DOI:
10.1039/C9DT03003D
(Communication)
Dalton Trans., 2019,
48, 12817-12821
Interrogating the steric outcome during H2 heterolysis: in-plane steric effects in the regioselective protonation of the PN3P-pincer ligand†
Received
22nd July 2019
, Accepted 2nd August 2019
First published on 5th August 2019
Abstract
H2 heterolysis to generate well-defined nickel hydride-proton complexes was achieved by the 2nd generation PN3P-pincer nickel platform. The regioselective protonation in the ligand framework was demonstrated for the first time to highlight the importance of in-plane hindrance during the H2 splitting process.
Heterolytic splitting of H2 to generate a proton (H+) and hydride (H−) has been considered to play a central role in H2 activation catalyzed by hydrogenase enzymes,1 which usually either contain a bimetallic core, [FeFe]/[FeNi], or a single Fe center as the active site.1a,b Investigations of the detailed structure of [FeFe]-hydrogenases have shown that a pendant amine base, acting as a proton relay, serves a vital role in facilitating the formation or heterolysis of H2.2 To mimic the catalysis of hydrogenases (Fig. 1A), a large number of Ni-containing models have been artificially synthesized and studied particularly by DuBois et al. (Fig. 1B).3 These [Ni(P2RN2R′)2]2+ hydrogenase models can mediate the oxidation and reproduction of H2. These systems involve either an oxidative addition of H2 by directly transferring its electrons to the central Ni, or a heterolytic (asymmetric) cleavage of H2 to produce the hydride-proton intermediate (Fig. 1C).3f,h Several investigations of heterolytic H2 cleavage by nickel complexes have supported the asymmetric transition state.3c,e,4 Heinekey and coworkers reported the first structurally well-defined nickel dihydrogen complex, in which the H2 molecule cleaves heterolytically in the presence of an external triethylamine as a base (Fig. 2).5 Caulton and co-workers showed that the cationic nickel η2-H2 complex implements H2 heterolysis facilitated by the internal amine base in the ligand skeleton (Fig. 2).4a Bullock et al. reported that the oxidation rate of H2 catalyzed by [Ni(PCy2NtBu2)2](BF4)2 increases with the decrease of the base size in the following order: NEt3 < tBuNH2 < nBuNH2.3m While heterolytic H2 cleavage reactions are highly relevant to enzymatic and organometallic hydrogenation processes, little is known to elucidate the influence of sterics on the protonation step. Herein, we demonstrate the metal–ligand cooperative heterolysis of H2 by a [(PN3P)Ni]+ species with site-selective protonation to feature the importance of the in-plane steric effects (Fig. 2).
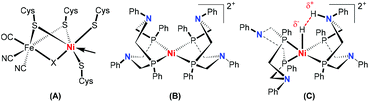 |
| | Fig. 1 Structures of hydrogenases and nickel mimics. (A) [NiFe] hydrogenase; (B) Ni hydrogenase mimic; (C) proposed transition state for production of H2 catalyzed by (B). | |
 |
| | Fig. 2 Heterolysis of H2 by cationic pincer nickel complexes. | |
We have recently reported a new class of nickel compounds supported by a PN3P-pincer ligand containing three nitrogen atoms in the ligand framework through a post-modification strategy (Fig. 3).6 Compared with the complexes derived from the conventional 2,6-diaminopyridine based ligands,7 these new complexes exhibited unparalleled thermal stability and distinct reactivity.6,8 In particular, a nickel hydroxide complex (PN3P)NiOH (1) was studied in details with respect to its strong basicity.8b Although hydrogenolysis of the M–OH bond to produce a M–H compound concomitant with water is a highly valuable reaction,9 unfortunately, our Ni–OH complex 1 showed no reaction toward H2 gas. This is in sharp contrast to other analogous Pd,9 Pt10 or Ru11 complexes, where the H2 molecule is readily cleaved by the M–OH moiety, presumably originating from the unfavorable formation of a (PN3P)Ni(H2) intermediate.4a Caulton and co-workers have reported that a free coordination site for H2 is important for the heterolysis to proceed in the pincer-type nickel compound.4a Consistent with their viewpoint, when one equivalent of B(C6F5)3 was introduced to the C6D6 solution of 1, the resulting borane adduct (PN3P)Ni(OH)B(C6F5)3 (2) could undergo the hydrogenolysis reaction. This is likely attributed to the property of anionic [(OH)B(C6F5)3]− as a better leaving group than the OH group. However, differently from what was reported by the groups Peters12 and Limberg,4b the [(PN3P)Ni(H2)]+ intermediate was not observed.4a,13 The hydrogenolysis reaction occurred immediately after the H2 was introduced into 2, as corroborated by the rapid color change from brown to light yellow. The 31P{1H} NMR showed significant downfield chemical shifts, in comparison with those from 2 (δ 103.5 and 103.2 ppm), with two sets of peaks observed at δ 147.5 and 144.9 ppm. The successful heterolysis of H2 to generate a proton and a hydride was further validated by the 1H NMR with a doublet of doublet peak at δ −16.20 ppm for the hydride and a signal at δ 9.41 ppm for the proton. The identity of product 3 was confirmed by X-ray crystallography (Fig. 4). Complex 3 consists of an anion [(OH)B(C6F5)3]− and a cation [(PN3HP)Ni(H)]+ in which the H−and H+ derived from the splitting of H2 bond to the nickel center and one of the nitrogen atoms, respectively (Scheme 1). Although various experiments on H2 heterolysis catalyzed by nickel compounds have been reported, to our knowledge, no well-defined nickel hydride-proton intermediate was obtained.
 |
| | Fig. 3 Synthesis of 2nd PN3P-pincer compounds through ligand post-modification strategy. | |
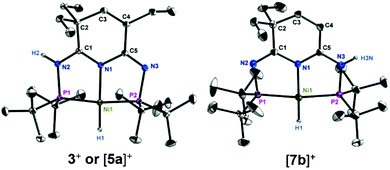 |
| | Fig. 4 Molecular structures of complexes 3+, [5a]+ and [7b]+. Thermal ellipsoids are shown at the 30% probability level; hydrogen atoms except Ni–H and N–H moieties are omitted for clarity. Selected bond lengths [Å] and angles [°]: for 3+: Ni(1)–N(1) 1.9015(11), Ni(1)–P(1) 2.1407(4), Ni(1)–P(2) 2.1401(4), Ni(1)–H(1) 1.351, N(2)–H(2) 0.85(1); N(1)–Ni(1)–H(1) 178.16, H(1)–Ni(1)–P(1) 91.33, H(1)–Ni(1)–P(2) 96.83, P(1)–Ni(1)–P(2) 171.776(15). For [5a]+: Ni(1)–N(1) 1.9017(7), Ni(1)–P(1) 2.1348(3), Ni(1)–P(2) 2.1300(3), Ni(1)–H(1) 1.374, N(2)–H(2) 0.863; N(1)–Ni(1)–H(1) 178.29, H(1)–Ni(1)–P(1) 95.05, H(1)–Ni(1)–P(2) 92.98, P(1)–Ni(1)–P(2) 171.738(9). For [7b]+: Ni(1)–N(1) 1.9022(14), Ni(1)–P(1) 2.1461(5), Ni(1)–P(2) 2.1431(5), Ni(1)–H(1) 1.37(3), N(3)–H(3N) 0.86(3); N(1)–Ni(1)–H(1) 178.3(12), H(1)–Ni(1)–P(1) 94.7(12), H(1)–Ni(1)–P(2) 93.9(12), P(1)–Ni(1)–P(2) 171.382(19). | |
 |
| | Scheme 1 Synthesis and H2 heterolysis of complex 2. | |
In order to further verify the importance of the leaving ability of ancillary ligand, we prepared a similar complex (4), (PN3P)NiOTf, containing a slightly stronger coordinating group, OTf, as the promoter of H2 heterolysis. The comparison of the crystal structures of 2 and 4 clearly showed that the length of the Ni–O bond (1.9169(11) Å) in complex 4 is shorter than that (2.0258(15) Å) in complex 2, suggesting that the leaving ability of (OH)B(C6F5)3 moiety is higher than that of the OTf group. Indeed, no reaction occurred for complex 4 in the presence of H2 at room temperature over 12 h. Only upon heating at 50 °C, a new product, 5a, was slowly formed. These results suggest that the displacement of OTf by H2 at an elevated temperature to generate [(PN3P)Ni(H2)]+ may play a vital role in promoting the activation of H2. The 1H NMR also shows protic and hydride signals at δ 10.16 and −16.19 ppm, respectively, similar to those in complex 3. The X-ray diffraction analysis revealed the structure of 5a to be [(PN3HP)Ni(H)]+[OTf]−(Fig. 4).
Both NMR and crystal data suggested that the proton from H2 heterolysis was transferred into one of the side arm nitrogen atoms of the PN3P ligand (Fig. 4). Caulton and co-workers have demonstrated that nitrogen atoms could potentially serve as a base to facilitate H2 heterolysis.4a As our ligand framework contains three nitrogen atoms, in principle there are three possible protonated products formed in this system (Fig. 5). 2D NOESY experiments were conducted to determine the structure of the Ni–H cation. The cross peak at 2.89/10.16 ppm was attributed to the correlation of the NH proton and the methylene protons of the two ethyl groups at the same side of the ligand (Fig. S10† for complex 5a). Likewise, the correlation of the NH proton and those of two equivalent tBu groups could also be observed at the position of 1.29/10.16 ppm, further suggesting that the proton was selectively added to one N atom (Ni1left), consistent with qualitative DFT calculations (Fig. 5). The counterpart Ni1right with the proton connecting to the N atom on the other side was found to be higher in energy. The relative Gibbs free energy of other structures Ni1H2, before H2 splitting, and Ni1mid were both found to be extremely unfavorable, indicative of the thermodynamic preference of the H2 cleavage process.
 |
| | Fig. 5 Structures of the probable intermediates and protonated products with relative Gibbs free energy data (the energetics in the first and second lines represent the energies in gas phase and in the experimental solvent (benzene) phase, respectively). | |
The selective protonation of the nitrogen atom may originate from the different electronic and/or steric effects on the nitrogen atoms. To further rationalize the selectivity, an analogous nickel triflate complex (6) containing only two ethyl groups on the PN3P framework was prepared. Interestingly, when complex 6 was used, both N side arms could be protonated to give a mixture of 7a and 7b in a ratio of approx. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 (Scheme 3). The major product 7b suggests that the steric effect is likely the dominating factor of the observed selectivity in the protonation step (Fig. 4), since 7a should experience similar electronic effects as 5a. This argument is further supported by very similar atomic charges from NPA of the two nitrogen sites in 4 (q(N2) = −0.616; q(N3) = −0.627). Analyzing the molecular structure of 5a, we notice that the diethyl groups are both out of the plane of the central heterocyclic ring, but the monoethyl group is in the plane, strongly indicating that the selectivity is caused by the in-plane hindrance. To further support this argument, two analogous complexes (8 and 10) were prepared via replacing the Et groups by Me and iPr groups and employed to conduct the heterolytic splitting of H2. It was found that the selectivity indeed remained the same (Scheme 2). The structures of 9a and 11a were also characterized by NMR and single crystal X-ray diffraction (Fig. 6). The distances between H and Me in 9a (H2–C6: 2.785 Å; H2–C7: 2.892 Å) are longer than those in 9b (H3A–C8: 2.505 Å), in agreement with the observation that protonation of the dimethyl side is more feasible (Fig. 7). When NMR experiments were conducted at an elevated temperature (110 °C), the rations of 5a/5b and 11a/11b changed (Fig. S33 and S35†), indicating that these complexes are interchangeable. These observations suggest that these regioselective products are likely the results of thermodynamic preference driven by the in-plane hindrance between the protonated N–H arm and the H or alkyl group at the sp2 carbon of the ring.
:6 (Scheme 3). The major product 7b suggests that the steric effect is likely the dominating factor of the observed selectivity in the protonation step (Fig. 4), since 7a should experience similar electronic effects as 5a. This argument is further supported by very similar atomic charges from NPA of the two nitrogen sites in 4 (q(N2) = −0.616; q(N3) = −0.627). Analyzing the molecular structure of 5a, we notice that the diethyl groups are both out of the plane of the central heterocyclic ring, but the monoethyl group is in the plane, strongly indicating that the selectivity is caused by the in-plane hindrance. To further support this argument, two analogous complexes (8 and 10) were prepared via replacing the Et groups by Me and iPr groups and employed to conduct the heterolytic splitting of H2. It was found that the selectivity indeed remained the same (Scheme 2). The structures of 9a and 11a were also characterized by NMR and single crystal X-ray diffraction (Fig. 6). The distances between H and Me in 9a (H2–C6: 2.785 Å; H2–C7: 2.892 Å) are longer than those in 9b (H3A–C8: 2.505 Å), in agreement with the observation that protonation of the dimethyl side is more feasible (Fig. 7). When NMR experiments were conducted at an elevated temperature (110 °C), the rations of 5a/5b and 11a/11b changed (Fig. S33 and S35†), indicating that these complexes are interchangeable. These observations suggest that these regioselective products are likely the results of thermodynamic preference driven by the in-plane hindrance between the protonated N–H arm and the H or alkyl group at the sp2 carbon of the ring.
 |
| | Scheme 2 Heterolysis of H2 by [Ni-OTf] complexes. | |
 |
| | Scheme 3 Heterolysis of H2 by complex 6. | |
 |
| | Fig. 6 Molecular structures of complexes [9a]+ and [11a]+. Thermal ellipsoids are shown at the 30% probability level; hydrogen atoms except Ni–H and N–H moieties are omitted for clarity. Selected bond lengths [Å] and angles [°]: for [9a]+: Ni(1)–N(1) 1.9037(11), Ni(1)–P(1) 2.1376(5), Ni(1)–P(2) 2.1379(5), Ni(1)–H(1) 1.28(3), N(2)–H(2) 0.79(2); N(1)–Ni(1)–H(1) 177.645, H(1)–Ni(1)–P(1) 95.299, H(1)–Ni(1)–P(2) 92.700, P(1)–Ni(1)–P(2) 171.994(16). For [11a]+: Ni(1)–N(1) 1.908(2), Ni(1)–P(1) 2.1229(7), Ni(1)–P(2) 2.1268(8), Ni(1)–H(1) 1.368, N(2)–H(2) 0.861; N(1)–Ni(1)–H(1) 176.323, H(1)–Ni(1)–P(1) 92.226, H(1)–Ni(1)–P(2) 95.120, P(1)–Ni(1)–P(2) 172.642. | |
 |
| | Fig. 7 The distances between H and Me in 9a and 9b. | |
In summary, we have demonstrated the importance of the leaving ability of coordinated anion for the H2 heterolysis process in the planar pincer geometry by comparing the effects of the (OH)B(C6F5)3 and the OTf moieties. We also presented a unique model for interrogating the in-plane interactions. Using two different 2nd generation PN3P-pincer nickel compounds (4 and 6), the H2 heterolysis and regioselective protonation of the in-plane less hindered nitrogen atoms in the ligand framework was observed. Together with both DFT calculations and control experiments, the significance of the steric factor was confirmed.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge the financial support from King Abdullah University of Science and Technology (KAUST).
Notes and references
-
(a) C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS PubMed;
(b) W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed;
(c) M. Winkler, M. Senger, J. Duan, J. Esselborn, F. Wittkamp, E. Hofmann, U.-P. Apfel, S. T. Stripp and T. Happe, Nat. Commun., 2017, 8, 16115 CrossRef CAS PubMed;
(d) M. Senger, K. Laun, F. Wittkamp, J. Duan, M. Haumann, T. Happe, M. Winkler, U.-P. Apfel and S. T. Stripp, Angew. Chem., Int. Ed., 2017, 56, 16503–16506 CrossRef CAS PubMed.
- M. R. DuBois and D. L. DuBois, Chem. Soc. Rev., 2009, 38, 62–72 RSC.
-
(a) C. J. Curtis, A. Miedaner, R. Ciancanelli, W. W. Ellis, B. C. Noll, M. Rakowski DuBois and D. L. DuBois, Inorg. Chem., 2003, 42, 216–227 CrossRef CAS PubMed;
(b) A. D. Wilson, R. H. Newell, M. J. McNevin, J. T. Muckerman, M. Rakowski DuBois and D. L. DuBois, J. Am. Chem. Soc., 2006, 128, 358–366 CrossRef CAS PubMed;
(c) A. D. Wilson, R. K. Shoemaker, A. Miedaner, J. T. Muckerman, D. L. DuBois and M. R. DuBois, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6951–6956 CrossRef CAS PubMed;
(d) M. R. DuBois and D. L. DuBois, C. R. Chim., 2008, 11, 805–817 CrossRef CAS;
(e) J. Y. Yang, R. M. Bullock, W. J. Shaw, B. Twamley, K. Fraze, M. R. DuBois and D. L. DuBois, J. Am. Chem. Soc., 2009, 131, 5935–5945 CrossRef CAS PubMed;
(f) A. Kachmar, V. Vetere, P. Maldivi and A. A. Franco, J. Phys. Chem. A, 2010, 114, 11861–11867 CrossRef CAS PubMed;
(g) J. Y. Yang, S. Chen, W. G. Dougherty, W. S. Kassel, R. M. Bullock, D. L. DuBois, S. Raugei, R. Rousseau, M. Dupuis and M. R. DuBois, Chem. Commun., 2010, 46, 8618–8620 RSC;
(h) M. Dupuis, S. Chen, S. Raugei, D. L. DuBois and R. M. Bullock, J. Phys. Chem. A, 2011, 115, 4861–4865 CrossRef CAS PubMed;
(i) M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed;
(j) T. Liu, S. Chen, M. J. O'Hagan, M. Rakowski DuBois, R. M. Bullock and D. L. DuBois, J. Am. Chem. Soc., 2012, 134, 6257–6272 CrossRef CAS PubMed;
(k) S. E. Smith, J. Y. Yang, D. L. DuBois and R. M. Bullock, Angew. Chem., Int. Ed., 2012, 51, 3152–3155 CrossRef CAS PubMed;
(l) S. Wiese, U. J. Kilgore, D. L. DuBois and R. M. Bullock, ACS Catal., 2012, 2, 720–727 CrossRef CAS;
(m) J. Y. Yang, S. E. Smith, T. Liu, W. G. Dougherty, W. A. Hoffert, W. S. Kassel, M. R. DuBois, D. L. DuBois and R. M. Bullock, J. Am. Chem. Soc., 2013, 135, 9700–9712 CrossRef CAS PubMed;
(n) A. Dutta, D. L. DuBois, J. A. S. Roberts and W. J. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16286–16291 CrossRef CAS PubMed;
(o) N. Priyadarshani, A. Dutta, B. Ginovska, G. W. Buchko, M. O'Hagan, S. Raugei and W. J. Shaw, ACS Catal., 2016, 6, 6037–6049 CrossRef CAS;
(p) E. B. Hulley, K. D. Welch, A. M. Appel, D. L. DuBois and R. M. Bullock, J. Am. Chem. Soc., 2013, 135, 11736–11739 CrossRef CAS PubMed.
-
(a) T. He, N. P. Tsvetkov, J. G. Andino, X. F. Gao, B. C. Fullmer and K. G. Caulton, J. Am. Chem. Soc., 2010, 132, 910–911 CrossRef CAS PubMed;
(b) H. Gehring, R. Metzinger, C. Herwig, J. Intemann, S. Harder and C. Limberg, Chem. – Eur. J., 2013, 19, 1629–1636 CrossRef CAS PubMed.
- S. J. Connelly, A. C. Zimmerman, W. Kaminsky and D. M. Heinekey, Chem. – Eur. J., 2012, 18, 15932–15934 CrossRef CAS PubMed.
-
(a) X. F. Wang, L. F. Yao, Y. P. Pan and K.-W. Huang, J. Organomet. Chem., 2017, 845, 25–29 CrossRef CAS;
(b) M.-H. Huang, J. Hu and K.-W. Huang, J. Chin. Chem. Soc., 2018, 65, 60–64 CrossRef CAS.
-
(a) H. Li, B. Zheng and K.-W. Huang, Coord. Chem. Rev., 2015, 293, 116–138 CrossRef;
(b) H. Li, T. P. Gonçalves, D. Lupp and K.-W. Huang, ACS Catal., 2019, 9, 1619–1629 CrossRef CAS;
(c) T. P. Gonçalves and K.-W. Huang, J. Am. Chem. Soc., 2017, 139, 13442–13449 CrossRef PubMed;
(d) H. Li, T. P. Gonçalves, Q. Zhao, D. Gong, Z. Lai, Z. Wang, J. Zheng and K.-W. Huang, Chem. Commun., 2018, 54, 11395–11398 RSC;
(e) H. Li, T. P. Gonçalves, J. Hu, Q. Zhao, D. Gong, Z. Lai, Z. Wang, J. Zheng and K.-W. Huang, J. Org. Chem., 2018, 83, 14969–14977 CrossRef CAS PubMed.
-
(a) C. Yao, X. Wang and K.-W. Huang, Chem. Commun., 2018, 54, 3940–3943 RSC;
(b) C. Yao, P. Chakraborty, E. Aresu, H. Li, C. Guan, C. Zhou, L.-C. Liang and K.-W. Huang, Dalton Trans., 2018, 47, 15997–16360 RSC.
- G. R. Fulmer, R. P. Muller, R. A. Kemp and K. I. Goldberg, J. Am. Chem. Soc., 2009, 131, 1346–1347 CrossRef CAS PubMed.
-
T. T. Wenzel, in Stud. Surf. Sci. Catal, ed. L. I. Simándi, Elsevier, 1991, vol. 66, pp. 545–554 Search PubMed.
- T. Matsumoto, Y. Nakaya, N. Itakura and K. Tatsumi, J. Am. Chem. Soc., 2008, 130, 2458–2459 CrossRef CAS PubMed.
- C. Tsay and J. C. Peters, Chem. Sci., 2012, 3, 1313–1318 RSC.
-
(a) T. Chen, H. Li, S. Qu, B. Zheng, L. He, Z. Lai, Z.-X. Wang and K.-W. Huang, Organometallics, 2014, 33, 4152–4155 CrossRef CAS;
(b) Y. Wang, B. Zheng, Y. P. Pan, C. L. Pan, L. P. He and K.-W. Huang, Dalton Trans., 2015, 44, 15111–15115 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed descriptions of the preparation and characterization of compounds 2–11; additional NMR spectra. CCDC 1891945–1891947, 1891951, 1891954, 1909960 and 1909961. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt03003d |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a









