
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The synthesis, characterisation and application of iron(III)–acetate complexes for cyclic carbonate formation and the polymerisation of lactide†
Oliver J.
Driscoll
,
Claudia H.
Hafford-Tear
,
Paul
McKeown
,
Jack A.
Stewart
,
Gabriele
Kociok-Köhn
,
Mary F.
Mahon
and
Matthew D.
Jones
 *
*
Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: mj205@bath.ac.uk
First published on 6th September 2019
Abstract
Herein, we report the preparation, characterisation and catalytic applications of air-stable Fe(III)–acetate complexes consisting of salan, salen and salalen ligand frameworks. Owing to the simple synthetic protocol employed, a wide range of complexes have been prepared and structure–activity-relationships investigated. X-ray diffraction confirmed the solid-state structures for eight of the complexes. These represent the first Fe(III)–acetate complexes applied for the selective coupling of CO2/epoxide and lactide polymerisation. The coupling of CO2 and challenging cyclohexene oxide substrate was performed under mild, solvent-free conditions (80 °C, 10 bar CO2) to selectively form the cis-cyclohexene carbonate as the exclusive product (selectivity >99%) with a metal loading of 0.08 mol%. A reduced aminopiperidine ligand backbone was found as the most active catalyst, and after investigating four co-catalysts, showed high functional group tolerance and robustness when applied to a broad, commercially available, terminal epoxide substrate scope with high conversions observed. The ring-opening polymerisation of rac-lactide was achieved using the Fe(III)OAc complexes using triethylamine and benzyl alcohol for initiation, interestingly isoselectivity was observed in some cases.
Introduction
With global population, energy and material demands increasing, sustainability in chemical processes is critical to address these issues.1,2 This has catalysed an impetus to explore Earth-abundant metal systems as catalysts, for example, iron.1–4 Recently, iron has re-attracted increasing attention due to its high abundance (4th most abundant element in the Earth's crust), low toxicity, low cost (both at commercial and industrial scale) and potential air-stability. A resurgence of iron catalysed reactions has been seen in the ring-opening polymerisation (ROP) of cyclic esters, such as lactide, and the catalytic transformation of CO2, such as with the coupling of epoxides.5–12Poly(lactic acid) (PLA) is a more sustainable, renewable, biodegradable and biocompatible alternative plastic compared to those derived from crude oil, with the potential for a ‘closed-loop’ lifecycle.13,14 PLA can be used in food packaging, drug delivery systems and biomedical applications.15–17 Industrially, PLA is formed via the ROP of lactide using Sn(II)–octanoate.14,18–20 With rac-LA, the stereoselectivity of the polymer tacticity is uncontrolled and causes diminished thermal properties. This can be solved by the use of metal initiators able to stereoselectively differentiate between the two enantiomers of a racemic mixture of lactide monomers (D/L).14,18 Control of the stereochemistry and tacticity enables control of the polymer microstructure and therefore the polymers’ bulk physical properties, such as flexibility, durability, thermal properties, crystallinity and biodegrability.21
Examples of Fe-mediated ROP in the literature are less prevalent despite the numerous benefits.22–37 Recently Duan et al. synthesised and applied air-stable Fe(III)–salen–chloride complexes to the ROP of caprolactone and lactide.12 Using propylene oxide (PO), as both the solvent and co-initiator, the postulated in situ generated Fe(III)–alkoxide species, formed via opening of PO by insertion of the Fe(III)–Cl bond, resulted in isotactic PLA (Pm = 0.53–0.78) with broad dispersities (Đ = 1.38–2.36). We recently reported a variety of Fe(III)–salalen complexes that followed this mechanism.10 Moderate isotacticity (Pm = 0.75–0.80) and good molecular weight control in PO were observed (Đ = 1.02–1.18). Batch kinetics using an Fe(III)–salalen complex indicated there was an induction period within the six hours, likely relating to the in situ generation of the Fe(III)–alkoxide species. Shaver and co-workers attempted to decrease the concentration of toxic PO used with Fe(III)– bis(phenoxyimine)–chloride complexes in toluene but found an excess was still required for controlled polymerisation and consistent initiation.11
Cyclic organic carbonates (COCs) are in high demand for a range of applications such as high boiling polar aprotic solvents, lithium-ion battery electrolytes, anti-foam additives, plasticisers, copolymerisation with cyclic esters and intermediates in both organic synthesis and industry.5,38 The traditional method of synthesis includes the use of toxic phosgene.38–41 A sustainable alternative is using CO2 as a renewable, non-toxic, abundant, cheap, ‘waste’ material and C1-synthon building block. Coupling this thermodynamically stable molecule with reactive epoxides enables the formation of either COCs and/or aliphatic polycarbonates (APCs) depending on the catalyst/co-catalyst system and reaction conditions employed which determines the selectivity and activity. There are only a few cases whereby the catalyst is able to produce both products and be switchable depending on conditions.41–44 There are a number of reviews on this area.38,40,41,45–49
There has been a significant focus on using Mg, Cr, Co, Zn and Al complexes, but only a limited number of mono- and dinuclear-iron examples have been reported for effective epoxide/CO2 coupling.5–8,42,50–53 A variety of ligand classes have been applied specifically to iron and recently these were reviewed by Della Monica et al.5 Salen, bidentate phenoxyimine (half salen) and, recently, salalen and salan ligands have all been successfully applied to the selective formation of COCs.54–58 Lamberti and co-workers reported the first such example of the application of Fe–salan and –salalen complexes, together with Fe–salen complexes for comparison, for the coupling of CO2 with propylene oxide (PO), cyclohexene oxide (CHO) and styrene oxide (SO).9 Varying the hybridisation of the nitrogen donor atoms, and maintaining the ethylene backbone, aromatic groups and chloride auxiliary group, it was observed that at 100 °C and 20 bar of CO2 the order of reactivity decreased from salan to salen to salalen. Recently, Kerton synthesised, studied and applied μ-oxo-bridged, dinuclear-Fe-amino-bis (phenolate) complexes to epoxide deoxygenation and CO2/epoxide coupling.7,59 The μ-oxo-bridged complexes with geometric preference values (τ) ranging from 0.22–0.31, indicate a square based pyramidal geometry, showed lower TOF values than the respective mononuclear Fe–chloride complexes and Lamberti's complexes.9 This implies the auxiliary ligand and the structure/flexibility of the bridging backbone is crucial for reactivity. Interestingly, it was proposed that the change of colour of the reaction mixture from purple to dark red during the CO2 reaction was due to a deoxygenation step.
In this work, we synthesised, characterised and explored a range of air-stable Fe(III)–acetate complexes with salalen, salan and salen ligand frameworks. To our surprise there are very few examples of Fe(III)–acetate complexes with ONNO ligands characterised in the solid-state. Examples herein represent the first Fe(III)–acetate complexes applied for polymerisation and CO2/epoxide coupling. The polymerisations were conducted using NEt3 and BnOH for initiation and isoselectivity was observed in certain cases. The formation of the cis-CHC product was observed when using CHO and, after investigating different co-catalysts, high functional group tolerance was observed when applied to a broad epoxide scope. With the ease of synthesis, the aryl and backbone substituents were modified and structure–activity-relationships investigated.
Results and discussion
Complex synthesis
The Fe(III)–acetate complexes were prepared, in air, from Fe(OAc)2 and ligand in ethanol. This method has been previously been used to make a μ-oxo-bridged Fe(III)–salen complex by Webster and co-workers,60 along with other examples of preparation and application of this complex in the literature.61–71 However, with the range of salalen, salan and salen ligands (X)72–76 used in this study, the Fe(X)OAc complex was consistently isolated (Scheme 1). All complexes were recrystallised or washed with cold ethanol and characterised by High-Resolution Mass Spectrometry (HR-MS), elemental analysis and Infra-Red spectroscopy (FT-IR). X-ray crystallography confirmed an acetate auxiliary group remained on the Fe-centre in the solid-state structure. Mass spectrometry confirmed the coordination of the ligand to the metal centre; [Fe(X)]+ ions were observed in all cases. Elemental analysis confirmed the Fe(III)–acetate was present and not the μ-oxo-bridged Fe(III) species in the bulk of the sample. | ||
| Scheme 1 The range of Fe(X)OAc complexes synthesised in this study. | ||
Using Evans’ NMR spectroscopic method, the effective magnetic moments for the Fe(1–12)OAc complexes generally ranged from 4.57–5.74 μB at 298 K in CDCl3, closer to the 5.92 μB spin-only value for high-spin d5 Fe(III) complexes (S = 5/2) than the 1.73 μB spin-only value for low-spin d5 Fe(III) complexes (S = 1/2). This is with the exception of Fe(9)OAc and Fe(12)OAc which had values of 3.29 μB and 2.10 μB respectively. This may be due to μ-oxo-bridged or Fe(OAc)2 impurities lowering the magnetic moments as reported in literature.59,77
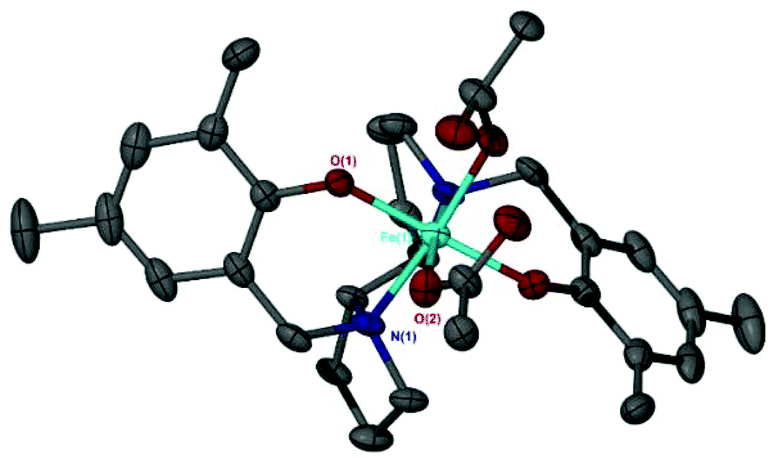
X-ray crystallography confirmed the solid-state structures for Fe(2/3/4/6meso/7meso/8/10)OAc to be distorted pseudo-trigonal bipyramidal geometries with the acetate auxiliary group occupying an equatorial site.
This geometry is analogous to the Fe(III)–chloride complexes previously reported.10 This is evident when comparing Fe(2)OAc and the analogous iron–chloride salalen complex {Fe(2)Cl}. Overlaying the solid-state structures shows the chloride and the central carbon of the acetate occupying the same equatorial position (Fig. 1). Focusing on this carbon atom of the acetate and disregarding the oxygen atoms, the geometric preference (τ) for Fe(2)OAc was similar to that of the Fe(2)Cl complex {Fe(2)OAc, τ = 0.65 vs. Fe(2)Cl, τ = 0.66} with a moderate preference for a trigonal bipyramidal (tbp) geometry (τ = 1) over the square based pyramidal (sbp) geometry (τ = 0). This preference decreases for the more symmetrical salen analogue Fe(10)OAc (τ = 0.59).78
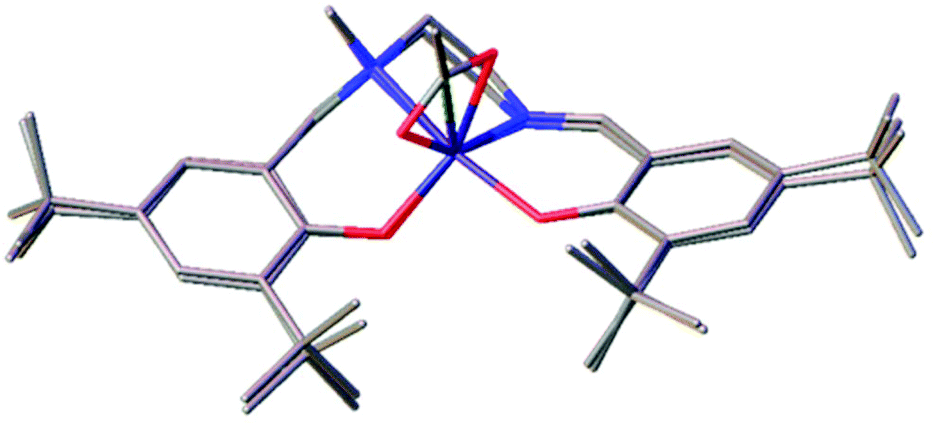 | ||
| Fig. 1 Solid-state structures of Fe(2)OAc and Fe(2)Cl overlayed with the acetate and chloride groups occupying the same position. | ||
For the other salalen complexes, this preference decreases further still when installing an electron withdrawing phenyl group on the amine {Fe(3)OAc, τ = 0.60} and when a more rigid aminopiperidine backbone was present {Fe(4)OAc, τ = 0.58} (Fig. 2). As was observed for the Fe(X)Cl complexes,10 in all salalen cases, the axial positions were occupied by O(2) and the secondary amine, N(1). The angles deviate slightly from the ideal 180°: {O(2)–Fe–N(1) = 165.35(6)° for Fe(2)OAc, =165.42(7)° for Fe(3)OAc, =165.13(6)° for Fe(4)OAc}. The largest equatorial angle, for all Fe-acetate complexes, was to the central acetate carbon {O(1)–Fe–C(acetate) = 126.20 for Fe(2)OAc, =129.28° for Fe(3)OAc, =130.56° for Fe(4)OAc}. This was with the exception of the Fe(10)OAc salen complex {O(1)–Fe–N(2) 123.48°} (Fig. 2). Fe(6meso)OAc, with a restricted bipyrrolidine salan backbone and methyl substituents on the aromatic rings, gave a small preference for the tbp geometry (τ = 0.58), however, increasing the steric bulk of these substituents to tBu groups forced the preference to increase moderately {Fe(7meso)OAc, τ = 0.63}. The lowest preference was observed with Fe(8)OAc (τ = 0.54) consisting of a rigid six-membered aminopiperidine ring on one nitrogen donor atom and an unsubstituted nitrogen in the backbone (Fig. 2). Fe(8)OAc is also observed to have a different configuration to Fe(4)OAc, with the piperidine ring in an equatorial site rather than axial. This is in agreement with the corresponding aluminium complexes.73,76
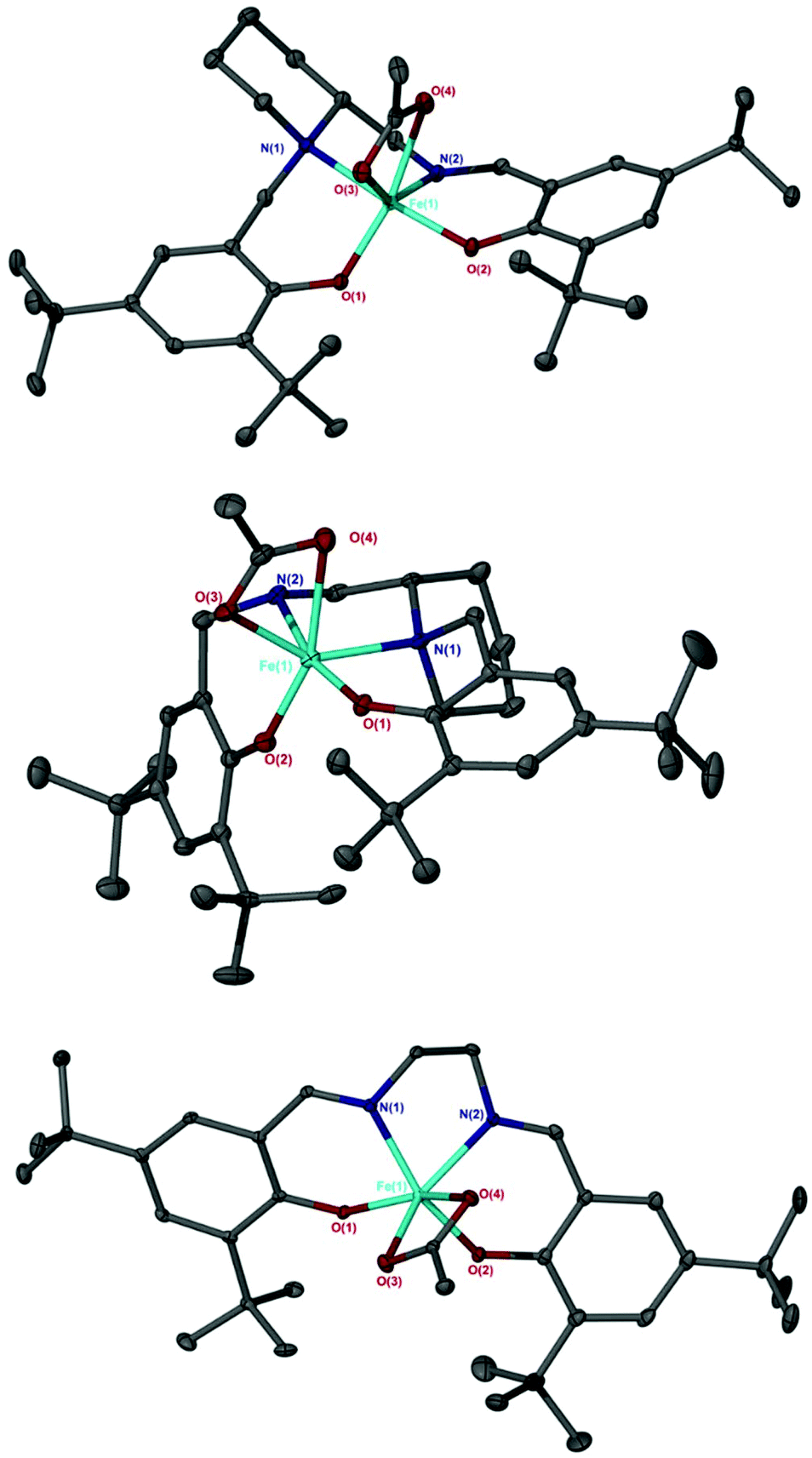 | ||
| Fig. 2 Solid-state structures of Fe(4)OAc (Top), Fe(8)OAc (middle) and Fe(10)OAc (bottom). Ellipsoids are shown at 30% probability level and all hydrogen atoms have been removed for clarity. | ||
The metal-to-acetate bond lengths to each oxygen donor atom were different however and not identical in all complexes. For Fe(2)OAc, these bond lengths varied by 0.1136 Å {Fe–O(3) = 2.0872(15) Å and Fe–O(4) = 2.2008(15) Å} highlighting asymmetry of the acetate geometry and bond delocalisation. In this particular example, there is an intermediate hydrogen bonding interaction between O(3) and an ethanol solvent of recrystallisation. The overlying trend was that the oxygen donor atom O(4) cis to the imine nitrogen N(2) had a longer metal-to-acetate bond length.
Crystals of suitable quality were not obtained for Fe(9)OAc to allow comparison with the solid-state structure of the analogous μ-oxo-bridged Fe(III)–salen complex {[Fe(9)]2O} confirmed by Webster.60 However, pXRD of Fe(9)OAc isolated in this study afforded a different diffraction pattern compared to that calculated from the μ-oxo-bridged single crystal X-ray data (see ESI†).60 This provided further evidence for the isolation of acetate complexes and not μ-oxo-bridged in this study.
Epoxide and CO2 coupling
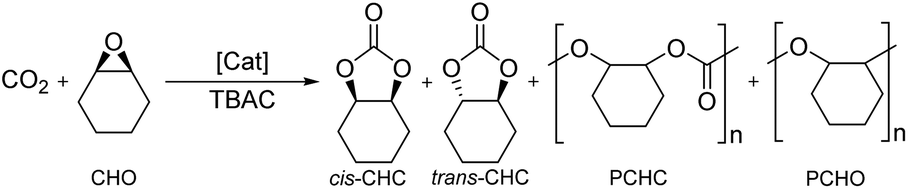
The Fe(X)OAc complexes were screened for the coupling of CO2 with distilled cyclohexene oxide (CHO) to study their activities and selectivity (Table 1). CHO, an internal epoxide, is a more challenging substrate compared to alternatives, such as PO, due to the high steric hindrance and the possibility of forming four products, reducing selectivity. Reactions were performed solvent-free with a 0.08 mol% catalyst loading and 0.64 mol% tetrabutylammonium chloride (TBAC) as a co-catalyst, following catalyst/co-catalyst amounts employed by Lamberti for Al(III)–salalen complexes,79 at 10 bar pressure of CO2 at 80 °C for 24 hours. Conversion, activity and product selectivity were determined by taking aliquots of the crude product mixture for 1H NMR spectroscopy and integrating observed methine proton resonance signals for cis-cyclohexene carbonate (cis-CHC), trans-cyclohexene carbonate (trans-CHC), polycyclohexene carbonate (PCHC) and polycyclohexene oxide (PCHO) respectively to CHO. In the majority of cases, cis-CHC was formed as the exclusive product, which is rare in the literature due to the bicyclic ring strain of CHC. The thermodynamic product is trans-CHC which is usually formed over cis-CHC via back-biting reactions of the PCHC product polymer chain.42 The formation of cis-CHC is encouraged by using excess co-catalyst to allow for the proposed double inversion reaction pathway. Lamberti observed that at 4 eq. or higher of co-catalyst, selectivity was at >99% and therefore 8 eq. was used here.79 PCHO, formed via polymerisation of the epoxide without insertion of the CO2, was the most common side-product observed in this study. The PCHC product is reported to appear at δ 4.65 ppm, close to the cis-CHC.42 ESI-MS confirmed that cyclic carbonate was indeed formed and not the PCHC, moreover no polymer was observed in the GPC analysis. Control reactions revealed that TBAC co-catalyst was required to open the epoxide and no reaction was observed with just [Fe] catalyst (Table 1, entry 6). Activity was observed using TBAC either with Fe(OAc)2 metal precursor or without an iron catalyst (Table 1, entries 1 and 2), however product selectivity was significantly lowered (76% and 83% respectively). Recently, North and co-workers reported that salen ligands without metal and co-catalyst were able to carry out the reaction at 10 bar pressure of CO2 at 120 °C via a ‘dual activation mechanism’.80 However, with ligand 2 and no TBAC co-catalyst showed 0% conversion and with TBAC co-catalyst was active but with a poor selectivity. (Table 1, entries 3 and 4). Overall, all control reactions demonstrated poor product selectivity (Table 1, entries 1–4 and 6).|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Conv.a/% | Selectivity for cis-CHCa/% |
cis-CHC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :trans-CHC:PCHC:PCHO ratioa/% :trans-CHC:PCHC:PCHO ratioa/% |
TOFa/h−1 |
| Conditions: [Fe] Catalyst (0.08 mol%, 1 eq.), TBAC (8 eq.), CHO (5.0 mL), 10 bar CO2, 80 °C, 24 h.a Determined via1H NMR spectroscopy using the methine resonances of cis-CHC (δ 4.66 ppm), trans-CHC (δ 3.99 ppm) and PCHO (δ 3.35 ppm).b No TBAC added.c Time = 72 h.d 20 bar CO2. | |||||
| 1 | Fe(OAc)2 | 48 | 76 | 76:1:0:23 |
26 |
| 2 | None | 43 | 83 | 83:0:0:17 |
22 |
| 3b | Ligand 2 | 0 | — | — | 0 |
| 4 | Ligand 2 | 47 | 84 | 84:2:0:14 |
24 |
| 5 | Fe(1)OAc | 40 | >99 | >99:0:0:0 |
21 |
| 6b | Fe(2)OAc | 0 | — | — | 0 |
| 7 | Fe(2)OAc | 45 | >99 | >99:0:0:0 |
23 |
| 8c | Fe(2)OAc | 52 | 96 | 96:4:0:0 |
9 |
| 9d | Fe(2)OAc | 47 | >99 | >99:0:0:0 |
24 |
| 10 | Fe(2)Cl | 41 | >99 | >99:0:0:0 |
21 |
| 11 | Fe(3)OAc | 38 | >99 | >99:0:0:0 |
20 |
| 12 | Fe(4)OAc | 46 | >99 | >99:0:0:0 |
24 |
| 13 | Fe(5)OAc | 53 | >99 | >99:0:0:0 |
28 |
| 14 | Fe(6meso)OAc | 30 | 57 | 57:6:0:37 |
16 |
| 15 | Fe(7meso)OAc | 45 | >99 | >99:0:0:0 |
23 |
| 16 | Fe(8)OAc | 66 | >99 | >99:0:0:0 |
34 |
| 17 | Fe(9)OAc | 28 | >99 | >99:0:0:0 |
15 |
| 18 | Fe(10)OAc | 43 | >99 | >99:0:0:0 |
22 |
| 19 | Fe(11)OAc | 52 | >99 | >99:0:0:0 |
27 |
| 20 | Fe(12)OAc | 59 | 84 | 84:0:0:16 |
31 |
The Fe(III)–salalen–acetate complexes were initially explored. The increase of steric bulk at R1, from H to tBu, increased conversion {Fe(1)OAc, R1 = H, 40% vs. Fe(2)OAc, R1 = tBu, 45%} while selectivity remained >99% for the cis-CHC. This increase could also be related to the increased electron donation of tBu compared to H. The Fe(III)–acetate complex was marginally more active than the corresponding analogous Fe(III)–chloride complex {Fe(2)OAc, R1 = tBu, 45% vs. Fe(2)Cl, 41%}. Increasing both the reaction time from 24 h to 72 h and pressure of CO2 from 10 bar to 20 bar led to a slight increase in the conversion (Table 1, entry 7 vs. entries 8 and 9). Modifying the substituent on the amine group (R2) from an electron donating Me group to an electron withdrawing phenyl moiety lowered the conversion to 38% (Table 1, entry 11). Changing the ethylene backbone to a more rigid aminopiperidine {Fe(4)OAc} maintained the same activity and a more flexible propyl backbone {Fe(5)OAc} increased conversion marginally to 53%. The increase of activity with an increase of flexibility agrees with that observed by Garden and Lamberti.9,54
The Fe(III)–salan–acetate complexes were generally more effective than the salalen complexes. Modifying the ethylene backbone to bipyrrolidine and changing the hybridisation of the nitrogen atom from imine to amine (salalen to salan) offered minimal improvement in activity. The product selectivity was maintained at >99% except for Fe(6meso)OAc where 37% of the product was PCHO and 6% of trans-CHC. Fe(6meso)OAc afforded a poor elemental analysis which is potentially related to unreacted Fe(OAc)2 which may explain the poor selectivity observed. The most effective catalyst in this study was Fe(8)OAc, containing both a rigid aminopiperidine ring on one nitrogen donor atom and a NH group. The conversion was 66% with cis-CHC the exclusive product.
North proposed intramolecular H-bonding interactions in the cyclic carbonate mechanism when an uncomplexed salen ligand was applied as the catalyst, with the hydrogen atoms of both phenol groups interacting with the imine nitrogen atoms and incoming epoxide/CO2.80 The NH of Fe(8)OAc could potentially be involved in H-bonding interactions, with the incoming epoxide/CO2, leading to the dramatic improvement in activity compared to the unreduced Fe(4)OAc {Fe(8)OAc, 66% vs. Fe(4)OAc, 46%}. The importance of H-bonding interactions has recently been showed by Romain for lactide polymerisation.81 For the Fe(III)–salen–acetate complexes, activity was increased by installing a planar phenyl ring into the Fe(III)–salen ethylene backbone {Fe(11)OAc, 52% vs. Fe(9)OAc, 28% and Fe(12)OAc, 59% vs. Fe(10)OAc, 43%}. Indeed Fe(12)OAc gave the second highest activity observed in this study, however, this was accompanied by a decrease in product selectivity to 84%.
Different co-catalysts were explored using the most effective catalyst, Fe(8)OAc (Table 2). It was found that moving away from TBAC had detrimental effects on activity. While cis-CHC remained the exclusive product for tetrabutylammonium bromide (TBAB) and bis (triphenylphosphine)iminium chloride (PPNCl) as the co-catalyst, the conversion was reduced. Tetrabutylammonium acetate (TBAAc), with the same acetate anion as the auxiliary ligand of the catalyst, was less active and selective. Solubility of the co-catalyst in CHO and solvent-free conditions was one possible reason for these observed results. TBAC was completely soluble at room temperature and also the most active.
| Entry | Co-catalyst | Conv.a/% | Selectivity for cis-CHCa/% |
cis-CHC:trans-CHC:PCHC:PCHO ratioa/% |
TOFa/h−1 |
|---|---|---|---|---|---|
| Conditions: [Fe] Catalyst (0.08 mol%, 1 eq.), co-catalyst (8 eq.), CHO (5.0 mL), 10 bar CO2, 80 °C, 24 h.a Determined via1H NMR Spectroscopy. | |||||
| 1 | TBAB | 57 | >99 | >99:0:0:0 |
30 |
| 2 | TBAAc | 32 | 64 | 64:17:0:19 |
17 |
| 3 | PPNCl | 58 | >99 | >99:0:0:0 |
30 |
The combination of Fe(8)OAc and TBAC showed high functional group tolerance and robustness when applied to a broad, terminal epoxide substrate scope (Table 3). Moderate to high conversions to the cyclic carbonate products were observed while varying the sterics and electronics of the epoxide. All products were analysed from the crude reaction mixtures using 1H NMR spectroscopy, ESI-MS to confirm the cyclic product (not polymer) was present and GPC analysis to confirm no polymer was produced. Styrene oxide (SO), with a sterically bulky phenyl group, showed similar reactivity to the sterically bulky, internal CHO and a modest 66% conversion. The sterically unhindered propylene oxide (PO) was more reactive as expected and resulted in a higher conversion (79%). Electron-withdrawing groups (EWGs), generally increased reactivity and epoxide ring-opening as expected. Epichlorohydrin (ECH) with a chloro-EWG showed modest conversion (75%) however, phenylglycidyl ether (PGE) and allylglycidyl ether (AGE) achieved high conversions (97% and 93% respectively). Due to the solidification of the phenoxymethyl ethylene carbonate product from PGE and inefficient mechanical stirring towards the end of the 24 hours, the reaction was repeated for 16 hours to afford a higher TOF value of 63 h−1.
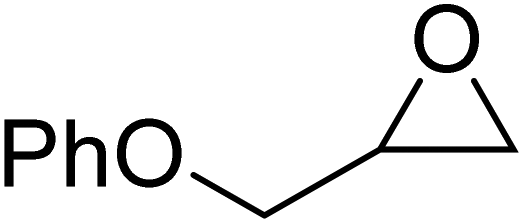
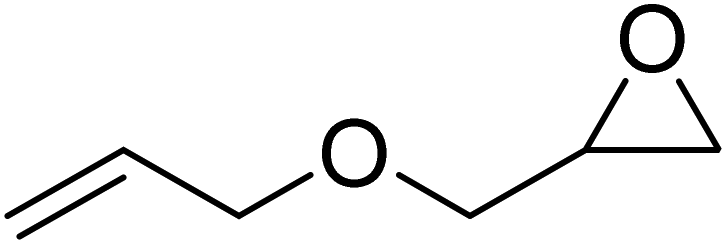
The chirality of the bipyrrolidine backbone of Fe(6meso)OAc was explored further by attempting to synthesise two other possible stereoisomers, Fe(6RR)OAc and Fe(6SS)OAc.
Changing the chirality could affect the structure and coordination around the metal centre, with potential for these new species to increase the activity for the asymmetric CO2/epoxide reaction.82 Applying the complexes to the coupling of CO2 with distilled CHO, conversion increased moderately for the (S,S) enantiomer compared to the meso enantiomer {Fe(6meso)OAc, 30% vs. Fe(6SS)OAc, 47%} (Table 4). However, there was a stark increase when applying the (R,R) enantiomer where conversion doubled to 60% {Fe(6meso)OAc, 30% vs. Fe(6RR)OAc, 60%}. Mass spectrometry confirmed the coordination of the ligand to the metal centre {[Fe(6RR)]+} and FT-IR also supported the formation of an Fe(III)–acetate complex. However, single-crystal X-ray crystallography confirmed the solid-state structure of the complex to be Fe(6RR)Y2 (Y = OAc, OEt, HOAc or HOEt) with a pseudo octahedral geometry (Fig. 3). An effective magnetic moment of 5.58 μB at 298 K in CDCl3 was observed for Fe(6RR)Y2, using Evans’ NMR spectroscopic method, which is more consistent with a high spin d5 Fe(III) center. Therefore one of the auxiliary groups (OEt/OAc) must be protonated in the solid-state structure but given the disorder and partial occupancy of these sites it is not possible to reliably locate a hydrogen atom.
| Entry | Catalyst | Conv.a/% | Selectivity for cis-CHCa/% |
Cis-CHC:trans-CHC:PCHC:PCHO ratioa/% |
TOFa/h−1 |
|---|---|---|---|---|---|
| Conditions: [Fe] Catalyst (0.08 mol%, 1 eq.), TBAC (8 eq.), CHO (5.0 mL), 10 bar CO2, 80 °C, 24 h.a Determined via1H NMR spectroscopy using the methine resonances of cis-CHC (δ 4.66 ppm), trans-CHC (δ 3.99 ppm) and PCHO (δ 3.35 ppm). | |||||
| 1 | Fe(6meso)OAc | 30 | 57 | 57:6:0:37 |
16 |
| 2 | Fe(6RR)OAc | 60 | >99 | >99:0:0:0 |
31 |
| 3 | Fe(6SS)OAc | 47 | >99 | >99:0:0:0 |
24 |
 | ||
| Fig. 3 Solid-state structure of Fe(6RR)Y2. Ellipsoids are shown at 30% probability level and all hydrogen atoms have been omitted for clarity. | ||
The elemental analysis of Fe(6RR)Y2 was in close agreement with the solid-state structure. Suitable crystals for crystallography were not realised for Fe(6SS)OAc, it is therefore conceivable that this complex has a similar structure to that of Fe(6RR)Y2.
Throughout this study, and especially while coupling with CHO, the reaction mixtures generally change colour from dark purple to dark red. Kerton has postulated this to be due to the formation of a μ-oxo-bridged Fe(III) species from the Fe(III) chloride complexes, via epoxide deoxygenation, which then proceeds via a less active catalytic cycle.59 UV-visible spectroscopy, in acetonitrile, was recorded between 300–800 nm in absorbance mode and showed diagnostic absorption bands at 445 nm and 505 nm for Fe(2)Cl and 515 nm with a small peak at 450 nm for Fe(2)OAc. These were attributed to being ligand-to-metal charge-transfer (LMCT) bands from the transitions between the phenolate oxygen to the high-spin Fe(III) center, which is generally accepted in the literature.7,9,59 This indicated the Fe(2)OAc was minimally more Lewis acidic than Fe(2)Cl as would be expected and, crucially, no absorption band at around 430 nm was observed, indicating μ-oxo-bridged Fe(III) complexes were not initially present in the bulk of the samples. These LMCT transitions are also causing the dark purple colour that is visibly observed. Absorption bands at 300 nm or lower are assigned as π → π * transitions between the phenolate moieties but are present with or without Fe metal and hence not diagnostic. After the reaction of CHO and CO2, with Fe(2)Cl or Fe(2)OAc, a hypsochromic shift is observed toward 430 nm and 425 nm respectively, which is proposed to be due to the formation of μ-oxo-bridged Fe(III) complexes in agreement with Kerton (Fig. 4).59 This implies that epoxide deoxygenation is still occurring despite moving from a chloride to a more stable bidentate acetate group. Recently, Della Monica reported the formation of an anionic Fe(III) metallate species by the addition of excess TBAB to a Fe(III)–chloride complex bearing a bis-thioether-diphenolate ligand. The combination of 8 eq. of TBAC to Fe(2)OAc, in acetonitrile, resulted in the solution remaining purple and the UV-visible spectroscopy profile unchanged, implying no metallate species were being formed in this study, and was not the cause for the observed colour change (see ESI†).5,83
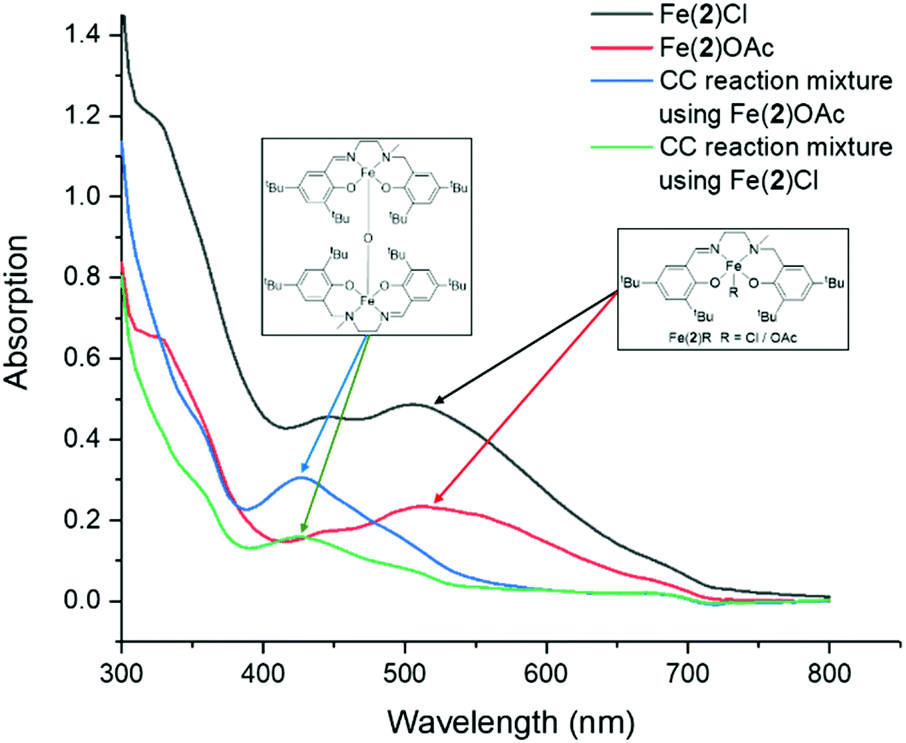 | ||
| Fig. 4 UV-visible absorption spectra for Fe(2)Cl, Fe(2)OAc and the crude CO2 coupling reaction mixtures using Fe(2)Cl and Fe(2)OAc. | ||
Ring-opening polymerisation
All iron acetate complexes were trialled for activity in the ROP of rac-LA. Initial experiments demonstrated the need for both Et3N and BnOH to facilitate the polymerisation. The former is presumably required to remove the OAc group and the latter to generate an active alkoxide species. Subsequent polymerisations were carried out at 100 °C for 24 hours, with a loading of 100:1:1:1 ([LA]:[Fe]:[BnOH]:[Et3N]) (Table 5).
| Catalyst | Conv.a/% | P m | M n,theo /g mol−1 | M n /g mol−1 | Đ | |
|---|---|---|---|---|---|---|
| Conditions: Toluene, 100 °C, [LA]:[Fe]:[BnOH]:[Et3N] = 100:1:1:1, 24 h.a Determined from 1H NMR spectroscopy.b Determined from 1H{1H} NMR spectroscopy.c Calculated based on polymer conversion {(%conv × Mn,LA) + Mn,BnOH}, rounded to the nearest 50.d Measured by GPC (THF, RI), rounded to the nearest 50. |
||||||
| Salalen | Fe(1)OAc | 26 | — | 3850 | 2000 | 1.17 |
| Fe(2)OAc | 57 | 0.53 | 8300 | 6550 | 1.13 | |
| Fe(3)OAc | 60 | 0.45 | 8750 | 7300 | 1.26 | |
| Fe(4)OAc | 5 | — | 800 | — | — | |
| Fe(5)OAc | 43 | 0.5 | 6300 | 6700 | 1.10 | |
| Salan | Fe(6meso)OAc | 92 | 0.58 | 13400 |
19900 |
1.30 |
| Fe(6RR)OAc | 21 | — | 3150 | 1850 | 1.27 | |
| Fe(6SS)OAc | 32 | — | 4700 | 2050 | 1.20 | |
| Fe(7meso)OAc | 51 | 0.49 | 7450 | 6600 | 1.09 | |
| Fe(8)OAc | 93 | 0.67 | 13500 |
8700 | 1.09 | |
| Salen | Fe(9)OAc | 95 | 0.56 | 13800 |
7500 | 1.49 |
| Fe(10)OAc | 67 | 0.71 | 9800 | 7750 | 1.16 | |
| Fe(11)OAc | 94 | 0.61 | 13600 |
8500 | 1.65 | |
| Fe(12)OAc | 89 | 0.71 | 12900 |
11700 |
1.46 | |
The salalen complexes typically gave low conversion and no selectivity. Increasing the steric bulk of the ethylene diamine salalen {Fe(1)OAc vs. Fe(2)OAc/Fe(3)OAc} gave a slight improvement in activity. Increasing the backbone to propylene caused a reduction in conversion. Molecular weights were observed to be lower than expected based on conversion. Where recorded, relatively narrow dispersities are observed for these complexes (Đ = 1.10–1.26). MALDI-ToF analysis for polymer derived from Fe(2/3)OAc revealed a symmetrical major series which had the expected BnO–/–H end groups. However, there was a minor series that exhibited ethoxide end groups.
For these complexes, ethanol molecules were observed in the unit cell of the solid-state structures and these would act as additional initiating groups accounting for the reduced molecular weights. The degree of transesterification is negligible. Compared to the chloride analogues,10 Fe(1–2)OAc were less active, requiring higher temperatures to achieve moderate conversion. The iron salan acetate complexes typically achieved higher conversion relative to the salalen complexes. For these complexes, reducing the steric contribution of the ligand had a positive impact on complex activity {Fe(6meso)OAc vs. Fe(7meso)OAc}. The chiral complexes, Fe(6RR/6SS)OAc performed less well compared to the meso form and this could be related to the differences in the solid-state structures. The reduced complex of 4, Fe(8)OAc, demonstrates a substantial improvement in activity compared to the salalen complex, Fe(4)OAc. This complex also imparts a slight isotactic preference on the polymerisation (Pm = 0.66). MALDI-TOF analysis of polymer derived from Fe(8)OAc showed two symmetrical series centered on 5300 g mol−1 (see ESI†). The major series has the expected end groups of BnO–/–H and the minor series was related to initiation by ethanol, the latter being the solid-state recrystallisation solvent.
There is no evidence of transesterification highlighting the control of Fe(8)OAc. The salen complexes also achieve higher conversion under these conditions. The increased planarity, due to a phenyl backbone, has little effect on the polymerisation. There is an isotactic bias for all of the salen complexes (Pm = 0.56–0.71). MALDI-ToF analysis of polymer derived from Fe(10)OAc showed there to be only the expected benzyl alkoxide end groups. However, a minor series demonstrated the operation of undesirable transesterification to give peak separations of 72 g mol−1. While less active than the previously reported analogous iron chloride complex,12 Fe(10)OAc was observed to give higher isotacticity.
The more active catalysts were also tested at 80 °C (Table 6). While the level of stereocontrol was not increased by this temperature decrease, high conversion was achievable and the dispersity was improved in all cases. MALDI-ToF analysis of polymer derived from Fe(6meso)OAc revealed BnO–/–H end groups with no transesterification. The molecular weight was, however, lower than expected potentially due to EtOH solvent in the recrystallised complex.
| Catalyst | Conv.a/% | P m | M n,theo /g mol−1 | M n /g mol−1 | Đ |
|---|---|---|---|---|---|
| Conditions: Toluene, 80 °C, [LA]:[Fe]:[BnOH]:[Et3N] = 100:1:1:1, 24 h.a Determined from 1H NMR spectroscopy.b Determined from 1H{1H} NMR spectroscopy.c Calculated based on polymer conversion {(%conv × Mn,LA) + Mn,BnOH}, rounded to the nearest 50.d Measured by GPC (THF, RI), rounded to the nearest 50. |
|||||
| Fe(6meso)OAc | 96 | 0.58 | 13950 |
8100 | 1.09 |
| Fe(8)OAca | 94 | 0.66 | 13600 |
9900 | 1.07 |
| Fe(11)OAca | 95 | 0.61 | 13800 |
10750 |
1.49 |
| Fe(12)OAca | 82 | 0.71 | 11900 |
11050 |
1.37 |
Conclusions
Fourteen air-stable Fe(III)–acetate complexes were synthesised and characterised by elemental analysis, ESI-MS, FT-IR spectroscopy and Evans’ NMR spectroscopic method. X-ray crystallography confirmed the solid-state structures for Fe(2/3/4/6meso/7meso/8/10)OAc to be distorted pseudo-trigonal bipyramidal geometries. Due to the simple synthetic method, a range of ligand frameworks were explored and structure–activity-relationships investigated, when the complexes were applied to the coupling of CO2/CHO and the ROP of rac-lactide. Fe(8)OAc, with the reduced aminopiperidine ligand backbone, was found to be the most active complex for the selective formation of cis-CHC from, the challenging internal epoxide, CHO with a TOF value of 34 h−1 at mild conditions (80 °C, 10 bar CO2, 24 h). High functional group tolerance was also demonstrated when applied to a broad, terminal epoxide substrate scope. Chirality of the ligand backbone was explored using Fe(6meso/6RR/6SS)OAc and the R,R enantiomer showed a vast improvement in activity, potentially due to the formation of an octahedral complex. The change of colour of the CO2/epoxide coupling reaction mixtures was attributed to the formation of μ-oxo-bridged Fe(III) complexes via UV-Vis spectroscopy. All Fe(III)–acetate complexes were trialled for the ROP of rac-LA using both Et3N and BnOH, which were shown to be needed for initiation. Fe(8)OAc and Fe(12)OAc were the most effective complexes for the ROP of rac-LA with a slight isotactic bias observed (Pm = 0.66 and Pm = 0.71 respectively).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the University of Bath and the EPRSC (EP/L016443/1 and EP/P016405/1) for funding. We also thank Isabel Thomlinson and Daniel Berry for help with the graphical abstract. MC2 are also acknowledged for the use of their analytical facilities.Notes and references
- P. Chirik and R. Morris, Acc. Chem. Res., 2015, 48, 2495–2495 CrossRef CAS.
- M. Albrecht, R. Bedford and B. Plietker, Organometallics, 2014, 33, 5619–5621 CrossRef CAS.
- C. Darcel and J.-B. Sortais, Isr. J. Chem., 2017, 57, 1069–1069 CrossRef CAS.
- Topics of Organometallic Chemistry, ed. B. Plietker, Springer, Iron Catal., 2011 Search PubMed.
- F. Della Monica, A. Buonerba and C. Capacchione, Adv. Synth. Catal., 2019, 361, 265–282 CrossRef CAS.
- F. Chen, N. Liu and B. Dai, ACS Sustainable Chem. Eng., 2017, 5, 9065–9075 CrossRef CAS.
- D. Alhashmialameer, J. Collins, K. Hattenhauer and F. M. Kerton, Catal. Sci. Technol., 2016, 6, 5364–5373 RSC.
- A. Buonerba, A. De Nisi, A. Grassi, S. Milione, C. Capacchione and B. Rieger, Catal. Sci. Technol., 2015, 5, 118–123 RSC.
- M. Cozzolino, V. Leo, C. Tedesco, M. Mazzeo and M. Lamberti, Dalton Trans., 2018, 47, 13229–13238 RSC.
- O. J. Driscoll, C. K. C. Leung, M. F. Mahon, P. McKeown and M. D. Jones, Eur. J. Inorg. Chem., 2018, 5129–5135 CrossRef.
- E. Fazekas, G. S. Nichol, J. A. Garden and M. P. Shaver, ACS Omega, 2018, 3, 16945–16953 CrossRef CAS.
- R. Duan, C. Hu, X. Li, X. Pang, Z. Sun, X. Chen and X. Wang, Macromolecules, 2017, 50, 9188–9195 CrossRef CAS.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS.
- J. Payne, P. Mckeown and M. D. Jones, Polym. Degrad. Stab., 2019, 165, 170–181 CrossRef CAS.
- A. C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS.
- H. Tsuji, Macromol. Biosci., 2005, 5, 569–597 CrossRef.
- R. A. M. Rabnawaz, I. Wyman and S. Cheng, Green Chem., 2017, 19, 4737–4753 RSC.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS.
- J. C. Buffet and J. Okuda, Polym. Chem., 2011, 2, 2758–2763 RSC.
- C. K. Williams and M. A. Hillmyer, Polym. Rev., 2008, 48, 1–10 CrossRef CAS.
- H. R. Kricheldorf and D. O. Damrau, Macromol. Chem. Phys., 1997, 198, 1767–1774 CrossRef CAS.
- B. J. O'Keefe, S. M. Monnier, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2001, 123, 339–340 CrossRef.
- U. Herber, K. Hegner, D. Wolters, R. Siris, K. Wrobel, A. Hoffmann, C. Lochenie, B. Weber, D. Kuckling and S. Herres-Pawlis, Eur. J. Inorg. Chem., 2017, 2017, 1341–1354 CrossRef CAS.
- Y. Y. Kang, H. R. Park, M. H. Lee, J. An, Y. Kim and J. Lee, Polyhedron, 2015, 95, 24–29 CrossRef CAS.
- P. Marin, M. J.-L. Tschan, F. Isnard, C. Robert, P. Haquette, X. Trivelli, L.-M. Chamoreau, V. Guérineau, I. del Rosal, L. Maron, V. Venditto and C. M. Thomas, Angew. Chem., Int. Ed., 2019, 58, 1–6 CrossRef.
- R. D. Rittinghaus, P. M. Schäfer, P. Albrecht, C. Conrads, A. Hoffmann, A. N. Ksiazkiewicz, O. Bienemann, A. Pich and S. Herres-Pawlis, ChemSusChem, 2019, 12, 2161–2165 CrossRef CAS.
- A. C. Silvino, A. L. C. Rodrigues and J. A. L. C. Resende, Inorg. Chem. Commun., 2015, 55, 39–42 CrossRef CAS.
- J. A. Stewart, P. Mckeown, O. J. Driscoll, M. F. Mahon, B. D. Ward and M. D. Jones, Macromolecules, 2019, 52, 5977–5984 CrossRef CAS.
- B. J. O'Keefe, L. E. Breyfogle, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2002, 124, 4384–4393 CrossRef.
- M. Stolt and A. Södergård, Macromolecules, 1999, 32, 6412–6417 CrossRef CAS.
- D. S. McGuinness, E. L. Marshall, V. C. Gibson and J. W. Steed, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3798–3803 CrossRef CAS.
- X. Wang, K. Liao, D. Quan and Q. Wu, Macromolecules, 2005, 38, 4611–4617 CrossRef CAS.
- V. C. Gibson, E. L. Marshall, D. Navarro-Llobet, A. J. P. White and D. J. Williams, J. Chem. Soc., Dalton Trans., 2002, 4321–4322 RSC.
- K. R. Delle Chiaie, A. B. Biernesser, M. A. Ortuño, B. Dereli, D. A. Iovan, M. J. T. Wilding, B. Li, C. J. Cramer and J. A. Byers, Dalton Trans., 2017, 46, 12971–12980 RSC.
- A. B. Biernesser, B. Li and J. A. Byers, J. Am. Chem. Soc., 2013, 135, 16553–16560 CrossRef CAS.
- A. B. Biernesser, K. R. D. Chiaie, J. B. Curley and J. A. Byers, Angew. Chem., Int. Ed., 2016, 55, 5251–5254 CrossRef CAS.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC.
- L. Cuesta-Aluja, A. Campos-Carrasco, J. Castilla, M. Reguero, A. M. Masdeu-Bultó and A. Aghmiz, J. CO2 Util., 2016, 14, 10–22 CrossRef CAS.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS.
- H. Büttner, L. Longwitz, J. Steinbauer, C. Wulf and T. Werner, Top. Curr. Chem., 2017, 375, 1–56 CrossRef PubMed.
- A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2011, 47, 212–214 RSC.
- M. Taherimehr, S. M. Al-Amsyar, C. J. Whiteoak, A. W. Kleij and P. P. Pescarmona, Green Chem., 2013, 15, 3083–3090 RSC.
- M. Taherimehr, J. P. C. C. Sertã, A. W. Kleij, C. J. Whiteoak and P. P. Pescarmona, ChemSusChem, 2015, 8, 1034–1042 CrossRef CAS PubMed.
- V. Besse, F. Camara, C. Voirin, R. Auvergne, S. Caillol and B. Boutevin, Polym. Chem., 2013, 4, 4545–4561 RSC.
- H. Zhang, H. Liu and J. Yue, Chem. Rev., 2014, 114, 883–898 CrossRef CAS.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312–1330 RSC.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef.
- C. J. Whiteoak, B. Gjoka, E. Martin, M. Mart, E. C. Escudero-ada, C. Zonta, G. Licini and A. W. Kleij, Inorg. Chem., 2012, 51, 10639–10649 CrossRef CAS.
- C. J. Whiteoak, E. Martin, M. Belmonte, J. Benet-buchholz and A. W. Kleij, Adv. Synth. Catal., 2012, 354, 469–476 CrossRef CAS.
- X. Sheng, L. Qiao, Y. Qin, X. Wang and F. Wang, Polyhedron, 2014, 74, 129–133 CrossRef CAS.
- M. A. Fuchs, T. A. Zevaco, E. Ember, O. Walter, I. Held and E. Dinjus, Dalton Trans., 2013, 42, 5322–5329 RSC.
- E. Fazekas, G. S. Nichol, M. P. Shaver and J. A. Garden, Dalton Trans., 2018, 47, 13106–13112 RSC.
- M. Sunjuk, A. S. Abu-Surrah, E. Al-Ramahi, A. K. Qaroush and A. Saleh, Transition Met. Chem., 2013, 38, 253–257 CrossRef CAS.
- A. S. Abu-Surrah, H. M. Abdel-Halim, H. A. N. Abu-Shehab and E. Al-Ramahi, Transition Met. Chem., 2017, 42, 117–122 CrossRef CAS.
- J. Peng, H. J. Yang, Y. Geng, Z. Wei, L. Wang and C. Y. Guo, J. CO2 Util., 2017, 17, 243–255 CrossRef CAS.
- A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822–9837 CrossRef CAS.
- K. A. Andrea, T. R. Brown, J. N. Murphy, D. Jagota, D. McKearney, C. M. Kozak and F. M. Kerton, Inorg. Chem., 2018, 57, 13494–13504 CrossRef CAS.
- K. J. Gallagher and R. L. Webster, Chem. Commun., 2014, 50, 12109–12111 RSC.
- S. K. Edulji and S. B. T. Nguyen, Organometallics, 2003, 22, 3374–3381 CrossRef CAS.
- G. Hilt, C. Walter and P. Bolze, Adv. Synth. Catal., 2006, 348, 1241–1247 CrossRef CAS.
- J. E. Davies and B. M. Gatehouse, Acta Crystallogr., Sect. B: Struct. Sci., 1973, 29, 1934–1942 CrossRef CAS.
- P. Muthupandi and G. Sekar, Org. Biomol. Chem., 2012, 10, 5347–5352 RSC.
- A. Jozwiuk, A. L. Ingram, D. R. Powell, B. Moubaraki, N. F. Chilton, K. S. Murray and R. P. Houser, J. Chem. Soc., Dalt. Trans., 2014, 43, 9740–9753 RSC.
- D. J. Darensbourg, C. G. Ortiz and D. R. Billodeaux, Inorg. Chim. Acta, 2004, 357, 2143–2149 CrossRef CAS.
- S. Koner, S. Iijima, M. Watanabe and M. Sato, J. Coord. Chem., 2003, 56, 103–111 CrossRef CAS.
- C. Floriani and G. Fachinetti, J. Chem. Soc., Chem. Commun., 1973, 17–18 RSC.
- P. Coggon, A. T. Mcphail, F. E. Mabbs and V. N. McLachlan, J. Chem. Soc. A Inorganic, Phys. Theor. Chem., 1971, 1014–1019 Search PubMed.
- M. Gerloch, E. D. McKenzie and A. D. C. Towl, J. Chem. Soc. A, 1969, 2850–2858 RSC.
- F. Calderazzo, C. Floriani, R. Henzi and F. L'Eplattenier, J. Chem. Soc. A, 1969, 1378–1386 RSC.
- E. L. Whitelaw, M. G. Davidson and M. D. Jones, Chem. Commun., 2011, 47, 10004–10006 RSC.
- P. McKeown, M. G. Davidson, G. Kociok-Köhn and M. D. Jones, Chem. Commun., 2016, 52, 10431–10434 RSC.
- E. L. Whitelaw, G. Loraine, M. F. Mahon and M. D. Jones, Dalton Trans., 2011, 40, 11469–11469 RSC.
- S. M. Kirk, G. Kociok-Köhn and M. D. Jones, Organometallics, 2016, 35, 3837–3843 CrossRef CAS.
- P. McKeown, M. G. Davidson, J. P. Lowe, M. F. Mahon, L. H. Thomas, T. J. Woodman and M. D. Jones, Dalton Trans., 2016, 45, 5374–5387 RSC.
- J. A. Bertrand, J. L. Breece and P. G. Eller, Inorg. Chem., 1974, 13, 125–131 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- M. Cozzolino, T. Rosen, I. Goldberg, M. Mazzeo and M. Lamberti, ChemSusChem, 2017, 10, 1217–1223 CrossRef CAS.
- X. Wu, C. Chen, Z. Guo, M. North and A. C. Whitwood, ACS Catal., 2019, 9, 1895–1906 CrossRef CAS.
- S. Gesslbauer, R. Savela, Y. Chen, A. J. P. White and C. Romain, ACS Catal., 2019, 7912–7920 CrossRef CAS.
- M. D. Jones, S. L. Hancock, P. McKeown, P. M. Schafer, A. Buchard, L. H. Thomas, M. F. Mahon and J. P. Lowe, Chem. Commun., 2014, 50, 15967–15970 RSC.
- F. Della Monica, B. Maity, T. Pehl, A. Buonerba, A. De Nisi, M. Monari, A. Grassi, B. Rieger, L. Cavallo and C. Capacchione, ACS Catal., 2018, 8, 6882–6893 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complex characterisation and representative spectra for catalysis. CCDC 1940647–1940654. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt03327k |
| This journal is © The Royal Society of Chemistry 2019 |