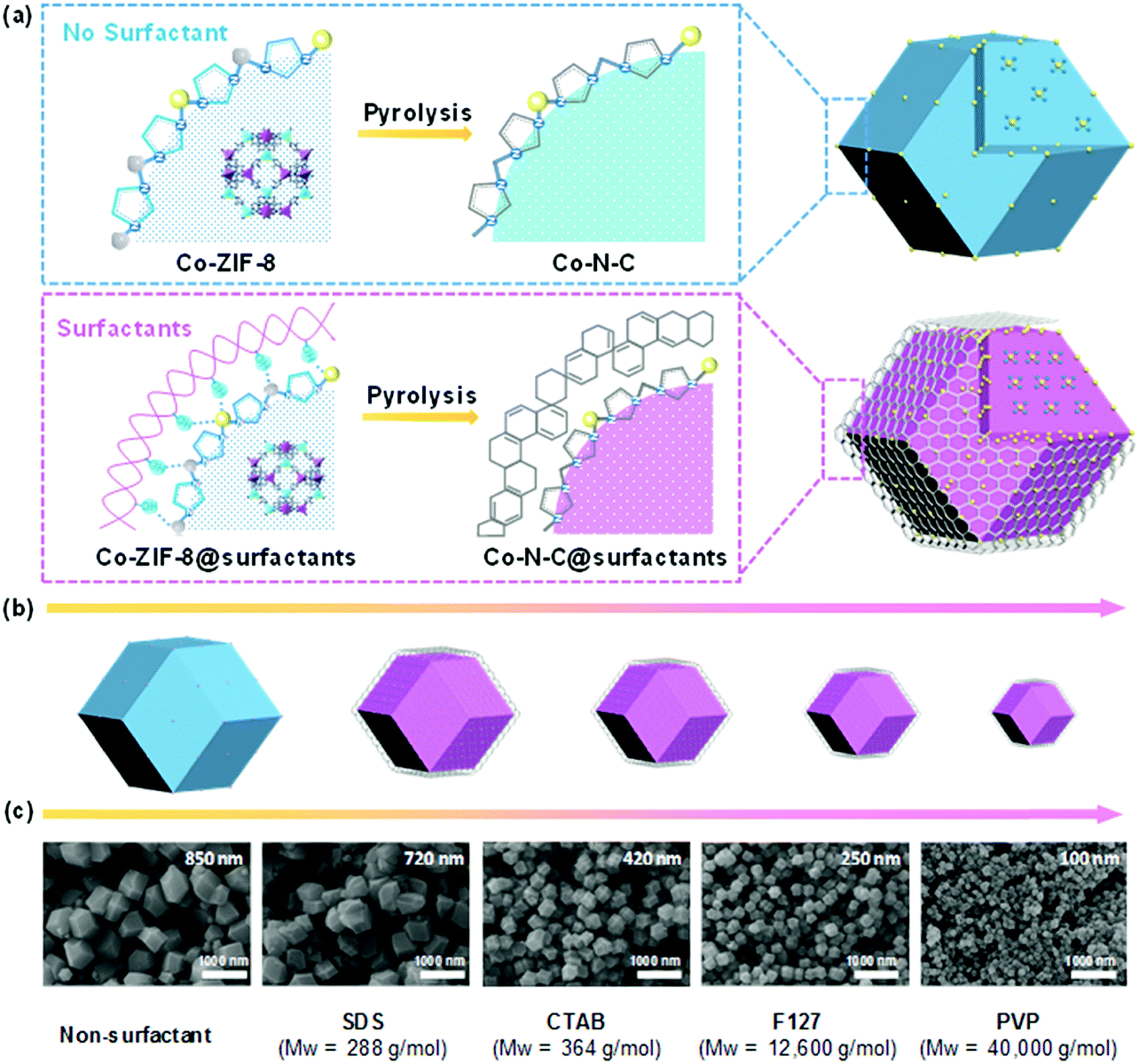
The synthesis procedures for the core–shell structured atomically dispersed Co–N–C catalysts are illustrated in Fig. 1a. The experimental details are given in the ESI.† It was initially driven by the organometallic reaction of Zn2+/Co2+ ions and 2-methylimidazole ligands to form Co doped ZIF-8 nanocrystals, then surfactants were added as the capping agents to regulate the crystallization. The surface of Co-ZIF-8 polyhedrons has abundant Zn2+ and Co2+ sites, which can be easily coordinated with the hydrophilic groups of the surfactants. This coordination effect can slow down the crystal growth rate and control the crystal size and morphology of Co-ZIF-8 crystals.45,46 During the subsequent pyrolysis, it can be speculated that the surfactant layers are the first one to carbonize, forming a carbon shell, coating on the Co-ZIF-8 polyhedrons.47 With increasing temperatures, the Co-ZIF-8 polyhedrons begin to carbonize. The strong cohesive interface interaction leads to a significant confinement effect, thus avoiding the collapse of the internal microporous carbon structures derived from Co-ZIF-8 polyhedrons while also mitigating the agglomeration of neighboring Co single atomic sites.48 As a result, the Co–N–C@surfactant catalyst has an abundantly-microporous structure and high density of CoN4 atomic sites. Four types of surfactants (e.g., anionic surfactant SDS, cationic surfactant CTAB, non-ionic triblock copolymer F127, and non-ionic surfactant PVP) were examined in terms of their effectiveness to tune catalyst morphologies and properties. The molecular formula of these surfactants are shown in Fig. S1 (ESI†). Typically, surfactant-free Co-ZIF-8 particles had a non-uniform rhombododecahedral shape with an average size of 850 nm, yet there were some particles with smaller sizes of approximately 100 nm (Fig. S2, ESI†). Due to the capping ability of different surfactants, the particle sizes of the Co-ZIF-8@suffactant particles proportionally decreased from 850 to 100 nm when molecular weights of the surfactants increase (Fig. 1b). In addition, all the Co-ZIF-8@surfactant nanocrystals presented uniform particle sizes. This is a result that the surfactants formed the micelles which evenly dispersed zinc and cobalt ions in the methanol solution.49 Although the use of PVP resulted in the smallest size of catalysts (down to 100 nm), particle fusing was observed. As for the Co-ZIF-8@F127 catalyst, nanoparticles showed isolated dispersion with an average diameter of 250 nm (Fig. S3, ESI†). High-resolution transmission electron microscopy (HR-TEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images were employed to further reveal the detailed structures and morphologies. Fig. 2 compares the surfactant-free Co-ZIF-8 and the Co-ZIF-8@F127 nanocrystal precursors and their corresponding catalysts after thermal activations. Compared to the rhombododecahedral shape of the Co-ZIF-8 precursor (Fig. 2A and Fig. S4, ESI†), a core–shell structure is observed in the Co-ZIF-8@F127 precursor (Fig. 2B and Fig. S5, ESI†), which consisted of a Co-ZIF-8 nanocrystal core and a surfactant F127 polymer shell. STEM-EDS elemental mapping results in Fig. 2 indicate that C, N, and Co are uniformly dispersed into the precursors regardless of the addition of the F127 surfactant. Fig. S6 (ESI†) shows X-ray diffraction (XRD) patterns of the Co-ZIF-8 and Co-ZIF-8@F127 precursors. Their diffraction patterns are identical, indicating that the addition of surfactants and the doping of Co ions did not influence the crystalline structure of the ZIF-8.
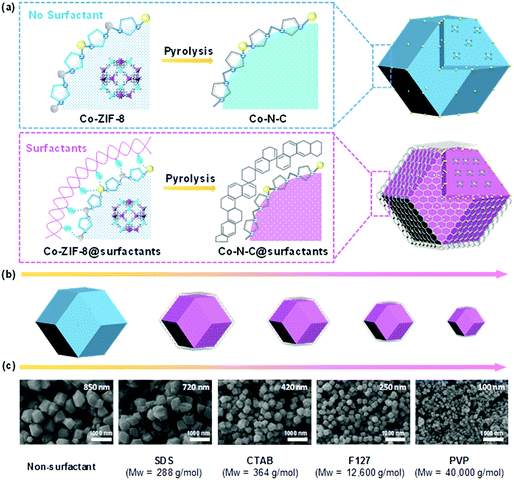 |
| | Fig. 1 (a) Proposed in situ confinement pyrolysis strategy to synthesize core–shell-structured Co–N–C@surfactants catalysts with increased active site density (The yellow, grey and blue balls represent Co, Zn and N atoms, respectively.). (b) Expected particle sizes and (c) SEM images to show the changes in the size and morphology of the catalysts with varying surfactants including SDS, CTAB, F127 and PVP. | |
 |
| | Fig. 2 HRTEM, HAADF-STEM and STEM-EDS elemental mappings for (A) surfactant-free Co-ZIF-8 precursor, (B) Co-ZIF-8@F127 precursor, (C) surfactant-free Co–N–C catalyst and (D) Co–N–C@F127 catalyst. | |
After a thermal treatment at 900 °C, the size and shape of the various Co–N–C catalysts were similar to their corresponding Co-ZIF-8 crystal precursors (Fig. S7, ESI†). The hydrocarbon networks in all Co-ZIF-8 crystals were completely carbonized as evidenced in the XRD patterns showing dominant peaks at 25° and 44° for the (002) and (101) planes of carbon, respectively (Fig. S8, ESI†). Based on HR-TEM and HAADF-STEM images (Fig. 2C and D), the carbon networks in both Co–N–C and Co–N–C@F127 catalysts were highly disordered due to the doping of the heteroatoms N, which led to the turbostratic stacking of graphite planes.50 Notably, the Co–N–C@F127 exhibited a typical core–shell structure, in which the core was derived from the Co-doped ZIF-8 nanocrystal and the carbon shell was from the surfactant F127 layers (Fig. 2D).51 The partially graphitized carbon shells could be clearly observed at the edge of the polyhedron, attributable to the graphitization of F127. Compared to the carbon structures in the shells, the carbon cores derived from the ZIF-8 precursors seems more amorphous and porous. Raman spectra in Fig. S9 and Table S2 (ESI†) indicate that, regardless of the type of surfactants, all the catalysts exhibited similar carbon structures with dominant D and G bands at 1350 and 1585 cm−1, associated with the disordered carbons and sp2 hybridized graphitic carbons, respectively.52 Among the samples studied, Co–N–C@F127 demonstrates a relatively high ID/IG value of 1.52, suggesting the largest amounts of defects. This finding is in good agreement with the X-ray photoelectron spectroscopy (XPS) results, discussed in detail later, which show this catalyst containing the highest content of N and Co dopants.50
The porosity of Co–N–C@surfactant nanocrystals prepared with different surfactants was quantified by N2 adsorption–desorption measurements (Fig. S10 and S11, ESI†). The surfactant-free Co–N–C catalyst revealed a type IV sorption isotherm. The increased adsorption volume at a low relative pressure indicates the existence of micropores, while the distinct hysteresis loop in the range of P/P0 0.4–0.8 is indicative of a mesoporous feature.16 In contrast, the isotherms of Co–N–C@F127 porous carbon revealed a type I sorption isotherm, an indication of the existence of only micropores in the absence of mesopores. The BET specific surface area and the total pore volume of the Co–N–C@F127 catalyst are 825 m2 g−1 and 0.54 cm3 g−1, respectively (Table S3, ESI†), which are much higher than those of the surfactant-free Co–N–C catalyst (324 m2 g−1 and 0.235 cm3 g−1). The comparison implies that direct carbonization of the Co-ZIF-8 without protection from F127 most likely led to the significant obstruction and the collapse of micropores within ZIF-8. It has been postulated that ORR active sites in M–N–C catalysts are likely located inside or around the micropores of carbon phases.53 Therefore, the richness of micropores in the Co–N–C@F127 catalyst is beneficial for accommodating a high density of active sites.
High-density atomically dispersed CoN4 sites
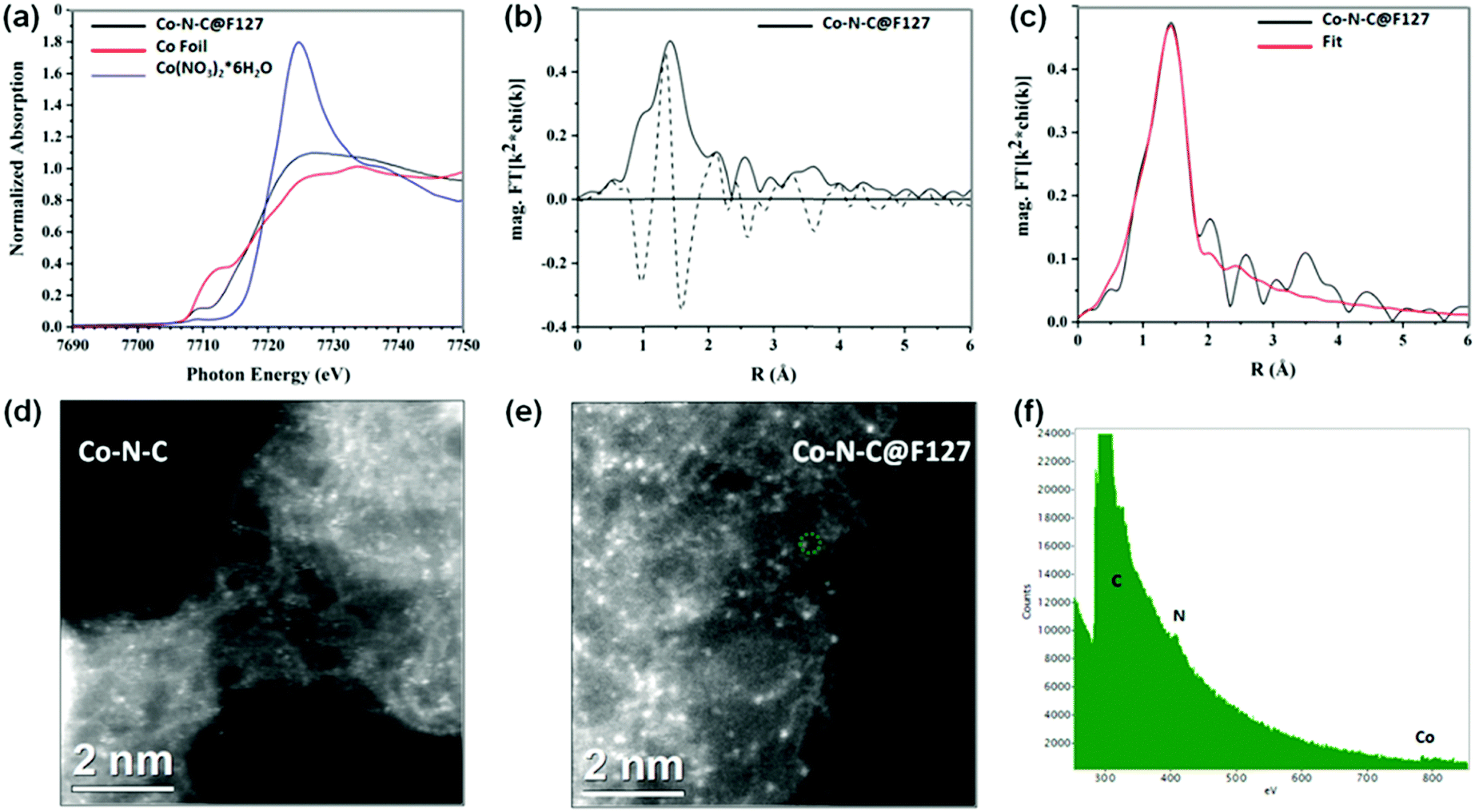
The N-coordinated Co sites, whose local coordination environments are similar to those of the CoN4 in Co-porphyrin structures, have been predicted as the possible ORR active sites in Co-based catalysts.54 To further determine the local chemical bonding of Co atoms in the Co–N–C@F127 catalyst, the catalyst was analyzed using Co K-edge X-ray absorption spectroscopy encompassing both the near-edge and extended energy regions, XANES and EXAFS, respectively. The results are presented in Fig. 3a–c. The increased XANES white line intensity of Co–N–C@F127 compared to the reference cobalt foil is consistent with the sample containing oxidized Co and the similar pre-edge energy of the catalyst and the Co(NO3)2 reference (Table S4, ESI†) indicates the Co is in the 2+ oxidation state. The pre-edge feature in the Co K-edge XANES spectrum (∼7708–7711 eV) arises from the a 1s to 3d transition and in general the intensity of this peak in the K-edge spectra of 3d metals is related to the extent of 3d–4p mixing, which increases with decreasing centrosymmetry of the metal coordination environment.55 The pre-edge peak intensity for the catalyst compared to that of the octahedral Co(NO3)2 reference suggests that the coordination of Co in the catalyst is less centrosymmetric than the octahedral complex (Fig. 3a). The magnitude and imaginary part of the Fourier transform of the k2-weighted EXAFS of Co–N–C@F127 are shown in Fig. 3b. A peak arising from light scattering (C/N/O) nearest-neighbors is observed at 1.6 Å (phase uncorrected distance) and was fit using a single Co–N scattering path.11,56 The fitted coordination number of 3.6 ± 0.6 is in agreement with the XANES with Co having a tetrahedral geometry. The Co–N bond length derived from the EXAFS fit was 1.94 ± 0.02 Å. No evidence of Co–Co scattering is observed in the EXAFS of the Co–N–C/F127 catalyst, which is consistent with an atomically dispersed species. Moreover, HAADF-STEM imaging coupled with electron energy loss spectroscopy (EELS) were performed. As depicted in Fig. 3d–f, well-dispersed isolated Co atomic sites are clearly observed in both the Co–N–C and the Co–N–C@F127 catalysts, located at both the edge sites and in the carbon matrix. EELS point spectra taken by focusing the electron beam on the bright dot in the HAADF-STEM image (green circle) in Fig. 3e and f indicates that Co atoms and N co-exist in the form of CoNx. This atomic level spectroscopic analysis agrees with the results from XANES and EXAFS, verifying that well-dispersed atomic Co sites are coordinated with N. Although it is challenging to obtain accurate sample composition via the EELS overall composition analysis, there is a clear tendency of more Co (perhaps 3× as much), as well as more N (2×) and slightly more O in the Co–N–C@F127 catalysts as compared to those in the surfactant-free Co–N–C catalyst (Tables S5–S8, ESI†). This suggests significantly increased density of CoN4 active sites due to the confined role of using the F127 surfactant.
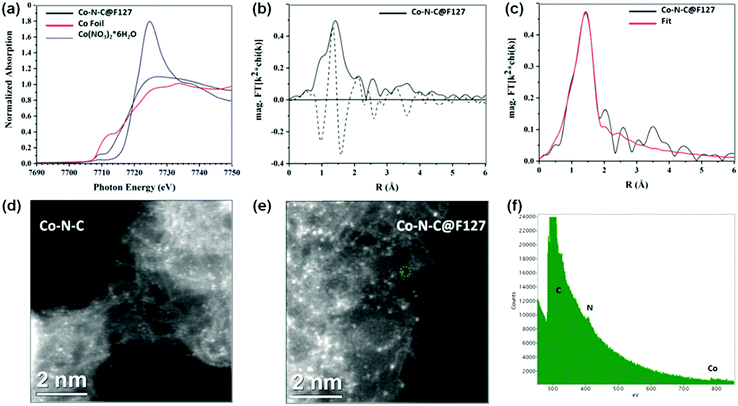 |
| | Fig. 3 (a) Co K edge XANES spectra. (b) The magnitude (solid black) and imaginary part (dashed black) of the Fourier transform of the k2-weighted EXAFS. (c) Fit of the magnitude of the Fourier transform of the k2-weighted EXAFS (data-black and fit-red) for the Co–N–C@F127 catalyst. Aberration-corrected HAADF-STEM images with accompanying EELS point spectra of (d) Co–N–C and (e and f) Co–N–C@F127 catalyst. | |
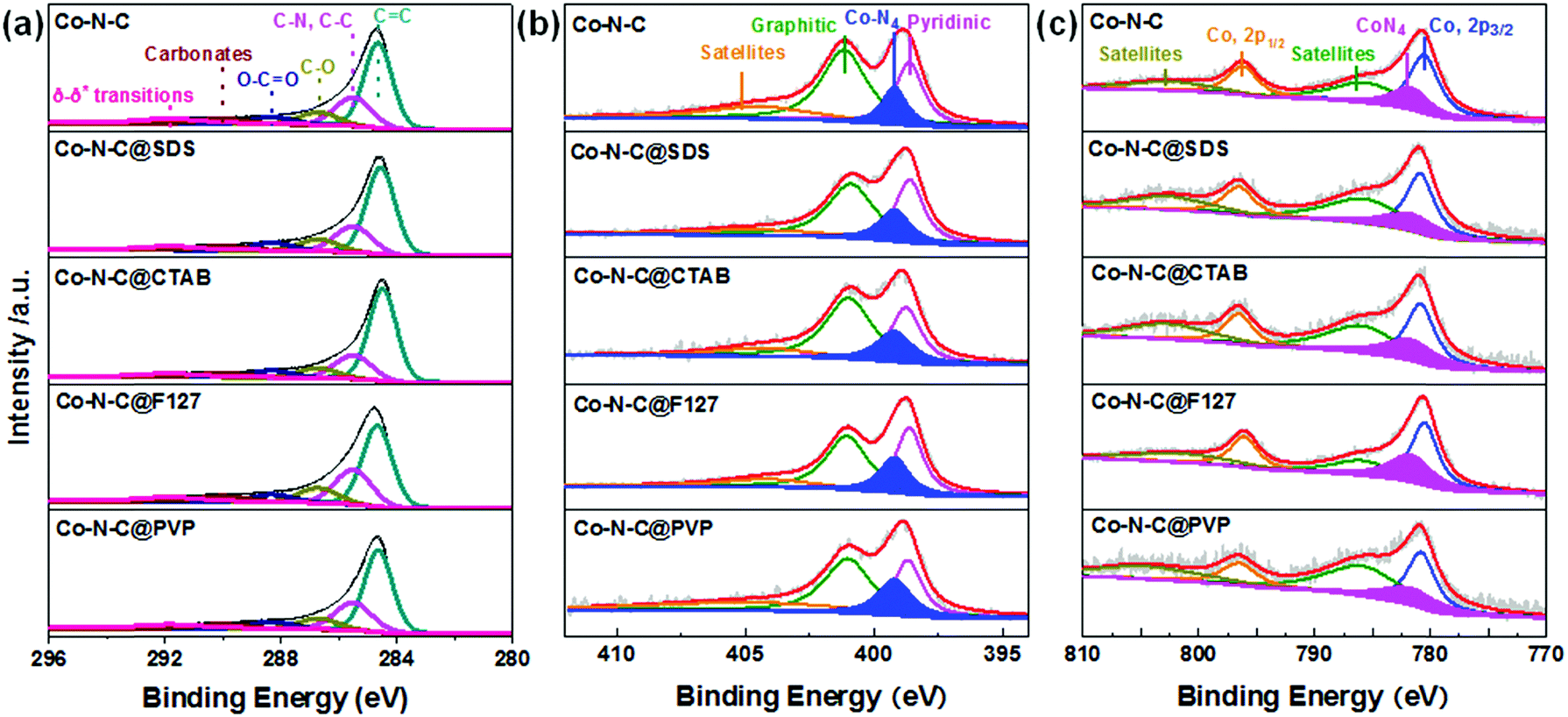
Elemental analysis and XPS measurements were conducted to probe the content and chemical composition of C, N, and Co in the surface layers (less than 10 nm) of these Co–N–C catalysts. With the addition of the surfactant F127, the content of Co is slightly increased from 0.9 at% for the surfactant-free Co–N–C to 1.0 at% for the Co–N–C@F127 (Table S7, ESI†). For other surfactants derived Co–N–C catalysts, they showed similar Co content. Since Co atoms under the porous carbon shells could not be accurately detected by using XPS, the Co content of the catalysts was further analyzed by X-ray fluorescence (XRF) and inductively coupled plasma atomic emission spectroscopy (ICP-AES) (Table S8, ESI†). These results also indicated that the Co content in the Co–N–C@F127 catalyst is the highest than any other catalysts, suggesting the beneficial role of F127 in maintaining more atomic Co sites within the carbon framework during pyrolysis. The samples obtained from different surfactants exhibits similar C 1s XPS spectra (Fig. 4a and Table S9, ESI†). The Co 2p3/2 (780.5 eV) and Co 2p1/2 (795.6 eV) peaks, as shown in Fig. 4b and Fig. S10 (ESI†), are typical features of Co2+ species,57,58 consistent with the XANES results. The N 1s XPS for all the samples (Fig. 4c and Fig. S11, ESI†) reveal three main components including pyridinic-N (398.4 eV), graphitic-N (401.1 eV), and oxidized graphitic-N (403–405 eV). The Co–N–C@F127 catalyst displays a significantly increased N content of 9.1 at% with respect to the 6.1 at% N for the surfactant-free Co–N–C catalyst and others. It should be noted that the N content reaches to the highest level compared to all of other such-synthesized M–N–C catalysts through high temperature approaches.7,8,59 Such a high N content is attributed to the possible confinement pyrolysis effect of F127 to retain N during high-temperature pyrolysis, which is beneficial for the formation and retention of high-density CoN4 active sites. An additional peak at 399.2 eV is likely assigned to N atoms bonding to atomic Co sites in the form of CoN4.60 Among all the catalysts studied, the Co–N–C@F127 catalyst presents the highest intensity of this peak, implying the largest fraction of CoN4 sites.
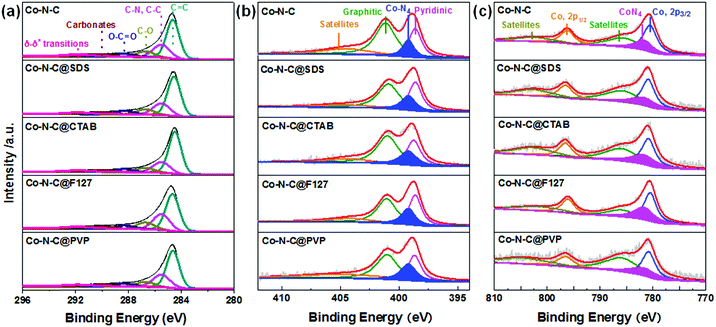 |
| | Fig. 4 XPS analysis of (a) C 1s, (b) N 1s and (c) Co 2p to elucidate the effect of surfactants for different catalysts including Co–N–C; Co–N–C@SDS, Co–N–C@CTAB; Co–N–C@F127 and Co–N–C@PVP catalysts. | |
Density functional theory (DFT) calculations
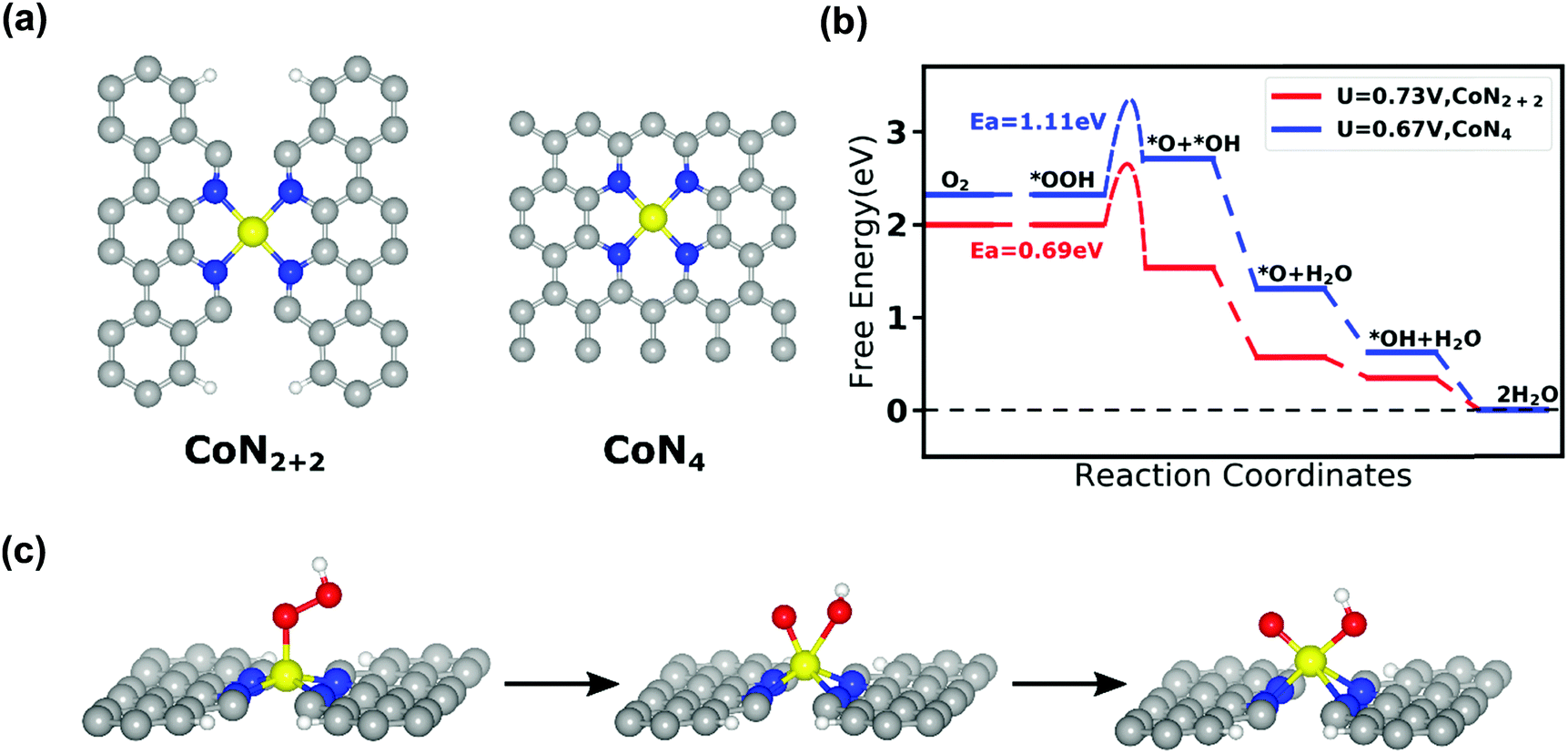
To theoretically evaluate the ORR activity and selectivity on the identified CoN4 sites in the Co–N–C catalysts, we performed first principles DFT calculations to study the adsorption energy, free energy evolution, and O–O bond breaking activation energy on possible CoNxCy active sites during the ORR. Based on the XANES analysis results (i.e., coordination number of N around Co is 3.6), we focused our present computational study on CoN4 (a Co–N4 moiety embedded in intact graphitic layer) and CoN2+2 (a Co–N4 moiety bridging over two adjacent armchair graphitic edge) sites, as depicted in Fig. 5a. From their optimized structures, the Co–N bond length is predicted to be 1.85 Å in the CoN4 site and 1.88 Å in the CoN2+2 site. Our theoretical prediction of the Co–N bond length is close to the value of 1.94 Å based on the aforementioned EXAFS analysis. It should be noted that these two types of the metal-N4 coordination geometries have been proposed as ORR active sites in Fe–N–C catalysts in a prior study.61 The four-electron (4e−) ORR on the Co–N–C catalysts starts with the adsorption of reactant O2 and ends with the release of product H2O. Consistent with this process, we have determined the adsorption configurations (Fig. S12, ESI†) and adsorption energy (Table S12, ESI†) of O2, OOH, O, OH, and H2O on the CoN4 and CoN2+2 sites. DFT results show that both CoN4 and CoN2+2 can bind O2 appropriately to initiate the ORR as well as bind H2O weakly to complete ORR. Moreover, we examined the thermodynamic free energy change for a 4e− ORR pathway, in which O2 molecule will be first adsorbed on the top of the central Co, then O2 will be protonated to form OOH, the OOH will dissociate into O and OH, and finally both O and OH will be pronated to form product H2O. Here, we employed the computational hydrogen electrode method developed by Norskov et al.62 and computed the free energies of every elementary steps as a function of electrode potential U with reference to reversible hydrogen electrode (RHE). Fig. 5b shows that the free energy change for these elementary ORR reactions on the CoN2+2 site become negative (i.e., exergonic reaction) when the electrode potential U is lower than a limiting potential of 0.73 V. Thus, the CoN2+2 site is predicted to be thermodynamically capable of catalyzing the 4e− ORR. In contrast, we predicted a free energy barrier of 0.39 eV for *OOH to dissociate to *O and *OH on the CoN4 site. This result suggests that it is thermodynamically difficult for the 4e− ORR to occur on this CoN4 site.
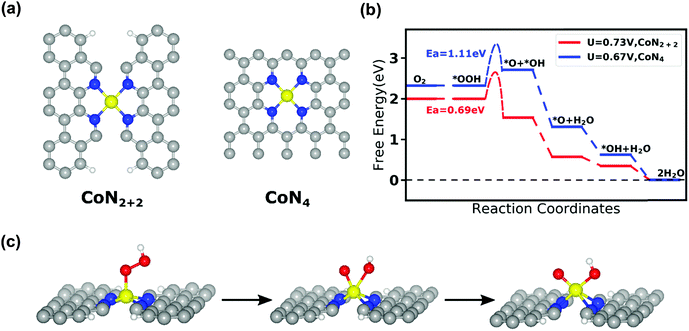 |
| | Fig. 5 (a) Atomistic structure of CoN2+2 and CoN4 active sites of the Co–N–C catalysts. (b) Calculated free energy evolution diagram for 4e− ORR pathway on the CoN2+2 site under a limiting electrode potential of U = 0.73 V and on CoN4 site under a limiting electrode potential of U = 0.67 V. (c) Atomistic structure of the initial state (left), transition state (middle), and final state (right) for OOH dissociation reaction on the CoN2+2 site. In this figure, the gray, blue, yellow, red and white balls represent C, N, Co, O, and H atoms, respectively. | |
In order to elaborate the ORR pathway on CoN4 and CoN2+2 sites, the climbing image nudged elastic band (CI-NEB) calculation was carried out63 to locate the transition state and predict the activation energies for the OOH dissociation reaction. This is the crucial step that breaks the O–O bond in the 4e− ORR pathway, on both the CoN4 and CoN2+2 sites. Fig. 5c shows the atomic details of this reaction on the CoN2+2 site. In the initial state, the OOH is adsorbed on the central Co atom; in the final state, both the dissociated O and OH are co-adsorbed on the central Co atom. Our DFT calculations indicate that the OOH dissociation reaction must overcome an activation energy of 0.69 eV on the CoN2+2 site and 1.11 eV on the CoN4 site (Fig. 5b). In comparison, our previous DFT study predicted the activation energy for the same OOH dissociation reaction to be 0.56 eV on FeN4 site, which is able to catalyze the 4e− ORR.64,65 Consequently, it can be inferred from these computational results that CoN2+2 can catalyze the 4e− ORR, similar to FeN4 whereas CoN4 cannot catalyze the 4e− ORR owing to the insurmountable activation energy for O–O bond breaking on this site.
Catalyst activity and stability
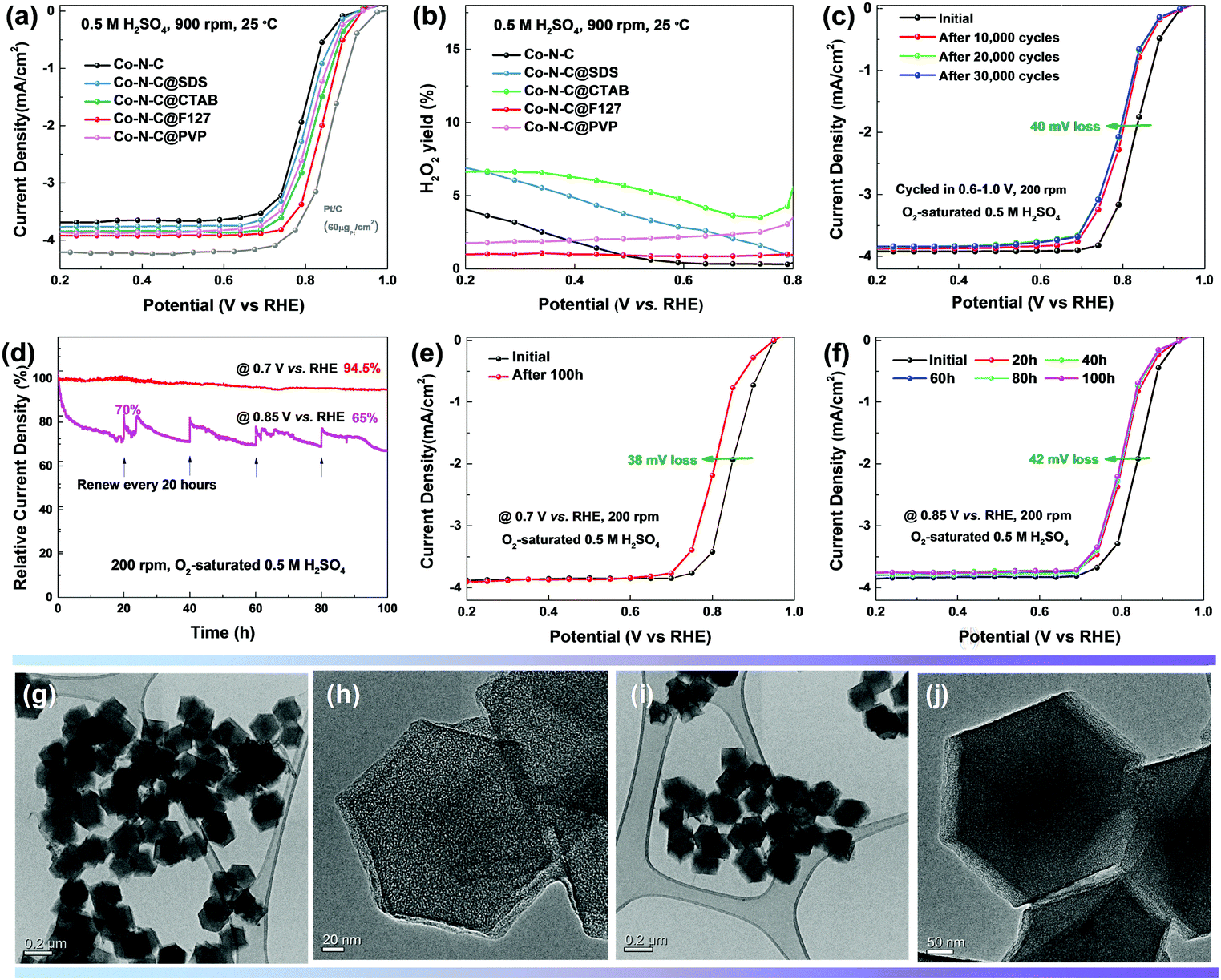
The oxygen reduction activity and four-electron selectivity (H2O2 yield) of various catalysts were evaluated in O2-saturated 0.5 M H2SO4 solution. Without Co doping, the N–C catalyst showed poor activity with a low onset potential (Eonset, defined as potential at which the current density reaches 0.1 mA cm−2) of 0.81 V and a half-wave potential (E1/2, defined as the potential at which the current reaches half the limiting current density) of 0.59 V vs. RHE (Fig. 6a and Fig. S13, ESI†). Co doping boosted the ORR performance, most likely associated with the formation of CoN4 sites which are more intrinsically active than the metal-free N activated C sites based on our theoretical predictions. The ORR activity was found to be dependent on the type of surfactants used. The highest ORR activity was measured for the Co–N–C@F127 catalyst, with a size of 250 nm, exhibiting an Eonset of 0.93 V and an E1/2 of 0.84 V vs. RHE. It should be noted that the correlation of doped Co content and the corresponding ORR activity follows a so-called “volcano plot” (Fig. S14, ESI†). Lower doping yields insufficient density of active sites, while higher doping leads to Co agglomeration and unfavorable carbon structures (i.e., less defect and porosity). This suggests that maximum atomic Co sites coordinated with N generate largest density of active sites corresponding to the best activity. This remarkable activity for the Co–N–C catalyst in acid is comparable to that of the state-of-the-art Fe–N–C (Eonset at 0.95 V and E1/2 at 0.85 V).15 This ORR activity in challenging acidic media is superior to that of previously known PGM-free and Fe-free catalysts (Table S1, ESI†), representing a new record. In addition, only negligible H2O2 was generated during the ORR on the Co–N–C@F127 catalyst (Fig. 6b), indicating a dominant 4e− reduction pathway. Combined with the DFT simulation results, it suggests that our synthesis method for Co–N–C catalysts increases the amount of CoN2+2 sites, which are on the edge of carbon layers and highly active for the 4e− ORR. The Co–N–C@F127 also demonstrates excellent stability during both potential cycling (0.6–1.0 V, 50 mV s−1) and holding at constant potentials (0.7 and 0.85 V for 100 hours) in O2-saturated 0.5 M H2SO4. There is a loss of only 40 mV in E1/2 after 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 potential cycles from 0.6 to 1.0 V (Fig. 6c). The corresponding CV profiles are compared in Fig. S15 (ESI†) during the potential cycling, indicating initial carbon oxidation during the first 10000 cycles in good agreement to the major loss occurred in the first 10000 cycles with a loss of 30 mV. This thus implies that the rapid degradation of unstable active sites is associated with the carbon oxidation at surfaces. The remaining active sites are more stable against degradation. Furthermore, constant potential tests were conducted for up to 100 hours at relatively high potentials of 0.7 and 0.85 V (Fig. 6d–f), respectively. Retention of initial activity up to 94.5% and 65% was determined at both potentials, respectively. At 0.85 V, the significant loss occurred at the initial 20 hours, which is good agreement with the potential cycling tests. To further elucidate the possible degradation likely due to carbon corrosion, the samples after the 30000 potential cycles and the 100 hour 0.85 V potential holds tests were analyzed by STEM imaging (Fig. 6g–j, Fig. S16 and S17, ESI†). The catalyst particles became rough at the surface after two long-term stability tests, which is probably associated with the initial activity loss. Overall, the carbon particle morphologies and structures were maintained and were similar to that observed in the pre-test catalyst samples. The observed structural stability may be attributed to the protective role of graphitized carbon layers,66,67 which prevents the continuous corrosion and oxidization from the external acidic environment of the electrolyte.
000 potential cycles from 0.6 to 1.0 V (Fig. 6c). The corresponding CV profiles are compared in Fig. S15 (ESI†) during the potential cycling, indicating initial carbon oxidation during the first 10000 cycles in good agreement to the major loss occurred in the first 10000 cycles with a loss of 30 mV. This thus implies that the rapid degradation of unstable active sites is associated with the carbon oxidation at surfaces. The remaining active sites are more stable against degradation. Furthermore, constant potential tests were conducted for up to 100 hours at relatively high potentials of 0.7 and 0.85 V (Fig. 6d–f), respectively. Retention of initial activity up to 94.5% and 65% was determined at both potentials, respectively. At 0.85 V, the significant loss occurred at the initial 20 hours, which is good agreement with the potential cycling tests. To further elucidate the possible degradation likely due to carbon corrosion, the samples after the 30000 potential cycles and the 100 hour 0.85 V potential holds tests were analyzed by STEM imaging (Fig. 6g–j, Fig. S16 and S17, ESI†). The catalyst particles became rough at the surface after two long-term stability tests, which is probably associated with the initial activity loss. Overall, the carbon particle morphologies and structures were maintained and were similar to that observed in the pre-test catalyst samples. The observed structural stability may be attributed to the protective role of graphitized carbon layers,66,67 which prevents the continuous corrosion and oxidization from the external acidic environment of the electrolyte.
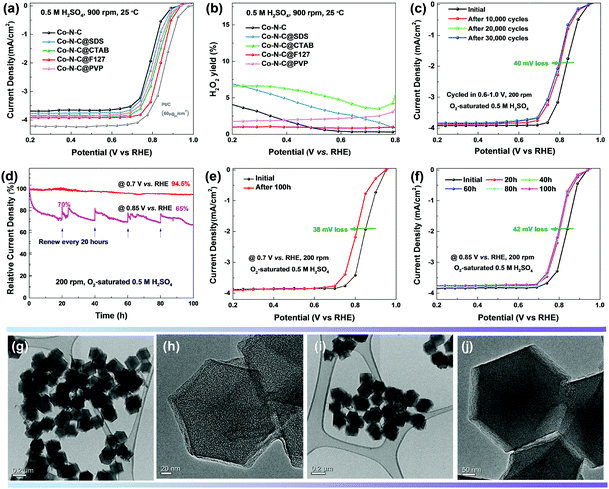 |
| | Fig. 6 (a) ORR polarization plots and (b) calculated H2O2 yield for different Co-ZIF-8@surfactants derived catalysts in 0.5 M H2SO4 at 25 °C and at 900 rpm. (c) Potential cycling (0.6–1.0 V) stability test of best Co–N–C@F127 catalysts in O2-saturated 0.5 M H2SO4 (the Pt/C catalyst was tested in 0.1 M HClO4) (d) 100 h chronoamperometry tests at (e) 0.7 V and (f) 0.85 V, respectively. Bright field and high resolution TEM images of Co–N–C@F127 catalysts (g and h) after 100 h chronoamperometry test at 0.85 V and (i and j) after 30000 cycling (0.6–1.0 V) stability test. | |
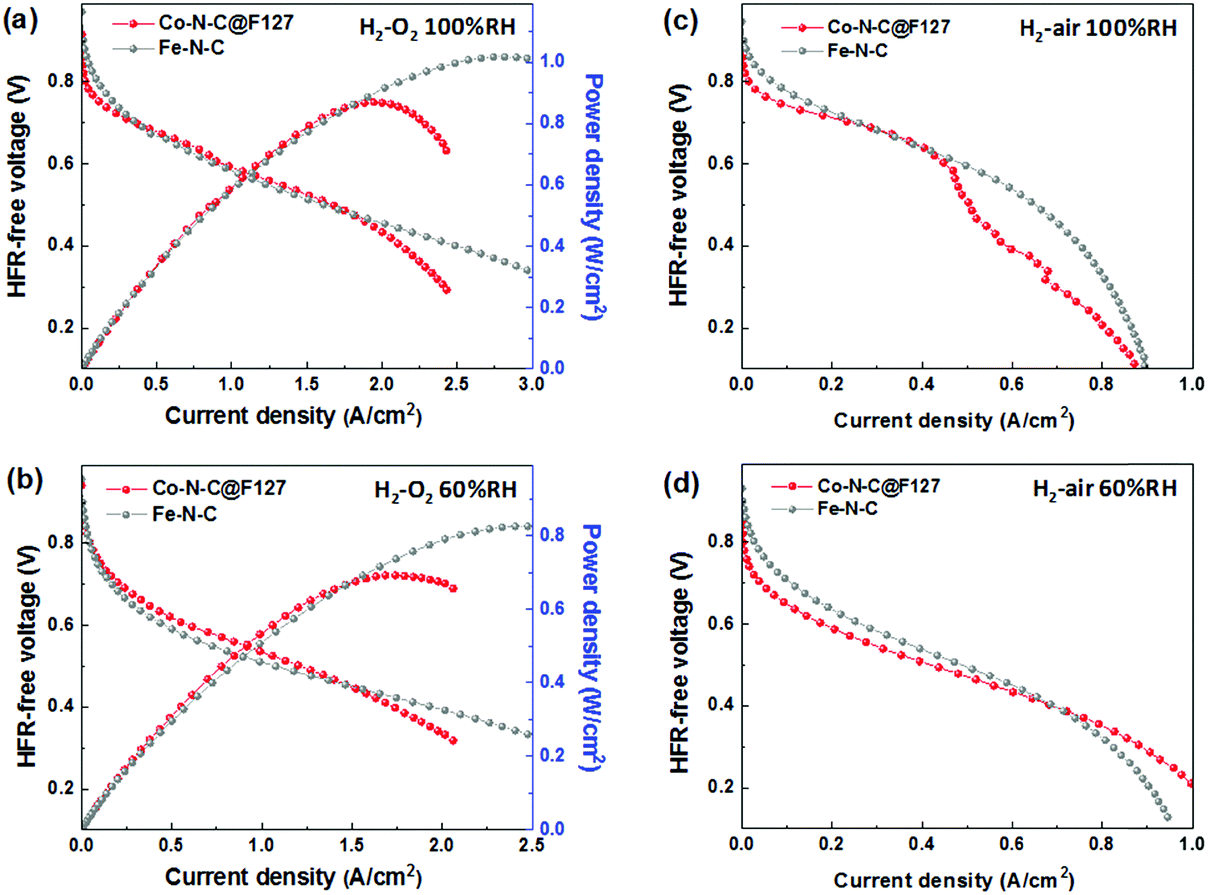
To examine the effectiveness of the Co–N–C@F127 catalyst as a practical PGM-free cathode in PEMFCs, the catalyst was incorporated into membrane-electrode assemblies (MEAs) with a total catalyst loading of 4.0 mg cm−2. When H2/O2 at 1.0 bar pressure was used (Fig. 7a, b and Fig. S18, ESI†), the cell exhibited an open-circuit voltage of 0.92 V and generated current densities of 30 mA cm−2 at 0.8 V and 2.2 A cm−2 at 0.4 V. It should be noted that, when a relative humidity (RH) of 100% was applied, the performance at high voltages (>0.7 V) was still inferior to that of the Fe–N–C catalysts. However, at moderate voltages (0.5–0.7 V) typical of PEMFC operation, the Co–N–C@F127 was able to generate comparable performance showing a high power density of 0.87 W cm−2. At a low RH (60%), the Co–N–C@F127 cathode exhibited slightly higher performance than a Fe–N–C catalyst, suggesting that water flooding is a serious issue of the Co–N–C@F127 cathode due to the catalyst's micropore feature. Fuel cell performance was then evaluated by using more practical H2/air at 1.0 bar. The polarization curves recorded at 100% RH indicates a significant mass transport loss associated with serious water flooding issue (Fig. 7c and Fig. S19, ESI†). However, at a relatively low RH of 60%, the Co–N–C@127 exhibited enhanced performance at all voltages studied (Fig. 7d). SEM images of a MEA cathode present very dense and aggregated morphologies (Fig. S20, ESI†), which is not favorable for mass transport. This suggests that further optimization of electrode structures is highly demanded to improve ionomer dispersion, facilitate mass transport, and mitigate the water flooding in the fuel cell electrodes. The durability of the Co–N–C@F127 catalyst in the MEA was further evaluated for 100 hours at a cell voltage of 0.7 V using H2 and air at 1.0 bar and two different RHs (Fig. S21, ESI†). Significant initial performance loss was observed, which is in good agreement with initial activity loss in RDE tests. Compared to other PGM-free and Fe-free cathodes in fuel cells,37,68,69 the performance durability at such a relatively high voltage (i.e., 0.7 V) was commendable. The performance loss is likely due to the possible surface oxidation of carbon along with degradation of three-phase interface within cathodes. Currently, performance durability especially at high voltages is still a grand challenge for PGM-free cathodes, which need increasing effort to address this issue.
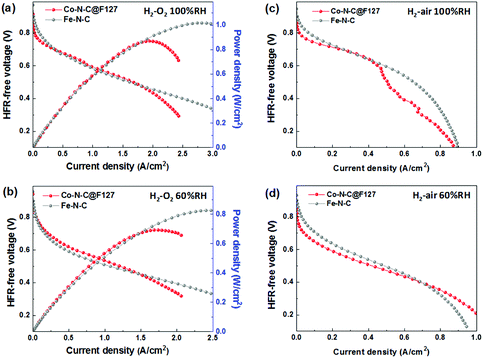 |
| | Fig. 7 Fuel cell performance before and after durability tests of best Co–N–C@F127 catalyst and Fe–N–C catalyst. (a and b) H2–O2 and (c and d) H2-air fuel cell polarization plots at different relative humidity (RH). Cell temperature: 80 °C; flow rate H2/O2 or air: 200/200 sccm, RH: 100% or 60%, 1 bar H2/O2 or air partial pressure. 4 mg cm−2, I/C = 0.6, Nafion 212. | |
Conclusion
In summary, we developed an effective surfactant-assisted confinement pyrolysis strategy to enable controlled synthesis of atomically dispersed CoN4 sites with increased density, therefore leading to significantly enhanced catalytic activity for the ORR in challenging acids for PEMFCs. Distinct from prior studies, Co-doped ZIF nanocrystal precursors were coated with a surfactant layer, which would be carbonized to graphitized carbon shells via a heat treatment. This carbon shell can effectively retain dominant micropores and high content of N in the carbon matrix, thus preventing the agglomeration of single atomic Co sites. Furthermore, we applied extensive physical characterization on the thus-synthesized catalysts to verify the atomic dispersion of CoN4 sites with increased density than the catalysts synthesized using conventional surfactant-free approach. Among four types of studied surfactants, the Co–N–C@F127 catalyst exhibited excellent ORR activity with the most positive E1/2 of 0.84 V (vs. RHE) along with a dominant 4e− ORR pathway in acidic media (H2O2 yield <2%). This superior ORR activity is the highest ever reported for PGM- and Fe-free catalysts. First principles DFT calculations further predicted that the CoN2+2 sites on the edge of carbon layers are able to catalyze the 4e− ORR showing comparable activity to FeN4 sites, whereas the conventional CoN4 sites embedded inside compact carbon layers could mainly catalyze the 2e− ORR. Unlike other Co catalysts, this new atomically dispersed Co–N–C catalyst derived from the surfactant-coated ZIF precursors is believed to contain substantial CoN2+2 sites and hence demonstrated highly active and selective for the desirable 4e− ORR in acids. Importantly, fuel cell tests further confirm the effectiveness of the Co–N–C@F127 cathode catalyst in PEMFCs with a power density of 0.87 W cm−2 comparable to that of a Fe–N–C catalyst-based cathode, especially at relatively low RH. Similar to Fe–N–C catalysts, initial performance degradation was observed with the Co–N–C catalyst in both RDE and fuel cell tests, which is likely due to carbon oxidation. One of our future focuses will be on the improvement of catalyst stability through engineering carbon structures in catalysts. The surfactant-assisted confinement strategy provides a new approach to synthesizing single metal site catalyst with significantly increased density of active sites for widespread electrochemical energy conversion applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
G. Wu thanks the financial support from the National Science Foundation (CBET-1604392, 1804326). G. Wu, G. F. Wang, and S. Litster acknowledge the support from U.S. DOE-EERE Fuel Cell Technologies Office (DE-EE0008076). Electron microscopy research was conducted at the Center for Functional Nanomaterials at Brookhaven National Laboratory (S. Hwang and D. Su, under contract No. DE-SC0012704) and the Center for Nanophase Materials Sciences of Oak Ridge National Laboratory (D. A. Cullen and K. L. More), which both are DOE Office of Science User Facilities. This research used resources of the Advanced Photon Source (APS), a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357.
References
- A. Morozan, B. Jousselme and S. Palacin, Energy Environ. Sci., 2011, 4, 1238–1254 RSC
 .
.
- F. Jaouen, E. Proietti, M. Lefèvre, R. Chenitz, J.-P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114–130 RSC .
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS PubMed .
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed .
- S. T. Thompson, A. R. Wilson, P. Zelenay, D. J. Myers, K. L. More, K. C. Neyerlin and D. Papageorgopoulos, Solid State Ionics, 2018, 319, 68–76 CrossRef CAS .
- G. Wu, Front. Energy, 2017, 11, 286–298 CrossRef .
- G. Wu and P. Zelenay, Acc. Chem. Res., 2013, 46, 1878–1889 CrossRef CAS PubMed .
- G. Wu, A. Santandreu, W. Kellogg, S. Gupta, O. Ogoke, H. Zhang, H.-L. Wang and L. Dai, Nano Energy, 2016, 29, 83–110 CrossRef CAS .
- J. Shui, M. Wang, F. Du and L. Dai, Sci. Adv., 2015, 1, e1400129 CrossRef PubMed .
- W. Gu, L. Hu, J. Li and E. Wang, Electroanalysis, 2018, 30, 1217–1228 CrossRef CAS .
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937 CrossRef CAS PubMed .
- H. Peng, F. Liu, X. Liu, S. Liao, C. You, X. Tian, H. Nan, F. Luo, H. Song and Z. Fu, ACS Catal., 2014, 4, 3797–3805 CrossRef CAS .
- J. Masa, A. Zhao, W. Xia, M. Muhler and W. Schuhmann, Electrochim. Acta, 2014, 128, 271–278 CrossRef CAS .
- G. Wu, M. Nelson, S. Ma, H. Meng, G. Cui and P. K. Shen, Carbon, 2011, 49, 3972–3982 CrossRef CAS .
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang, D. Su, Y. Shao and G. Wu, J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS PubMed .
- S. Gupta, S. Zhao, O. Ogoke, Y. Lin, H. Xu and G. Wu, ChemSusChem, 2017, 10, 774–785 CrossRef CAS PubMed .
- Z. Qiao, H. Zhang, S. Karakalos, S. Hwang, J. Xue, M. Chen, D. Su and G. Wu, Appl. Catal., B, 2017, 219, 629–639 CrossRef CAS .
- X. Wang, H. Zhang, H. Lin, S. Gupta, C. Wang, Z. Tao, H. Fu, T. Wang, J. Zheng, G. Wu and X. Li, Nano Energy, 2016, 25, 110–119 CrossRef CAS .
- D. Banham, S. Ye, K. Pei, J.-I. Ozaki, T. Kishimoto and Y. Imashiro, J. Power Sources, 2015, 285, 334–348 CrossRef CAS .
- V. Goellner, V. Armel, A. Zitolo, E. Fonda and F. Jaouen, J. Electrochem. Soc., 2015, 162, H403–H414 CrossRef CAS .
- J. A. Varnell, C. Edmund, C. E. Schulz, T. T. Fister, R. T. Haasch, J. Timoshenko, A. I. Frenkel and A. A. Gewirth, Nat. Commun., 2016, 7, 12582 CrossRef CAS PubMed .
- G. Zhang, R. Chenitz, M. Lefèvre, S. Sun and J.-P. Dodelet, Nano Energy, 2016, 29, 111–125 CrossRef CAS .
- C. H. Choi, C. Baldizzone, G. Polymeros, E. Pizzutilo, O. Kasian, A. K. Schuppert, N. Ranjbar Sahraie, M.-T. Sougrati, K. J. J. Mayrhofer and F. Jaouen, ACS Catal., 2016, 6, 3136–3146 CrossRef CAS .
- C. H. Choi, C. Baldizzone, J.-P. Grote, A. K. Schuppert, F. Jaouen and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2015, 54, 12753–12757 CrossRef CAS PubMed .
- J. Herranz, F. Jaouen, M. Lefevre, U. I. Kramm, E. Proietti, J.-P. Dodelet, P. Bogdanoff, S. Fiechter, I. Abs-Wurmbach and P. Bertrand, J. Phys. Chem. C, 2011, 115, 16087–16097 CrossRef CAS PubMed .
- C. H. Choi, H.-K. Lim, M. W. Chung, G. Chon, N. Ranjbar Sahraie, A. Altin, M.-T. Sougrati, L. Stievano, H. S. Oh, E. S. Park, F. Luo, P. Strasser, G. Dražić, K. J. J. Mayrhofer, H. Kim and F. Jaouen, Energy Environ. Sci., 2018 10.1039/C8EE01855C .
- S.-F. Kang and H.-M. Chang, Water Sci. Technol., 1997, 36, 215–222 CrossRef CAS .
- T. Sun, Q. Wu, R. Che, Y. Bu, Y. Jiang, Y. Li, L. Yang, X. Wang and Z. Hu, ACS Catal., 2015, 5, 1857–1862 CrossRef CAS .
- R. Zhou, M. Jaroniec and S. Z. Qiao, ChemCatChem, 2015, 7, 3808–3817 CrossRef CAS .
- S. Ma, G. A. Goenaga, A. V. Call and D. J. Liu, Chem. – Eur. J., 2011, 17, 2063–2067 CrossRef CAS PubMed .
- L. Chong, G. A. Goenaga, K. Williams, H. M. Barkholtz, L. R. Grabstanowicz, J. A. Brooksbank, A. B. Papandrew, R. Elzein, R. Schlaf and T. A. Zawodzinski Jr, ChemElectroChem, 2016, 3, 1541–1545 CrossRef CAS .
- Y. Han, Y.-G. Wang, W. Chen, R. Xu, L. Zheng, J. Zhang, J. Luo, R.-A. Shen, Y. Zhu and W.-C. Cheong, J. Am. Chem. Soc., 2017, 139, 17269–17272 CrossRef CAS PubMed .
- A. Kong, Y. Kong, X. Zhu, Z. Han and Y. Shan, Carbon, 2014, 78, 49–59 CrossRef CAS .
- H.-W. Liang, W. Wei, Z.-S. Wu, X. Feng and K. Müllen, J. Am. Chem. Soc., 2013, 135, 16002–16005 CrossRef CAS PubMed .
- L. Osmieri, A. H. Monteverde Videla, P. Ocón and S. Specchia, J. Phys. Chem. C, 2017, 121, 17796–17817 CrossRef CAS .
- S.-T. Chang, H.-C. Huang, H.-C. Wang, H.-C. Hsu, J.-F. Lee and C.-H. Wang, Int. J. Hydrogen Energy, 2014, 39, 934–941 CrossRef CAS .
- X. X. Wang, D. A. Cullen, Y. T. Pan, S. Hwang, M. Wang, Z. Feng, J. Wang, M. H. Engelhard, H. Zhang and Y. He, Adv. Mater., 2018, 30, 1706758 CrossRef PubMed .
- B. You, N. Jiang, M. Sheng, W. S. Drisdell, J. Yano and Y. Sun, ACS Catal., 2015, 5, 7068–7076 CrossRef CAS .
- Q. Cheng, S. Han, K. Mao, C. Chen, L. Yang, Z. Zou, M. Gu, Z. Hu and H. Yang, Nano Energy, 2018, 52, 485–493 CrossRef CAS .
- X. X. Wang, S. Hwang, Y.-T. Pan, K. Chen, Y. He, S. Karakalos, H. Zhang, J. S. Spendelow, D. Su and G. Wu, Nano Lett., 2018, 18, 4163–4171 CrossRef CAS PubMed .
- B. Y. Xia, Y. Yan, N. Li, H. B. Wu, X. W. D. Lou and X. Wang, Nat. Energy, 2016, 1, 15006 CrossRef CAS .
- J. Gao, K. Ye, L. Yang, W.-W. Xiong, L. Ye, Y. Wang and Q. Zhang, Inorg. Chem., 2013, 53, 691–693 CrossRef PubMed .
- I. S. Amiinu, X. Liu, Z. Pu, W. Li, Q. Li, J. Zhang, H. Tang, H. Zhang and S. Mu, Adv. Funct. Mater., 2018, 28, 1704638 CrossRef .
- T. Xing, Y. Lou, Q. Bao and J. Chen, CrystEngComm, 2014, 16, 8994–9000 RSC .
- Y. Pan, D. Heryadi, F. Zhou, L. Zhao, G. Lestari, H. Su and Z. Lai, CrystEngComm, 2011, 13, 6937–6940 RSC .
- J. Zhao, Y. Wang, W. Dong, Y. Wu, D. Li, B. Liu and Q. Zhang, Chem. Commun., 2015, 51, 9479–9482 RSC .
- G. Yang, F. Li, L. Wang, K. H. Row, H. Liu, L. Bai, W. Cao and T. Zhu, Chromatographia, 2008, 68, 27–31 CrossRef CAS .
- Q. Lai, Y. Zhao, Y. Liang, J. He and J. Chen, Adv. Funct. Mater., 2016, 26, 8334–8344 CrossRef CAS .
- M. S. Bakshi, Cryst. Growth Des., 2015, 16, 1104–1133 CrossRef .
- Y. Chang, F. Hong, C. He, Q. Zhang and J. Liu, Adv. Mater., 2013, 25, 4794–4799 CrossRef CAS PubMed .
- M. Jiang, X. Cao, D. Zhu, Y. Duan and J. Zhang, Electrochim. Acta, 2016, 196, 699–707 CrossRef CAS .
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Pöschl, Carbon, 2005, 43, 1731–1742 CrossRef CAS .
- E. Proietti, F. Jaouen, M. Lefèvre, N. Larouche, J. Tian, J. Herranz and J.-P. Dodelet, Nat. Commun., 2011, 2, 416 CrossRef PubMed .
- S. Liu, C. Deng, L. Yao, H. Zhong and H. Zhang, J. Power Sources, 2014, 269, 225–235 CrossRef CAS .
- T. E. Westre, P. Kennepohl, J. G. DeWitt, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119, 6297–6314 CrossRef CAS .
- W. Liu, L. Zhang, W. Yan, X. Liu, X. Yang, S. Miao, W. Wang, A. Wang and T. Zhang, Chem. Sci., 2016, 7, 5758–5764 RSC .
- D. Xu, P. Lu, P. Dai, H. Wang and S. Ji, J. Phys. Chem. C, 2012, 116, 3405–3413 CrossRef CAS .
- H. Jing, X. Song, S. Ren, Y. Shi, Y. An, Y. Yang, M. Feng, S. Ma and C. Hao, Electrochim. Acta, 2016, 213, 252–259 CrossRef CAS .
- H. Zhang, H. Osgood, X. Xie, Y. Shao and G. Wu, Nano Energy, 2017, 31, 331–350 CrossRef CAS .
- Y. Shao, S. Zhang, M. H. Engelhard, G. Li, G. Shao, Y. Wang, J. Liu, I. A. Aksay and Y. Lin, J. Mater. Chem., 2010, 20, 7491–7496 RSC .
- K. X. Liu, G. Wu and G. F. Wang, J. Phys. Chem. C, 2017, 121, 11319–11324 CrossRef CAS .
- J. K. Norskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef CAS .
- G. Henkelman, B. P. Uberuaga and H. Jonsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS .
- K. X. Liu, S. Kattel, V. Mao and G. F. Wang, J. Phys. Chem. C, 2016, 120, 1586–1596 CrossRef CAS .
- S. Kattel and G. F. Wang, J. Phys. Chem. Lett., 2014, 5, 452–456 CrossRef CAS PubMed .
- S. Gupta, S. Zhao, X. X. Wang, S. Hwang, S. Karakalos, S. V. Devaguptapu, S. Mukherjee, D. Su, H. Xu and G. Wu, ACS Catal., 2017, 7, 8386–8393 CrossRef CAS .
- S. Gupta, L. Qiao, S. Zhao, H. Xu, Y. Lin, S. V. Devaguptapu, X. Wang, M. T. Swihart and G. Wu, Adv. Energy Mater., 2016, 6, 1601198 CrossRef .
- G. Wu, K. Artyushkova, M. Ferrandon, A. J. Kropf, D. Myers and P. Zelenay, ECS Trans., 2009, 25, 1299–1311 CAS .
- M. Ferrandon, X. Wang, A. J. Kropf, D. J. Myers, G. Wu, C. M. Johnston and P. Zelenay, Electrochim. Acta, 2013, 110, 282–291 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ee02694g |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Sooyeon
Hwang
b,
David A.
Cullen
c,
M. Aman
Uddin
d,
Lisa
Langhorst
d,
Boyang
Li
e,
Stavros
Karakalos
a,
Sooyeon
Hwang
b,
David A.
Cullen
c,
M. Aman
Uddin
d,
Lisa
Langhorst
d,
Boyang
Li
e,
Stavros
Karakalos