
Two-dimensional materials in semiconductor photoelectrocatalytic systems for water splitting†
Monireh
Faraji‡
ab,
Mahdieh
Yousefi‡
c,
Samira
Yousefzadeh
d,
Mohammad
Zirak
e,
Naimeh
Naseri
a,
Tae Hwa
Jeon
 f,
Wonyong
Choi
*f and
Alireza Z.
Moshfegh
*ac
f,
Wonyong
Choi
*f and
Alireza Z.
Moshfegh
*ac
aDepartment of Physics, Sharif University of Technology, Tehran 11155-9161, Iran
bDepartment of Chemistry North Tehran Branch, Islamic Azad University, P.O. Box 16511-53311, Tehran, Iran
cInstitute for Nanoscience and Nanotechnology, Sharif University of Technology, P.O. Box 14588-8969, Tehran, Iran. E-mail: moshfegh@sharif.edu
dDepartment of Physics, Faculty of Science, Sahand University of Technology, P.O. Box 51335-1996, Tabriz, Iran
eDepartment of Physics, Hakim Sabzevari University, P.O. Box 961797648, Sabzevar, Iran
fDivison of Environmental Science and Engineering, Pohang University of Science and Technology (POSTECH), Pohang, 37673, Korea. E-mail: wchoi@postech.edu
First published on 10th October 2018
Abstract
Hydrogen (H2) production via solar water splitting is one of the most ideal strategies for providing sustainable fuel because this requires only water and sunlight. In achieving high-yield production of hydrogen as a recyclable energy carrier, the nanoscale design of semiconductor (SC) materials plays a pivotal role in both photoelectrochemical (PEC) and photocatalytic (PC) water splitting reactions. In this context, the advent of two-dimensional (2D) materials with remarkable electronic and optical characteristics has attracted great attention for their application to PEC/PC systems. The elaborate design of combined 2D layered materials interfaced with other SCs can markedly enhance the PEC/PC efficiencies via bandgap alteration and heterojunction formation. Three classes of 2D materials including graphene, transition metal dichalcogenides (TMDs), and graphitic carbon nitride (g-C3N4), and their main roles in the photoelectrocatalytic production of H2, are discussed in detail herein. We highlight the various roles of these 2D materials, such as enhanced light harvesting, suitable band edge alignment, facilitated charge separation, and stability during the water splitting reaction, in various SC/2D photoelectrode and photocatalytic systems. The roles of emerging 2D nanomaterials, such as 2D metal oxyhalides, 2D metal oxides, and layered double hydroxides, in PEC H2 production are also discussed.
 Top row from left to right: Monireh Faraji, Mahdieh Yousefi, Samira Yousefzadeh, Mohammad Zirak; bottom row from left to right: Naimeh Naseri, Tae Hwa Jeon, Wonyong Choi, Alireza Zaker Moshfegh | This review paper presents the comprehensive work of a multidisciplinary team at Sharif University of Technology and Pohang University of Science and Technology, including the NEST (Nano-Energy-Surface-Thin film) group of Alireza Zaker Moshfegh (bottom row) in Physics and the Eco-friendly Photoenergy Application group of Wonyong Choi (bottom row) in engineering, respectively. Individual expertise was provided by Dr Monireh Faraji (PhD, Tarbiat Modarres University, 2012, and assistant professor in North Tehran Branch, Islamic Azad University) in electrocatalysts for fuel cell electrodes and theoretical studies of PEC water splitting on SCs for solar hydrogen production, Mahdieh Yousefi (PhD student, Sharif University of Technology, 2014-present) in electrocatalysts for SC electrodes and experimental as well as theoretical studies of PEC water splitting on SCs for solar hydrogen production, Dr Samira Yousefzadeh (PhD, Sharif University of Technology, 2016, and assistant professor in Sahand University of Technology) in carbon based material (CNTs, graphene)–SC composite nanostructures and their applications in PEC water splitting processes, Dr Mohammad Zirak (PhD, Sharif University of Technology, 2016 and assistant professor in Hakim Sabzevari University) in two-dimensional materials, nano-photocatalysts, and nanomaterials for solar energy conversion and environmental applications, Dr Naimeh Naseri (PhD, Sharif University of Technology, 2012 and assistant professor in Sharif University of Technology) in solar hydrogen production, photo/electro catalysts, as well as PEC biosensor materials; Dr Tae Hwa Jeon (PhD, Pohang University of Science and Technology, 2017) in photoelectrochemistry for water splitting and environmental applications, Professor Wonyong Choi (PhD, California Institute of Technology, 1996) in SC photocatalysis and photochemistry for solar energy conversion and environmental applications, advanced oxidation processes, and environmental chemistry; Professor Alireza Zaker Moshfegh (PhD, University of Houston, 1990) in the synthesis and characterization of low-dimensional materials, in particular, novel 2D transition metal dichalcogenides for clean energy and environmental applications. |
Broader contextMassive consumption of fossil fuels has led to serious pollution of various environmental media and the accumulation of atmospheric CO2 that induces climate change. Sustainable production of hydrogen as a recyclable solar energy carrier from water splitting has been intensively investigated as a potential solution to solve such problems. Nanostructured semiconductor photoelectrodes or photocatalysts that enable solar hydrogen production are required to have the characteristics such as strong absorption of sunlight, suitable band gap and band energy levels, fast charge transport, good stability, high density of active sites, low material cost, and eco-friendliness. To meet such requirements, semiconductor(s) are being combined with various emerging 2D materials having unique structures and properties. In this context, the elaborate design of 2D-layered materials hybridized with semiconductor(s) can enhance the hydrogen production efficiency markedly. These 2D materials include graphene, transition metal dichalcogenides (TMDs) and graphitic carbon nitrides (g-C3N4), which form interfacial heterojunction with semiconductor(s) to improve their photoelectrocatalytic performance for water splitting. However, these materials have many drawbacks for practical applications mainly because of their thin-layered structure. We review and discuss many elaborate designs of the combined 2D-layered materials interfaced with other semiconductor(s), aiming to understand the main roles of these 2D structures in enhanced production of H2. |
1. Introduction
The significant dependence of the global economy on non-renewable and geopolitically sensitive fossil fuel energies has led to the requirement for breakthrough technologies to secure alternative clean and renewable energy supplies.1 Among the various renewable energy sources (i.e., wind, hydroelectric, tidal and ocean currents, geothermal, biomass, and solar), solar energy is by far the most abundant, inexpensive, non-polluting, and sustainable. Although the total solar energy incident on the earth over one hour is larger than the annual global energy consumption, the most critical challenge remains the collection and storage of this very diffuse form of energy to enable practical and continuous fuel supply.2 Hydrogen (H2), which has the highest energy density, is considered one of the promising energy carriers for storing solar energy in the form of the chemical bond energy between two H atoms.3 Among the various methods for the conversion of water and sunlight into H2, photoelectrochemical (PEC) water splitting with semiconductor (SC) photoelectrodes has attracted most interest owing to three main advantages: (i) generation of O2 and H2 at separate electrodes, which eliminates the separation issue; (ii) potential for operation under ambient conditions; and (iii) potential for construction of a system that includes only stable and abundant inorganic materials.1In 1972, Honda and Fujishima4 first demonstrated that a PEC cell comprising a single-crystalline TiO2(rutile) anode and a Pt cathode under ultraviolet (UV) irradiation achieved hydrogen production from water with an external bias. It is now recognized that water splitting into H2 and O2 occurs on SC surfaces when the absorption of photons with an energy larger than the bandgap energy (Eg) of SC generates electrons in the conduction band (CB) and leaves holes in the valence band (VB). The photo-generated electrons and holes may either recombine with each other or participate in chemical reactions with water to produce H2 and O2.5,6 Selecting an appropriate SC for water splitting requires the CB position to be more negative than the H+/H2 potential (0.0 VNHE) and the VB position to be more positive than the O2/H2O potential (1.23 VNHE).7–12 Numerous SC materials have been investigated as potential photocatalysts (e.g., WO3 and BiVO4). Meeting the requirements of both a sufficiently negative CB for H2 production and an acceptable narrow bandgap for visible-light absorption is a challenge because the VB (mostly composed of O 2p orbitals) of oxide SCs is highly positive. Although some narrow bandgap non-oxide SCs (e.g., CdS) have appropriate band levels for water splitting, the material itself is not sufficiently stable and is therefore subject to photocorrosion under light illumination.13 This restricts the practical application of such SCs, thus requiring various surface modifications.14
Designing visible-light active SCs for water splitting requires a suitable bandgap and band alignment, effective charge separation, fast charge transfer, and long-term durability in aqueous environments.15–18 These selection criteria are depicted in Fig. 1 and have been the main focus of active research in past decades. An interesting design strategy for meeting such requirements is to combine two-dimensional (2D) materials (e.g., graphene, MoS2, g-C3N4) with appropriate SCs. Fig. 2 shows the latest trend in the number of publications in this field. From a historical view point, research on 2D materials started in 1859.19 It is one of the most widely studied research areas owing to the novel properties and potential multi-purpose applications of such materials. The 2D materials are generally composed of strong covalent bonds leading to in-plane stability and weak van der Waals bonds, which sustain the stacked layer structure. Following the discovery of graphene in 2004, a new horizon has opened up for exploring other 2D layered materials such as transition metal dichalcogenides (TMD), transition metal oxides, graphitic carbon nitride (GCN), and hexagonal boron nitride (h-BN). These 2D materials can be integrated with a three-dimensional (3D) SC material as a new building block to fabricate interfacial heterostructures. Such structures have been applied to photovoltaic devices, hydrogen evolution catalysts, transistors, photodetectors, DNA detection, and lithium ion batteries.20
 | ||
| Fig. 1 Essential requirements for a suitable semiconducting material as a photoelectrode/photocatalyst for water splitting reactions. | ||
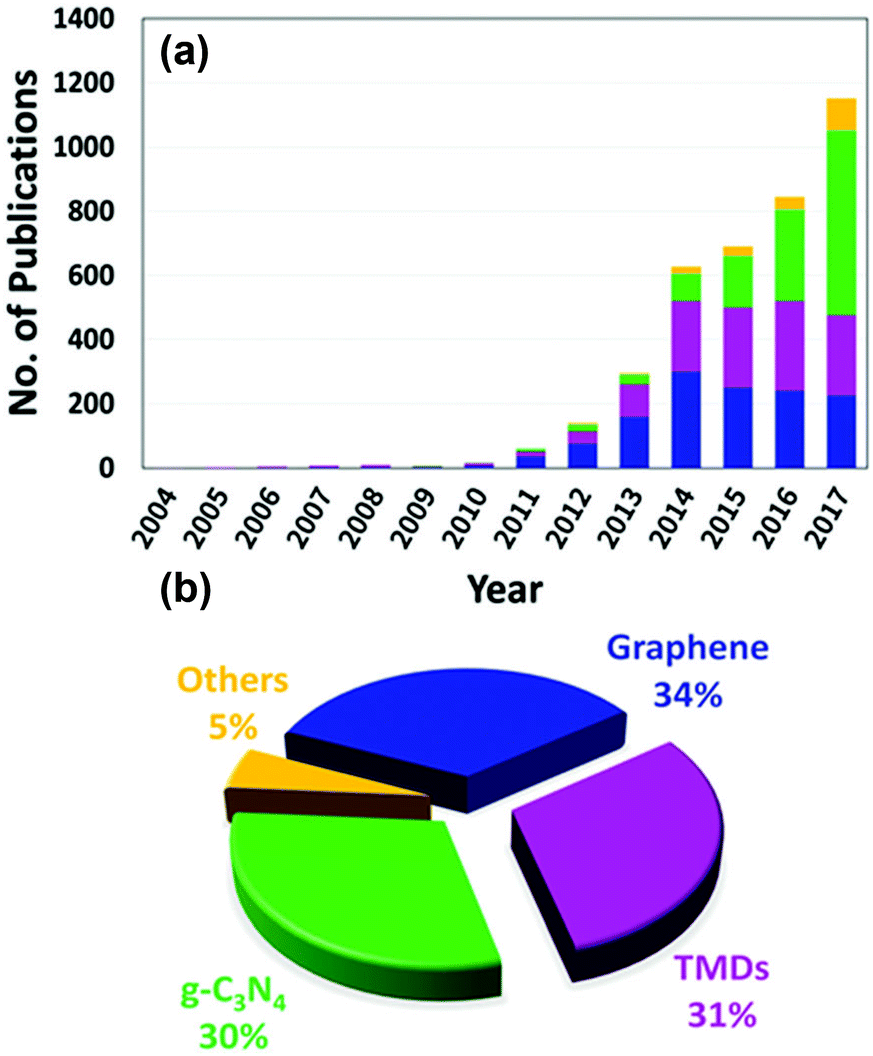 | ||
| Fig. 2 (a) The number of publications on composite 2D/SC materials applied to water splitting by photocatalysis and PEC processes between 2004 and 2017 (from Scopus) and (b) the contribution of each 2D material to the publication activity. | ||
The emergence of nanomaterials with adjustable shapes and dimensions ensure progress in PEC/photocatalytic water splitting. The enhanced hydrogen generation of such 2D/SC hybrids can be ascribed to the unique properties of the 2D materials, i.e., (i) 2D layered materials provide more reaction sites for catalytic reactions because of their larger specific surface area as compared with their bulk structures11,21–28 and (ii) the 2D nature of these materials prolongs the separation of photo-generated electrons and holes by minimizing the distance through which the electrons have to migrate before reaching the solid/water interface.11,29 Therefore, the utilization of 2D materials in photocatalysis/PEC can lead to better yields in photoconversion processes.30–34 In many cases, the 2D materials themselves are neither photocatalysts nor photoelectrodes; however, these materials have been successfully applied as sensitizers, electron mediators, co-catalysts, and protective layers in combination with other SC materials. The 2D/SC hybrid materials can induce synergetic effects and ultimately improve the electrical, optical, and PEC properties of 2D/SC electrodes.
This review article summarizes and discusses recent advances in the 2D/SC systems applied to water splitting. The main focus is on three representative 2D materials (graphene, TMD, and GCN) interfaced with SCs (Sections 2–4). The advent of new emerging 2D materials and their potential applications in water splitting are discussed in Section 5. In the final section, we conclude and summarize the future perspectives of 2D/SC research. The main principles of the PEC water splitting reaction and a comprehensive tool box for characterizing the optical, structural, electronic, and PEC properties of 2D/SC systems (as summarized in Scheme 1) are described in detail in the ESI.† A brief introduction to 2D materials, including their crystal structure and optical and electronic properties, is also included in the ESI.†
 | ||
| Scheme 1 Schematic presentation of a diagnostic tool box for studying 2D interfaced SCs. | ||
2. Graphene
2.1. Graphene as a conducting platform
When SC/graphene composites are irradiated, photoexcitation occurs on the SC side and graphene is usually involved in the subsequent charge-transfer processes. The main electronic role of graphene at the SC/graphene interface is to serve as an electron acceptor, transporter, and mediator in the graphene-based composites owing to its conductive 2D structure.35–42 As a unique 2D electron conductive platform with low Fermi level, graphene promotes electron–hole separation and electron transport in different photoelectrodes. The application of 2D material to photocatalytic systems has been widely investigated and the charge transfer between photoactive component materials (e.g., SC) and graphene has been confirmed via analysis.43 The design, fabrication, and application of graphene-based composite structures in photoelectrodes for overall water splitting have emerged as active research fields. The electronic roles of graphene and its derivatives are mainly two-fold: (i) charge separation/transport and (ii) charge-transfer mediation.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D vs. time (Fig. 3b) using eqn (S4) (ESI†), transient time constant (τ) was calculated to be 2.8 and 7.6 s for BiVO4 and BiVO4/rGO, respectively. Although a negligible amount of H2 and O2 gas was produced on pure BiVO4, the gas evolution rate was markedly increased with BiVO4/rGO. This improvement was ascribed to the enhanced electron–hole separation across the rGO interface, resulting in a longer electron lifetime. In the BiVO4/rGO film, photo-induced charge separation occurred within BiVO4 particles, followed by the rapid transfer of electrons to the rGO sheets and then to the collecting electrode (FTO).
D vs. time (Fig. 3b) using eqn (S4) (ESI†), transient time constant (τ) was calculated to be 2.8 and 7.6 s for BiVO4 and BiVO4/rGO, respectively. Although a negligible amount of H2 and O2 gas was produced on pure BiVO4, the gas evolution rate was markedly increased with BiVO4/rGO. This improvement was ascribed to the enhanced electron–hole separation across the rGO interface, resulting in a longer electron lifetime. In the BiVO4/rGO film, photo-induced charge separation occurred within BiVO4 particles, followed by the rapid transfer of electrons to the rGO sheets and then to the collecting electrode (FTO).
 | ||
| Fig. 3 (a) Photocurrent–voltage curves for BiVO4, BiVO4–rGO (under visible-light), and TiO2 (under UV illumination) for comparison. (b) Normalized LnD vs. time plots for both BiVO4 and BiVO4–rGO electrodes. (c) Electron transport in a PEC cell based on BiVO4 and rGO. Reproduced with permission from ref. 44 (Copyright 2010 American Chemical Society). Photocurrent vs. time and stability measurement of (d) G-1.0/Cu2O/Cu mesh and (e) Cu2O/Cu mesh. (f) The proposed charge-transfer mechanism of the graphene/Cu2O/Cu mesh nanocomposite electrode. Reproduced with permission from ref. 62 (Copyright 2014 The Royal Society of Chemistry). (g) The open-circuit potential (OCP) decay vs. time after UV irradiation was turned off, and (h) EIS Nyquist plots (under constant applied voltage of +0.7 V vs. Ag/AgCl) for TiO2 NF and GO (2 wt%)–TiO2 NF electrodes. The PEC experimental conditions were examined in 0.1 M KOH under continuous Ar purging, and λ > 320 nm irradiation. (i) H2 production in an aqueous suspension of TiO2 NF, GO (2 wt%)–TiO2 NF, and GO(s) (2 wt%)–TiO2 NF with Pt loading (0.1 wt%) in the presence of a catalyst (0.5 g L−1), [methanol]0 = 10 vol%, Ar purging, and λ > 320 nm irradiation. Reproduced with permission from ref. 63 (Copyright 2013 Elsevier). | ||
Photo-electrons (e−) can then flow through an external circuit to the counter electrode (Pt wire) on which H2O is reduced to H2, whereas the photo-generated holes (h+) cause water oxidation on the SC photoanode (Fig. 3c). In addition, an improved carrier diffusion length, improved mobility, and a reduced recombination rate for the BiVO4/rGO photoanode were confirmed by a digital simulation study, indicating that rGO acts as an electron acceptor and transporter.45 Similar behavior was observed for graphene in conjunction with other visible-light active SCs such as CdS,46–48 Fe2O3,49–53 InGaZn,54 Zn0.5Cd0.5S,55 BiMo0.03V0.97O4,56 and Zn1−xAgxO57 as the photoanode in PEC water splitting. Moreover, the charge-carrier separation and transport in visible active p-type SC materials can be changed using graphene in the photocathode. For example, Cu2O as a p-type SC with a relatively narrow bandgap energy (∼2 eV) is a promising and attractive photocathode material for solar hydrogen production.58 However, the downside is its poor stability in aqueous solution and its relatively short electron diffusion length vs. the light absorption penetration depth.59,60 Therefore, the combination of graphene with Cu2O can improve charge separation/transport and photostability. According to Tran et al.,61 a Cu2O nanoparticle/rGO composite was used as a photocatalyst for hydrogen generation. An enhanced hydrogen evolution efficiency and stable photocurrent density of −0.12 mA cm−2 at −0.4 V (vs. Ag/AgCl) for a Cu2O/rGO composite were observed owing to the presence of rGO acting as an electron acceptor and transporter. A previous study62 reported the preparation of a graphene/Cu2O composite through the electrochemical anodization of Cu metal to produce a Cu(OH)2 nanowire array (NWA)/Cu construction, followed by dip coating of GO at 0.25, 0.50, 0.75, 1.0, 1.5, and 3.0 mg mL−1 concentrations and then thermal reduction. Based on the results, optimum PEC performance was achieved at a 1.0 mg mL−1 concentration of graphene (G-1.0/Cu2O/Cu photocathode) (Fig. 3d). The report indicated a photocurrent density of −4.8 mA cm−2 at 0 V (vs. a reversible hydrogen electrode (RHE)) with a photostability of 83.3% after 20 minutes for the G-1.0/Cu2O/Cu mesh photocathode, which was two-fold higher compared with a bare Cu2O/Cu mesh (Fig. 3e). In addition, under dark and illuminated conditions, the G-1.0/Cu2O/Cu mesh photocathode exhibited a smaller semicircle diameter in the Nyquist curves, with a lower charge-transfer resistance (Rct). These results were attributed to the incorporation of graphene in the Cu2O/Cu mesh, which facilitated charge transfer from Cu2O to the electrolyte, thus increasing electron–hole separation and improving the electron conductivity and photoresponsivity of the system. Fig. 3f shows the mechanism of the PEC process for the G–Cu2O/Cu photocathode. According to analysis of the conduction and VB levels, Cu2O was excited under visible-light absorption and electrons and holes were generated in the conduction and VBs, respectively. The photo-generated electrons in the Cu2O were transferred to the graphene and participated in reducing H+ ions to produce H2, whereas the holes were transported to the surface of the Pt electrode resulting in O2 evolution reaction. In addition, graphene prevented any photocorrosion of Cu2O and improved the stability of the G–Cu2O/Cu photocathode as compared with the Cu2O/Cu photoelectrode (Fig. 3e).
For wide bandgap SCs, graphene can also improve photo-generated carrier separation and transportation. The charge-transfer mechanism between a wide bandgap SC and graphene has been widely studied using different forms of analyses. Most studies have focused on graphene/TiO2 composite photoanodes. For example, Kim et al.63 prepared one-dimensional TiO2 nanofibers (NFs) using an electrospinning method and embedded GO sheets inside the NFs. By applying open-circuit potential (OCP), decay kinetics, and electrochemical impedance spectroscopy (EIS) measurements, the charge transport and recombination properties of TiO2 NF and GO–TiO2 NF electrodes were evaluated and compared. Fig. 3g shows the OCP decay profiles of the TiO2 NF and GO–TiO2 NF after no UV irradiation. The recombination rate constant for GO–TiO2 NF was about 1.88 × 10−3 s−1, which was much smaller than that for TiO2 NF (9.84 × 10−3 s−1), implying that the charge recombination was inhibited by the incorporation of GO into TiO2 NF. These results were further supported by EIS measurements and Nyquist plots of the TiO2 NF and GO–TiO2 NF (Fig. 3h). The arc shape and charge-transfer resistance (Rct) values represent the charge transport and recombination kinetics in the system. As shown in Section 1.3.2 (ESI†) the calculation from the Nyquist plots indicated that the Rct values of TiO2 NF in the absence and presence of illumination were about 2 and 1.6 times higher, respectively, than those of GO–TiO2 NF. In addition, hydrogen production over the GO–TiO2 NF increased by 1.7 and 8.5 times, respectively, compared with bare TiO2 NF (Fig. 3i). Similar charge-transfer mechanisms were reported for graphene incorporated with wide bandgap SCs, including ZnO,64–69 WO3,70,71 and BiPO4.72 Therefore, graphene plays an important role as a charge acceptor and separator at SC/graphene composite interface and improves the water splitting rate. Several factors can influence the charge separator/transporter role of graphene, including the graphene content of the composite, interfacial effects, contact nature, and graphene doping; these are discussed in the following paragraphs.
By varying the graphene content, H2 production can be controlled in graphene/SC-based photoelectrodes. A graphene-based composite with an appropriate amount of graphene can decrease the recombination of electron–hole pairs and thus improve the photoactivity of these materials. Rai et al.53 studied the morphological, optical, and PEC properties of a Fe2O3–graphene nanoplate (GNP) composite thin film with GNP weight percentages of 0, 0.1, 0.2, 1, and 2 wt% (denoted as 4H, 4H:0.1G, 4H:0.2G, 4H:1G, and 4H:2G, respectively). The Fe2O3 composite photoelectrode with 0.2 wt% GNP (4H:0.2G) exhibited the highest photocurrent density of 2.5 mA cm−2 at 0.75 V (vs. a saturated calomel electrode (SCE)) under 150 mW cm−2 illumination (Fig. 4a) and a maximum solar-to-hydrogen conversion efficiency of 1.8% (Fig. 4b). In addition, the presence of highly conducting graphene reduced the electrical resistance of the Fe2O3–GNP composite (Fig. 4c). A sharp increase in the resistance of the Fe2O3–GNP composite at high concentrations (1 and 2 wt% GNP) can be ascribed to restacking of the graphene sheets.
 | ||
| Fig. 4 (a) Photocurrent density vs. applied potential curves for Fe2O3 and the Fe2O3–GNP composite thin film under visible-light illumination. (b) Solar-to-hydrogen conversion efficiency values of α-Fe2O3 and Fe2O3–GNP composite thin films. (c) Resistance vs. GNP concentration curve for the Fe2O3–GNP composite thin films. Reproduced with permission from ref. 53 (Copyright 2014 The Royal Society of Chemistry). (d) XPS high-resolution spectrum of C 1s core level for GO and TiO2–graphene nanocomposites. (e) Time-resolved PL spectra for TiO2 nanocrystals and TiO2–graphene nanocomposites. (f) CB and VB positions of the TiO2 nanocrystals and TiO2–graphene nanocomposites. Reproduced with permission from ref. 74 (Copyright 2014 Wiley-VCH). | ||
Another main parameter that determines the interfacial charge-transfer rate between SC and graphene is SC morphology and its interface with graphene. Various atomic structures of different crystal facets affect the SC–graphene composite interfaces.73 As an example, TiO2–graphene composites with different exposed TiO2 crystal facets ({101}, {100}, and {001} facets) were synthesized using a hydrothermal method (denoted as TiO2-101–G, TiO2-100–G and TiO2-001–G, respectively).74 According to the XPS analysis, an extra peak was observed and assigned to Ti–C bonds between the {100} facets and graphene, whereas {101} and {001} facets were connected only with graphene through Ti–O–C bonds (Fig. 4d). In addition, the interfacial charge transfer between different TiO2 crystal facets and graphene was investigated using a photoluminescence (PL) technique (Fig. 4e) excited by 350 nm pulsed laser. By fitting PL curves and calculating the half-life of PL signals (τ), electron transfer (ET) rate (kET) was obtained as follows:
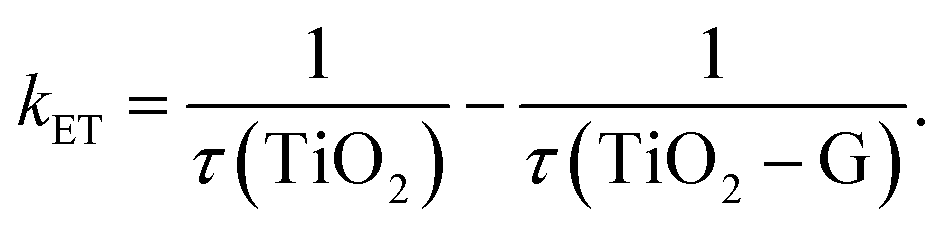 | (1) |
In addition to experimental studies, the interfacial structure and visible photoresponse activity of hybrid TiO2/graphene nanocomposites have been extensively investigated using first-principles calculations such as density functional theory (DFT).75–78 Li et al.78 reported that charge transfer occurred from graphene sheets to anatase TiO2 in the ground electronic state. This process resulted in the accumulation of holes in the graphene sheet.
The combination of graphene and TiO2 altered the electronic band structure of TiO2, including new transitions between (C 2p) states, and other energy states emerged in the middle bandgap of TiO2. Under visible-light illumination, electrons from the VBs of graphene (C 2p) can be excited to the CB in anatase TiO2 (Ti 3d). Therefore, electron–hole separation is efficiently enhanced and significantly inhibits the recombination rate. The differences in electronic structures and interfacial connections were primarily due to the different atomic structures of the different TiO2 crystal facets. By engineering the structures of TiO2–graphene interfaces, the photocatalytic activities of TiO2–graphene composites can be adjusted according to need.79 In addition to the TiO2–graphene system, Kang et al.65 investigated the effects of ZnO morphologies such as nanoparticles, nanosheets, nanospheres, and nanorods in rGO/ZnO composites in terms of degree of light absorption abilities, charge separation efficiencies, and photocatalytic H2 production performances. rGO sheets on vertically grown ZnO nanorods exhibited the highest H2 production rate for the following reasons: (i) higher ET rate from ZnO to rGO, (ii) lower recombination rate of the hole–electron pairs of ZnO, and (iii) strong chemical interaction.
The electron acceptor and transporter role of graphene can also be influenced by the geometry of contact between graphene and SC in graphene/SC composites.80 With the simplest geometry, SC particles are distributed on graphene sheets on which the particles contact the graphene sheets (Fig. 5a). This low contact area limits charge transfer from the SC to the graphene and the graphene–SC contact can be hindered at pH values higher than the zero point charge of the SC.81 The charge transfer can be enhanced by constructing a core–shell structure (Fig. 5b); however, under higher graphene loading, active sites on SC nanoparticles are blocked and light absorption is reduced under this condition. In another study, GO was embedded into TiO2 NFs (Fig. 5c and d). Based on the results, TiO2–NF, which contained densely packed TiO2 nanoparticles, could facilitate inter-particle charge transfer and better separation in the NF matrix. In addition, the presence of GO sheets eased the connection between densely packed TiO2 nanoparticles, leading to improved electron–hole pair separation.63 This phenomenon was confirmed by a low charge recombination rate and enhancement in PEC activity.
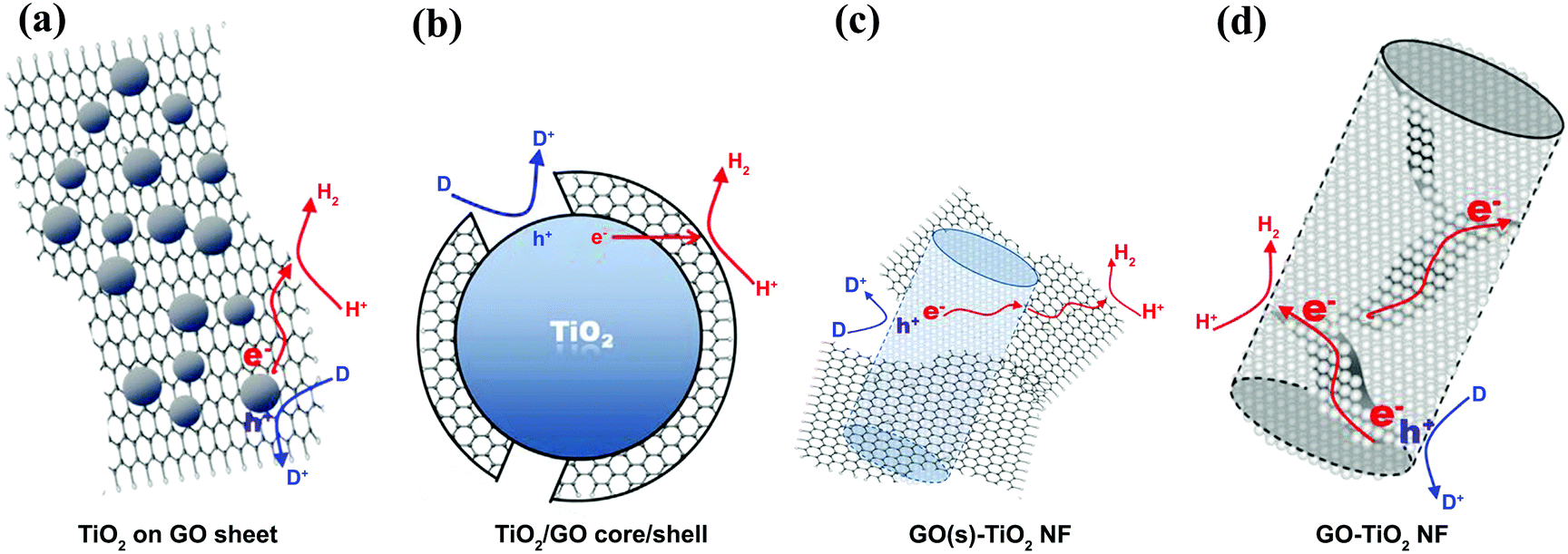 | ||
| Fig. 5 The different composite structures of graphene sheets combined with TiO2 and the related charge transfers for hydrogen evolution: (a) TiO2 nanoparticles on a graphene sheet, (b) the core (TiO2 nanoparticles)–shell (graphene sheet) structure, (c) graphene sheets linked to the external surface of TiO2 nanofibers, and (d) the graphene sheets embedded into the matrix of TiO2 nanofibers. Reproduced with permission from ref. 63 (Copyright 2014 Elsevier). | ||
The electronic properties of graphene can be adjusted via surface modification to improve PEC water splitting. The nitrogen doping of graphene is a common approach that can facilitate charge transport in graphene and is a method employed in various graphene-based photoelectrodes. Jia et al.46 used nitrogen-doped graphene as a supporting matrix for CdS and synthesized a series of N-doped graphene/CdS nanocomposites with different N-doped graphene loadings. The H2 evolution rate among the various graphene-based nanocomposites was optimized at a concentration of 2 wt% N-doped graphene. There was five-fold increase in H2 production as compared with pure CdS. The enhancement was due to the creation of a heterojunction between N-doped graphene and CdS, leading to more efficient ET. The increased and stable photocurrent implied that N-doped graphene in the nanocomposite acted as a protective layer to prevent the photocorrosion of CdS.46
To understand the enhancement mechanism, the electron lifetime (τe) was calculated based on Bode plots (Fig. 6a). Using the low frequency peak (fmax) of the Bode plot (high frequency peak is not shown), the electron lifetime can be estimated from the following equation:
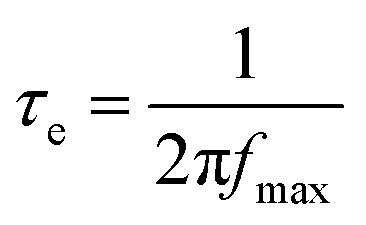 | (2) |
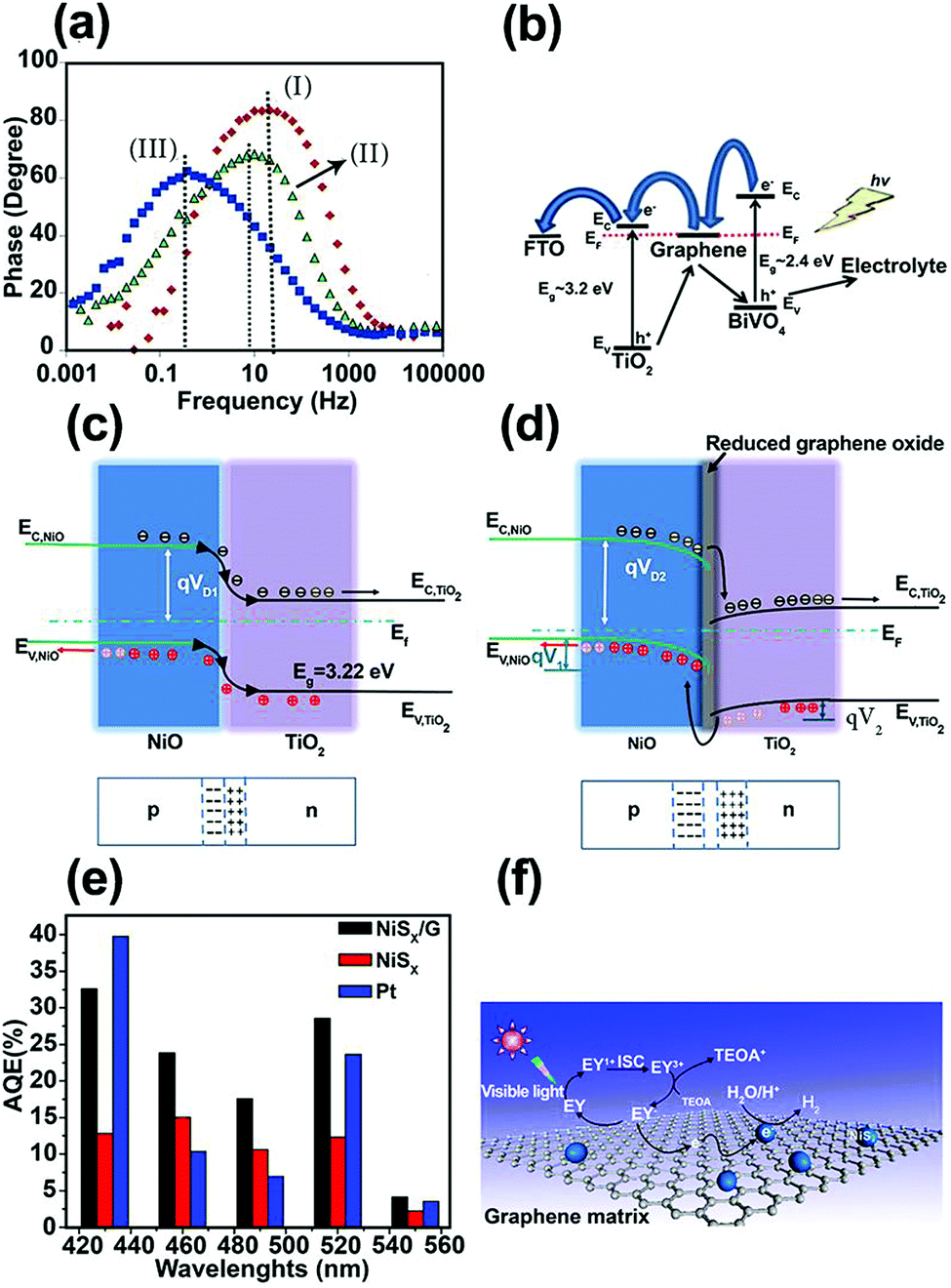 | ||
| Fig. 6 (a) Bode phase plots for the nanocomposite photoanodes. (b) Charge-carrier separation and transfer mechanism for the BiVO4/graphene/TiO2 nanocomposite photoelectrode under irradiation. Reproduced with permission from ref. 86 (Copyright 2016 Elsevier). Schematic energy diagrams of (c) NiO/TiO2 (p–n) nanojunction, and (d) NiO/rGO/TiO2 heterostructured coaxial nanocable systems. Reproduced with permission from ref. 87 (Copyright 2015 Elsevier). (e) Apparent quantum efficiencies (AQEs) of H2 evolution in 100 mL of a 10% (v/v) aqueous triethanolamine (TEOA) solution under light irradiation (λ ≥ 420 nm) for EY (eosin Y) (1.0 × 10−3 mol L−1)-photosensitized systems using NiSx (2.8 mg), Pt (2.8 mg), and the NiSx/G nanohybrid (NiSx, 2.8 mg; graphene, 6 mg) catalysts. (f) The proposed mechanism of hydrogen evolution by the EY–NiSx/G photocatalyst under visible-light irradiation. Reproduced with permission from ref. 91 (Copyright 2016 American Chemical Society). | ||
One of the most efficient charge separation approaches for maximizing the photoconversion activity is to form a p–n junction with a built-in electric field between two SC materials. However, charge accumulation during irradiation can reduce the built-in electric field. Graphene can play an important role as a conductor and separator at the interface of two n- and p-type SCs. Based on this concept, Yu et al.87 deposited rGO film on electrospun p-type NiO NFs and then assembled flower-like TiO2 on a NiO/rGO nanostructure, thus forming a NiO/rGO/TiO2 p–n heterostructured coaxial nanocable. This hierarchical heterostructure was able to produce more H2 in photocatalytic water splitting compared with NiO/rGO, NiO NF, flower-like TiO2, and NiO/TiO2 structures. According to transient photocurrent measurements, a photocurrent density of ∼7.0 μA cm−2 was reported for NiO/rGO/TiO2, which was higher than that for the NiO/rGO NF (0.2 μA cm−2), flower-like TiO2 nanowire (3.0 μA cm−2), and NiO/TiO2 heterostructure (5.0 μA cm−2). This enhancement was attributed to improved charge separation. A smaller semicircle in the EIS curves further confirmed that the rGO enhanced ET at the electrolyte/electrode interface and gave better separation of photo-generated e−–h+ pairs owing to the excellent electrical conductivity and “bridge-like” structure. Fig. 6c and d demonstrate the electronic band structure of NiO/TiO2 and NiO/rGO/TiO2 heterojunctions, respectively. The introduction of rGO between the NiO and TiO2 contact caused the Fermi level of the NiO to move downward by qV1, whereas the Fermi level of the TiO2 moved simultaneously upward by qV2. Thus, the barrier height under this condition ((qV1 + qV2) > 0) further increased the built-in electric field, space charge region, and ET to the electrolyte for efficient H+/H2 reduction.
In dye-sensitized photocatalytic systems, dye is a main component for enhancing visible-light absorption in wide bandgap SCs and photoexcited electrons transfer efficiently from the dye to the photocatalyst CB. This dye sensitization process can play an important role in hydrogen production. However, in actual experimental conditions, hydrogen production efficiency is low due to poor electron transport between the dye and the photocatalyst. Graphene may help overcome this problem. Many studies have applied graphene to different dye-sensitized photocatalysts. Several examples are eosin Y (EY) sensitized rGO with Pt nanoparticles,88,89 Co/graphene and CoSx/graphene,89 nickel/graphene,90 and NiSx/graphene.91 NiSx-decorated graphene (NiSx/G) as a noble-metal-free co-catalyst was synthesized using a successive ionic layer adsorption and reaction (SILAR) method to form NiSx on graphene and was sensitized by EY for hydrogen production under visible-light irradiation.91 The results showed a highest apparent quantum efficiency (AQE) of 32.5% at 430 nm (Fig. 6e). The overall mechanism of photocatalytic H2 generation in the EY–NiSx/G system is illustrated in Fig. 6f. Under visible-light irradiation, the adsorbed EY in the NiSx/G system is excited to the singlet excited state EY1* and subsequently to the lowest-lying triplet excited state EY3* via an intersystem crossing (ISC). When EY3* is quenched reductively by the sacrificial donor triethanolamine (TEOA), the produced EY−˙ species transfer their electrons to graphene, and subsequently to the NiSx co-catalyst for H2 generation. The role of graphene in electron separation and transport is in good agreement with the reduced PL intensity in this system.91
2.2. Graphene as a light harvester
To extend the light absorption edge and enhance the light absorption intensity for the graphene/SC composite, chemical bonding between SC and graphene can occur. Chandrasekaran et al. fabricated γ-Fe2O3/rGO for high-performance water splitting in the PEC process.92 To evaluate the optical bandgap of pristine γ-Fe2O3/rGO nanocomposites calcined at various temperatures, UV-vis absorption spectra was used to obtain a Tauc plot: (αhν)1/2vs. hν, where α is the absorption coefficient. The modification of γ-Fe2O3 with rGO and subsequent calcination at 300, 400, and 500 °C narrowed the bandgap (Fig. 7a). This reduction was ascribed to the formation of Fe–O–C chemical bonds in γ-Fe2O3/rGO (rGO-M) nanocomposites, as supported by XPS analysis through the shift of the O 2p to a higher energy with increasing calcination temperature (Fig. 7b).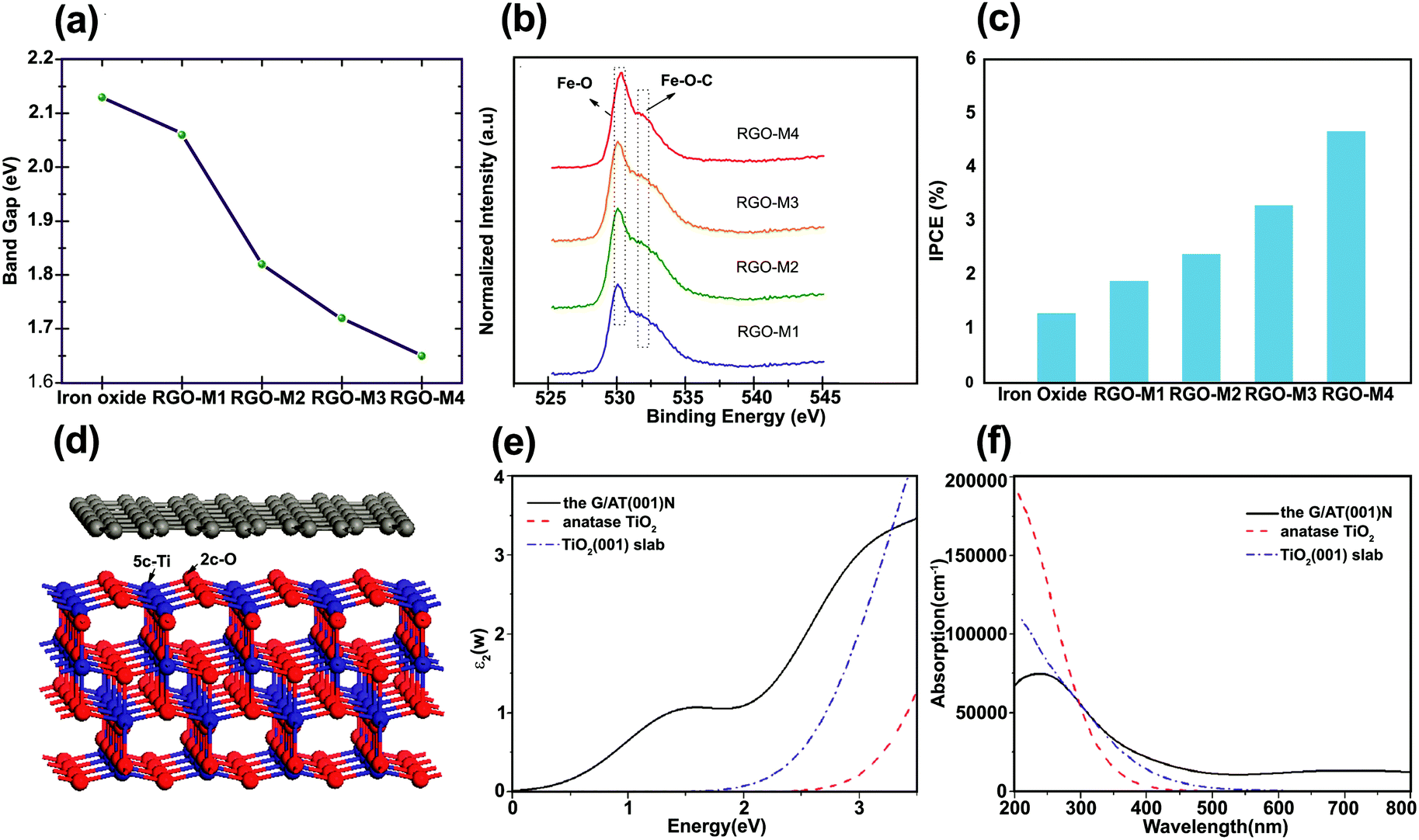 | ||
| Fig. 7 (a) Bandgap values of iron oxide and rGO/g-Fe2O3 samples (for sample identification the as-prepared, 300 °C, 400 °C, and 500 °C calcined samples are denoted as rGO-M1, rGO-M2, rGO-M3, and rGO-M4, respectively), (b) XPS spectra (O2p state) of rGO/g-Fe2O3 samples, (c) chemical energy conversion efficiency of pristine iron oxide and rGO/g-Fe2O3 samples (scan rate of 20 mV s in 1.0 M NaOH), and (d) incident-photon-to-current conversion efficiency of iron oxide and rGO/g-Fe2O3 nanocomposites. Reproduced with permission from ref. 92 (Copyright 2015 The Royal Society of Chemistry). (d) Model for simulating the interface between graphene and the anatase TiO2(001) surface calculated the imaginary part of (e) the dielectric function and (f) the UV-vis absorption spectra of bulk TiO2, anatase TiO2(001) slab, and graphene/TiO2(001) for the polarization vector perpendicular to the surface slab. Reproduced with permission from ref. 77 (Copyright 2013 American Chemical Society). | ||
The incident-photon-to-current efficiency (IPCE) value for rGO-M4 (the sample annealed at 500 °C) was enhanced approximately four times compared with that of the pure γ-Fe2O3 (Fig. 7c). In addition, the incorporation of rGO into γ-Fe2O3 with thermal treatment not only reduced the bandgap but also enhanced the photoconversion efficiency (defined as (1.23 V)/Jlight × 100, where Jlight is the incident light power) up to 0.76% at 1.8 V (vs. RHE) compared with pure iron oxide.92 The observation of progressive red shift in the absorbance onset with increasing rGO loading was also reported in another study.93 The formation of a metal–O–C chemical bond seems to be responsible for the red shift of a SC bandgap after graphene incorporation.93–97 Gupta et al.96 encapsulated Bi2Ti2O7 (BTO) with rGO to enhance the photocatalytic and photoelectrocatalytic activity of BTO and the optical bandgap was reduced from 2.8 (rGO free BTO) to 2.26 eV (with rGO (1 at%)–BTO) owing to Ti–O–C bond formation between the Ti–OH of BTO and the C from rGO. A similar behavior was observed for graphene-modified TiO2 photoanodes.97 The absolute absorbance of graphene–TiO2 was higher than bare TiO2 in the range 350–550 nm. On the other hand, Dubale et al. studied the synergetic effect of graphene in a graphene/Cu2O/Cu nanocomposite as a stable photocathode for hydrogen production.62 In this case, the absorption edge was not changed by the introduction of graphene, indicating that graphene was not incorporated into the lattice of the Cu2O/Cu mesh. The same findings were reported for ZnO/graphene98 and BiOIO3/rGO99 composites.
An electrochemical reduction (ER) treatment was used to significantly enhance absorption properties in the ultra violet and visible regions for TiO2 nanotubes (NT)/rGO photoanodes.100 Based on the UV-vis spectroscopic analysis, a narrower bandgap for the electrochemically treated TiO2 NTs samples was related to the Ti3+ self-doping. An extended long absorption from 400 to 600 nm was observed with the incorporation of electrochemically reduced GO into TiO2 NTs. The ER treated GO–TiO2 NTs showed notable photoactivity improvement of 96.2% at 350 nm compared to that of the bare TiO2 NTs (∼48%). The improvement decreased to 3.14% at 400 nm in IPCE measurements. Wang et al.101 fabricated graphene sheets on TiO2 nanotube arrays (TNAs) using a simple electrochemical method and showed that the rGO/TNAs exhibited the highest absorption intensity with a narrowed bandgap.
To understand the photoenhancement effect, Gao et al.77 conducted DFT calculations to examine the optical response of graphene/TiO2 (Fig. 7d). The imaginary component of the dielectric function was calculated to obtain the optical absorption spectra for the graphene/TiO2 composite. The calculated UV-vis absorption spectra of bulk TiO2, anatase TiO2(001) slab, and graphene/TiO2(001) are shown in Fig. 7e and f. The optical absorption edge of the graphene/TiO2(001) nanocomposite was shifted to a longer wavelength region with a markedly enhanced absorption in the visible region. This could possibly be due to the appropriate connection between the graphene sheets and the anatase TiO2(001) facet. The visible-light response of graphene/TiO2(001) can be attributed to ET from the anatase TiO2(001) slab to the graphene layer.77
Together with electrochemically reduced GO, Pan et al. investigated the effects of three different reduction routes, i.e., hydrothermal, hydrazine reduction, and UV-assisted photoreduction methods, to synthesize efficient BiPO4/rGO photocatalyst for hydrogen evolution.72 The sample containing 2 wt% GO exhibited the highest H2-production rate (30.6 μmol h−1) and was approximately two times higher than that of bare BiPO4. Although the BiPO4/rGO-2 nanocomposite showed the highest H2-production rate, the BiPO4/rGO-5 sample exhibited the strongest light absorption intensity and narrowest bandgap. It seems that an excessive loading rGO shields light and hinders the SC photoexcitation.72 The BiPO4/rGO nanocomposites (2 wt% rGO) were prepared using different methods and their activity for H2 production rate decreased in the following order: BiPO4/rGO-hydrothermal > BiPO4/rGO-photoreduction > BiPO4/rGO-hydrazine. This order was in good agreement with the optical band edge calculations. A shading effect was also reported for the anatase/graphene/rutile heterojunction.102 This study investigated the influence of the relative amount of anatase, rutile, and graphene on the H2 production rate and found that the optimum anatase/rutile (A/R) ratio was 7:3 and the optimal amount of graphene was 2 wt% (Fig. 8a). When 2 wt% graphene was introduced into the sample, a strong absorption above 420 nm was obtained and the photocatalytic H2 production rate increased from 1.1 to 1.7 mmol h−1 (Fig. 8b). However, further increase in the graphene loading beyond 2 wt% reduced the H2 production rate, as in the previous case of the BiPO4/rGO system. Liu et al. fabricated TiO2–graphene nanocomposites with adjustable TiO2 crystal facets.103 The role of different crystal facets on the cut-off wavelength of the nanocomposites was considered and TiO2-100–G demonstrated prominent red shift, indicating stronger interaction between TiO2-100 and graphene.
 | ||
| Fig. 8 (a) UV-vis absorption spectra of pure rutile, pure anatase, AR7/3, and AR7/3–2 wt% G samples, and (b) H2 production rates of different samples with various graphene content (at the same A/R ratio). Reproduced with permission from ref. 102 (Copyright 2014 American Chemical Society). (c) UV-vis absorption spectra for micro-RGO, nano-RGO, and QD–RGO, (d) UV-vis absorption spectra for pure CdSe, CdSe/micro-RGO, CdSe/nano-RGO, and CdSe/QD–RGO. Reproduced with permission from ref. 112 (Copyright 2014 Elsevier). | ||
An enhanced absorbance in the visible-light region can be obtained with doped graphene. Wang et al. prepared visible-light responsive hybrid nanosheets that comprised CdS and N-doped rGO.104 Compared with pure CdS, CdS/N-rGO exhibited a significant absorption enhancement in the visible region (550–800 nm). Recent studies incorporated graphene in binary systems and investigated their influences on PEC/photocatalytic activity. Wang et al. observed the visible-light sensitivity of graphene/CdS/Ag2S sandwich nanofilms for PEC water splitting.48 The results showed absorption enhancement in the visible-light region when graphene and Ag2S were introduced into CdS. An estimated optical bandgap energy of 2.13 eV was observed for the graphene/CdS/Ag2S and this structure was more appropriate for water splitting compared with bare CdS, graphene/CdS, and graphene/Ag2S/CdS. The energy band structure of graphene/CdS/Ag2S was suitable for efficient charge injection, separation, and transfer of photo-induced electrons and holes. Ullah et al. fabricated AgI-functionalized graphene (FG)–TiO2 with enhanced optical absorption.105 Possible explanations for the observed enhanced absorption and improved photocatalytic hydrogen evolution (230 μmol h−1) in this ternary system could be (i) an optimum loading effect, (ii) greater interfacial contact between FG and attached nanoparticles, and (iii) homogenous distribution of nanoparticles. Han et al.106 synthesized a novel 3D aerogel consisting of graphene, TiO2 nanoparticles, and MoS2 nanosheets. Owing to the excellent light absorption properties of graphene and narrower bandgap of MoS2, the ternary MoS2/TiO2/graphene aerogel demonstrated a profound enhancement in absorption intensity and distinct red shift in absorption edge between 385 and 405 nm. Hou et al. achieved highly efficient hydrogen production through the design of a ternary 3D architecture of CdS quantum dot (QD)/graphene/ZnIn2S4 heterostructures, which exhibited an enhanced absorption in the visible-light region compared with CdS QD/ZnIn2S4.107 Although graphene is commonly considered as an electron acceptor, the potential role of graphene as a photosensitizer of SCs has been also demonstrated, which excites electrons from graphene to SC. ZnWO4/graphene hybrids with different graphene contents were shown to enhance UV photocatalytic activity due to graphene sensitization.108 Zeng et al.109 demonstrated the effect of graphene incorporation as a visible-light sensitizer of TiO2 for hydrogen production, where the photoexcited graphene injects electrons into the SC CB and subsequently induces visible-light activity.108,110,111
Graphene in the form of multilayer sheets with a thickness of several hundred nanometers to micrometers can cause a light-shielding effect. Breaking graphene into graphene quantum dots (GQDs) can mitigate this effect.113 Tsai et al.112 investigated the PEC properties of three different types of rGO/CdSe, namely micro-rGO/CdSe, nano-rGO/CdSe, and QD–rGO/CdSe. In Fig. 8c and d, the absorption spectra indicate that the nano-rGO and QD–rGO (unlike the micro-rGO) had a well-defined HOMO–LUMO energy gap that enabled efficient light absorption and distinctive PL emission (not shown). This unique optical feature may diversify the applications of nano-rGO and QD–rGO, particularly in SC-based photocatalysis as they can contribute to overall photon harvesting as well as mediating the interfacial charge transfer.
The IPCE measurements of the pure CdSe and the three CdSe/rGO samples showed that the CdSe/QD–rGO exhibited the highest value. The IPCE improvement was observed in the near-UV region (350–400 nm) for the CdSe/nano-rGO and CdSe/QD–rGO, which matched the HOMO–LUMO absorption of the nano-rGO and QD–rGO, resulting in an enhancement in the overall photoconversion efficiency by absorbing additional photons in the UV region.112 The IPCE measurements of the pure CdSe and the three CdSe/rGO samples showed that the CdSe/QD–rGO exhibited the highest value. The enhanced IPCE was ascribed to facilitated charge pair separation (ET from QD–rGO LUMO to CdSe CB and concurrent hole transfer from CdSe VB to QD–rGO HOMO).112
2.3. Graphene as a high surface area host and support
A high degree of crystalline structure and large surface area are key prerequisites for reducing the recombination rate of photo-generated charge carriers.114 In this sense, mesoporous materials could be applied as photocatalysts since they supply continuous porous channels with high surface area and short charge-carrier migration distances within the mesoporous structure.40,115 Graphene nanosheets are also well known for their extra-large specific surface area (2600 m2 g−1). An atom-thick structure of graphene contains the highest specific surface area among all materials. As a result, the application of graphene as a support for various SC nanoparticle assemblies has been extensively studied in the last decade.116–119 Padhi et al.120 reported a novel photocatalytic system by hybridizing graphene with N-doped GaZn under a facile hydrothermal route for hydrogen production. The synthesized rGO/N-GaZn nanocomposites with different amounts of rGO (1, 3, 4, and 5 wt%) were denoted as 1rGO/N-GZ, 3rGO/N-GZ, 4rGO/N-GZ, and 5rGO/N-GZ, respectively. Fig. 9a and b compare the TEM images of N-GZ and rGO/N-GZ, clearly showing the dispersion of N-GZ on rGO sheets. The selected area electron diffraction (SAED) pattern of rGO/N-GZ exhibits the multiple bright continuous concentric rings corresponding to the diffraction of the (220), (400), (511), and (440) planes of polycrystalline N-GZ, which is consistent with XRD data. The arrangement of N-GZ nanoparticles on rGO prevented the restacking of graphene sheets and improved the stability of individual graphene sheets. The flat 2D-surface of graphene could act as a conductor of photoexcited electrons through a π-conjugated network to inhibit electron–hole recombination and enhance the photocatalytic activity of N-GZ nanoparticles. It should be also noted that the BET specific surface area was markedly enhanced when N-GZ nanoparticles were loaded on rGO sheets (Fig. 9c and d); the bare N-GZ surface area was 48 m2 g−1 and that of the rGO/N-GZ was in the range of 52–98 m2 g−1. This indicates that the presence of rGO as a support inhibited the agglomeration of N-GZ nanoparticles, thereby enhancing the surface area. This was further confirmed by TEM image analysis, which showed that N-GZ nanoparticles have an average particle size of 20 ± 0.9 nm, whereas that of rGO/N-GZ (16 ± 1 nm) is smaller.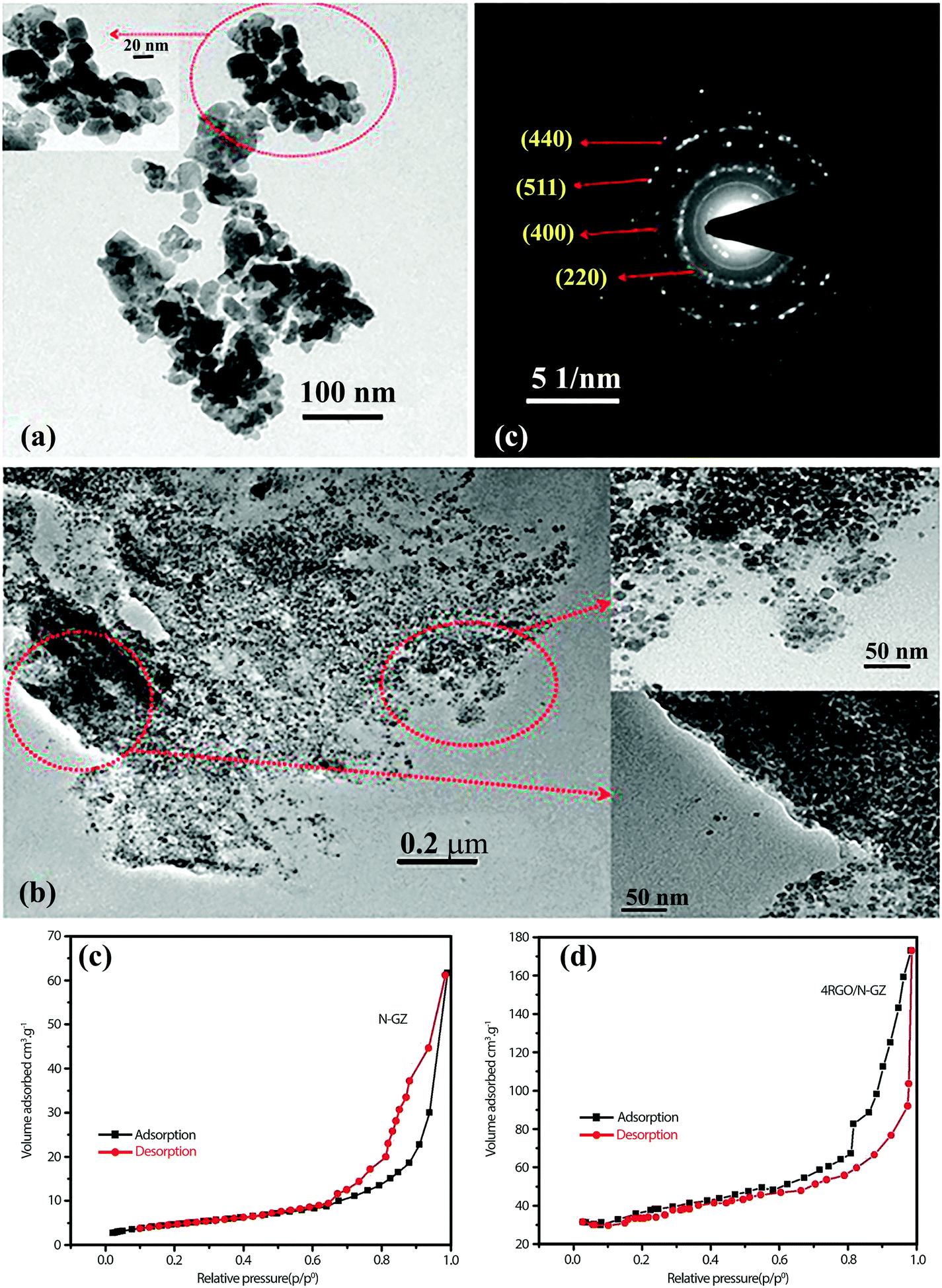 | ||
| Fig. 9 TEM images of (a) N-GZ, (b) rGO/N-GZ nanocomposite, and (c) SAED pattern of the rGO/N-GZ. (d) and (e) N2 adsorption isotherm of both composites. Reproduced with permission from ref. 120 (Copyright 2015 American Chemical Society). | ||
Despite the large surface area of graphene, the restacking of graphene sheets and formation of irreversible agglomerates during the assembly and drying processes can limit the access of the electrolyte ion to the active surface sites. To overcome these problems, 3D graphene frameworks (hydrogels and aerogels) have been developed recently.106,121,122 The physical properties of 3D graphene aerogel (e.g., volume, shape, and density) can be adjusted by the preparation methods.51,122,123 Han et al.124 reported the fabrication of TiO2 (P25) and CdS nanoparticles on 3D graphene-based aerogel via a facile one-pot hydrothermal process in a Teflon autoclave. The free-standing CdS/P25/graphene hydrogel was converted to aerogel during freeze-drying. An interconnected, micrometer-size, 3D porous network structure was observed for the CdS/P25/graphene aerogel, which contained CdS and TiO2 nanoparticles densely loaded on the graphene sheet supports. A summary of photocatalysts based on graphene and graphene derivatives for water splitting can be found in Table S1 (ESI†).
2.4. Status and prospects of graphene
Graphene has attracted wide attention, primarily owing to its improved charge-transfer kinetics with high electrical conductivity. In addition, the fact that graphene consists of only the earth-abundant carbon element and is relatively stable and chemically inert makes it a good candidate for practical applications that require mass production. Although graphene has various merits as a 2D material for PEC water splitting, it has some limitations for practical PEC water splitting applications. One of the main demerits of graphene in water splitting is its lack of photoactivity owing to the absence of a bandgap. As a result, graphene has been mainly employed as an electron transfer medium that is hybridized with photoactive SC materials in various ways in PEC and photocatalytic systems, which has been summarized in this section. Although graphene has been widely used in many PEC systems, there are few studies that have attempted to correlate the PEC activities with the intrinsic properties of the graphene/SC interface. This calls for more systematic investigation of (i) the interface between graphene and photoactive components at the molecular level, (ii) how the interfacial properties control the overall photoconversion processes, and (iii) ultimately, how we can design the most efficient graphene-containing hybrids with the highest photoactivity of water splitting. Another notable demerit of graphene is the poor catalytic activity of its pristine form, which makes it unsuitable as a hydrogen evolving catalyst. This is why graphene hybrids often need co-catalysts such as noble metal nanoparticles, although researchers aim to employ graphene as an alternative catalytic material to replace expensive noble metal catalysts. Therefore, urgent efforts in graphene research should be directed toward understanding, modifying, and controlling the intrinsic catalytic activities of graphene materials. Finally, the most severe shortcoming of graphene-based materials is the lack of long-term stability. Although graphene components have demonstrated relatively stable performances in their initial property evaluation across diverse applications, their real long-term stability under practical commercial application conditions has been never confirmed. Without overcoming this problem, graphene will remain only a novel laboratory material.3. Transition metal dichalcogenides (TMDs)
TMDs have attracted much attention due to their optical, mechanical, and electrical properties and have been studied across a wide range of applications such as catalysis, biosensors, photodetectors, transistors, solid lubricants, memory devices, lithium battery cathodes, photovoltaics, and photocatalytic and PEC conversions.125–138 The TMDs (e.g., MoS2, WS2, and TiSe2) shown in Fig. S8 (ESI†), are a large group of layered materials with the general formula MX2, where M is a transition metal element of group 4–10 ((Ti, Zr, Hf), (V, Nb, Ta), and (Mo, W)) and X is the chalcogen atom (S, Se, Te).139,140 TMD nanosheets can play different roles in PEC and PC applications. They can act as a photosensitizer by increasing light harvesting in the visible region of the solar spectrum, a charge separator through suitable energy band alignment, and a charge transporter. The exact role of 2D nanosheets depends on the use of the reaction system.141–149 Further detailed information on the role of TMD materials is discussed below.3.1. TMDs as a light harvester
2D WS2 and MoS2 nanosheets have appropriate energy bandgaps (Eg) for solar absorption. These can be tuned within the range 1.2–2 eV depending on thickness. Owing to the quantum confinement effect, different bandgaps can be obtained in 2D TMD nanosheets by controlling thickness and lateral size.150,151 These narrow bandgap materials can be used as a sensitizer to extend absorption of other SCs in the visible region.141–143,152,153 For instance, UV-vis absorption spectra showed that the bandgap energy of CdS nanoparticles decreased from 2.5 to 2.1 eV after depositing WS2 nanosheets on the CdS/ITO (Fig. 10a).143 As for MoS2 2D nanosheets, the loading of MoS2 on TiO2 also enhanced light absorption (Fig. 10b). The heterojunction of TMDs with the base SC material enhanced the visible-light absorption efficiency.106,141,143,154–156 | ||
| Fig. 10 (a) Tauc-plots of CdS/WS2/ITO thin films. The inset shows the corresponding absorption spectra.143 (b) UV-visible absorption spectra of (1) TiO2 nanofibers, (2) TiO2@MoS2 heterostructures, and (3) bare MoS2 nanosheets. Insets show the corresponding Tauc-plots of TiO2 and TiO2@MoS2 heterostructures to determine their bandgap values. Reproduced with permission from ref. 154 (Copyright 2014 Elsevier). (c) Schematic illustration of S atoms with mono-coordination, bi-coordination, and tri-coordination in a MoS2 sheet. (d) Co-catalytic mechanism of MoS2 sheet for H2 generation in lactic acid solution. Reproduced with permission from ref. 159 (Copyright 2014 American Chemical Society). | ||
3.2. TMD as a co-catalyst
Platinum (Pt) is the most efficient and commonly used co-catalyst in photocatalytic applications. Nevertheless, because of its high cost and low abundance, attempts have been made to identify alternative materials to substitute for Pt. Studies on 2D materials (e.g., graphene and TMDs and their composites) have opened new opportunities to find a suitable replacement for Pt. It has been verified that ΔGH* (the Gibbs free energy for atomic hydrogen adsorption) on the edge sites of nanoscale MoS2 is comparable to that of Pt.157 Based on DFT calculations, the surface bonding energy of atomic hydrogen on the MoS2 surface is close to zero, which is similar to that of Pt.158 Therefore, great efforts have been devoted to enhancing the catalytic activity of TMDs, particularly for H2 production.The CB position of bulk MoS2 is not adequate for H+ reduction but that of the nanosized 2D MoS2 structure is due to the quantum confinement effect.160 It has been shown that WS2 and MoS2 nanosheets can act as an efficient co-catalyst for photocatalytic H2 production with even higher activity than Pt.159,161–163 The superior co-catalytic activity of MoS2 has been confirmed experimentally by many studies. Annealing amorphous MoS2 nanosheets in junction with a TiO2 layer on a GaInP2 electrode resulted in a MoSx–TiO2 interfacial layer whose PEC activity was higher than that of a PtRu/GaInP2 electrode. The annealed MoS2/TiO2–GaInP2 electrode showed IPCE up to 75% across the visible-light range, which was about 10% higher than the PtRu-modified electrode.163 The maximum visible photocatalytic H2 evolution rate of Mn0.25Cd0.75S with a 0.7 wt% MoS2 loading was 12.5 mmol g−1 h−1, which was higher than that of Mn0.25Cd0.75S/Pt (1.0 wt%) (10.9 mmol g−1 h−1).161 These results indicate that MoS2 can be a low-cost co-catalyst comparable to Pt.
Fig. 10d presents the co-catalytic mechanism of H2 generation on a MoS2 nanosheet surface. The active S atoms on the exposed edges of MoS2 can increase its activity for H2 production. The activity of S atoms in MoS2 nanosheets depends on their coordination. The S atoms with mono- and bi-coordination (colored in red and dark brown in Fig. 10c and d) show a higher activity than tri-coordinated S for H2 production. Both mono- and bi-coordinated S atoms are unsaturated and can form strong bonds with H+ ions in solution. As a result, H+ ions are easily reduced to H2 by electrons. In contrast, there is no activity for saturated S atoms on the basal plane with tri-coordination. Therefore, the nanosized few-layer MoS2 and WS2 with more exposed edges and unsaturated active S atoms exhibit more activity toward hydrogen evolution. This mechanism has been verified experimentally by many studies. Linear scan voltammograms obtained under dark conditions for a MoS2 nanosheet decorated with p-type Cu2O (MoS2@Cu2O) showed that the proton reduction potential changed from −1.21 V (vs. SCE) for the Cu2O electrode to −0.72 V (vs. SCE) for the MoS2@Cu2O electrode. The reduction in overpotential was attributed to available active sites on the MoS2 nanosheets.164 Decorating TiO2–GaInP2 electrode with amorphous MoS2, which contained unsaturated S atoms, induced a saturated photocurrent of −11 mA cm−2 (at 0 V vs. a RHE in 0.5 M H2SO4 under 1 sun illumination), which was higher than that obtained for a PtRu/GaInP2 electrode (−10 mA cm−2). This was ascribed to the unsaturated S atoms on amorphous MoS2, which adsorbed H+ ions.163
3.3. TMD as a charge separator and transporter
The 2H-WS2 and 1T-WS2 phases also exhibit similar behaviors to their MoS2 counterparts. The combination of these two WS2 nanostructured polymorphs with TiO2 nanoparticles was investigated for photocatalytic water splitting.166 In the TiO2–2H-WS2 heterojunction, photo-generated electrons are transferred from 2H-WS2 to TiO2, whereas in the TiO2–1T-WS2 system, photo-generated electrons in TiO2 are transferred to 1T-WS2 (Fig. 11a and b).166 Clearly, the 1T-WS2 phase exhibited higher co-catalytic activity for H2 production due to higher charge mobility and more active reduction sites. 1T-WS2 is also a metastable phase and should not be considered as a stable co-catalyst for long-term performance. These two phases behave differently in charge generation, separation, and transportation.
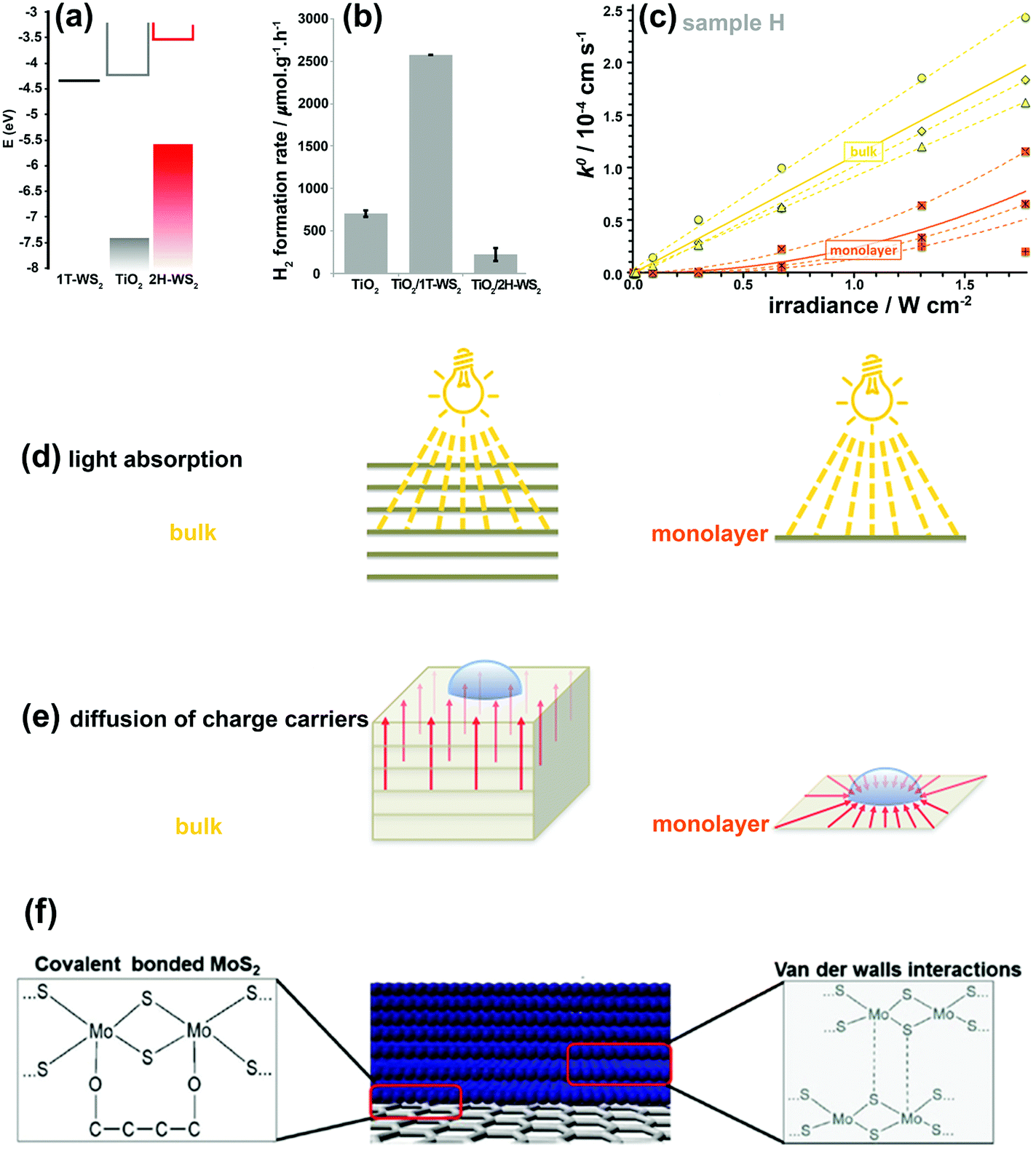 | ||
| Fig. 11 (a) Electronic band positions of 1T-WS2, TiO2, and 2H-WS2 nanostructures. (b) Photocatalytic H2 production rates for bare TiO2 and its composites with 1T-WS2 and 2H-WS2 nanostructures. Reproduced with permission from ref. 166. (Copyright 2014 American Chemical Society). (c) k0versus irradiance for monolayer and bulk MoS2. The dashed and solid lines show the best fits for the individual measurements and the averaged response, respectively. Schematic illustration demonstrating various light absorption efficiencies (d) and the different charge-carrier diffusion profiles (e) in monolayer and bulk MoS2. Reproduced with permission from ref. 180 (Copyright 2014 American Chemical Society). (f) Interfacial interactions between MoS2/MoS2 layers and MoS2/graphene. Reproduced with permission from ref. 181 (Copyright 2014 Wiley-VCH). | ||
The photo-generated electron–hole pair lifetime is a key factor influencing the efficiency of PC and PEC systems. The charge pair lifetime is very short in bare 2H-MoS2 and 2H-WS2 nanosheets. However, by constructing an appropriate architecture of 2D 2H-WS2 and 2H-MoS2 nanosheets combined with other SCs and charge-carrier materials, their lifetime can be significantly increased. For example, when the TiO2–MoS2 interface is irradiated, the electrons are excited from TiO2 VB to CB, and subsequently transferred to 2H-MoS2 CB for H2 evolution because the TiO2 CB position is more negative than in 2H-MoS2 CB.154 The same mechanism can be applied to SrZrO3–MoS2, MoS2–ZnIn2S4, Bi2S3/WS2, Ag2S@MoS2, and Zn0.5Cd0.5S/WS2 heterojunction interfaces.167–172 DFT calculations elucidate the charge-transfer mechanism between two SCs. Faraji et al.173 studied the interface of the Mo1−xWxS2/TiO2 heterostructure to understand how the presence of 2D dichalcogenide alloys enhances PEC activity under visible-light irradiation. The electrons transfer from TiO2 to Mo1−xWxS2, which induces better charge separation. In addition, the Mo1−xWxS2/TiO2 heterostructures exhibited higher effective mass ratios (mass of hole to mass of electron), which induced better charge separation, faster charge transfer to the surface, and consequently higher PEC activity.173
1T-MoS2 and 1T-WS2 counterparts have also been employed as co-catalysts and charge separators in PC and PEC applications. 1T-MoS2 nanosheets can act as co-catalysts and offer the following advantages: (i) noble-metal-free, (ii) high mobility for charge transport (semi-metallic behavior), (iii) high density of active sites for H2 evolution on basal planes, and (iv) high light transparency.174 2H-MoS2 and 1T-MoS2 were compared as co-catalysts for TiO2. The 1T-MoS2 nanosheets provide an electron delivery pathway with higher mobility and more catalytic H+ reduction sites on their basal plane. Thus, the photo-generated electrons on TiO2 nanocrystals do not need to migrate to the catalyst edge sites. The basal plane of the 2H phase shows no catalytic activity for H+ reduction and may block active sites on the bottom surface of TiO2 nanocrystals. Therefore, both basal reduction sites and shorter charge travel length can lead to excellent H2 production activity in TiO2–MoS2 (1T). However, it should be noted that 1T-MoS2 is a thermodynamically metastable phase and can be transformed back to 2H-MoS2.174 Thus, the successful application of 1T-MoS2 as an efficient co-catalyst is hindered, particularly in slow kinetics reactions. 1T-MoS2 can be stabilized by intercalation of ammonium175 and CO2165 and through the substitutional doping of several electron donors such as rhenium176 and vanadium177 atoms. It is also reported that 1T-MoS2 sheets can form chemical bonds to oxygen functional groups on GO, thus stabilizing the 1T phase in the 1T-MoS2/GO composite.178 Despite such attempts, the 1T phase still suffers from difficulties in its preparation process.
The linear dependence of k0 on light intensity for bulk MoS2 indicates that a linear diffusion profile is responsible between the deepest light-absorbing layer and the MoS2 surface after light penetrates the bulk MoS2 (Fig. 11d and e). Therefore, effective interlayer charge-carrier transport occurs between individual S–Mo–S layers. In monolayer MoS2, the photo-generated charge carriers are restricted and diffuse only through a single 2D sheet, which is a radial charge diffusion profile (Fig. 11d and e). Hence, a nonlinear (parabolic) dependence of k0 on irradiance intensity is observed in Fig. 11c. The normalized k0 for thickness highlights that PEC properties are limited only to the top layer of MoS2 and that the bulk layers can only contribute to the interlayer transport of photo-generated charge carriers without participating in the PEC reactions.
Considering the issues discussed relating to the catalytic activity of 2D TMD nanosheets under both dark and illuminated conditions, an optimum choice should be made between bulk and monolayer systems to achieve the best performance for energy storage/conversion applications. As such, the best texture with the most active sites, high conductivity, high surface area, and strong light absorption is offered by few-layered TMD nanosheets (5 layers in average) with edge-defected sites, which are strongly anchored vertically to the conductive substrate.134,180,182
In addition to PL and TRPL analyses, other electrochemical measurements can also be used to investigate the effect of interfacial contacts on charge transport. Tang et al.153 prepared a layered MoS2 coupled with a metal organic framework (MOF)-derived dual-phase TiO2 (MDT) electrode for PEC water splitting. Sn4+-exchanged MIL-125(Ti) and pure MIL-125(Ti) (donated as DT and 0DT, respectively) were synthesized and a MoS2 layer was deposited on DT to obtain xMDT samples (x represents the MoS2/TiO2 mol%). The PL intensity of the optimum xMTD samples was reduced as compared with the DT and 0DT samples, indicating that MoS2 nanosheets could effectively suppress photo-induced carrier recombination through interfacial charge separation. The Mott–Schottky plots (Fig. 12a) revealed that the flat band potential underwent a negative shift after decoration of DT samples with MoS2 nanosheets, proving that the heterojunction between TiO2 and MoS2 could hinder charge-carrier recombination. From the slope of the Mott–Schottky plots, the carrier density of the 0.5MDT sample was 5.5 times higher than that of 0DT, confirming the higher electron–hole separation efficiency. In addition, the lifetime (τe) of interfacial electrons was determined using Bode phase diagrams (Fig. 12b). τe was estimated to be 7.95 and 22.94 ms for 0DT and 0.5MDT, respectively. The larger τe for the MDT samples implied better atomic contact at the electrode/electrolyte interface and more efficient suppression of back-reaction electrons in the electrolyte. The results confirmed that the MoS2 loading could effectively facilitate interfacial carrier transfer processes.153
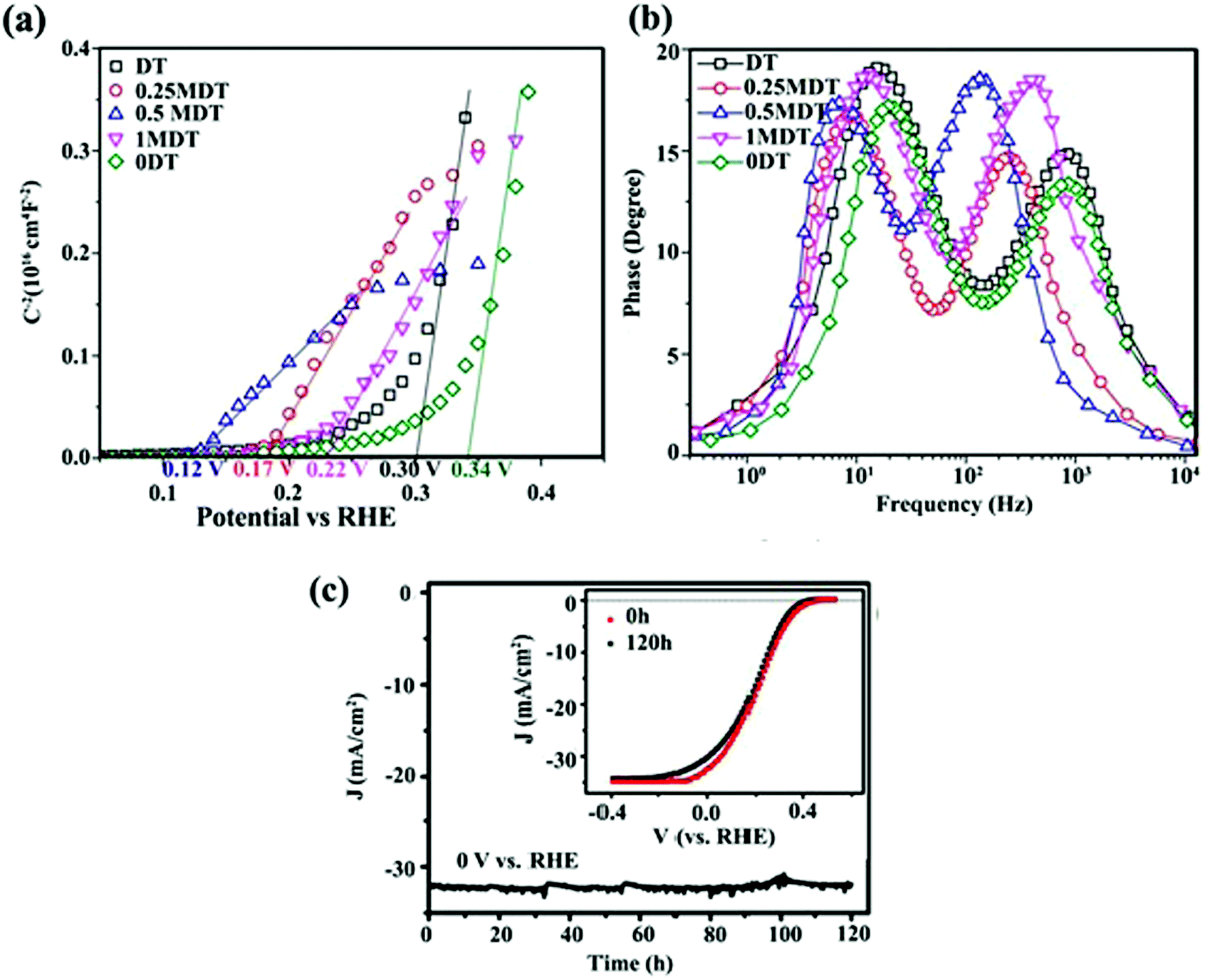 | ||
| Fig. 12 (a) Mott–Schottky plots and (b) Bode phase plots of 0DT, DT, and xMDT samples. Reproduced with permission from ref. 153 (Copyright 2017 The Royal Society of Chemistry). (c) Photocurrent stability of the MoS2/Al2O3/n+p-Si electrode during H2 generation. The inset shows initial J–V curve and that after and 120 h Xe lamp irradiation. Reproduced with permission from ref. 189 (Copyright 2017 American Chemical Society). | ||
Characterizing the photoresponse behavior of SC materials under irradiation is also critically important. In an ideal photo-responsive material, there is a linear relationship between measured photocurrent density (J) and light intensity (I) (J–I). However, under real conditions, the relation should presented as a power law dependence J–Iβ (0 ≤ β ≤ 1). A larger β value (closer to 1) implies a better photoactivity of the sample. When β = 1, there is no nonlinear effect, such as direct recombination of electron–hole pairs in the layer; therefore, separated free carriers dominate the process. In addition, scaling of the exponent close to 1 is expected for the layer when the electron and hole transports are comparably efficient. This is a good parameter for best charge separation and transportation in the samples.143 Zirak et al.143 studied the interfacial effect of CdS and WS2 nanosheets via the scaling exponent and obtained β for a CdS/ITO electrode and CdS/WS2/ITO heterojunction thin films. The β for the CdS/ITO electrode was 0.88, which was less than the value (β = 0.91) for the optimized CdS/2H-WS2/ITO (Fig. 13a). These results suggest that the bimolecular recombination of charge carriers decreased in the WS2/CdS heterojunction, leading to enhancement of the power conversion efficiency. This observation indicates that the photo-electron generation process is dominated by the electron injection step in photoabsorption.
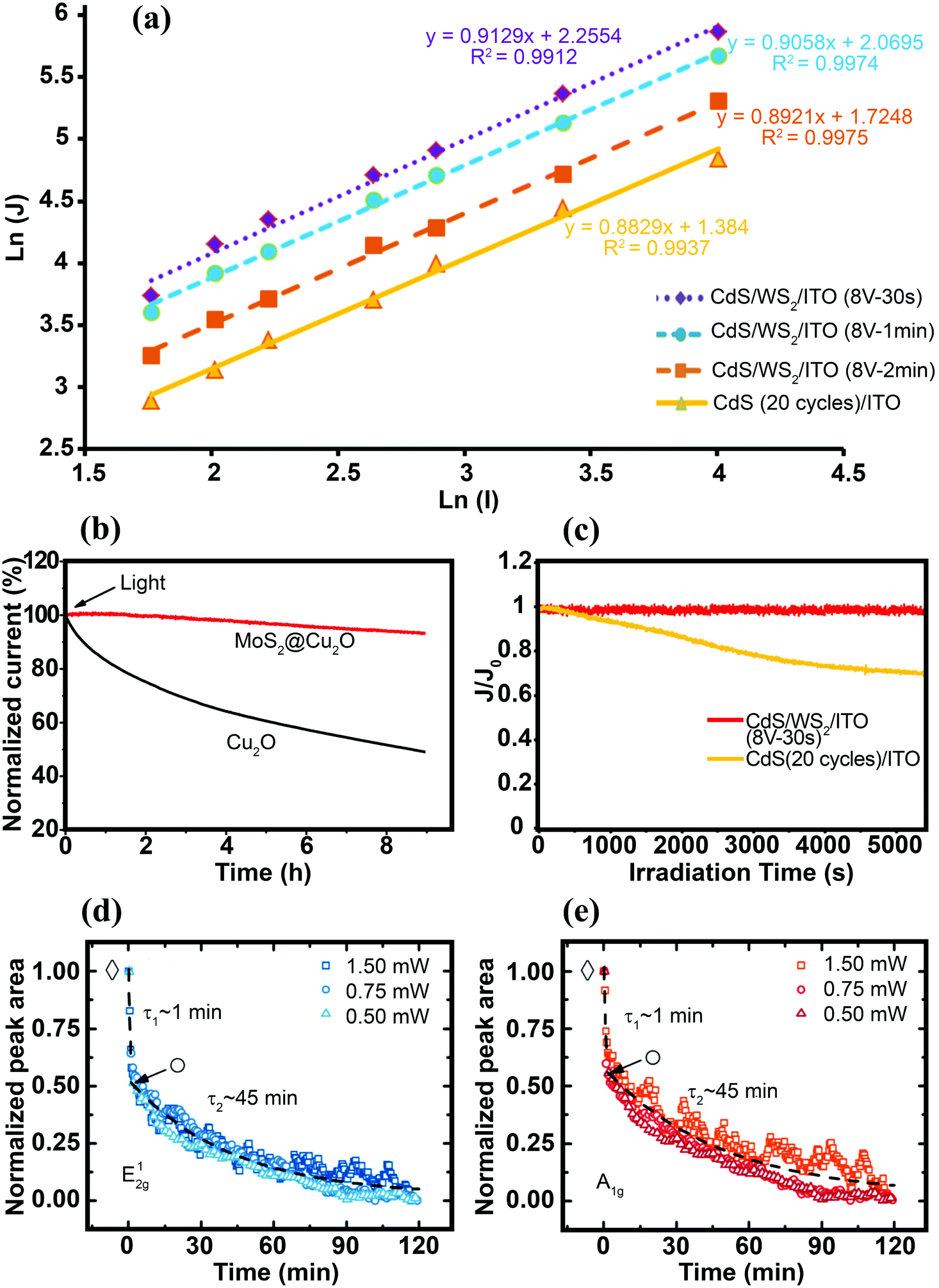 | ||
| Fig. 13 (a) Photocurrent density logarithm (ln(J)) as a function of light intensity logarithm (ln(I)) for the CdS/WS2/ITO thin films. For better presentation, the data for CdS/WS2/ITO (8 V-2 min) have been vertically shifted by −0.3 on the scale.143 (b) Stability of the bare Cu2O and MoS2@Cu2O electrodes under continuous light irradiation (λ = 480 nm, applied bias = −0.1 V vs. SCE). Reproduced with permission from ref. 164 (Copyright 2014 American Chemical Society). (c) Variation in the normalized photocurrent density (J/J0, where J0 is the photocurrent density at t = 0 s) vs. time obtained for CdS/ITO and CdS/WS2/ITO electrodes under continuous Xe lamp irradiation.143 Time dependence of normalized Raman peak area intensities of the E12g (d) and A1g (e) modes obtained for the MoS2 nanosheets immersed in DI water, under continuous laser illumination with various light intensities. The fits show different decay rates (τ1 and τ2) for bilayer and the remaining monolayer flake, respectively. Reproduced with permission from ref. 193 (Copyright 2015 American Chemical Society). | ||
3.4. TMDs as a stabilizer
Charge separation at the interface of the 2D materials (e.g., MoS2 and WS2 nanosheets) with other SCs is an important process in PEC water splitting. One of the most important results is photo-stabilization of non-stable photocatalysts such as Cu2O, CdS and GaInP2.163,164,189–191 p-Cu2O decorated with optimized MoS2 showed a seven-fold higher photocurrent density than did bare Cu2O. In addition, MoS2@Cu2O exhibited good photostability with a loss of only 7% from its original photocurrent after 9 h of continuous photo-irradiation.164 Excellent photocurrent stability was observed for a Al2O3/n+p-Si photocathode after the deposition of vertically aligned MoS2 nanosheets at the surface of photocathode.189 An optimized MoS2/Al2O3/n+p-Si photocathode exhibited high photocurrent (36 mA cm−2) with almost no loss after 120 h of illumination (Fig. 12c). The deposition of MoS2 nanosheets lowered the hydrogen evolution overpotential (0.02 V), suggesting that MoS2 nanosheets facilitated interfacial charge transfer and prevented photocorrosion of the silicon matrix.189The oxidation/reduction potential of materials is important to understanding how MoS2 and WS2 can protect materials against photocorrosion and provide good stability. For example, the oxidation and reduction potentials of Cu2O lie exactly within the bandgap of Cu2O. Therefore, photoexcited electrons and holes may react with the lattice ions before the successful interfacial charge transfer to redox electrolytes. As a result, self-photocorrosion occurs at the surface during PEC and PC experiments, causing damage to the Cu2O electrode.192 The same phenomenon is also responsible for CdS photocorrosion. The introduction of MoS2 and WS2 as co-catalysts, and the formation of an interfacial junction between Cu2O (or CdS) with TMD nanosheets, enhances the charge separation efficiency and prevents photo-generated electrons and holes from attacking the host material (Cu2O or CdS).164,190Fig. 13b and c clearly demonstrate that MoS2 and WS2 can stabilize the photoactivity of unstable Cu2O and CdS, respectively.
In addition to the positive effect of TMDs in making other SCs stable, the photostability of a bare TMD 2D nanosheet itself is an important issue, particularly in an aqueous environment. The photostability of exfoliated single- and few-layer MoS2 immersed in water was investigated using μ-Raman spectroscopy.193 The unaffected and non-photodegraded crystalline volume was determined in situ by real-time Raman spectroscopy through monitoring the time evolution of phonon-mode energies and changes in the integrated intensity of individual modes as fingerprints for the number of layers. The measured A1g and E12g phonon modes of MoS2 bilayers irradiated by laser beam (Elaser = 2.54 eV, more than the direct bandgap of MoS2) showed that transition occurred from the MoS2 bilayer to the MoS2 monolayer after ∼1 minute of light exposure (Fig. 13d and e). Long time laser irradiation times (various intensities) and monitoring Raman modes revealed that both of the phonon modes contained two distinct degradation rates with exponential decay rates. The initial fast decay rate (τ1 ≈ 1 min) was interpreted as stability at the bilayer edge site, whereas the slower decay rate (τ2 ≈ 45 min) was due to stability of the monolayer. The overall exponential decay was independent of laser input power between 0.5 and 1.5 mW, implying that the laser power (0.5 mW) saturated the corrosion process.193 In contrast to MoS2 edge sites, no flake corrosion was observed during laser irradiation for the basal plane of trilayer MoS2 immersed in water. In addition, when the edge and basal sites were irradiated with Elaser = 1.59 eV (lower energy than the Eg of few-layer MoS2), no corrosion was observed either on the MoS2 edge or the basal planes, even at a high laser intensity (1.5 W). Therefore, the photoexcitation of electron–hole pairs is essential for the photodegradation process. The role of reactive species in the electrolyte is also important to the process. The degradation rate of edge sites was significantly reduced when oxygen was removed from the electrolyte, and the MoS2 terrace sites immersed in water were stable under an extreme irradiation intensity of P ≈ 10 mW μm−2 (equivalent to 107 suns).193 The effects of various MoS2 and WS2165 interface structures on PC and PEC H2 production are summarized in Table S2 (ESI†).
3.5. Status and prospects of TMDs
TMD is a newly emerging material for PEC water splitting because it possesses suitable bandgap values (1–2 eV) for efficient visible-light absorption and active catalytic sites for hydrogen evolution. TMDs have various compositions, which enable flexible tuning of the bandgap via modification. However, the practical application of TMDs is not yet a reality as many critical problems need to be overcome. The most important issue is the low efficiency and durability of TMDs for PEC water splitting application. Since only the edge sites of the 2H phase of TMDs are catalytically active, it is highly desired to develop 2D morphologies of TMDs with higher active edge sites. More efforts should be devoted to producing chemically active basal planes of 2H-TMDs and to increase the stability of 1T-TMD nanosheets. The PEC activity of bare TMDs is not high enough for practical applications, mainly because of the low electrical conductivity of the 2H phase and the low stability of the 1T phase. To overcome this, the relationships between the PEC activities and the relevant electronic, optical, and surface-related properties of TMDs as a function of the number of 2D nanosheet layers should be understood. In addition, the effects of structural and surface/interface modifications on the PEC-related phenomena should be more thoroughly investigated. However, the synthesis and modification of TMDs with controllable size, surface defects, and a crystalline phase is highly challenging. More facile synthesis methods for TMDs are yet to be developed for practical and large-scale PEC applications.TMDs can also serve as hydrogen evolution catalysts in PEC system but they need to be more efficient. To make TMD co-catalysts more competitive with Pt, it is necessary to shift the ΔGH* toward zero on the TMD surface. To do this, the electronic and surface properties of TMDs should be controlled in a systematic manner to modify the hydrogen bonding energy to the catalytically active sites. This can be achieved via alloying and doping (replacing the metal (Mo and W) and/or chalcogen atoms), defect engineering, phase (2H, 1T or a mixture of the two), and strain engineering. On the other hand, the most common strategy for employing TMDs in PEC water splitting is to hybridize them with other SC materials. In particular, the development of a interfacial junction between TMDs and SC with good stability is critical for practical applications. Since the deposition of thin layers of TMD on SC is difficult, better methods to minimize the defects between the two junction materials need to be developed.
4. Polymeric graphitic carbon nitride (g-C3N4)
4.1. g-C3N4 as a stabilizer
Polymeric graphitic carbon nitride (g-C3N4:GCN) is receiving intense interest because of its good thermal and chemical stabilities.194 Yang et al.195 prepared a GCN/CuInS2 (CIS) composite thin film as a photocathode for hydrogen production. There is a work function difference between GCN film on a CIS substrate (4.3 eV) and an unmodified CIS substrate (5.5 eV), which indicates the formation of an electric field at the interface, which should facilitate the separation of charge carriers under irradiation. Fig. 14a shows the photocurrent–time course for a GCN/CIS photocathode in acidic aqueous solution under continuous illumination (λ > 400 nm) at a constant potential of −0.5 (vs. Ag/AgCl). The time-profile of the photocurrent is divided into two parts: (i) the initial period of 4 h with a decreasing current and (ii) the following period of 18 h with a constant photocurrent. The initial decrease in photocurrent could be attributed to surface recombination of photo-induced charge carriers. The continuing photocurrent showed long-term stability, which implies that incorporation of GCN protected the CIS photocathode and that there was a negligible direct contact between the CIS and the acidic solution. The role of GCN as a passivation layer reduced the surface and interface states. Similar behavior was reported by Wang et al., who fabricated GCN/ZnO nanotube arrays.196 In PL analysis, the lower emission intensity of the GCN/ZnO/FTO photocathode indicates a slower recombination rate of photo-generated charge carriers as compared with pristine GCN (Fig. 14b). The EIS of the samples revealed a negative shift in flat band potential, a higher donor density (Nd), and a lower charge-transfer resistance when GCN was incorporated into ZnO. According to Wang et al.,196 two factors may contribute to the enhanced PEC properties of GCN/ZnO. Since a type II heterojunction was formed in the GCN/ZnO photoelectrode, the excited electrons on GCN were transferred to the FTO substrate through the CB of ZnO, facilitating the separation of the charge carriers (Fig. 14c). The formation of an external electric field across the interface under the applied potential could further promote the transfer and separation of photo-generated charge carriers. GCN also serves as a protection layer for CdS, which is subject to photooxidative corrosion by holes. Zheng et al.197 fabricated uniform hollow carbon nitride spheres (HCNS) with diameters of ∼320 nm and loaded CdS QDs (5–7 nm in size) onto the shell of the HCNS (Fig. 14d). PL analysis showed that the PL emission intensity was reduced when CdS QDs were loaded on the HCNS surface, which can be attributed to charge separation through the CdS/HCNS interface. EIS Nyquist plot analysis showed that the semicircle for CdS–HCNS is smaller than that for HCNS (Fig. 14e), which also supports that the hypothesis that the favorable band alignment at the interface (as illustrated in Fig. 14f) facilitates the interfacial charge transfer. | ||
| Fig. 14 (a) Time course of the photocurrent for the GCN/CIS photocathode in 0.1 M H2SO4 aqueous solution (pH = 1) under continuous illumination of visible light. Reproduced with permission from ref. 195 (Copyright 2013 The Royal Society of Chemistry). (b) Photoluminescence spectra of the GCN and GCN/ZnO/FTO. (c) Proposed mechanism of charge separation and transfer between GCN and ZnO nanotube arrays under visible-light irradiation. Reproduced with permission from ref. 196 (Copyright 2014 Springer). (d) Scanning electron microscopy, (e) EIS Nyquist of CdS–HCNS sample, and (f) schematic illustration of CdS–HCNS heterojunction system. Reproduced with permission from ref. 197 (Copyright 2015 Elsevier). | ||
4.2. Modification of GCN
The intrinsic photocatalytic activity of GCN in PEC water splitting requires significant improvement because of its low surface area, high recombination rate, and poor optical absorption above 420 nm. To overcome these problems, various modification approaches have been proposed.198–202 These strategies will be discussed below. | ||
| Fig. 15 (a) Typical AFM of the as-prepared GCN nanosheets and corresponding line scan of different regions. (b) Valence band XPS spectra of layered GCN and GCN nanosheet. Reproduced with permission from ref. 204 (Copyright 2015 American Chemical Society). (c) EIS response of bulk GCN ((i) under dark conditions and (ii) under visible-light irradiation) and GCN monolayer ((iii) under dark conditions and (iv) under visible-light irradiation). Reproduced with permission from ref. 205 (Copyright 2013 The Royal Society of Chemistry). (d) Bode phase plots and (e) H2 production from aqueous solution of bulk GCN and nanosheet–GCN. Reproduced with permission from ref. 206 (Copyright 2016 Elsevier). | ||
The effect of GCN exfoliation on the charge-transfer mechanism can be investigated by EIS. Fig. 15c presents the EIS Nyquist plots of single atomic layer GCN and bulk GCN electrodes under dark and visible-light illumination conditions. The value of Rct decreased from 2571 to 980 Ω when bulk GCN was exfoliated into nanosheets, implying more efficient charge transfer in the GCN monolayer because of electron–phonon interaction, which enhances the conductivity of material. Lin et al. carried out analysis for periodic on/off photocurrent responses and demonstrated similar trends.205 The GCN nanosheets exhibited higher photocurrents as compared with the bulk GCN, which implies that electrons are more easily transferred on the GCN nanosheets. The enhanced activity of the GCN nanosheets can be ascribed to different factors, including: (i) a higher surface area leading to more reactive sites and (ii) enhanced charge separation and transfer by shortening the migration distance between the charge generation sites and the interfacial transfer sites.205 One of the conventional methods to increase surface area is to generate porosity in the structure. Ma et al. prepared high density porous GCN nanosheets206 of high surface area. The contact between the charge carriers and the water molecules was enhanced due to the porous nature of the prepared GCN. In addition, Bode plots of the samples shown in Fig. 15d indicated that electron lifetime in the GCN porous nanosheets increased to 32.4 ms (from 17.3 ms in the bulk GCN). As a result, the hydrogen production rate on the GCN nanosheets was approximately six times higher than that of the bulk GCN (Fig. 15e).
In recent studies, the synthesis of GCN QDs has attracted much attention owing to their features, including: (i) narrow size distribution and (ii) high fluorescence emission.207 Li et al.208 fabricated and grafted GCN QDs onto the inner wall of single-crystalline TiO2 NTAs, which improved absorption in the wide visible region as well as the charge separation efficiency. This can be ascribed to the formation of a heterojunction at the interface between the GCN QDs and the TiO2 NTA walls. The sample containing an optimal amount of GCN QDs exhibited a lower resistance and a high photocurrent (Fig. 16a). However, a higher GCN QD loading reduced the photocurrent response. This is ascribed to the agglomeration of GCN QDs, which reduces the light absorption and the interfacial interaction at the GCN QD/TiO2 NTA heterojunction. The proposed mechanism of charge separation and transfer in GCN QDs/TiO2 NTAs is illustrated in Fig. 16b. The multi-reflections of light inside the TiO2 NTAs resulted in more photo-electron generation. A similar study was conducted by Su et al. on a GCN QD-modified TiO2 NTA photoelectrode,209 which presented a higher photoconversion efficiency compared with pristine TiO2 NTAs (Fig. 16c).
 | ||
| Fig. 16 (a) Photocurrent responses under visible-light (λ > 420 nm) illumination (the as-obtained g-C3N4/TiO2-NTAs electrodes are labeled CTX, where X represents the mass (X = 0, 1, 3, 5, or 7 g) of melamine in the crucible before the CVD process). (b) Schematic illustration of charge separation and transfer in GCN/TiO2-NTAs under visible-light irradiation. Reproduced with permission from ref. 208 (Copyright 2016 Elsevier). (c) Corresponding photoconversion efficiency of TiO2 and GCN QDs/TiO2 NTAs. Reproduced with permission from ref. 209 (Copyright 2016 Elsevier). | ||
The incorporation of elements and impurities into the GCN framework is an intriguing approach for promoting the electrical, optical, and surface properties of GCN. For example, P-doping causes an increase in electrical conductivity, leading to a higher charge-carrier density; thus, the photocurrent can be enhanced by a factor of five.211 Ran and co-workers fabricated a porous P-doped GCN nanosheet (PCN-S) with a wide pore size distribution using thermal exfoliation.212 Among the different synthesized samples, including bulk GCN (CN-B), GCN nanosheets (CN-S), bulk P-doped GCN (PCN-B), and PCN-S, PCN-S exhibited the largest surface area. XPS analysis confirmed the substitution of C by P to form P–N bonds. In Fig. 17a, the PCN-S sample shows a very long absorption tail, attributed to the formation of mid-gap states owing to the hybridization of C2s2p, N2s2p, and P3s3p below the CB of GCN, which further facilitated photoexcitation of electrons from the VB (Fig. 17b). They suggested that PCN-S showed the highest hydrogen production rate owing to several factors: (i) extension of visible-light absorption up to 557 nm owing to the existence of mid-gap states; (ii) the role of mid-gap states as charge separation centers, which suppress the recombination rate; (iii) increase in optical path via multiple scattering effects in the macroporous structure of PCN-S; and (iv) significant reduction in charge diffusion path. Aside from phosphorus, numerous studies have attempted the doping of non-metal elements such as carbon,213 sulfur,214 and oxygen215 into GCN to alter the electrical and optical properties and enhance the photocatalytic performance. Ruan et al.216 reported the fabrication of a photoanode comprised of nanojunction structures on carbon nitride (CN) film with a boron-doped carbon nitride (BCN) monolayer. This exhibited a photocurrent density of ∼100 μA cm−2 at 1.23 V (vs. RHE) under 1 sun irradiation, which was 10 times higher than that of a bulk CN photoanode. Co-doped GCN nanosheets (e.g. B/F) have also received interest due to the low recombination rate of charge carriers and the high H2 generation rate.217
 | ||
| Fig. 17 (a) DRS spectra and (b) electronic band structures. Reproduced with permission from ref. 212 (Copyright 2015 American Chemical Society). (c) Photocurrent vs. time plots of Fe2O3 and GCN/Fe2O3 electrodes at the applied potential of 0.23 V (vs. Ag/AgCl). (d) Schematic representation for electron–hole separation and transfer at the GCN/Fe2O3 photocatalyst interface. Reproduced with permission from ref. 221 (Copyright 2014 Elsevier). (e) SC–SC all-solid-state Z-scheme structure. Reproduced with permission from ref. 223 (Copyright 2015 Thee Royal Society of Chemistry). | ||
A popular metal oxide photocatalyst, TiO2, has been considered as an appropriate SC to form a heterojunction with GCN. GCN/TiO2 heterojunctions have been synthesized with different facets of TiO2. The different crystal facets have various surface states, different atomic arrangements, and termination of bonding networks with different surface energies. Huang et al.224 investigated the direct Z-schemes heterojunction between GCN and a TiO2 hollow nanobox with co-exposed (001) and (101) facets. Charge-carrier migration occurs at the interface of two SCs in conventional (e.g. type II) designs, resulting in energy loss due to band alignment. A Z-scheme system has been developed in which two SCs with a staggered alignment are brought into contact with each other. In Fig. 17e, the photo-generated electrons residing at the CB edge of SC 1 can recombine with photo-generated holes residing at the VB edge of SC 2. The remaining holes from SC 1 and electrons from SC 2 can be used for oxidation and reduction reactions, respectively.223 Therefore, the enhanced photocatalytic activity of the TiO2-hollow nanobox/GCN heterojunction can be attributed to: (i) hindered charge recombination owing to the spatial separation of electrons and holes at the interface of GCN and TiO2 and (ii) improved spatial charge separation within a single TiO2 nanobox because of the formation of an internal electric field across surface heterojunction between different crystal facets of anatase (i.e., (101) and (001)).224
The metal-core@SC-shell structure has received much attention owing to: (i) the protection of metal against corrosion; (ii) the maximized interaction between metal and SC, thus favoring the transfer of plasmonic energy; and (iii) penetration of the local electromagnetic field into the shell. Bai et al.226 developed core–shell Ag@GCN nanocomposites (see Fig. 18a) and a continuous well-defined interface was observed between Ag nanoparticles and GCN shells. According to UV-vis absorption curves, core–shell Ag@GCN demonstrated weak and broad plasmon absorption peaks at around 407 and 600 nm, which showed intensity dependence on Ag contents (Fig. 18b). The peak intensity at 334 nm decreased when Ag contents increased, reflecting in the LSPR response of the Ag cores. In addition, Ag@GCN demonstrated significant enhancement in hydrogen generation compared with pristine GCN. The core metal Ag acted as a sink for photo-electrons, resulting in longer charge-carrier lifetimes.
 | ||
| Fig. 18 (a) HRTEM images of Ag@GCN-0.5 wt% photocatalyst and (b) UV-vis absorption curves of the GCN and Ag@GCN photocatalysts. Reproduced with permission from ref. 226 (Copyright 2014 Elsevier). (c) HRTEM image of GL-MoS2/GCN4 (2.0%). Reproduced with permission from ref. 230 (Copyright 2016 Wiley-VCH). (d) Proposed photocatalytic mechanisms of charge transfer in the MoS2–GCN heterojunction system under visible-light irradiation. Reproduced with permission from ref. 231 (Copyright 2013 Elsevier). (e) The charge-transfer mechanism of the mt-GCN/MoS2 sample in the case of p–n-type contact. Reproduced with permission from ref. 232 (Copyright 2016 American Chemical Society). (f) I–V curves for WO3 NSAs and WO3/g-C3N4 NSAs. Reproduced with permission from ref. 233 (Copyright 2013 The Royal Society of Chemistry). | ||
The 2D MoS2 nanosheet has received priority consideration owing to: (i) a suitable bandgap of 1.9 eV; (ii) a large specific surface area; and (iii) a pivotal role as a co-catalyst. In addition, the lattice mismatch difference between GCN and graphene such as MoS2 (GL-MoS2) is small; therefore, an intimate contact could form, thus promoting photo-induced charge separation across the interfacial contact. Under this condition and considering the above advantages of MoS2, Yan et al.230 synthesized a GL-MoS2/GCN heterojunction with different GL-MoS2 concentrations using a facile solvo-thermal method.
The HRTEM was applied to confirm the existence of a heterojunction between GCN and GL-MoS2. As is illustrated in Fig. 18c, an interface with a well-formed contact led to improvement in the transfer of photo-generated carriers. Moreover, a reduced optical bandgap, as well as an enhanced absorption intensity in the visible region, resulted from the light-harvesting feature of the GL-MoS2. Furthermore, EIS measurements revealed that the introduction of GL-MoS2 decreased the charge-transfer resistance, which prolonged electron lifetime. Fig. 18d sheds light on a possible mechanism for the enhancement of charge separation. As shown below, the layered MoS2 provided hydrogen-generating catalytic sites that could accept photo-induced electrons.231
A different mechanism was proposed for the observed improved photocatalytic activity of the MoS2–GCN heterojunction structure. Depending on preparation method, MoS2 shows either an n-type or p-type characteristic. In this regard, Ye et al. prepared a visible active MoS2 (p-type)/S-doped GCN (S/GCN)(n-type) heterojunction using a combination of CVD and hydrothermal growth methods.232 According to their optical measurements, an enlargement in bandgap with respect to GCN was attributed to the quantum confinement effect and homogenous doping of S atoms. However, the introduction of MoS2 into S–GCN caused a bandgap reduction, thus absorbing more photons. This led to the generation of more photoexcited charge carriers. The photocurrent–time analysis of S–GCN and MoS2/S–GCN were measured at an applied bias of 0.5 V (vs. Ag/AgCl) under visible-light illumination. In comparison with S–GCN, the deposition of the MoS2 layer on S–GCN increased the anodic photocurrent to a significant degree. It should also be noted that different analyses, such as the OCP transient tests and EIS measurements (Mott–Schottky plots are presented in the ESI†), were performed to reveal all prepared samples showing n-type characteristic behavior. Fig. 18e depicts the possible mechanism behind the observed PEC enhancement. After the contact between S–GCN and MoS2, the bands of MoS2 exhibited a negative (upward) shift, whereas, the energy bands of S–GCN shifted positively (downward). In this situation, photo-induced charge separation was promoted owing to electron migration from the CB of MoS2 to the CB of the S–GCN and hole migration from the VB band of S–GCN to the VB band of MoS2.232
Li et al.233 successfully fabricated 2D/2D WO3/g-C3N4 nanosheet arrays (WO3/g-C3N4 NSAs) for the splitting of seawater. According to the results, shown in Fig. 18f, the WO3/g-C3N4 NSAs exhibited a two-fold higher photocurrent density than that of WO3 NSAs. A similar result was reported by Y. Ma et al.234 for Bi2MoO6 nanosheet arrays modified with ultrathin GCN monolayers as co-catalysts to enhance PEC activity. This unique photoanode yielded a photocurrent density of 520 mA cm−2 at 0.8 V (vs. SCE) under visible-light irradiation, which was 2.3 times higher than that of the pure Bi2MoO6 nanosheet film.
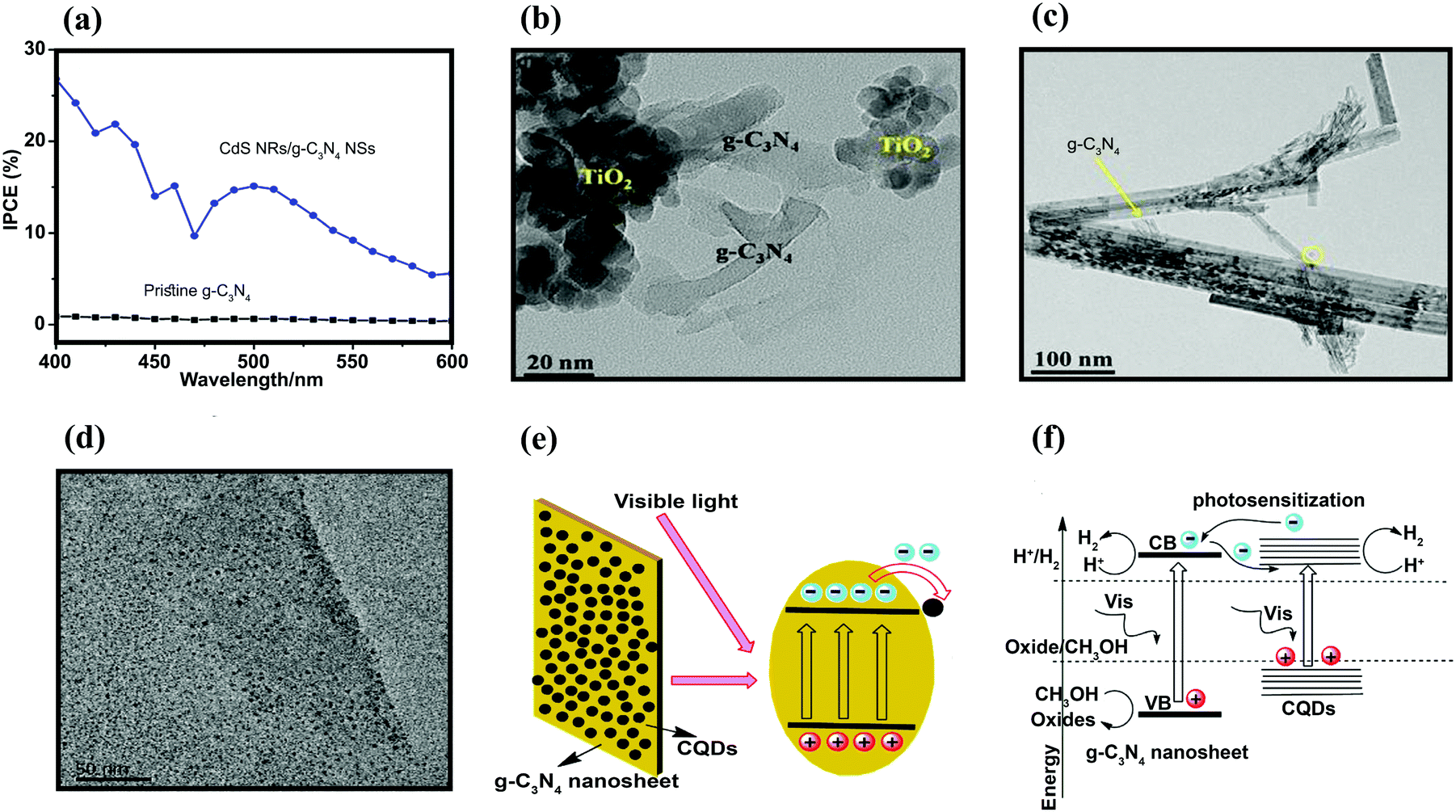 | ||
| Fig. 19 (a) IPCE plots of pristine GCN and CdS NRs/GCN NS. Reproduced with permission from ref. 235 (Copyright 2016 Springer). (b and c) TEM images of 10%-GCN/TiO2 NPs and 10%-GCN/TiO2 nanotubes, respectively. Reproduced with permission from ref. 236 (Copyright 2015 Elsevier). (d) TEM images of CQDs-0.2%/GCN nanosheets, (e) illustration of the photocatalytic process for GCN/CQDs, and (f) photocatalysts under visible-light illumination (λ > 420 nm). Reproduced with permission from ref. 202 (Copyright 2016 Elsevier). | ||
The formation of a strong interfacial connection between TiO2 NTs/TiO2 NPs and GCN was confirmed by TEM analysis (Fig. 19b and c). The interfacial interaction between GCN and TiO2 (NTs and NPs) was also confirmed by XPS analysis through shifting the N 1s spectrum toward higher binding energies when incorporating GCN into TiO2 (NTs or NPs). The 3%-GCN/TiO2 nanotubes exhibited the highest photocurrent density and were consistent with the PL spectrum. The 3%-GCN/TiO2 NTs indicated the lowest peak intensity of all TiO2 samples with different GCN contents. This was because direct Z-3%-GCN/TiO2 NTs showed more efficient Z-scheme type transfer of the photo-induced charge.236 In addition, GCN was deposited onto ZnO NR arrays, forming a core–shell heterostructure in a thermal annealing process. Different analyses further confirmed that GCN/ZnO NRs exhibited better PEC performance. In the DRS spectra, GCN/ZnO NRs significantly enhanced absorption intensity, indicating a low recombination rate of photoexcited electron–hole pairs. Based on PEC measurements, the addition of ZnO NRs into GCN led to significant enhancement in photo-induced charge separation across the formed heterojunction. The enhanced PEC activity could be due to appropriate band edge potentials, resulting in ET from GCN to ZnO. This ET was further improved by applying a bias potential owing to the formation of an external electric field between GCN and ZnO NRs.237 Xiao et al. showed similar results for a ternary GCN/Pt/ZnO NW photoanode.238 In addition, Li et al.239 synthesized 2D/1D GCN/WO3 heterojunction photoanodes by impregnating GCN nanosheets into WO3 NRs to achieve exceptional PEC performance in the visible-light region. The IPCE value of the GCN/WO3 photoanode was 57.8% at 330 nm and 8.4% at 430 nm, which was higher than that for a pristine WO3-NR photoanode (43.8% at 330 nm and 2.3% at 430 nm). The facilitated charge separation at the interface was considered the main reason for the enhanced PEC activity.
4.3. Status and prospects of GCN
GCN is a metal-free photocatalyst that has suitable bandgap and band edge positions for both hydrogen production and water oxidation. GCN has attracted intensive interest from scientists owing to its facile synthesis, low material cost, and visible-light activity. In particular, the diversity of precursor molecules and the ease of synthetic modification have led to the preparation of a number of GCN-based materials. However, GCN has some noticeable demerits for PC and PEC applications such as fast charge recombination, a relatively large bandgap (∼2.6 eV) that does not allow sufficient solar absorption, and low surface area. Among these, low light absorption (λ < 460 nm) is the most critical issue as it limits the theoretical maximum efficiency of GCN. Therefore, GCN has been modified to improve its light absorption efficiency using the following approaches: (1) hybridization of GCN with other SCs with suitable bandgaps, (2) doping with different metal and non-metal elements for higher visible-light absorption, and (3) noble metal nanoparticle (e.g., Au, Ag, and Pt) loading to enhance visible-light absorption through the SPR effect. Another approach to improving the photocatalytic activity of GCN is nanostructuring of its bulk material to increase the effective surface area and reduce the charge recombination rate. Various synthetic approaches have been employed to prepare a number of modified GCN compositions and structures with enhanced photoactivity. Despite extensive efforts in GCN research, GCN as a solar conversion material is still far from usable in practical applications. For example, fabricating stable GCN electrodes with the desired morphology and activity has not been well established for PEC applications. Therefore, innovative approaches for improving the photo-efficiency and stability of GCN and the development of a facile fabrication method for GCN electrodes are urgently needed for PEC applications.5. Emerging 2D materials
In addition to graphene, TMDs, and GCN, which have been discussed above, elemental 2D materials, layered metal oxides, oxyhalides, and layered double hydroxides are being actively investigated for their potential application to PEC water splitting.5.1. Elemental 2D materials for PEC applications
The specific features of graphene have prompted researchers to seek other elemental 2D materials. For example, (h-BN), silicene, germanene, stanine, and phosphorene have been developed.244 In these newly discovered 2D materials, a monolayer of black phosphorous (BP)245 and titled phosphorene have been fabricated using a sticky-tape method.246 Phosphorene has various advantages such as (i) quantum confinement and improved electronic/optical properties; (ii) low carrier recombination due to surface passivation without dangling bonds; (iii) matching with other 2D materials to form heterostructures; (iv) large lateral size with extremely high specific surface area; and (v) a thickness-dependent adjustable direct bandgap. These properties make phosphorene a promising candidate in many photocatalytic applications.245,247,248 In spite of these advantages, the practical application of phosphorene remains limited owing to the potential for its degradation in air and water. Several studies have attempted to tackle these problems in order to produce air-, water-, and light-stable phosphorene. Wang et al.249 produced water- and light-stable phosphorene nanosheets and applied them as a photosensitizer for singlet oxygen evolution. The result is useful for the future application of 2D phosphorene in catalysis. Sa et al.246 carried out DFT calculations and reported that the VBM of pristine phosphorene was not suitable for H2O oxidation under ambient conditions (25 °C); however, its CBM showed appropriate potential to reduce H+ to H2 at pH = 0, implying that phosphorene could perform half of the water splitting reaction. Nevertheless, the redox potential of water was dependent on solution pH. The investigations also revealed that at pH = 8, the VBM and CBM of phosphorene were at the appropriate condition for the redox potential of water.246 These results indicate that phosphorene could be used as a photocatalyst for solar water splitting. In addition, phosphorene could be applied as a co-catalyst for modifying other photocatalysts such as CdS, ZnS,250 ZnxCd1−xS,251 and g-C3N4.252 Ran et al.250 computationally and experimentally studied the effects of the mechanical mixing of phosphorene with CdS, ZnS, and Zn0.8Cd0.2S. The coupling of phosphorene with CdS resulted in a significantly improved photocatalytic H2-production activity of 11192 mol h−1 g−1 and a promising quantum yield of 34.7% at 420 nm. This quantum yield was higher than that of CdS coupled with a metal-free co-catalyst. The results further indicate that this enhanced efficiency was due to the proper electronic interaction between CdS and phosphorene, together with a favorable band structure, superior charge transfer, and copious active sites on phosphorene.250
Boron nitride (BN) is also considered an elemental 2D material. BN contains different structures including amorphous BN, hexagonal BN, cubic BN, and rare wurtzite BN.253 Hexagonal BN (h-BN) is the most stable crystalline form among all these structures; the layered structure, arranged with boron and nitrogen atoms in a 2-dimensional plane, shows a honeycomb lattice structure, which is similar to the structure of graphene.254 Although boron and nitrogen atoms are strongly bound within a layer, the layers are piled together via weak van der Waals force and this can be easily exfoliated into thin layer/monolayer. h-BN can be used as complementary material to graphene owing to their similar properties.255 h-BN shows high thermal stability and conductivity, high mechanical strength, and excellent lubricating properties. These properties can be applied in many applications such as thermoelectric devices and microelectronics.256 h-BN is also a SC with a large bandgap (over 5 eV), and this is not suitable for use as a photocatalyst. However, h-BN-based materials are widely applied in PC and PEC systems by modifying their properties, such as elemental doping and surface functionalization, with other SC materials (e.g. TiO2 and WO3).256–258 He et al.259 synthesized the heterostructure of carbon nitride and h-BN nanosheets (CN/BN) using an annealing mixture of h-BN and urea (as a precursor of carbon nitride).259 The bandgap of the hybrid CN/BN varied from 3.84 to 2.67 eV under increased loadings of carbon nitride, which improved the light absorption efficiency of the h-BN. The CN/BN heterostructure exhibited enhanced photocatalytic productions of H2 and H2O2 of 2.4 and 59.8 μmol h−1, respectively. The results were comparable to Pt-loaded h-BN. Huang et al.260 prepared a material with a ternary component that doped carbon into BN nanosheets (h-BCN). The bandgap of h-BN was reduced to 2.08 eV by the doping of carbon into h-BN, enabling photocatalytic H2 generation (∼4 μmol h−1) under visible-light irradiation.
5.2. 2D metal oxide nanomaterial-based photocatalysts
Over recent years, several metal oxides such as TiO2, Fe2O3, ZnO, SnO2, and WO3 have been widely used as photocatalysts for solar water splitting. Despite increasing interest in these 2D materials, non-layered oxide nanosheets (TiO2 and Fe2O3) have been extensively studied and compared with layered 2D nanosheets such as graphene and TMD.261 The synthesis of 2D non-layered nanomaterials remains challenging owing to the strong interaction between metal cations and oxygen anions. Several non-layered 2D nanosheets such as TiO2, WO3, and SnO2 have been fabricated and used in different applications.262–266 In these 2D non-layered oxide nanosheet photocatalysts, TiO2 based 2D nanosheets have been comprehensively studied owing to their good stability, nontoxicity, low cost, and available sources in nature.267–269 Sasaki et al.270 investigated 2D TiO2 in 1990. A previous study synthesized a facet-controlled TiO2 nanosheet in which TiO2 nanoparticles were grown in preferential directions6,271 to achieve special properties.272 The decreased thickness of TiO2 resulted in bandgap enlargement due to a quantum confinement effect. A large bandgap limits the photocatalytic application of TiO2 nanosheets owing to their inability to absorb visible light. Techniques such as anion/cation doping have been investigated to reduce the bandgap of TiO2 nanosheets.273–277 However, anion/cation doping into the TiO2 nanosheet structure leads to a faster recombination rate of electron–hole carriers, resulting in a reduction in photon conversion. An alternative co-doping approach is used to resolve this phenomenon.275,2782D metal oxide nanosheets have been widely used in PEC water splitting reactions. Yao et al.263 constructed a porous hybrid structure containing Fe2O3 nanothorn/TiO2 nanosheet (Fe2O3NT/TiO2NS) photoanodes that exhibited high PEC activity under visible light. The photocurrent density (2.50 mA cm−2) was six times higher than that of bare TiO2 photoanodes. Wang et al.265 used the layer by layer (LBL) assembly method to synthesize coated ultrathin TiO2 films on FTO substrates. The LBL-deposited TiO2 films were found to improve the PEC water splitting performance of hematite films by obstructing the back transfer of electrons from the FTO to the hematite films. Best performance was identified for the two-layer TiO2 with a thickness below 1.5 nm. Zhang et al.264 fabricated γ-monoclinic WO3 multilayer nanosheets with exposed (002) facets using a facile solvo-thermal route. The electrode consisted of multilayered WO3 and exhibited a photocurrent density of 1.62 mA cm−2 at 1.25 V (vs. Ag/AgCl) (pH 6.8). In addition, a photoconversion efficiency of 0.154% was achieved under AM 1.5G owing to preferentially exposed highly active (002) facets.
Hou et al.266 fabricated a 3D hybrid WO3/C3N4/CoOx comprised of a branched WO3 2D nanosheet heterojunction with 2D C3N4 decorated with CoOx nanoparticles. This exhibited superior PEC performance for water oxidation. EIS measurements were performed to study interface charge transfer in the hybrid system. A smaller arc radius was observed. This this indicates lower charge-transfer resistance and efficient photo-generated electron–hole pairs separation for three-dimensionally branched (3DB) WO3-NA/C3N4-NS/CoOx under dark and light-irradiated conditions, as compared with WO3-NA/C3N4-NS and WO3-NA.266
5.3. 2D Metal oxyhalide-based photocatalysts
Bismuth oxyhalides (BiOX (X = Cl, Br, or I)) have attracted much interest owing to their distinctive layered structure and good optical/electrical properties.279 Bismuth oxyhalides are formed by [Bi2O2]2+ layers packed between two slabs of halogen ions, resulting in the formation of self-built internal static electric fields and outstanding photocatalytic activity.280–289 The unique features BiOXs can be used as photocatalysts for water splitting.290–296 Ye et al.286 prepared BiOI/BiVO4 photoanodes via an electrodeposition method followed by a calcination process. The BiOI/BiVO4 photoanodes showed enhanced PEC performance under visible-light illumination due to a synergistic effect. XRD, Raman, TEM, and XPS investigations proved the formation of p–n junctions, which resulted in expanded light absorption and facilitated photo-generated electron–hole separation. The p–n junction caused a synergistic effect in BiOI/BiVO4 PEC activity. Fu et al.282 fabricated a composite system of TiO2−x/BiOCl and these visible active photocatalysts reduced the recombination rate of photoexcited carriers. In addition, these catalysts were fabricated under different Bi/Ti molar ratios. Ultraviolet-visible DRS (UV-vis DRS) revealed the presence of Ti3+, Ti2+, and oxygen vacancies in TiO2−x, which could increase light absorption. Linear scan voltammetry and EIS showed that faster ET occurred in heterojunctions with an optimized Bi/Ti molar ratio. Therefore, the reduced TiO2−x/BiOCl heterojunction with an optimized Bi/Ti molar ratio showed a higher photocurrent density (0.755 mA cm−2) with a photoconversion efficiency (up to 0.634%) that was 10.5 and 22.6 times larger than those of pure TiO2 and BiOCl, respectively.Shan et al.280 prepared BiOI nanosheets with exposed {001} facets by chemical transformation at 25 °C. The photocatalytic activity measurement for PEC water splitting revealed that the performance of tetragonal BiOI was better than that of monoclinic Bi2O3 under visible-light irradiation. A comparative study of the VB between α-Bi2O3 and BiOI showed that the holes in the VB of BiOI contained a higher oxidation potential than α-Bi2O3. In the preferential growth direction, BiOI showed vigorous visible-light absorbance, which could be attributed to photoenhancement activity. In addition, the concise VB energy position and oxygen generation capability of BiOI nanosheets were confirmed by ultraviolet photoemission spectroscopy (UPS) analysis. Further information on applications of UPS are described in the ESI.†
5.4. Layered double hydroxides-based photocatalysts
The layered double hydroxides (LDHs) or bimetallic hydroxides with the general formula [M1−x2+Mx3+(OH)2][Ax/n]·mH2O are 2D materials used as co-catalysts in PEC water splitting systems.297–300 Different LDHs can be synthesized by adjusting the x fraction, resulting in different chemical compositions and structural morphologies.301–313 Qi et al.301 applied the modified Cu2O/NiFe-LDH electrodes and observed a significant seven-fold increase in photocurrent intensity under an applied voltage of −0.2V (vs. Ag/AgCl). This enhancement was due to better ET toward the electrolyte in the NiFe-LDH overlayer owing to suitable energy level alignment. Extended photostability investigations suggested that the Cu2O/NiFe-LDH photocathodes exhibited no reduction in photocurrent measurement after 40hours of light operation at −0.2V (vs. Ag/AgCl) (biased condition). This enhancement made Cu2O/NiFe-LDH a suitable photocathode material for low biased PEC H2 generation. After 8hours of H2 evolution under −0.2V (vs. Ag/AgCl), the PEC cell reached 78% faradaic performance. This result confirmed Cu2O/NiFe-LDH as a promising photocatalyst for self-biased PEC water splitting under sunlight irradiation.
Fan et al.314 constructed hierarchical WO3@NiFe-LDH nanoarrays by combining a hydrothermal preparation of WO3 nanorod arrays and electrochemical deposition of layer-structured NiFe-LDH. The WO3@NiFe-LDH photoanodes exhibited excellent PEC activity with lower bias potential and higher anodic photocurrent density than a pristine WO3 photoanode. The addition of ultrathin NiFe-LDH nanoflakes with an optimum fraction of WO3 led to a 0.6 V negative shift of onset potential and improved the photocurrent density to ∼1.10 mA cm−2 at a bias potential of 1.20 V (vs. SCE). This superior performance was related to light-harvesting characteristic and the co-catalytic role of the NiFe-LDH for oxygen evolution and retardation of the electron–hole recombination process.
6. Conclusions and perspectives
The development of practical PEC and PC devices to produce H2 fuel is one of the most important scientific and technological challenges for a sustainable green society. To achieve this, ideal photoelectrodes and photocatalysts should satisfy the following characteristics: adequate absorption of sunlight, suitable bandgap (Eg), appropriate band energy level alignments, high charge transport ability, good stability, highly accessible active sites, low material cost, and eco-friendliness. To fulfill all these criteria, 2D materials such as graphene, TMDs, and GCN have demonstrated great potential for solar-driven hydrogen production through water splitting. Although these materials have exhibited excellent properties for photo(electro)catalytic activities to certain extent, they also suffer from many setbacks in practical applications. As light absorption of such thin 2D materials is low, they are not an efficient photocatalyst; they need to be coupled with other SC photocatalytic materials. The overall performance of PEC water splitting depends on numerous factors such as efficient light-harvesting ability, charge separation ability, catalytic activity for hydrogen evolution and water oxidation, surface area, and stability; these factors are closely interconnected. These issues present key problems and great challenges for 2D/SC materials in PEC and PC applications. Research on 2D materials is in its initial stage; hence, such materials are far from ready practical use. In particular, it should be noted that the intrinsic thermodynamic instability of 2D materials presents the biggest challenge for real-world applications. However, the long-term stability issue is not seriously addressed in most published studies despite it critical importance and the PEC and PC performances of the 2D/SC systems have been estimated and reported only for the as-synthesized fresh materials. The widespread utilization of emerging 2D materials critically depends on such issues. The merits and demerits of 2D materials for PEC water splitting are summarized in Table 1 and schematically illustrated in Fig. 20.| Graphene | TMD | GCN (g-C3N4) | |
|---|---|---|---|
| Merits | Facilitates charge-transfer kinetics because of its excellent electrical conductivity (106 S cm−1)110 | An active light absorption component (direct bandgap) | Suitable bandgap and band positions for visible-light driven water splitting315 |
| Composed of earth-abundant carbon element only | Suitable and tunable bandgap for solar light absorption (various compositions) | Facile synthesis and a variety of chemical modification methods | |
| Chemically inert | A good hydrogen evolving co-catalyst | Low cost, metal-free, and stable photocatalytic material | |
| High surface area | Chemical stability | ||
| Demerits | Absence of electronic bandgap (semi-metal) | Difficult synthesis with controllable size, defects, and crystal phase | Fast charge recombination316 |
| Poor catalytic activity of pristine graphene | Low conductivity | Limited light absorption (λ < 460 nm)317 | |
| Subject to gradual oxidation in the long-term operation (intrinsic instability) | Unstable 1T-phase | Low surface area (10–15 m2 g−1)318 | |
| Poor catalytic activity of pristine GCN for water splitting | |||
| Highest efficiency reported for water splitting (selected) | Pt/NG/Si photocathode 319 | MoS 2 /Al 2 O 3 /Si photocathode 189 | CoO x /C 3 N 4 /Ba-TaON photoanode 324 |
| J ph = −10 mA cm−2 (@ 0.25 V vs. RHE), STH = 3.05% | J ph = −32 mA cm−2 (@ 0 V vs. RHE) | J ph = 4.57 mA cm−2 (@ 1.23 V vs. RHE), IPCE = 62% (λ = 400 nm @ 1.23 V vs. RHE) | |
| N-GQS/Si photocathode 320 | MoS x Cl y /Si photocathode 322 | CQDs/C 3 N 4 /TiO 2 photoanode 325 | |
| J ph = −35 mA cm−2 (@ 0 V vs. RHE) | J ph = −43 mA cm−2 (@ 0 V vs. RHE) | J ph = 2.2 mA cm−2 (@ 1.23 V vs. RHE), IPCE = 60% (λ = 380 nm @ 1.23 V vs. RHE), STH = 1.45% | |
| GQDs/CdSe/P25 photoanode 321 | S:MoP/Si photocathode 323 | ||
| J ph = 14.16 mA cm−2 (@ −0.6 V vs. RHE), IPCE = 72% (λ = 480 nm @ −0.6 V vs. RHE) | J ph = −33.1 mA cm−2 (@ 0 V vs. RHE), IPCE = 80% (λ = 480 nm @ 0 V vs. RHE) | ||
| Problems to be solved | Hybridization with other photoactive components due to the absence of bandgap | Improvement of low efficiency and stability for PEC activity | Hybridization, bandgap engineering by doping (improve charge transfer and light absorption) |
| Modification (e.g., doping, surface functionalization) to enhance the catalytic activity | Systematical in-depth studies on the interface between TMDs and SCs | Morphology design (increase the surface area) | |
| Better understanding and control of graphene/SC interfaces | Facile and controllable production of low-dimensional and defect-rich morphology and abundant exposed active edge activation of basal planes | ||
| Stable junction with SC | |||
 | ||
| Fig. 20 Schematic representation of relative functional roles of various 2D materials employed in photoelectrocatalytic water splitting reactions. | ||
Being composed of carbon element only on the 2D sheet, graphene has a great potential as a functional nanomaterial with excellent conductivity, high surface area, and low cost. As for graphene, the design, synthesis, and control of suitable compositions, structures, and morphologies to optimize light absorption and charge transport properties are challenges in fabricating SC/graphene composite for PEC and PC applications. The optical properties and electronic structure of graphene-based materials can be adjusted by modifying the dimensions (e.g., 3D graphene, 2D graphene sheets, 1D graphene nanoribbon, and 0D GQDs). These materials, each with specific properties, can be successfully incorporated into different SCs to improve solar light absorption, effective surface area, and charge-carrier separation/transfer during PEC water splitting processes. Since graphene itself with semi-metal property has no electronic bandgap, it has to be hybridized with photoactive (light harvesting) materials for PEC/PC applications. Therefore, systematic studies to understand the nature of the interface between graphene and SCs are essential, which is poorly understood. In addition, pristine graphene has little catalytic activity for water splitting reaction and should be loaded with noble metal cocatalysts and modified by various methods such as surface functionalization and foreign element doping. Graphene has been frequently tested as a platform of many catalytic nanoparticles but the support effects of graphene on the catalytic activity of water splitting should be understood at the molecular level.
In the case of TMDs, an urgent challenge is to improve the conductivity of nanosheets. Phase engineering (i.e., 2H to 1T) and combination with better conductive materials such as graphene can possibly overcome this drawback. In addition, the problem of forming a suitable interface between TMDs and different metal oxide SCs (e.g., TiO2) need to be resolved to achieve high PEC performance. Moreover, since the defects or edges act as the active sites of TMDs for catalytic reaction, facile synthesis of the controlling morphology, structure, and chemical composition of TMDs or TMD/SC hybrid systems is essential to overcome these issues. Considering all of the discussed issues related to the catalytic activity of 2D nanosheets of TMDs under both dark and illuminated conditions, an optimum choice should be selected between bulk and monolayer systems to achieve best performance for energy storage/conversion applications. The best structure, incorporating more active sites, desired conductivity, high surface area, and high light absorption ability, is that of few-layered TMD nanosheets (5 layers on average) with edge-defect sites that are anchored vertically to the conductive substrate.
As an emerging 2D material, GCN has received tremendous attentions as an organic photoactive material for solar energy utilization. The synthetic simplicity and the diversity of chemical modification methods of GCN-based materials have accelerated the explosive growth of this research area. Despite the poor catalytic activity of pure GCN for hydrogen production, GCN can be employed in water splitting systems as both a photocatalyst and a modifier in conjunction with other SCs. GCN can serve as a protective layer of a reactive SC surface as well. The main role of GCN in GCN/SC systems is to retard electron–hole recombination, which has been frequently confirmed by a reduced PL intensity, reduced charge-transfer resistance in EIS analysis, and enhanced photocurrent density. However, the photocatalytic activity of GCN-based systems is often limited owing to its low effective surface area, poor optical absorption above 420 nm, and significant charge recombination. Different approaches such as dimensional tuning, metal/non-metal doping, and heterostructure/interfacial formation can be used to overcome these problems.
Finally, various experimental and theoretical studies have proposed several new 2D materials such as TaS2, TiS2, WSe2, MoSe2; single-layer group-III monochalcogenides (e.g., GaSe, GaTe, InS, InSe, InTe); and single-layer metal–phosphorus-trichalcogenides (i.e., MPX3 (M = Zn, Mg, Ag0.5Sc0.5, Ag0.5In0.5 and X = S, Se)); each of which exhibits suitable electronic properties for PEC water splitting. However, to move beyond the laboratory scale, large-scale production and industrial applications of 2D/SC photoelectrodes require cost-effective fabrication processes.
Although remarkable progress has been made to improve the electronic and optical properties of 2D materials, the above mentioned roles of 2D materials in 2D/SC composites are not fully understood owing to a lack of experimental evidence and insufficient analyses. Therefore, comprehensive studies to understand such mechanisms can help design high-performance 2D-based PC and PEC systems, particularly for water splitting applications. Due to the limitations of available experimental technologies, theoretical calculations and modeling are imperative and can be used to predict the PEC efficiency of 2D/SC photoelectrodes in order to understand their physical/chemical properties. To accelerate the progress of the 2D/SC PC and PEC systems for water splitting, the material preparation and performance demonstration should go beyond the laboratory scale, which should present the real challenge. The overall solution needs not only the breakthroughs in efficient and durable 2D/SC materials but also economical and scalable solar systems and engineering for commercialization. Despite the tremendous efforts and interests in the materials development, relatively little attention has been paid to the practical aspects and engineering problems. This calls for more balanced and practical strategy and methodology in future research dealing with 2D/SC solar water splitting system.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors would like to thank Mr Navid Sarikhani for his discussion about the manuscript. We highly appreciate the efforts from Dr Omid Bavi and Mr Amir Hossein Sanayei for their preparation of schemes and figures. Authors would like to thank Ms Amene Naseri for useful discussion. We also thank Dr Ka Hei Lui for his careful English editing of the manuscript. This work was financially supported by Research and Technology Council of Sharif University of Technology (The Grant Program, Grant No. G930206), Iran National Science Foundation (Research Chair Award of Surface and Interface Physics, Grant No. 940009) and the Iran Science Elites Federation (Grant of the top 100 national science elites). We are also grateful for the partial financial supports from the Iran Nanotechnology Initiative Council (INIC) and the National Research Foundation of Korea (NRF, MSICT) through the Global Research Laboratory (GRL) Program (No. NRF-2014K1A1A2041044) and KCAP (Sogang Univ.) (No. 2009-0093880).References
- M. G. R. Van de Krol, Photoelectrochemical Hydrogen Production, Springer, 2012 Search PubMed.
- N. S. Lewis, Science, 2007, 315, 798–801 CrossRef CAS PubMed.
- R. Liu, Z. Zheng, J. Spurgeon and X. Yang, Energy Environ. Sci., 2014, 7, 2504–2517 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- M. Reza Gholipour, C. T. Dinh, F. Béland and T. O. Do, Nanoscale, 2015, 7, 8187–8208 RSC.
- J. Li and N. Wu, Catal. Sci. Technol., 2015, 5, 1360–1384 RSC.
- R. Sathre, J. B. Greenblatt, K. Walczak, I. D. Sharp, J. C. Stevens, J. W. Ager and F. A. Houle, Energy Environ. Sci., 2016, 9, 803–819 RSC.
- H. Döscher, J. L. Young, J. F. Geisz, J. A. Turner and T. G. Deutsch, Energy Environ. Sci., 2016, 9, 74–80 RSC.
- J. W. Ager, M. R. Shaner, K. A. Walczak, I. D. Sharp and S. Ardo, Energy Environ. Sci., 2015, 8, 2811–2824 RSC.
- S. Hu, C. Xiang, S. Haussener, A. D. Berger and N. S. Lewis, Energy Environ. Sci., 2013, 6, 2984–2993 RSC.
- J. Low, S. Cao, J. Yu and S. Wageh, Chem. Commun., 2014, 50, 10768–10777 RSC.
- D. Chen, H. Zhang, Y. Liu and J. Li, Energy Environ. Sci., 2013, 6, 1362–1387 RSC.
- V. J. Babu, S. Vempati, T. Uyar and S. Ramakrishna, Phys. Chem. Chem. Phys., 2015, 17, 2960–2986 RSC.
- W. Zhen, X. Ning, B. Yang, Y. Wu, Z. Li and G. Lu, Appl. Catal., B, 2018, 221, 243–257 CrossRef CAS.
- V. Etacheri, C. Di Valentin, J. Schneider, D. Bahnemann and S. C. Pillai, J. Photochem. Photobiol., C, 2015, 25, 1–29 CrossRef CAS.
- S. Haussener, C. Xiang, J. M. Spurgeon, S. Ardo, N. S. Lewis and A. Z. Weber, Energy Environ. Sci., 2012, 5, 9922–9935 RSC.
- H. G. Park and J. K. Holt, Energy Environ. Sci., 2010, 3, 1028–1036 RSC.
- M. Qorbani, N. Naseri, O. Moradlou, R. Azimirad and A. Moshfegh, Appl. Catal., B, 2015, 162, 210–216 CrossRef CAS.
- B. C. Brodie, Philos. Trans. R. Soc. London, 1859, 149, 249–259 CrossRef.
- G. R. Bhimanapati, Z. Lin, V. Meunier, Y. Jung, J. Cha, S. Das, D. Xiao, Y. Son, M. S. Strano and V. R. Cooper, ACS Nano, 2015, 9, 11509–11539 CrossRef CAS PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2012, 41, 782–796 RSC.
- D. M. Andoshe, J. M. Jeon, S. Y. Kim and H. W. Jang, Electron. Mater. Lett., 2015, 11, 323–335 CrossRef CAS.
- S. Cao and J. Yu, J. Phys. Chem. Lett., 2014, 5, 2101–2107 CrossRef CAS PubMed.
- H. Tang, C. M. Hessel, J. Wang, N. Yang, R. Yu, H. Zhao and D. Wang, Chem. Soc. Rev., 2014, 43, 4281–4299 RSC.
- S. Sarkar, M. L. Moser, X. Tian, X. Zhang, Y. F. Al-Hadeethi and R. C. Haddon, Chem. Mater., 2014, 26, 184–195 CrossRef CAS.
- T. F. Yeh, J. Cihlář, C. Y. Chang, C. Cheng and H. Teng, Mater. Today, 2013, 16, 78–84 CrossRef CAS.
- S. Cao and J. Yu, J. Photochem. Photobiol., C, 2016, 27, 72–99 CrossRef CAS.
- F. Bonaccorso, L. Colombo, G. Yu, M. Stoller, V. Tozzini, A. C. Ferrari, R. S. Ruoff and V. Pellegrini, Science, 2015, 347, 1246501 CrossRef PubMed.
- A. K. Singh, K. Mathew, H. L. Zhuang and R. G. Hennig, J. Phys. Chem. Lett., 2015, 6, 1087–1098 CrossRef CAS PubMed.
- Y. Sun, Q. Wu and G. Shi, Energy Environ. Sci., 2011, 4, 1113–1132 RSC.
- D. Mateo, J. Albero and H. García, Energy Environ. Sci., 2017, 10, 2392–2400 RSC.
- C. Huang, C. Li and G. Shi, Energy Environ. Sci., 2012, 5, 8848–8868 RSC.
- J. L. Gunjakar, I. Y. Kim, J. M. Lee, N. S. Lee and S. J. Hwang, Energy Environ. Sci., 2013, 6, 1008–1017 RSC.
- V. Chabot, D. Higgins, A. Yu, X. Xiao, Z. Chen and J. Zhang, Energy Environ. Sci., 2014, 7, 1564–1596 RSC.
- Y.-J. Cho, H.-i. Kim, S. Lee and W. Choi, J. Catal., 2015, 330, 387–395 CrossRef CAS.
- D. K. Lee, K. S. Han, W. H. Shin, J. W. Lee, J. H. Choi, K. M. Choi, Y. Lee, H.-i. Kim, W. Choi and J. K. Kang, J. Mater. Chem. A, 2013, 1, 203–207 RSC.
- G.-h. Moon, H.-i. Kim, Y. Shin and W. Choi, RSC Adv., 2012, 2, 2205–2207 RSC.
- T. Soltani, A. Tayyebi and B.-K. Lee, Sol. Energy Mater. Sol. Cells, 2018, 185, 325–332 CrossRef CAS.
- M. Ghorbani, H. Abdizadeh, M. Taheri and M. R. Golobostanfard, Int. J. Hydrogen Energy, 2018, 43, 7754–7763 CrossRef CAS.
- C.-J. Chang, Y.-G. Lin, H.-T. Weng and Y.-H. Wei, Appl. Surf. Sci., 2018, 451, 198–206 CrossRef CAS.
- F. Opoku, K. K. Govender, C. G. C. E. van Sittert and P. P. Govender, Carbon, 2018, 136, 187–195 CrossRef CAS.
- W. Xue, X. Hu, E. Liu and J. Fan, Appl. Surf. Sci., 2018, 447, 783–794 CrossRef CAS.
- H. Chang and H. Wu, Energy Environ. Sci., 2013, 6, 3483–3507 RSC.
- Y. H. Ng, A. Iwase, A. Kudo and R. Amal, J. Phys. Chem. Lett., 2010, 1, 2607–2612 CrossRef CAS.
- H. S. Park, H. W. Ha, R. S. Ruoff and A. J. Bard, J. Electroanal. Chem., 2014, 716, 8–15 CrossRef CAS.
- L. Jia, D. H. Wang, Y. X. Huang, A. W. Xu and H. Q. Yu, J. Phys. Chem. C, 2011, 115, 11466–11473 CrossRef CAS.
- X. Zhang, F. Xu, B. Zhao, X. Ji, Y. Yao, D. Wu, Z. Gao and K. Jiang, Electrochim. Acta, 2014, 133, 615–622 CrossRef CAS.
- B. Wang, Z. Liu, J. Han, T. Hong, J. Zhang, Y. Li and T. Cui, Electrochim. Acta, 2015, 176, 334–343 CrossRef CAS.
- A. G. Tamirat, W. N. Su, A. A. Dubale, C. J. Pan, H. M. Chen, D. W. Ayele, J. F. Lee and B. J. Hwang, J. Power Sources, 2015, 287, 119–128 CrossRef CAS.
- Q. Wu, J. Zhao, K. Liu, H. Wang, Z. Sun, P. Li and S. Xue, Int. J. Hydrogen Energy, 2015, 40, 6763–6770 CrossRef CAS.
- K. Y. Yoon, J. S. Lee, K. Kim, C. H. Bak, S. I. Kim, J. B. Kim and J. H. Jang, ACS Appl. Mater. Interfaces, 2014, 6, 22634–22639 CrossRef CAS PubMed.
- F. Meng, J. Li, S. K. Cushing, J. Bright, M. Zhi, J. D. Rowley, Z. Hong, A. Manivannan, A. D. Bristow and N. Wu, ACS Catal., 2013, 3, 746–751 CrossRef CAS.
- S. Rai, A. Ikram, S. Sahai, S. Dass, R. Shrivastav and V. R. Satsangi, RSC Adv., 2014, 4, 17671–17679 RSC.
- S. Martha, D. K. Padhi and K. Parida, ChemSusChem, 2014, 7, 585–597 CrossRef CAS PubMed.
- J. Zhang, W. Zhao, Y. Xu, H. Xu and B. Zhang, Int. J. Hydrogen Energy, 2014, 39, 702–710 CrossRef CAS.
- W. Li, J. Yue, Y. Bu and Z. Chen, RSC Adv., 2015, 5, 77823–77830 RSC.
- S. Upadhyay, S. Bagheri and S. B. Abd Hamid, Int. J. Hydrogen Energy, 2014, 39, 11027–11034 CrossRef CAS.
- L. Wu, L.-k. Tsui, N. Swami and G. Zangari, J. Phys. Chem. C, 2010, 114, 11551–11556 CrossRef CAS.
- S. Hacialioglu, F. Meng and S. Jin, Chem. Commun., 2012, 48, 1174–1176 RSC.
- P. E. de Jongh, D. Vanmaekelbergh and J. J. Kelly, J. Electrochem. Soc., 2000, 147, 486–489 CrossRef CAS.
- P. D. Tran, S. K. Batabyal, S. S. Pramana, J. Barber, L. H. Wong and S. C. J. Loo, Nanoscale, 2012, 4, 3875–3878 RSC.
- A. A. Dubale, W. N. Su, A. G. Tamirat, C. J. Pan, B. A. Aragaw, H. M. Chen, C. H. Chen and B. J. Hwang, J. Mater. Chem. A, 2014, 2, 18383–18397 RSC.
- H. I. Kim, S. Kim, J. K. Kang and W. Choi, J. Catal., 2014, 309, 49–57 CrossRef CAS.
- R. Lv, X. Wang, W. Lv, Y. Xu, Y. Ge, H. He, G. Li, X. Wu, X. Li and Q. Li, J. Chem. Technol. Biotechnol., 2015, 90, 550–558 CrossRef CAS.
- W. Kang, X. Jimeng and W. Xitao, Appl. Surf. Sci., 2016, 360, 270–275 CrossRef CAS.
- S. J. Teh, C. W. Lai and S. B. A. Hamid, J. Energy Chem., 2016, 25, 336–344 CrossRef.
- S. Chandrasekaran, J. S. Chung, E. J. Kim and S. H. Hur, Chem. Eng. J., 2016, 290, 465–476 CrossRef CAS.
- S. Bera, A. Naskar, M. Pal and S. Jana, RSC Adv., 2016, 6, 36058–36068 RSC.
- I. Khan, A. A. M. Ibrahim, M. Sohail and A. Qurashi, Ultrason. Sonochem., 2017, 37, 669–675 CrossRef CAS PubMed.
- J. Lin, P. Hu, Y. Zhang, M. Fan, Z. He, C. K. Ngaw, J. S. C. Loo, D. Liao and T. T. Y. Tan, RSC Adv., 2013, 3, 9330–9336 RSC.
- M. E. Khan, M. M. Khan and M. H. Cho, RSC Adv., 2016, 25, 20824–20833 RSC.
- B. Pan, Y. Wang, Y. Liang, S. Luo, W. Su and X. Wang, Int. J. Hydrogen Energy, 2014, 39, 13527–13533 CrossRef CAS.
- F. Nunzi, E. Mosconi, L. Storchi, E. Ronca, A. Selloni, M. Grätzel and F. De Angelis, Energy Environ. Sci., 2013, 6, 1221–1229 RSC.
- L. Liu, Z. Liu, A. Liu, X. Gu, C. Ge, F. Gao and L. Dong, ChemSusChem, 2014, 7, 618–626 CrossRef CAS PubMed.
- S. Ayissi, P. A. Charpentier, N. Farhangi, J. A. Wood, K. Palotás and W. A. Hofer, J. Phys. Chem. C, 2013, 117, 25424–25432 CrossRef CAS.
- R. Long, N. J. English and O. V. Prezhdo, J. Am. Chem. Soc., 2012, 134, 14238–14248 CrossRef CAS PubMed.
- H. Gao, X. Li, J. Lv and G. Liu, J. Phys. Chem. C, 2013, 117, 16022–16027 CrossRef CAS.
- X. Li, H. Gao and G. Liu, Comput. Theor. Chem., 2013, 1025, 30–34 CrossRef CAS.
- G.-h. Moon, D.-h. Kim, H.-i. Kim, A. D. Bokare and W. Choi, Environ. Sci. Technol. Lett., 2014, 1, 185–190 CrossRef CAS.
- I. Y. Kim, J. M. Lee, T. W. Kim, H. N. Kim, H.-i. Kim, W. Choi and S.-J. Hwang, Small, 2012, 8, 1038–1048 CrossRef CAS PubMed.
- J.-W. Jang, S. Cho, G.-h. Moon, K. Ihm, J. Y. Kim, D. H. Youn, S. Lee, Y. h. Lee, W. Choi, K.-H. Lee and J. S. Lee, Chem. – Eur. J., 2012, 18, 2762–2767 CrossRef CAS PubMed.
- Y. Hou, F. Zuo, A. Dagg and P. Feng, Nano Lett., 2012, 12, 6464–6473 CrossRef CAS PubMed.
- J. Selvaraj, S. Gupta, S. Delacruz and V. Subramanian, ChemPhysChem, 2014, 15, 2010–2018 CrossRef CAS PubMed.
- H. Li, Z. Xia, J. Chen, L. Lei and J. Xing, Appl. Catal., B, 2015, 168-169, 105–113 CrossRef CAS.
- X. Wang, J. Xie and C. M. Li, J. Mater. Chem. A, 2015, 3, 1235–1242 RSC.
- S. Yousefzadeh, M. Faraji and A. Z. Moshfegh, J. Electroanal. Chem., 2016, 763, 1–9 CrossRef CAS.
- X. Yu, J. Zhang, Z. Zhao, W. Guo, J. Qiu, X. Mou, A. Li, J. P. Claverie and H. Liu, Nano Energy, 2015, 16, 207–217 CrossRef CAS.
- S. Min and G. Lu, Int. J. Hydrogen Energy, 2012, 37, 10564–10574 CrossRef CAS.
- C. Kong, S. Min and G. Lu, Int. J. Hydrogen Energy, 2014, 39, 4836–4844 CrossRef CAS.
- W. Zhang, Y. Li, X. Zeng and S. Peng, Sci. Rep., 2015, 5, 10589 CrossRef CAS PubMed.
- C. Kong, S. Min and G. Lu, ACS Catal., 2014, 4, 2763–2769 CrossRef CAS.
- S. Chandrasekaran, S. H. Hur, E. J. Kim, B. Rajagopalan, K. F. Babu, V. Senthilkumar, J. S. Chung, W. M. Choi and Y. S. Kim, RSC Adv., 2015, 5, 29159–29166 RSC.
- B. Gupta, A. A. Melvin, T. Matthews, S. Dhara, S. Dash and A. K. Tyagi, Int. J. Hydrogen Energy, 2015, 40, 5815–5823 CrossRef CAS.
- T.-D. Nguyen-Phan, V. H. Pham, E. W. Shin, H.-D. Pham, S. Kim, J. S. Chung, E. J. Kim and S. H. Hur, Chem. Eng. J., 2011, 170, 226–232 CrossRef CAS.
- F.-X. Xiao, J. Miao and B. Liu, J. Am. Chem. Soc., 2014, 136, 1559–1569 CrossRef CAS PubMed.
- S. Gupta and V. Subramanian, ACS Appl. Mater. Interfaces, 2014, 6, 18597–18608 CrossRef CAS PubMed.
- M. Ibadurrohman and K. Hellgardt, Int. J. Hydrogen Energy, 2014, 39, 18204–18215 CrossRef CAS.
- T. Xu, L. Zhang, H. Cheng and Y. Zhu, Appl. Catal., B, 2011, 101, 382–387 CrossRef CAS.
- T. Xiong, F. Dong, Y. Zhou, M. Fu and W.-K. Ho, J. Colloid Interface Sci., 2015, 447, 16–24 CrossRef CAS PubMed.
- X. Zhang, B. Zhang, D. Huang, H. Yuan, M. Wang and Y. Shen, Carbon, 2014, 80, 591–598 CrossRef CAS.
- Y. Wang, Z. Li, Y. Tian, W. Zhao, X. Liu and J. Yang, J. Mater. Sci., 2014, 49, 7991–7999 CrossRef CAS.
- Y. Yan, C. Wang, X. Yan, L. Xiao, J. He, W. Gu and W. Shi, J. Phys. Chem. C, 2014, 118, 23519–23526 CrossRef CAS.
- L. Liu, Z. Liu, A. Liu, X. Gu, C. Ge, F. Gao and L. Dong, ChemSusChem, 2014, 7, 618–626 CrossRef CAS PubMed.
- S. Wang, J. Li, X. Zhou, C. Zheng, J. Ning, Y. Zhong and Y. Hu, J. Mater. Chem. A, 2014, 2, 19815–19821 RSC.
- K. Ullah, A. Ullah, A. Aldalbahi, J. D. Chung and W. C. Oh, J. Mol. Catal. A: Chem., 2015, 410, 242–252 CrossRef CAS.
- W. Han, C. Zang, Z. Huang, H. Zhang, L. Ren, X. Qi and J. Zhong, Int. J. Hydrogen Energy, 2014, 39, 19502–19512 CrossRef CAS.
- J. Hou, C. Yang, H. Cheng, Z. Wang, S. Jiao and H. Zhu, Phys. Chem. Chem. Phys., 2013, 15, 15660–15668 RSC.
- X. Bai, L. Wang and Y. Zhu, ACS Catal., 2012, 2, 2769–2778 CrossRef CAS.
- P. Zeng, Q. Zhang, X. Zhang and T. Peng, J. Alloys Compd., 2012, 516, 85–90 CrossRef CAS.
- G. Xie, K. Zhang, B. Guo, Q. Liu, L. Fang and J. R. Gong, Adv. Mater., 2013, 25, 3820–3839 CrossRef CAS PubMed.
- M.-Q. Yang and Y.-J. Xu, J. Phys. Chem. C, 2013, 117, 21724–21734 CrossRef CAS.
- K. A. Tsai and Y. J. Hsu, Appl. Catal., B, 2015, 164, 271–278 CrossRef CAS.
- Y. Yu, J. Ren and M. Meng, Int. J. Hydrogen Energy, 2013, 38, 12266–12272 CrossRef CAS.
- H. Huang, Z. Yue, G. Li, X. Wang, J. Huang, Y. Du and P. Yang, J. Mater. Chem. A, 2013, 1, 15110–15116 RSC.
- G.-h. Moon, Y. Shin, D. Choi, B. W. Arey, G. J. Exarhos, C. Wang, W. Choi and J. Liu, Nanoscale, 2013, 5, 6291–6296 RSC.
- X. Y. Zhang, H. P. Li, X. L. Cui and Y. Lin, J. Mater. Chem., 2010, 20, 2801–2806 RSC.
- M. J. Zhou, N. Zhang and Z. H. Hou, Int. J. Photoenergy, 2014, 2014, 1–6 Search PubMed.
- G. Nagaraju, G. Ebeling, R. V. Gonçalves, S. R. Teixeira, D. E. Weibel and J. Dupont, J. Mol. Catal. A: Chem., 2013, 378, 213–220 CrossRef CAS.
- C.-J. Chang, K.-W. Chu, M.-H. Hsu and C.-Y. Chen, Int. J. Hydrogen Energy, 2015, 40, 14498–14506 CrossRef CAS.
- D. K. Padhi, K. Parida and S. K. Singh, J. Phys. Chem. C, 2015, 119, 6634–6646 CrossRef CAS.
- J. Sun, M. A. Memon, W. Bai, L. Xiao, B. Zhang, Y. Jin, Y. Huang and J. Geng, Adv. Funct. Mater., 2015, 25, 4334–4343 CrossRef CAS.
- B. Qiu, M. Xing and J. Zhang, J. Am. Chem. Soc., 2014, 136, 5852–5855 CrossRef CAS PubMed.
- M. Q. Yang, N. Zhang, M. Pagliaro and Y. J. Xu, Chem. Soc. Rev., 2014, 43, 8240–8254 RSC.
- W. Han, L. Ren, L. Gong, X. Qi, Y. Liu, L. Yang, X. Wei and J. Zhong, ACS Sustainable Chem. Eng., 2014, 2, 741–748 CrossRef CAS.
- Q. Lu, Y. Yu, Q. Ma, B. Chen and H. Zhang, Adv. Mater., 2016, 28, 1917–1933 CrossRef CAS PubMed.
- D. Voiry, J. Yang and M. Chhowalla, Adv. Mater., 2016, 28, 6197–6206 CrossRef CAS PubMed.
- Y. Chen, C. Tan, H. Zhang and L. Wang, Chem. Soc. Rev., 2015, 44, 2681–2701 RSC.
- F. Wang, T. A. Shifa, X. Zhan, Y. Huang, K. Liu, Z. Cheng, C. Jiang and J. He, Nanoscale, 2015, 7, 19764–19788 RSC.
- R. Lv, J. A. Robinson, R. E. Schaak, D. Sun, Y. Sun, T. E. Mallouk and M. Terrones, Acc. Chem. Res., 2015, 48, 56–64 CrossRef CAS PubMed.
- M. Xu, T. Liang, M. Shi and H. Chen, Chem. Rev., 2013, 113, 3766–3798 CrossRef CAS PubMed.
- S. Z. Butler, S. M. Hollen, L. Cao, Y. Cui, J. A. Gupta, H. R. Gutiérrez, T. F. Heinz, S. S. Hong, J. Huang and A. F. Ismach, ACS Nano, 2013, 7, 2898–2926 CrossRef CAS PubMed.
- A. Eftekhari, Appl. Mater. Today, 2017, 8, 1–17 CrossRef.
- M. Zhao, M.-J. Chang, Q. Wang, Z.-T. Zhu, X.-P. Zhai, M. Zirak, A. Z. Moshfegh, Y.-L. Song and H.-L. Zhang, Chem. Commun., 2015, 51, 12262–12265 RSC.
- M. Samadi, N. Sarikhani, M. Zirak, H. Zhang, H.-L. Zhang and A. Z. Moshfegh, Nanoscale Horiz., 2018, 3, 90–204 RSC.
- H. Li, Y. Shi and L.-J. Li, Carbon, 2018, 127, 602–610 CrossRef CAS.
- H. Xu, J. Yi, X. She, Q. Liu, L. Song, S. Chen, Y. Yang, Y. Song, R. Vajtai and J. Lou, Appl. Catal., B, 2018, 220, 379–385 CrossRef CAS.
- B. Chen, Y. Meng, J. Sha, C. Zhong, W. Hu and N. Zhao, Nanoscale, 2018, 10, 34–68 RSC.
- C. Cong, J. Shang, Y. Wang and T. Yu, Adv. Opt. Mater., 2018, 6, 1700767 CrossRef.
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- X. Duan, C. Wang, A. Pan, R. Yu and X. Duan, Chem. Soc. Rev., 2015, 44, 8859–8876 RSC.
- W. Zhao, Y. Liu, Z. Wei, S. Yang, H. He and C. Sun, Appl. Catal., B, 2016, 185, 242–252 CrossRef CAS.
- L. Zheng, S. Han, H. Liu, P. Yu and X. Fang, Small, 2016, 12, 1527–1536 CrossRef CAS PubMed.
- M. Zirak, M. Zhao, O. Moradlou, M. Samadi, N. Sarikhani, Q. Wang, H.-L. Zhang and A. Moshfegh, Sol. Energy Mater. Sol. Cells, 2015, 141, 260–269 CrossRef CAS.
- J. Choi, D. Amaranatha Reddy, N. S. Han, S. Jeong, S. Hong, D. Praveen Kumar, J. K. Song and T. K. Kim, Catal.: Sci. Technol., 2017, 7, 641–649 RSC.
- S. Bellani, L. Najafi, A. Capasso, A. E. Del Rio Castillo, M. R. Antognazza and F. Bonaccorso, J. Mater. Chem. A, 2017, 5, 4384–4396 RSC.
- M. C. Hsiao, C. Y. Chang, L. J. Niu, F. Bai, L. J. Li, H. H. Shen, J. Y. Lin and T. W. Lin, J. Power Sources, 2017, 345, 156–164 CrossRef CAS.
- X. Chen, D. McAteer, C. McGuinness, I. Godwin, J. N. Coleman and A. R. McDonald, Chem. – Eur. J., 2018, 24, 351–355 CrossRef CAS PubMed.
- J. Zhao, P. Zhang, J. Fan, J. Hu and G. Shao, Appl. Surf. Sci., 2018, 430, 466–474 CrossRef CAS.
- T. N. Trung, D.-B. Seo, N. D. Quang, D. Kim and E.-T. Kim, Electrochim. Acta, 2018, 260, 150–156 CrossRef CAS.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699 CrossRef CAS PubMed.
- J. A. Wilson and A. Yoffe, Adv. Phys., 1969, 18, 193–335 CrossRef CAS.
- S. Ma, J. Xie, J. Wen, K. He, X. Li, W. Liu and X. Zhang, Appl. Surf. Sci., 2017, 391, 580–591 CrossRef CAS.
- R. Tang, R. Yin, S. Zhou, T. Ge, Z. Yuan, L. Zhang and L. Yin, J. Mater. Chem. A, 2017, 5, 4962–4971 RSC.
- C. Liu, L. Wang, Y. Tang, S. Luo, Y. Liu, S. Zhang, Y. Zeng and Y. Xu, Appl. Catal., B, 2015, 164, 1–9 CrossRef CAS.
- B. Chai, M. Xu, J. Yan and Z. Ren, Appl. Surf. Sci., 2018, 430, 523–530 CrossRef CAS.
- P. Xiao, J. Lou, H. Zhang, W. Song, X.-L. Wu, H. Lin, J. Chen, S. Liu and X. Wang, Catal. Sci. Technol., 2018, 8, 201–209 RSC.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- K. Chang, Z. Mei, T. Wang, Q. Kang, S. Ouyang and J. Ye, ACS Nano, 2014, 8, 7078–7087 CrossRef CAS PubMed.
- F. Meng, J. Li, S. K. Cushing, M. Zhi and N. Wu, J. Am. Chem. Soc., 2013, 135, 10286–10289 CrossRef CAS PubMed.
- Q. Z. Huang, Y. Xiong, Q. Zhang, H. C. Yao and Z. J. Li, Appl. Catal., B, 2017, 209, 514–522 CrossRef CAS.
- R. Raja, P. Sudhagar, A. Devadoss, C. Terashima, L. K. Shrestha, K. Nakata, R. Jayavel, K. Ariga and A. Fujishima, Chem. Commun., 2015, 51, 522–525 RSC.
- J. Gu, J. A. Aguiar, S. Ferrere, K. X. Steirer, Y. Yan, C. Xiao, J. L. Young, M. Al-Jassim, N. R. Neale and J. A. Turner, Nat. Energy, 2017, 2, 16192 CrossRef CAS.
- Y. F. Zhao, Z. Y. Yang, Y. X. Zhang, L. Jing, X. Guo, Z. Ke, P. Hu, G. Wang, Y. M. Yan and K. N. Sun, J. Phys. Chem. C, 2014, 118, 14238–14245 CrossRef CAS.
- Y. Qi, Q. Xu, Y. Wang, B. Yan, Y. Ren and Z. Chen, ACS Nano, 2016, 10, 2903–2909 CrossRef CAS PubMed.
- B. Mahler, V. Hoepfner, K. Liao and G. A. Ozin, J. Am. Chem. Soc., 2014, 136, 14121–14127 CrossRef CAS PubMed.
- L. Wei, Y. Chen, Y. Lin, H. Wu, R. Yuan and Z. Li, Appl. Catal., B, 2013, 144, 521–527 CrossRef.
- H. Zhao, R. Sun, X. Li and X. Sun, Mater. Sci. Semicond. Process., 2017, 59, 68–75 CrossRef CAS.
- Q. Tian, L. Zhang, J. Liu, N. Li, Q. Ma, J. Zhou and Y. Sun, RSC Adv., 2015, 5, 734–739 RSC.
- S. P. Vattikuti, J. Shim and C. Byon, J. Solid State Chem., 2018, 258, 526–535 CrossRef CAS.
- C. Liu, B. Chai, C. Wang, J. Yan and Z. Ren, Int. J. Hydrogen Energy, 2018, 43, 6977–6986 CrossRef CAS.
- L. Bai, X. Cai, J. Lu, L. Li, S. Zhong, L. Wu, P. Gong, J. Chen and S. Bai, ChemCatChem, 2018, 10, 2107–2114 CrossRef CAS.
- M. Faraji, M. Sabzali, S. Yousefzadeh, N. Sarikhani, A. Ziashahabi, M. Zirak and A. Z. Moshfegh, RSC Adv., 2015, 5, 28460–28466 RSC.
- S. Bai, L. Wang, X. Chen, J. Du and Y. Xiong, Nano Res., 2014, 8, 175–183 CrossRef.
- Q. Liu, X. Li, Q. He, A. Khalil, D. Liu, T. Xiang, X. Wu and L. Song, Small, 2015, 11, 5556–5564 CrossRef CAS PubMed.
- A. N. Enyashin, L. Yadgarov, L. Houben, I. Popov, M. Weidenbach, R. Tenne, M. Bar-Sadan and G. Seifert, J. Phys. Chem. C, 2011, 115, 24586–24591 CrossRef CAS.
- X. Sun, J. Dai, Y. Guo, C. Wu, F. Hu, J. Zhao, X. Zeng and Y. Xie, Nanoscale, 2014, 6, 8359–8367 RSC.
- S. Byun, D. M. Sim, J. Yu and J. J. Yoo, ChemElectroChem, 2015, 2, 1938–1946 CrossRef CAS.
- Y. Yu, S.-Y. Huang, Y. Li, S. N. Steinmann, W. Yang and L. Cao, Nano Lett., 2014, 14, 553–558 CrossRef CAS PubMed.
- M. Velický, M. A. Bissett, C. R. Woods, P. S. Toth, T. Georgiou, I. A. Kinloch, K. S. Novoselov and R. A. W. Dryfe, Nano Lett., 2016, 16, 2023–2032 CrossRef PubMed.
- E. G. da Silveira Firmiano, A. C. Rabelo, C. J. Dalmaschio, A. N. Pinheiro, E. C. Pereira, W. H. Schreiner and E. R. Leite, Adv. Energy Mater., 2014, 4, 1301380 CrossRef.
- A. Bayat, M. Zirak and E. Saievar Iranizad, ACS Sustainable Chem. Eng., 2018, 6, 8374–8382 CrossRef CAS.
- J. Bai, T. Meng, D. Guo, S. Wang, B. Mao and M. Cao, ACS Appl. Mater. Interfaces, 2018, 10, 1678–1689 CrossRef CAS PubMed.
- H. Zhu, J. Zhang, R. Yanzhang, M. Du, Q. Wang, G. Gao, J. Wu, G. Wu, M. Zhang and B. Liu, Adv. Mater., 2015, 27, 4752–4759 CrossRef CAS PubMed.
- Q. Wang, S. Dong, D. Zhang, C. Yu, J. Lu, D. Wang and J. Sun, J. Mater. Sci., 2018, 53, 1135–1147 CrossRef CAS.
- T. Jia, A. Kolpin, C. Ma, R. C. T. Chan, W. M. Kwok and S. C. E. Tsang, Chem. Commun., 2014, 50, 1185–1188 RSC.
- A. Khademi, R. Azimirad, A. A. Zavarian and A. Z. Moshfegh, J. Phys. Chem. C, 2009, 113, 19298–19304 CrossRef CAS.
- W. Qin, Y. Li, Y. Teng and T. Qin, J. Colloid Interface Sci., 2018, 512, 826–833 CrossRef CAS PubMed.
- R. Fan, J. Mao, Z. Yin, J. Jie, W. Dong, L. Fang, F. Zheng and M. Shen, ACS Appl. Mater. Interfaces, 2017, 9, 6123–6129 CrossRef CAS PubMed.
- Y. Li, L. Wang, T. Cai, S. Zhang, Y. Liu, Y. Song, X. Dong and L. Hu, Chem. Eng. J., 2017, 321, 366–374 CrossRef CAS.
- Y. Liu, H. Niu, W. Gu, X. Cai, B. Mao, D. Li and W. Shi, Chem. Eng. J., 2018, 339, 117–124 CrossRef CAS.
- T. Bak, J. Nowotny, M. Rekas and C. Sorrell, Int. J. Hydrogen Energy, 2002, 27, 991–1022 CrossRef CAS.
- E. Parzinger, B. Miller, B. Blaschke, J. A. Garrido, J. W. Ager, A. Holleitner and U. Wurstbauer, ACS Nano, 2015, 9, 11302–11309 CrossRef CAS PubMed.
- J. Zhang, Y. Chen and X. Wang, Energy Environ. Sci., 2015, 8, 3092–3108 RSC.
- F. Yang, V. Kuznietsov, M. Lublow, C. Merschjann, A. Steigert, J. Klaer, A. Thomas and T. Schedel-Niedrig, J. Mater. Chem. A, 2013, 1, 6407–6415 CAS.
- J. Wang, F.-Y. Su and W.-D. Zhang, J. Solid State Electrochem., 2014, 18, 2921–2929 CrossRef CAS.
- D. Zheng, G. Zhang and X. Wang, Appl. Catal., B, 2015, 179, 479–488 CrossRef CAS.
- H. Ou, P. Yang, L. Lin, M. Anpo and X. Wang, Angew. Chem., Int. Ed., 2017, 56, 10905–10910 CrossRef CAS PubMed.
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. Wang and M. J. Bojdys, Functional carbon nitride materials – design strategies for electrochemical devices, Macmillan Publishers Limited, 2017 Search PubMed.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- H. Bian, Y. Ji, J. Yan, P. Li, L. Li, Y. Li and S. Liu, Small, 2018, 14, 1703003 CrossRef PubMed.
- Q. Liu, T. Chen, Y. Guo, Z. Zhang and X. Fang, Appl. Catal., B, 2016, 193, 248–258 CrossRef CAS.
- S. Thaweesak, M. Lyu, P. Peerakiatkhajohn, T. Butburee, B. Luo, H. Chen and L. Wang, Appl. Catal., B, 2017, 202, 184–190 CrossRef CAS.
- J. Xu, L. Zhang, R. Shi and Y. Zhu, J. Mater. Chem. A, 2013, 1, 14766–14772 RSC.
- Q. Lin, L. Li, S. Liang, M. Liu, J. Bi and L. Wu, Appl. Catal., B, 2015, 163, 135–142 CrossRef CAS.
- L. Ma, H. Fan, J. Wang, Y. Zhao, H. Tian and G. Dong, Appl. Catal., B, 2016, 190, 93–102 CrossRef CAS.
- A. Bandyopadhyay, D. Ghosh, N. M. Kaley and S. K. Pati, J. Phys. Chem. C, 2017, 121, 1982–1989 CrossRef CAS.
- G. Li, Z. Lian, W. Wang, D. Zhang and H. Li, Nano Energy, 2016, 19, 446–454 CrossRef CAS.
- J. Su, L. Zhu and G. Chen, Appl. Catal., B, 2016, 186, 127–135 CrossRef CAS.
- Y. Hou, Z. Wen, S. Cui, X. Guo and J. Chen, Adv. Mater., 2013, 25, 6291–6297 CrossRef CAS PubMed.
- Y. Zhang, T. Mori, J. Ye and M. Antonietti, J. Am. Chem. Soc., 2010, 132, 6294–6295 CrossRef CAS PubMed.
- J. Ran, T. Y. Ma, G. Gao, X. W. Du and S. Z. Qiao, Energy Environ. Sci., 2015, 8, 3708–3717 RSC.
- G. Dong, K. Zhao and L. Zhang, Chem. Commun., 2012, 48, 6178–6180 RSC.
- S. Stolbov and S. Zuluaga, J. Phys.: Condens. Matter, 2013, 25 Search PubMed.
- J. Li, B. Shen, Z. Hong, B. Lin, B. Gao and Y. Chen, Chem. Commun., 2012, 48, 12017–12019 RSC.
- Q. Ruan, W. Luo, J. Xie, Y. Wang, X. Liu, Z. Bai, C. J. Carmalt and J. Tang, Angew. Chem., Int. Ed., 2017, 56, 8221–8225 CrossRef CAS PubMed.
- H. Wang, C. Yang, M. Li, F. Chen and Y. Cui, Mater. Lett., 2018, 212, 319–322 CrossRef CAS.
- X. An, C. Hu, H. Lan, H. Liu and J. Qu, ACS Appl. Mater. Interfaces, 2018, 10, 6424–6432 CrossRef CAS PubMed.
- Z. Yajun, S. Jian-Wen, M. Dandan, F. Zhaoyang, C. Linhao, S. Diankun, W. Zeyan and N. Chunming, ChemSusChem, 2018, 11, 1187–1197 CrossRef PubMed.
- M. Ning, Z. Chen, L. Li, Q. Meng, Z. Chen, Y. Zhang, M. Jin, Z. Zhang, M. Yuan, X. Wang and G. Zhou, Electrochem. Commun., 2018, 87, 13–17 CrossRef CAS.
- Y. Liu, Y. X. Yu and W. D. Zhang, Int. J. Hydrogen Energy, 2014, 39, 9105–9113 CrossRef CAS.
- R. Cheng, L. Zhang, X. Fan, M. Wang, M. Li and J. Shi, Carbon, 2016, 101, 62–70 CrossRef CAS.
- S. Bai, J. Jiang, Q. Zhang and Y. Xiong, Chem. Soc. Rev., 2015, 44, 2893–2939 RSC.
- Z. a. Huang, Q. Sun, K. Lv, Z. Zhang, M. Li and B. Li, Appl. Catal., B, 2015, 164, 420–427 CrossRef CAS.
- S. Tonda, S. Kumar and V. Shanker, Mater. Res. Bull., 2016, 75, 51–58 CrossRef CAS.
- X. Bai, R. Zong, C. Li, D. Liu, Y. Liu and Y. Zhu, Appl. Catal., B, 2014, 147, 82–91 CrossRef CAS.
- W.-J. Ong, Front. Mater., 2017, 4, 11 Search PubMed.
- Y. Hou, A. B. Laursen, J. Zhang, G. Zhang, Y. Zhu, X. Wang, S. Dahl and I. Chorkendorff, Angew. Chem., Int. Ed., 2013, 52, 3621–3625 CrossRef CAS PubMed.
- H. Cheng, J. Hou, O. Takeda, X.-M. Guo and H. Zhu, J. Mater. Chem. A, 2015, 3, 11006–11013 RSC.
- J. Yan, Z. Chen, H. Ji, Z. Liu, X. Wang, Y. Xu, X. She, L. Huang, L. Xu, H. Xu and H. Li, Chem. – Eur. J., 2016, 22, 4764–4773 CrossRef CAS PubMed.
- L. Ge, C. Han, X. Xiao and L. Guo, Int. J. Hydrogen Energy, 2013, 38, 6960–6969 CrossRef CAS.
- L. Ye, D. Wang and S. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 5280–5289 CrossRef CAS PubMed.
- Y. Li, X. Wei, X. Yan, J. Cai, A. Zhou, M. Yang and K. Liu, Phys. Chem. Chem. Phys., 2016, 18, 10255–10261 RSC.
- Y. Ma, Z. Wang, Y. Jia, L. Wang, M. Yang, Y. Qi and Y. Bi, Carbon, 2017, 114, 591–600 CrossRef CAS.
- Z. Li, Z. Liu, B. Li, D. Li, C. Ge and Y. Fang, J. Mater. Sci.: Mater. Electron., 2016, 27, 2904–2913 CrossRef CAS.
- W.-K. Jo and T. S. Natarajan, Chem. Eng. J., 2015, 281, 549–565 CrossRef CAS.
- P.-Y. Kuang, Y.-Z. Su, G.-F. Chen, Z. Luo, S.-Y. Xing, N. Li and Z.-Q. Liu, Appl. Surf. Sci., 2015, 358, 296–303 CrossRef CAS.
- J. Xiao, X. Zhang and Y. Li, Int. J. Hydrogen Energy, 2015, 40, 9080–9087 CrossRef CAS.
- H. Li, F. Zhao, J. Zhang, L. Luo, X. Xiao, Y. Huang, H. Ji and Y. Tong, Mater. Chem. Front., 2017, 1, 338–342 RSC.
- D. Zeng, W. Xu, W.-J. Ong, J. Xu, H. Ren, Y. Chen, H. Zheng and D.-L. Peng, Appl. Catal., B, 2018, 221, 47–55 CrossRef CAS.
- Q. Fan, Y. Huang, C. Zhang, J. Liu, L. Piao, Y. Yu, S. Zuo and B. Li, Catal. Today, 2016, 264, 250–256 CrossRef CAS.
- C. Xu, Q. Han, Y. Zhao, L. Wang, Y. Li and L. Qu, J. Mater. Chem. A, 2015, 3, 1841–1846 RSC.
- Y. Li, X. Feng, Z. Lu, H. Yin, F. Liu and Q. Xiang, J. Colloid Interface Sci., 2018, 513, 866–876 CrossRef CAS PubMed.
- A. Bandyopadhyay, D. Ghosh and S. K. Pati, J. Phys., Lett., 2018, 9, 1605–1612 CAS.
- P. Jinbo, B. Alicja, Y. Yin, T. Barbara, Z. Liang, F. Lei, R. G. Mendes, T. Gemming, Z. Liu and M. H. Rummeli, Adv. Energy Mater., 2018, 8, 1702093 CrossRef.
- B. Sa, Y. L. Li, J. Qi, R. Ahuja and Z. Sun, J. Phys. Chem. C, 2014, 118, 26560–26568 CrossRef CAS.
- M. Z. Rahman, C. W. Kwong, K. Davey and S. Z. Qiao, Energy Environ. Sci., 2016, 9, 709–728 RSC.
- J. Hu, Z. Guo, P. E. McWilliams, J. E. Darges, D. L. Druffel, A. M. Moran and S. C. Warren, Nano Lett., 2016, 16, 74–79 CrossRef CAS PubMed.
- H. Wang, X. Yang, W. Shao, S. Chen, J. Xie, X. Zhang, J. Wang and Y. Xie, J. Am. Chem. Soc., 2015, 137, 11376–11382 CrossRef CAS PubMed.
- J. Ran, B. Zhu and S.-Z. Qiao, Angew. Chem., Int. Ed., 2017, 56, 10373–10377 CrossRef CAS PubMed.
- J. Ran, X. Wang, B. Zhu and S.-Z. Qiao, Chem. Commun., 2017, 53, 9882–9885 RSC.
- J. Ran, W. Guo, H. Wang, B. Zhu, J. Yu and S.-Z. Qiao, Adv. Mater., 2018, 30, 1800128 CrossRef PubMed.
- L. Song, Z. Liu, A. L. M. Reddy, N. T. Narayanan, J. Taha-Tijerina, J. Peng, G. Gao, J. Lou, R. Vajtai and P. M. Ajayan, Adv. Mater., 2012, 24, 4878–4895 CrossRef CAS PubMed.
- J. Wang, F. Ma and M. Sun, RSC Adv., 2017, 7, 16801–16822 RSC.
- Z. Liu, L. Song, S. Zhao, J. Huang, L. Ma, J. Zhang, J. Lou and P. M. Ajayan, Nano Lett., 2011, 11, 2032–2037 CrossRef CAS PubMed.
- Q. Weng, X. Wang, X. Wang, Y. Bando and D. Golberg, Chem. Soc. Rev., 2016, 45, 3989–4012 RSC.
- J. Zhao and Z. Chen, J. Phys. Chem. C, 2015, 119, 26348–26354 CrossRef CAS.
- X. Fu, Y. Hu, Y. Yang, W. Liu and S. Chen, J. Hazard. Mater., 2013, 244–245, 102–110 CrossRef CAS PubMed.
- Z. He, C. Kim, L. Lin, T. H. Jeon, S. Lin, X. Wang and W. Choi, Nano Energy, 2017, 42, 58–68 CrossRef CAS.
- C. Huang, C. Chen, M. Zhang, L. Lin, X. Ye, S. Lin, M. Antonietti and X. Wang, Nat. Commun., 2015, 6, 7698 CrossRef PubMed.
- D. Jun, X. Jun, L. Huaming and L. Zheng, Adv. Mater., 2018, 30, 1704548 CrossRef PubMed.
- Y. Liu, Z. Kang, H. Si, P. Li, S. Cao, S. Liu, Y. Li, S. Zhang, Z. Zhang, Q. Liao, L. Wang and Y. Zhang, Nano Energy, 2017, 35, 189–198 CrossRef CAS.
- H. Yao, L. Liu, W. Fu, H. Yang and Y. Shi, FlatChem, 2017, 3, 1–7 CrossRef CAS.
- J. Zhang, P. Zhang, T. Wang and J. Gong, Nano Energy, 2015, 11, 189–195 CrossRef CAS.
- D. Wang, X. T. Zhang, P. P. Sun, S. Lu, L. L. Wang, Y. A. Wei and Y. C. Liu, Int. J. Hydrogen Energy, 2014, 39, 16212–16219 CrossRef CAS.
- Y. Hou, F. Zuo, A. P. Dagg, J. Liu and P. Feng, Adv. Mater., 2014, 26, 5043–5049 CrossRef CAS PubMed.
- L. Wang and T. Sasaki, Chem. Rev., 2014, 114, 9455–9486 CrossRef CAS PubMed.
- C. Li, H. Wang, D. Lu, W. Wu, J. Ding, X. Zhao, R. Xiong, M. Yang, P. Wu, F. Chen and P. Fang, J. Alloys Compd., 2017, 699, 183–192 CrossRef CAS.
- R. Edy, Y. Zhao, G. S. Huang, J. J. Shi, J. Zhang, A. A. Solovev and Y. Mei, Prog. Nat. Sci.: Mater. Int., 2016, 26, 493–497 CrossRef CAS.
- T. Sasaki and M. Watanabe, J. Am. Chem. Soc., 1998, 120, 4682–4689 CrossRef CAS.
- 12th Russia/CIS/Baltic/Japan Symposium on Ferroelectricity, RCBJSF 2014 and 9th International Conference on Functional Materials and Nanotechnologies, FM and NT 2014, ed. A. Sternberg, L. Grinberga, A. Sarakovskis and M. Rutkis, Institute of Physics Publishing, 2015, p. 275.
- X.-Q. Gong and A. Selloni, J. Phys. Chem. B, 2005, 109, 19560–19562 CrossRef CAS PubMed.
- W.-J. Yin, S.-H. Wei, M. M. Al-Jassim and Y. Yan, Phys. Rev. Lett., 2011, 106, 066801 CrossRef PubMed.
- Y. Gai, J. Li, S.-S. Li, J.-B. Xia and S.-H. Wei, Phys. Rev. Lett., 2009, 102, 036402 CrossRef PubMed.
- Y. Liu, W. Zhou and P. Wu, Mater. Chem. Phys., 2017, 186, 333–340 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- S. Rehman, R. Ullah, A. Butt and N. Gohar, J. Hazard. Mater., 2009, 170, 560–569 CrossRef CAS PubMed.
- W. Zhu, X. Qiu, V. Iancu, X.-Q. Chen, H. Pan, W. Wang, N. M. Dimitrijevic, T. Rajh, H. M. Meyer, M. P. Paranthaman, G. M. Stocks, H. H. Weitering, B. Gu, G. Eres and Z. Zhang, Phys. Rev. Lett., 2009, 103, 226401 CrossRef PubMed.
- Y. Zhang, C. Lin, Q. Lin, Y. Jin, Y. Wang, Z. Zhang, H. Lin, J. Long and X. Wang, Appl. Catal., B, 2018, 235, 238–245 CrossRef CAS.
- L. W. Shan, L. Q. He, J. Suriyaprakash and L. X. Yang, J. Alloys Compd., 2016, 665, 158–164 CrossRef CAS.
- P. Y. Kuang, J. R. Ran, Z. Q. Liu, H. J. Wang, N. Li, Y. Z. Su, Y. G. Jin and S. Z. Qiao, Chem. – Eur. J., 2015, 21, 15360–15368 CrossRef CAS PubMed.
- R. Fu, X. Zeng, L. Ma, S. Gao, Q. Wang, Z. Wang, B. Huang, Y. Dai and J. Lu, J. Power Sources, 2016, 312, 12–22 CrossRef CAS.
- W. Q. Fan, X. Q. Yu, S. Y. Song, H. Y. Bai, C. Zhang, D. Yan, C. B. Liu, Q. Wang and W. D. Shi, CrystEngComm, 2014, 16, 820–825 RSC.
- X. Zhang, H. Yang, B. Zhang, Y. Shen and M. Wang, Adv. Mater. Interfaces, 2016, 3, 1500273 CrossRef.
- Y. X. Yu, W. X. Ouyang and W. D. Zhang, J. Solid State Electrochem., 2014, 18, 1743–1750 CrossRef CAS.
- K. H. Ye, Z. Chai, J. Gu, X. Yu, C. Zhao, Y. Zhang and W. Mai, Nano Energy, 2015, 18, 222–231 CrossRef CAS.
- H. Ma, J. Zhang and Z. Liu, Appl. Surf. Sci., 2017, 423, 63–70 CrossRef CAS.
- Y. Bao and K. Chen, Appl. Surf. Sci., 2018, 437, 51–61 CrossRef CAS.
- J. Di, J. Xia, H. Li, S. Guo and S. Dai, Nano Energy, 2017, 41, 172–192 CrossRef CAS.
- H. Li and L. Zhang, Curr. Opin. Green Sustainable Chem., 2017, 6, 48–56 CrossRef.
- W. Fan, C. Li, H. Bai, Y. Zhao, B. Luo, Y. Li, Y. Ge, W. Shi and H. Li, J. Mater. Chem. A, 2017, 5, 4894–4903 RSC.
- Y. Mi, L. Wen, Z. Wang, D. Cao, R. Xu, Y. Fang, Y. Zhou and Y. Lei, Nano Energy, 2016, 30, 109–117 CrossRef CAS.
- L. Hao, L. Jie, A. Zhihui, J. Falong and Z. Lizhi, Angew. Chem., Int. Ed., 2018, 57, 122–138 CrossRef PubMed.
- J. Wu and X. Cao, Electrochim. Acta, 2017, 247, 646–656 CrossRef CAS.
- C. Zhai, J. Hu, M. Sun and M. Zhu, Appl. Surf. Sci., 2018, 430, 578–584 CrossRef CAS.
- A. Malathi, P. Arunachalam, A. N. Grace, J. Madhavan and A. M. Al-Mayouf, Appl. Surf. Sci., 2017, 412, 85–95 CrossRef CAS.
- R. A. Sayed, S. E. Abd El Hafiz, N. Gamal, Y. GadelHak and W. M. A. El Rouby, J. Alloys Compd., 2017, 728, 1171–1179 CrossRef CAS.
- C. Weijian, W. Taotao, X. Jiawei, L. Shikuo, W. Zidan and S. Song, Small, 2017, 13, 1602420 CrossRef PubMed.
- X. Zhang, R. Wang, F. Li, Z. An, M. Pu and X. Xiang, Ind. Eng. Chem. Res., 2017, 56, 10711–10719 CrossRef CAS.
- R. Chong, B. Wang, C. Su, D. Li, L. Mao, Z. Chang and L. Zhang, J. Mater. Chem. A, 2017, 5, 8583–8590 RSC.
- H. Qi, J. Wolfe, D. Fichou and Z. Chen, Sci. Rep., 2016, 6, 30882 CrossRef CAS PubMed.
- J. Guo, C. Mao, R. Zhang, M. Shao, M. Wei and P. Feng, J. Mater. Chem. A, 2017, 5, 11016–11025 RSC.
- S. Azuma, G. Kawamura, H. Muto, N. Kakuta and A. Matsuda, Key Eng. Mater., 2014, 129–133 CAS.
- S. Zheng, J. Lu, D. Yan, Y. Qin, H. Li, D. G. Evans and X. Duan, Sci. Rep., 2015, 5, 12170 CrossRef CAS PubMed.
- R. Zhang, M. Shao, S. Xu, F. Ning, L. Zhou and M. Wei, Nano Energy, 2017, 33, 21–28 CrossRef CAS.
- D. P. Sahoo, S. Nayak, K. H. Reddy, S. Martha and K. Parida, Inorg. Chem., 2018, 57, 3840–3854 CrossRef CAS PubMed.
- M. S. Islam, M. Kim, X. Jin, S. M. Oh, N.-S. Lee, H. Kim and S.-J. Hwang, ACS Energy Lett., 2018, 3, 952–960 CrossRef CAS.
- X. Lv, X. Xiao, M. Cao, Y. Bu, C. Wang, M. Wang and Y. Shen, Appl. Surf. Sci., 2018, 439, 1065–1071 CrossRef CAS.
- T. S. Sinclair, H. B. Gray and A. M. Müller, Eur. J. Inorg. Chem., 2018, 2018, 1060–1067 CrossRef CAS.
- J. Di, J. Xia, H. Li and Z. Liu, Nano Energy, 2017, 35, 79–91 CrossRef CAS.
- S. Bai, H. Chu, X. Xiang, R. Luo, J. He and A. Chen, Chem. Eng. J., 2018, 350, 148–156 CrossRef CAS.
- L. Yang, L. Chen, D. Yang, X. Yu, H. Xue and L. Feng, J. Power Sources, 2018, 392, 23–32 CrossRef CAS.
- W. Shanpeng, W. Jian, Y. Junwen, H. Qi, G. Dongsheng and L. Li-Min, ChemElectroChem, 2018, 5, 1357–1363 CrossRef.
- X. Fan, B. Gao, T. Wang, X. Huang, H. Gong, H. Xue, H. Guo, L. Song, W. Xia and J. He, Appl. Catal., A, 2016, 528, 52–58 CrossRef CAS.
- Y. Yang, S. Wang, Y. Li, J. Wang and L. Wang, Chem. – Asian J., 2017, 12, 1421–1434 CrossRef CAS PubMed.
- O. Elbanna, M. Fujitsuka and T. Majima, ACS Appl. Mater. Interfaces, 2017, 9, 34844–34854 CrossRef CAS PubMed.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- A. Naseri, M. Samadi, A. Pourjavadi, A. Z. Moshfegh and S. Ramakrishna, J. Mater. Chem. A, 2017, 5, 23406–23433 RSC.
- U. Sim, T. Y. Yang, J. Moon, J. An, J. Hwang, J. H. Seo, J. Lee, K. Y. Kim, J. Lee, S. Han, B. H. Hong and K. T. Nam, Energy Environ. Sci., 2013, 6, 3658–3664 RSC.
- U. Sim, J. Moon, J. An, J. H. Kang, S. E. Jerng, J. Moon, S. P. Cho, B. H. Hong and K. T. Nam, Energy Environ. Sci., 2015, 8, 1329–1338 RSC.
- K. Y. Yoon, H. J. Ahn, M. J. Kwak, P. Thiyagarajan and J. H. Jang, Adv. Opt. Mater., 2015, 3, 907–912 CrossRef CAS.
- Q. Ding, J. Zhai, M. Cabán-Acevedo, M. J. Shearer, L. Li, H. C. Chang, M. L. Tsai, D. Ma, X. Zhang, R. J. Hamers, J.-H. He and S. Jin, Adv. Mater., 2015, 27, 6511–6518 CrossRef CAS PubMed.
- K. C. Kwon, S. Choi, J. Lee, K. Hong, W. Sohn, D. M. Andoshe, K. S. Choi, Y. Kim, S. Han, S. Y. Kim and H. W. Jang, J. Mater. Chem. A, 2017, 5, 15534–15542 RSC.
- J. Hou, H. Cheng, O. Takeda and H. Zhu, Energy Environ. Sci., 2015, 8, 1348–1357 RSC.
- Q. Wei, X. Yan, Z. Kang, Z. Zhang, S. Cao, Y. Liu and Y. Zhang, J. Electrochem. Soc., 2017, 164, H515–H520 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ee00886h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |