
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2 hydrogenation by phosphorus–nitrogen PN3P-pincer iridium hydride complexes: elucidation of the deactivation pathway†
Yupeng
Pan
ab,
Chao
Guan
a,
Huaifeng
Li
a,
Priyanka
Chakraborty
a,
Chunhui
Zhou
a and
Kuo-Wei
Huang
 *a
*a
aKAUST Catalysis Center and Division of Physical Sciences and Engineering, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia. E-mail: hkw@kaust.edu.sa
bShenzhen Grubbs Institute, Southern University of Science and Technology (SUSTech), Shenzhen, 518055, P. R. China
First published on 24th July 2019
Abstract
PN3P–Ir pincer hydride complexes were synthesized and characterized as catalysts and key intermediates in the direct hydrogenation of CO2 to formate under mild conditions. The formation of a dearomatized PN3P*–Ir(I)–CO species was identified as a plausible key process accountable for the loss of catalytic activity in the CO2 hydrogenation.
Carbon capture and sequestration (CCS) has been the subject of extensive research and commercial efforts.1–5 The selective hydrogenation of carbon dioxide (CO2) under mild conditions represents an economical and sustainable method of preparing valuable products and fuels from CO2.6,7 In this regard, significant achievements have been made using single-site homogeneous catalysts in CO2 transformation, especially in the selective conversion of CO2 and H2 to formic acid or formate.8,9 While the hydrogenation of CO2 to HCOOH is thermodynamically favored in the aqueous phase, the energy barrier is high. As a result, high reaction pressures and temperatures are generally needed. A number of transition-metal complexes, such as rhodium,10–15 ruthenium16–27 and iridium,28–37 have been investigated for the hydrogenation of CO2. Among these metals, iridium complexes appear very promising.
In recent years, highly efficient and selective hydrogen transfer reactions and dehydrogenation reactions mediated by pincer-ligated complexes have been reported.38,39 In addition, the catalytic conversion of n-alkanes to alkylaromatics using pincer supported iridium complexes40–44 and of glycerol to lactic acid45–47 have been well developed. For the reduction of CO2, Himeda and Fujita et al. used a dinuclear proton-switchable iridium catalyst bearing an N,N′-type ligand, 4,4′,6,6′-tetrahydroxy-2,2′-bipyrimidine, as a bridging group, to afford an average TOF of 53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 h−1 in the presence of KHCO3 at 80 °C under 50 bar.32 When a strong electron-donating PNP ligand was introduced into the CO2 hydrogenation, high activities of Ir(III) complexes towards CO2 hydrogenation were demonstrated by the Nozaki group (Fig. 1), and an unprecedentedly high TON of 3500000 was achieved.29 Analogous “saturated” systems by Hazari et al. also showed a high TON of 348000,30 although a much higher temperature and pressure were necessary. Recent mechanistic studies on homogeneous iridium catalytic systems in CO2 hydrogenation suggested that the CO2 activation pathways are dependent on the nature of the Ir catalysts and that a hydrido ligand on an Ir(III) center is essential.48–51
800 h−1 in the presence of KHCO3 at 80 °C under 50 bar.32 When a strong electron-donating PNP ligand was introduced into the CO2 hydrogenation, high activities of Ir(III) complexes towards CO2 hydrogenation were demonstrated by the Nozaki group (Fig. 1), and an unprecedentedly high TON of 3500000 was achieved.29 Analogous “saturated” systems by Hazari et al. also showed a high TON of 348000,30 although a much higher temperature and pressure were necessary. Recent mechanistic studies on homogeneous iridium catalytic systems in CO2 hydrogenation suggested that the CO2 activation pathways are dependent on the nature of the Ir catalysts and that a hydrido ligand on an Ir(III) center is essential.48–51
 | ||
| Fig. 1 Examples of PNXP pincer Ir(III) trihydride catalysts for CO2 hydrogenation. | ||
We recently demonstrated that PN3(P)-pincer complexes show unique properties in various challenging transformations in organic synthesis and catalytic studies,52–57 showing diverse catalytic activities and different thermodynamic and kinetic properties.58,59 Driven by the potential advantages of employing more electron-rich pincer complexes, herein, we present our development on PN3P–Ir(III) trihydride complexes in CO2 hydrogenation to formate. A probable catalyst-deactivation step in the formation of a dearomatized PN3P*Ir–CO complex is also discussed.
As has been suggested that Ir(III) trihydride complexes serve as effective catalysts in the hydrogenation of CO2,29 we developed a general route for the synthesis of the Ir(III) complexes based on PN3P ligands (Scheme 1). Cationic iridium(I) complexes 1a and 1b were synthesized by coordinating the PN3PR ligands (R = cyclopentyl (cPe) and t-butyl (tBu)) with a stoichiometric amount of [Ir(coe)2Cl]2, similar to the procedure provided in the literature.60 We then introduced H2 to eliminate the coordinating coe moiety resulting in the formation of cationic iridium(III) complexes 2a and 2b after the oxidative addition of one equivalent of H2. The NMR data of 2b collected in CDCl3 at room temperature showed only one N–H (2H, δ 9.27 ppm) signal and one Ir–H (2H, δ −26.87 ppm) signal in the 1H NMR spectrum and a signal at δ 126.2 ppm in the 31P NMR spectrum, suggesting a symmetric conformation enforced by the PN3P pincer ligand. In addition, the solid state molecular structure of 2b was consistent with the NMR details where two hydrides are equivalent (Fig. 2). The structure shows a square pyramidal configuration with a chloride as a dissociated counter anion.
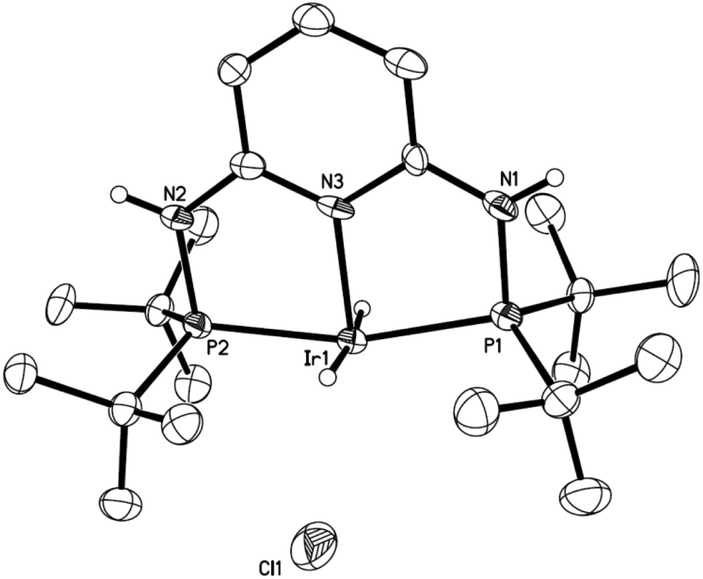 | ||
| Fig. 2 X-ray structure of complex 2b with 40% probability of thermal ellipsoids. The solvent and hydrogen atoms, except N–H and hydrides, were omitted for clarity. Selected bond lengths (Å): Ir1–N3 2.110(6), Ir1–P1 2.284(2), and Ir1–P2 2.279(2). Selected bond angles (°): (2b) P1–Ir1–P2 163.64(7). | ||
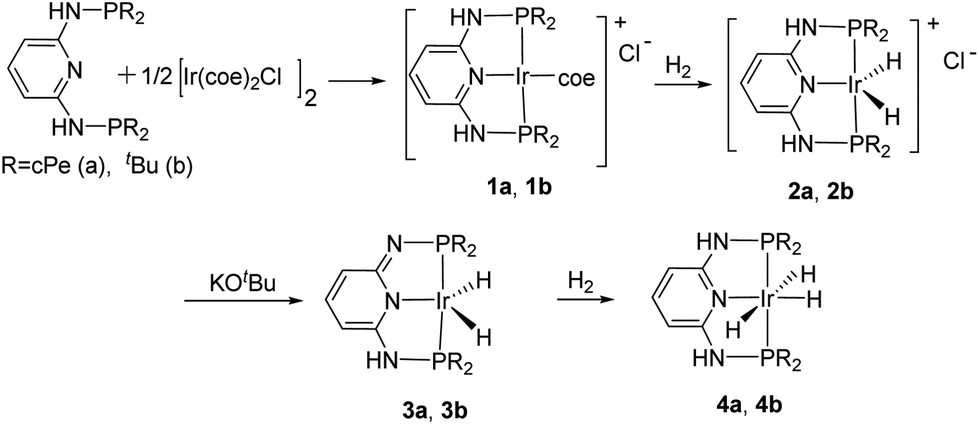 | ||
| Scheme 1 Synthesis of PN3P–Ir(III) trihydride complexes. | ||
The neutral dearomatized complexes 3a and 3b were obtained by employing one equivalent of KOtBu to deprotonate one of the NH arms in the PN3P pincer ligand.61,62 Unlike their –CH2 analogs,62 these dearomatized PN3P* complexes were stable enough to be isolated at room temperature. The 1H NMR spectrum of 3b in C6D6 shows three sets of sp2 C–H signals at δ 5.21 (d), 6.85 (d), and 6.92 (t) ppm, in agreement with the dearomatization of the central pyridine ring. As expected, two sets of doublet phosphorus signals were observed in the 31P NMR spectrum (2JP–P = 299 Hz), with an apparent triplet for a hydride (2JP–H = 12.0 Hz) at δ −24.93 ppm in the 1H NMR spectrum.38,52–56,58,59 The solid-state molecular structure of 3b was obtained with two peaks in the Fourier map, indicative of two hydrides forming a distorted trigonal bipyramidal configuration (Fig. 3).
 | ||
| Fig. 3 X-ray structure of complex 3b with 40% probability of thermal ellipsoids. The solvent and hydrogen atoms, except N–H and hydrides, were omitted for clarity. Selected bond lengths (Å): (3b) Ir1–N3 2.063(6), Ir1–P1 2.2725(8), and Ir1–P2 2.2955(8). Selected bond angles (°): (3b) P1–Ir1–P2 162.79(3). | ||
Finally, the trihydride complexes 4a and 4b were obtained by treating 3a and 3b in the presence of H2 in THF. The 1H NMR spectrum of complex 4a in C6D6 shows two sets of hydride signals, at δ −18.15 ppm and −11.76 ppm with an integral intensity ratio of 2:1, and two sets of signals for three protons in the sp2 region (5.60 (d) and 6.81 (t) ppm), indicating the rearomatization of the central pyridine ring. Complex 4a was confirmed by the X-ray diffraction analysis (Fig. 4). The solid-state molecular structure of 4a was consistent with the NMR details. Trihydride 4a has an octahedral geometry with three peaks in the Fourier map assigned to hydrides on the Ir center, consistent with the reported iPr–PNP–Ir trihydride compound.29
 | ||
| Fig. 4 X-ray structures of complex 4a with 40% probability of thermal ellipsoids. The solvent and hydrogen atoms, except N–H and hydrides, were omitted for clarity. Selected bond lengths (Å): (4a) Ir1–N3 2.117(3). Selected bond angles (°): (4a) P1–Ir1–P2 164.07(3). | ||
Several pincer-ligated iridium hydride complexes29,30 are well known to catalyze CO2 hydrogenation. Therefore, the catalytic activity of complexes 4a and 4b was examined accordingly in THF/H2O solution (Table 1). According to the results of entries 1 and 2, the performance of 4b (yield 17.6%) is better than that of 4a (yield 6.3%) under the same conditions. 4b was thus chosen to evaluate the influence of the reaction conditions, such as temperature and reaction time, for the CO2 hydrogenation reaction. The results of entries 2–4 show that the yield increases (17.6% to 49.6%) in the beginning with increasing temperature (120–130 °C), but then decreases with temperature increasing continually (130–140 °C). These observations suggest that 4b is not stable at high temperature. Prolonging the reaction time to 18 hours did not improve the yield of formate (49.6% to 51.0%, entry 5), indicating that 4b is likely to be deactivated within 12 hours.
| Entry | Cat. | T (°C) | P (psi) | Time (h) | Yielda (%) | TON | TOF (h−1) |
|---|---|---|---|---|---|---|---|
|
a Total pressure at room temperature: 120 psi (H2:CO2 = 1:1). Yields: Calculated by 1H NMR analysis using sodium 3-(trimethylsilyl)-1-propanesulfonate as an internal standard, based on the added KOH base (1.0 × 10−3 mol). Catalyst loading: 1.0 × 10−7 mol. [THF]/[H2O] = 1/4 (ratio by volume).
|
|||||||
| 1 | 4a | 120 | 120 | 12 | 6.3 | 630 | 53 |
| 2 | 4b | 120 | 120 | 12 | 17.6 | 1760 | 147 |
| 3 | 4b | 130 | 120 | 12 | 49.6 | 4960 | 413 |
| 4 | 4b | 140 | 120 | 12 | 30.6 | 3060 | 255 |
| 5 | 4b | 130 | 120 | 18 | 51.0 | 5100 | 283 |
| 6 | 5b | 130 | 120 | 18 | — | — | — |
To gain more insight into the catalyst deactivation, additional experiments were carried out. The treatment of 4b with H2 and CO2 (1:1, 120 psi) at 140 °C for 24 hours afforded 5b in 78% yield after recrystallization (Scheme 2), and in this process, the water should be generated as a by-product (Scheme 2). Furthermore, the hydrogenation of CO2 which is catalyzed by complex 5b showed no activity (Table 1, entry 6). The 31P NMR spectrum of 5b indicated the presence of two nonequivalent P atoms. In the 1H NMR spectrum, three sets of sp2 C–H signals at 5.11(d), 6.74 (d), and 6.83 (t) ppm were observed, indicating the dearomatization of the central pyridine ring. The molecular structure of 5b shows a slightly distorted square-planar coordination geometry (Fig. 5). The metal–carbon bond distance is 1.837(6) Å, just slightly longer than that in the PNP–Ir–CO complex (Ir–CO: 1.818(2) Å).62 The Ir–N bond distance of 5b (2.076(4) Å) and that in the PNP–Ir–CO complex (2.083(2) Å) were practically identical. Consistent with our results, 5b was unable to catalyze the CO2 hydrogenation under the same conditions shown in Table 1. The formation of the CO ligand implies that the reverse water gas shift (RWGS) reaction also takes place in the presence of the PN3P–Ir complex,62–66 and this process is likely responsible for the loss of catalytic activity.
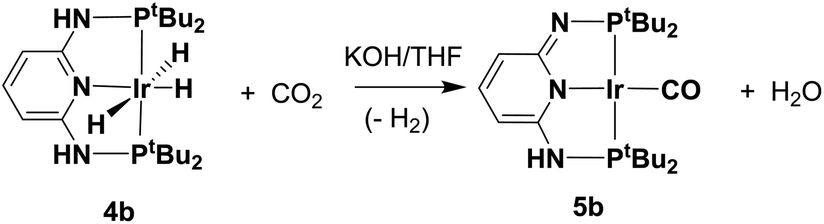 | ||
| Scheme 2 Production complex 5b from complex 4b in the presence of CO2/H2. | ||
 | ||
| Fig. 5 X-ray structure of 5b with 40% probability of thermal ellipsoids. The solvent and hydrogen atoms, except N–H, were omitted for clarity. Selected bond lengths (Å): (5b) Ir1–N3 2.076(4), Ir1–C22 1.837(6), Ir1–P1 2.3000(14), and Ir1–P2 2.3008(13). Selected bond angles (°): (5b) P1–Ir1–P2 163.33(5) and C(22)–Ir(1)–N(1) 179.3(2). | ||
In summary, we have developed a general method for synthesizing PN3P pincer Ir(III) trihydride complexes and obtained several key structures involved in the CO2 hydrogenation process. While the Ir complexes showed favorable activity (a turnover number up to 5100), the formation of a dearomatized PN3P*–Ir(I)–CO species was identified as a plausible key pathway for the catalyst deactivation. Our work reveals that the RWGS should be taken into account for future catalyst design to avoid this unfavorable process to maximize the catalyst activity and lifetime.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funding from the King Abdullah University of Science and Technology (KAUST).Notes and references
- P. G. Jessop, F. Joó and C.-C. Tai, Coord. Chem. Rev., 2004, 248, 2425 CrossRef CAS.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed.
- C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024 CrossRef CAS PubMed.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2017, 118, 434 CrossRef PubMed.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo and L. A. Hackett, Energy Environ. Sci., 2018, 11, 1062 RSC.
- M. Aresta, Carbon dioxide as chemical feedstock, John Wiley & Sons, 2010 Search PubMed.
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510 CrossRef CAS PubMed.
- S. Moret, P. J. Dyson and G. Laurenczy, Nat. Commun., 2014, 5, 4017 CrossRef CAS PubMed.
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2017, 118, 372 CrossRef PubMed.
- J. C. Tsai and K. M. Nicholas, J. Am. Chem. Soc., 1992, 114, 5117 CrossRef CAS.
- T. Burgemeister, F. Kastner and W. Leitner, Angew. Chem., Int. Ed. Engl., 1993, 32, 739 CrossRef.
- F. Gassner and W. Leitner, J. Chem. Soc., Chem. Commun., 1993, 19, 1465 RSC.
- W. Leitner, E. Dinjus and F. Gaßner, J. Organomet. Chem., 1994, 475, 257 CrossRef CAS.
- R. Fornika, H. Gorls, B. Seemann and W. Leitner, J. Chem. Soc., Chem. Commun., 1995, 14, 1479 RSC.
- Y.-N. Li, L.-N. He, A.-H. Liu, X.-D. Lang, Z.-Z. Yang, B. Yu and C.-R. Luan, Green Chem., 2013, 15, 2825 RSC.
- P. G. Jessop, T. Ikariya and R. Noyori, Nature, 1994, 368, 231 CrossRef CAS.
- P. G. Jessop, Y. Hsiao, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 344 CrossRef CAS.
- C. Yin, Z. Xu, S.-Y. Yang, S. M. Ng, K. Y. Wong, Z. Lin and C. P. Lau, Organometallics, 2001, 20, 1216 CrossRef CAS.
- P. Munshi, A. D. Main, J. C. Linehan, C.-C. Tai and P. G. Jessop, J. Am. Chem. Soc., 2002, 124, 7963 CrossRef CAS PubMed.
- J. Elek, L. Nádasdi, G. Papp, G. Laurenczy and F. Joó, Appl. Catal., A, 2003, 255, 59 CrossRef CAS.
- C. Federsel, R. Jackstell, A. Boddien, G. Laurenczy and M. Beller, ChemSusChem, 2010, 3, 1048 CrossRef CAS PubMed.
- G. A. Filonenko, R. van Putten, E. N. Schulpen, E. J. Hensen and E. A. Pidko, ChemCatChem, 2014, 6, 1526 CrossRef CAS.
- J. Kothandaraman, M. Czaun, A. Goeppert, R. Haiges, J. P. Jones, R. B. May, G. S. Prakash and G. A. Olah, ChemSusChem, 2015, 8, 1442 CrossRef CAS PubMed.
- K. Rohmann, J. Kothe, M. W. Haenel, U. Englert, M. Hölscher and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 8966 CrossRef CAS PubMed.
- G. H. Gunasekar, J. Shin, K.-D. Jung, K. Park and S. Yoon, ACS Catal., 2018, 8, 4346 CrossRef CAS.
- P. Hu, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2016, 55, 1061 CrossRef CAS PubMed.
- C. Guan, Y. Pan, E. P. L. Ang, J. Hu, C. Yao, M.-H. Huang, H. Li, Z. Lai and K.-W. Huang, Green Chem., 2018, 20, 4201 RSC.
- Y. Himeda, N. Onozawa-Komatsuzaki, H. Sugihara and K. Kasuga, Organometallics, 2007, 26, 702 CrossRef CAS.
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168 CrossRef CAS PubMed.
- T. J. Schmeier, G. E. Dobereiner, R. H. Crabtree and N. Hazari, J. Am. Chem. Soc., 2011, 133, 9274 CrossRef CAS PubMed.
- R. Tanaka, M. Yamashita, L. W. Chung, K. Morokuma and K. Nozaki, Organometallics, 2011, 30, 6742 CrossRef CAS.
- J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383 CrossRef CAS PubMed.
- W.-H. Wang, J. F. Hull, J. T. Muckerman, E. Fujita and Y. Himeda, Energy Environ. Sci., 2012, 5, 7923 RSC.
- W.-H. Wang, J. T. Muckerman, E. Fujita and Y. Himeda, ACS Catal., 2013, 3, 856 CrossRef CAS.
- S. Xu, N. Onishi, A. Tsurusaki, Y. Manaka, W. H. Wang, J. T. Muckerman, E. Fujita and Y. Himeda, Eur. J. Inorg. Chem., 2015, 2015, 5591 CrossRef CAS.
- R. Puerta-Oteo, M. Hölscher, M. V. Jiménez, W. Leitner, V. Passarelli and J. s. J. Pérez-Torrente, Organometallics, 2017, 37, 684 CrossRef.
- Z. Lu, V. Cherepakhin, I. Demianets, P. J. Lauridsen and T. Williams, Chem. Commun., 2018, 54, 7711 RSC.
- H. Li, B. Zheng and K.-W. Huang, Coord. Chem. Rev., 2015, 293–294, 116 CrossRef CAS.
- H. Li, T. P. Gonçalves, D. Lupp and K.-W. Huang, ACS Catal., 2019, 9, 1619 CrossRef CAS.
- J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761 CrossRef CAS PubMed.
- M. C. Haibach, S. Kundu, M. Brookhart and A. S. Goldman, Acc. Chem. Res., 2012, 45, 947 CrossRef CAS PubMed.
- C. Cheng, B. G. Kim, D. Guironnet, M. Brookhart, C. Guan, D. Y. Wang, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2014, 136, 6672 CrossRef CAS PubMed.
- M. C. Haibach, N. Lease and A. S. Goldman, Angew. Chem., Int. Ed., 2014, 53, 10160 CrossRef CAS PubMed.
- S. Fang, H. Chen and H. Wei, RSC Adv., 2018, 8, 9232 RSC.
- J. Campos, U. Hintermair, T. P. Brewster, M. K. Takase and R. H. Crabtree, ACS Catal., 2014, 4, 973 CrossRef CAS.
- L. S. Sharninghausen, J. Campos, M. G. Manas and R. H. Crabtree, Nat. Chem., 2014, 5, 973 Search PubMed.
- M. Iglesias and L. A. Oro, Chem. Soc. Rev., 2018, 47, 2772 RSC.
- A. Azua, S. Sanz and E. Peris, Chem. – Eur. J., 2011, 17, 3963 CrossRef CAS PubMed.
- F. J. Fernández-Alvarez, M. Iglesias, L. A. Oro and V. Polo, ChemCatChem, 2013, 5, 3481 CrossRef.
- Y. Suna, M. Z. Ertem, W.-H. Wang, H. Kambayashi, Y. Manaka, J. T. Muckerman, E. Fujita and Y. Himeda, Organometallics, 2014, 33, 6519 CrossRef CAS.
- Y. Suna, Y. Himeda, E. Fujita, J. T. Muckerman and M. Z. Ertem, ChemSusChem, 2017, 10, 4535 CrossRef CAS PubMed.
- T. Chen, L.-P. He, D. Gong, L. Yang, X. Miao, J. Eppinger and K.-W. Huang, Tetrahedron Lett., 2012, 53, 4409 CrossRef CAS.
- L.-P. He, T. Chen, D. Gong, Z. Lai and K.-W. Huang, Organometallics, 2012, 31, 5208 CrossRef CAS.
- L.-P. He, T. Chen, D.-X. Xue, M. Eddaoudi and K.-W. Huang, J. Organomet. Chem., 2012, 700, 202 CrossRef CAS.
- T. Chen, H. Li, S. Qu, B. Zheng, L. He, Z. Lai, Z.-X. Wang and K.-W. Huang, Organometallics, 2014, 33, 4152 CrossRef CAS.
- D. Gong, W. Liu, T. Chen, Z.-R. Chen and K.-W. Huang, J. Mol. Catal. A: Chem., 2014, 395, 100 CrossRef CAS.
- H. Li, Y. Wang, Z. Lai and K.-W. Huang, ACS Catal., 2017, 7, 4446 CrossRef CAS.
- Y. Wang, B. Zheng, Y. Pan, C. Pan, L. He and K.-W. Huang, Dalton Trans., 2015, 44, 15111 RSC.
- Y. Pan, C. L. Pan, Y. Zhang, H. Li, S. Min, X. Guo, B. Zheng, H. Chen, A. Anders, Z. Lai and K.-W. Huang, Chem. – Asian J., 2016, 11, 1357 CrossRef CAS PubMed.
- C. Zhou, J. Hu, Y. Wang, C. Yao, P. Chakraborty, H. Li, C. Guan, M.-H. Huang and K.-W. Huang, Org. Chem. Front., 2019, 6, 721 RSC.
- M. Gupta, C. Hagen, W. C. Kaska, R. E. Cramer and C. M. Jensen, J. Am. Chem. Soc., 1997, 119, 840 CrossRef CAS.
- M. Feller, U. Gellrich, A. Anaby, Y. Diskin-Posner and D. Milstein, J. Am. Chem. Soc., 2016, 138, 6445 CrossRef CAS PubMed.
- A. Anaby, M. Feller, Y. Ben-David, G. Leitus, Y. Diskin-Posner, L. J. Shimon and D. Milstein, J. Am. Chem. Soc., 2016, 138, 9941 CrossRef CAS PubMed.
- W. C. Kaska, S. Nemeh, A. Shirazi and S. Potuznik, Organometallics, 1988, 7, 13 CrossRef CAS.
- S. Nemeh, C. Jensen, E. Binamira-Soriaga and W. C. Kaska, Organometallics, 1983, 2, 1442 CrossRef CAS.
- K.-W. Huang, J. H. Han, C. B. Musgrave and E. Fujita, Organometallics, 2007, 26, 508 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and 1H and 13C NMR spectra for the compounds. CCDC 1423984 (4a), 1423985 (2b), 1482628 (5b) and 1914804 (3b). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt01319a |
| This journal is © The Royal Society of Chemistry 2019 |