
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSlow magnetic relaxation in Ni–Ln (Ln = Ce, Gd, Dy) dinuclear complexes†
Anna
Vráblová
 ab,
Milagros
Tomás
*c,
Larry R.
Falvello
b,
Ľubor
Dlháň
d,
Ján
Titiš
e,
Juraj
Černák
*a and
Roman
Boča
e
ab,
Milagros
Tomás
*c,
Larry R.
Falvello
b,
Ľubor
Dlháň
d,
Ján
Titiš
e,
Juraj
Černák
*a and
Roman
Boča
e
aDepartment of Inorganic Chemistry, Institute of Chemistry, P. J. Šafárik University in Košice, Moyzesova 11, Košice, Slovakia. E-mail: juraj.cernak@upjs.sk
bInstituto de Ciencia de Materiales de Aragón (ICMA), Departamento de Química Inorgánica, University of Zaragoza–CSIC, E-50009 Zaragoza, Spain
cCentro de Quimica y Materiales de Aragón (CEQMA-ISQCH), Departamento de Química Inorgánica, Pedro Cerbuna 12, University of Zaragoza–CSIC, E-50009 Zaragoza, Spain. E-mail: milagros.t@csic.es
dInstitute of Inorganic Chemistry, Slovak University of Technology, 812 37 Bratislava, Slovakia
eDepartment of Chemistry, FPV, University of SS Cyril and Methodius, 917 01 Trnava, Slovakia
First published on 23rd August 2019
Abstract
Three new isomorphous complexes [Ni(o-van-en)LnCl3(H2O)] [H2(o-van-en) = N,N′-ethylene-bis(3-methoxysalicylaldiminate; Ln = Ce (1), Gd (2), Dy (3)] were prepared by a stepwise reaction using mild conditions and were structurally characterised as dinuclear molecules in which Ni and Ln are coordinated by the compartmental Schiff base ligand (o-van-en)= and doubly bridged by O atoms. While the nickel(II) centre is diamagnetic within the N2O2 square-planar coordination of the Schiff base ligand, the lanthanide atoms are octa-coordinated to give an {LnCl3O5} chromophore with a fac-arrangement of the chlorido ligands. AC magnetic measurements revealed that all three complexes, including the nominally isotropic Gd(III) system, show field induced slow magnetic relaxation with two or three relaxation channels: at T = 1.9 K the low-frequency relaxation time is τLF(1) = 0.060 s at BDC = 0.5 T, τLF(2) = 0.37 s at BDC = 0.3 T, and τLF(3) = 1.29 s at BDC = 0.15 T.
Introduction
Since the identification of the first single-molecule magnet (SMM) {Mn12} in 1993,1 research on this phenomenon has continued apace and has also been extended to lanthanide based materials.2 As early as 2004, heteronuclear 3d–4f complexes were observed to display slow magnetic relaxation.3 The advantage of transition metal and lanthanide complexes lies in a fusion of the properties of both. In fact, the magnetic study of complexes comprising transition metals and lanthanides dates back to 1985, when Gatteschi et al. observed unexpected ferromagnetic coupling between copper(II) and gadolinium(III) ions.4More recent research has yielded a few examples of a significant reduction in quantum tunnelling magnetization (QTM) due to non-negligible magnetic exchange interactions between lanthanide and transition metal ions.5 A recent study on 3d–4f complexes with diamagnetic 3d metal ions such as zinc(II) or cobalt(III) showed an enhancement of the Ueff barrier in comparison to their mononuclear lanthanide analogues;6 it was suggested that the presence of a diamagnetic 3d cation near the lanthanide central atom, with a monatomic O bridge, induces a large charge polarisation on the bridging oxygen atom that favours an increase in the Ueff barrier. This observation suggests a new strategy in designing 3d–4f complexes with diamagnetic 3d ions.5,7–10
Regarding the 4f elements, heavy lanthanide ions were widely used for the preparation of SMM materials due to their large angular momentum J and large magnetic anisotropy.11–13 However, a recent study of light lanthanide ions revealed surprising magnetic behaviour for cerium(III).14 Despite the small magnetic moment, which arises from its small angular momentum J and small Landé g factor, the spin–orbit coupling is strong enough to create significant magnetic anisotropy and thus an energy barrier between the two orientations of the magnetization. The first cerium(III) based SMM was reported in 2013![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15 and later other cerium(III) complexes were reported to be SMMs.16–19
15 and later other cerium(III) complexes were reported to be SMMs.16–19
Among the lanthanide ions, the case of gadolinium(III) complexes represents another breakthrough in the theory of slow relaxation of magnetization. Despite its essentially isotropic ground state with L = 0, several materials have been reported for which magnetic anisotropy around the gadolinium(III) centre was induced by local coordination, magnetic exchange coupling or electron density donation.20–29 These materials containing gadolinium(III) may be considered as members of a new class of SMMs with the highest known spin state (4f7) for a single ion.
Some considerable effort has been extended to produce strictly dinuclear 3d–4f molecular complexes30–32 to serve as building blocks for the further preparation and study of polynuclear clusters33–36 and polymers.37–39 A related fact in the context of the present work is that salen-type Schiff bases originating from o-vanillin and related aldehydes have been used in coordination chemistry due to their ability to form complexes with a rich variety of geometries and structural dimensions.40
In what follows we describe the preparation, chemical and structural characterization and magnetic properties of three isomorphous dinuclear [Ni(o-van-en)Ln(H2O)Cl3] complexes [Ln = Ce (1), Gd (2), Dy (3); H2L = H2(o-van-en) = N,N′-ethylene-bis(3-methoxysalicylaldiminate)]. The choice of Ce(III), Dy(III) and Gd(III) was designed to expose the main differences in the magnetic behaviours of a light lanthanide ion, a heavy lanthanide ion and the lanthanide ion with a spherical orbital ground state, respectively.
Results and discussion
Synthesis and identification
The three complexes [Ni(o-van-en)LnCl3(H2O)] [Ln = Ce (1), Gd (2), Dy (3)] were first synthesized in microcrystalline form using mild solution techniques in a modification of the method of Costes and co-workers.41 The synthetic procedure is depicted in Scheme 1. The products were characterized using chemical analysis and IR spectroscopy; the observed absorption bands are listed in the ESI.† The imine group absorption, ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), observed slightly above 1600 cm−1, can be considered as a characteristic band. In the IR spectrum of the ligand, this vibration was observed at 1631 cm−1 which agrees with the reported value of 1628 cm−1 reported for the same Schiff base,42 and is similar to the value of 1625 cm−1 for a related Schiff base L [L = N,N′-bis((E)-3-phenylallylidene)-3-methyl-1,2-phenylenediamine].43 The ligand bonded in the complex [CdLX2] (X = Cl, Br, I, SCN) exhibits a shift of 1–7 cm−1 to lower wavenumbers. In the spectra of the complexes 1, 2 and 3 the vibration ν(CN) was observed at 1622–1623 cm−1.
N), observed slightly above 1600 cm−1, can be considered as a characteristic band. In the IR spectrum of the ligand, this vibration was observed at 1631 cm−1 which agrees with the reported value of 1628 cm−1 reported for the same Schiff base,42 and is similar to the value of 1625 cm−1 for a related Schiff base L [L = N,N′-bis((E)-3-phenylallylidene)-3-methyl-1,2-phenylenediamine].43 The ligand bonded in the complex [CdLX2] (X = Cl, Br, I, SCN) exhibits a shift of 1–7 cm−1 to lower wavenumbers. In the spectra of the complexes 1, 2 and 3 the vibration ν(CN) was observed at 1622–1623 cm−1.
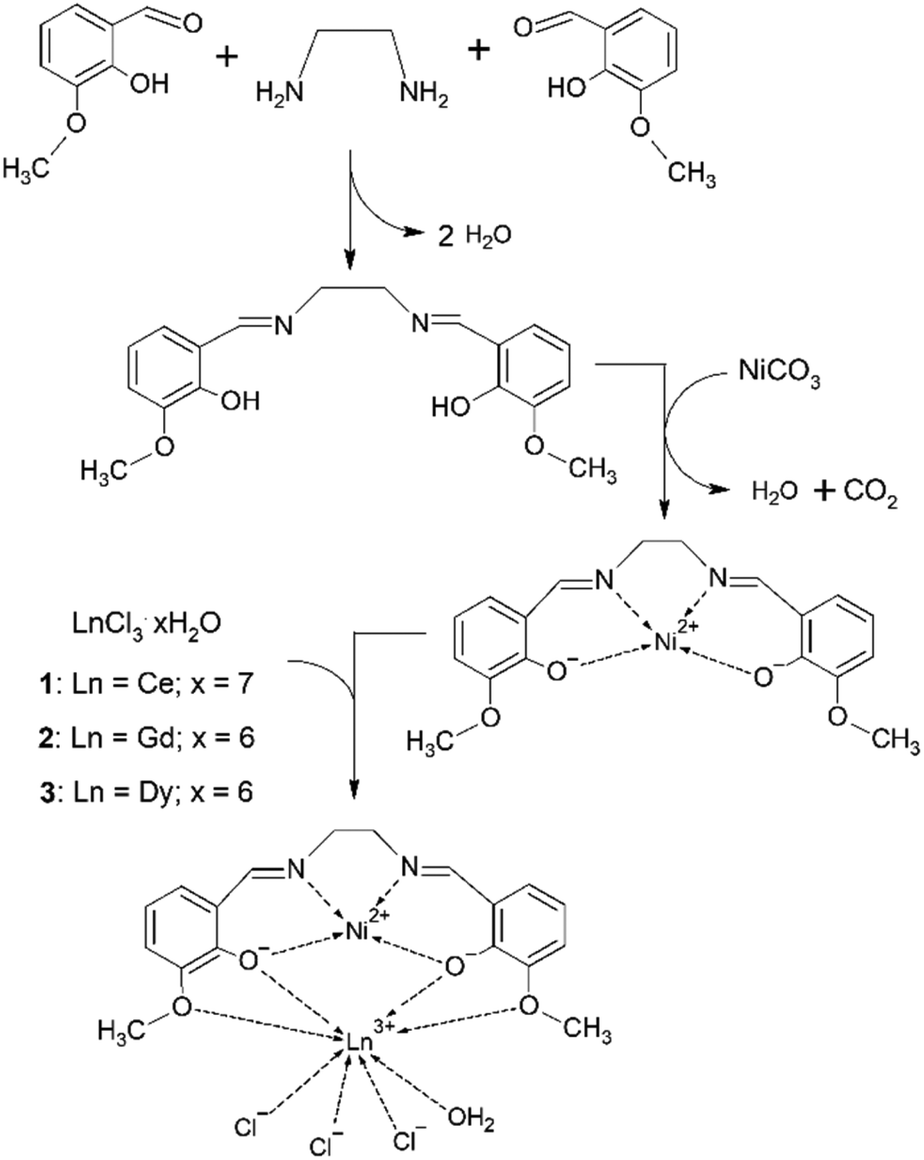 | ||
| Scheme 1 Schematic representation of the synthetic route for preparation of 1, 2 and 3. | ||
Recrystallization of these microcrystalline products or reaction by diffusion techniques yielded single crystals of 1–3. The phase identities of the microcrystalline products with the corresponding single-crystal samples were established by LeBail refinement using the powder diffraction patterns for all three samples (see Fig. S1†).
Crystal structures
Compounds 1–3 are isomorphous, so we will limit our description and discussion to complex 1 with the corresponding geometric parameters of 2 and 3 given in parentheses. We note that all three crystal structures were studied at room temperature. The crystal structure of 1 consists of neutral heterodinuclear molecules of [Ni(o-van-en)Ce(H2O)Cl3] (Fig. 1). The Ni(II) atom is coordinated in square-planar form by the N2O2 donor set of the potentially ditopic ligand (o-van-en)2−. The Ce(III) central atom is octa-coordinated by four oxygen atoms from the (o-van-en)2− ligand, one aqua and three chlorido ligands, yielding an {LnCl3O5} chromophore (Fig. 1). Calculations using the program SHAPE44 indicate that the coordination polyhedron is close to a bicapped (O1 and O4) trigonal prism [Fig. S2;† the same holds for the Dy(III) and Gd(III) complexes]. It is significant (vide infra) that the three chlorido ligands form a single face of the polyhedron.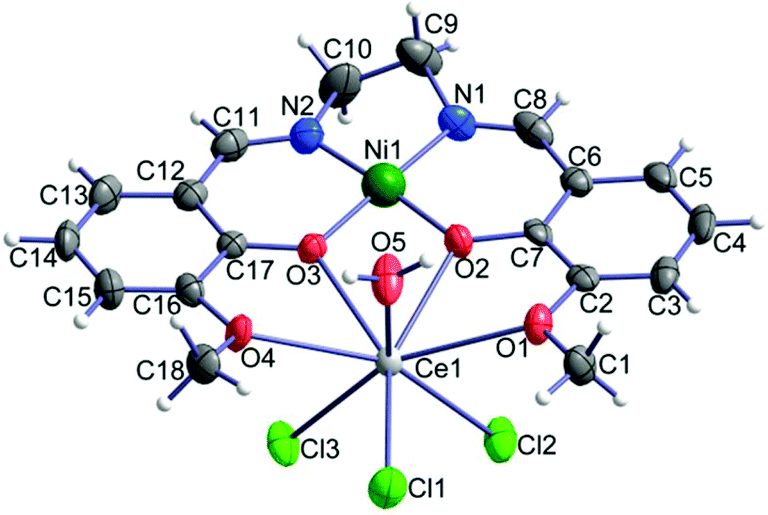 | ||
| Fig. 1 Molecular structure of [Ni(o-van-en)Ce(H2O)Cl3]; thermal ellipsoids are drawn with 50% probability. | ||
Analogous [Ni(o-van-en)Ln(NO3)3]·(CH3)2CO heterodinuclear complexes with auxiliary nitrato ligands and containing various lanthanide atoms have been reported;45,46 but to our knowledge no analogous complexes with other 3d elements with auxiliary chlorido ligands have been studied. On the other hand, eight analogous heterodinuclear complexes containing chlorido ligands were reported with modified Schiff base ligands.34,47–51 An interesting example is the complex [NiCl2(o-van-bu)YbCl(H2O)3] with a hexacoordinated Ni(II) central atom (donor set Cl2O2N2).48
The observed Ni–N and Ni–O as well as the Ce–O bond distances are similar to those reported in the analogous complexes [Ni2(o-van-en)2Ce(NO3)2](NO3),52 ([Cu(o-van-en)Gd(NO3)3]·0.25H2O,53 [Co(o-van-en)(CH3COO)2Dy(NO3)2]54) (Table 1). The Ln–Cl bond distances in 1 are in the range of 2.717(2)–2.802(2) Å [2.630(3)–2.714(3) Å for 2 and 2.6005(10)–2.6955(9) Å for 3], which can be compared to the average Ce–Cl distance of 2.7467 Å (2.6829 Å for Gd–Cl and 2.6357 Å for Dy–Cl) found in the CSD.55 The CSD further reveals that in all Ni–Ln (Ln = Ce, Gd, Dy) with Ln–Ni distances shorter than 5 Å the two metal atoms are bridged by monatomic O bridges, and the mean values of the Ni–Ln distances are 3.58 Å, 4.53 and 4.45 Å for Ce–Ni, Gd–Ni and Dy–Ni, respectively. In our compounds 1–3 the observed Ln–Ni distances are significantly shorter than the corresponding calculated mean values (Table 1). The remaining geometric parameters in all three complexes exhibit expected values.
| 1 | 2 | 3 | |
|---|---|---|---|
| Ln–Cl1 | 2.717(2) | 2.630(3) | 2.6005(10) |
| Ln–Cl2 | 2.802(2) | 2.714(3) | 2.6940(9) |
| Ln–Cl3 | 2.789(2) | 2.709(3) | 2.6955(9) |
| Ln–O1 | 2.710(5) | 2.658(7) | 2.654(2) |
| Ln–O2 | 2.485(5) | 2.396(7) | 2.364(3) |
| Ln–O3 | 2.469(5) | 2.385(7) | 2.343(2) |
| Ln–O4 | 2.697(5) | 2.650(7) | 2.648(2) |
| Ln–O5 | 2.518(5) | 2.393(6) | 2.376(3) |
| Ni–O2 | 1.843(5) | 1.842(7) | 1.843(2) |
| Ni–O3 | 1.843(5) | 1.833(7) | 1.842(2) |
| Ni–N1 | 1.827(6) | 1.816(9) | 1.834(3) |
| Ni–N2 | 1.829(7) | 1.825(9) | 1.836(3) |
| Ln⋯Ni | 3.4876(10) | 3.3959(14) | 3.3735(5) |
Although molecular compounds with Ln–Cl bonds are known, these represent only about 6% of all molecular lanthanide complexes.55 Those in which the lanthanide is bonded to three terminal chlorido ligands are just around 1%. For comparison, the number of molecular lanthanide compounds with three coordinated NO3− groups is more than five times that with three Cl− ligands. Nevertheless, given the large number of molecular Ln compounds that have been characterized, there are some 400 compounds in the latter class – Ln–Cl3(terminal). The only eight heterodinuclear Tr–Ln complexes with Schiff bases similar to the one in compounds 1–3 and with chlorido ligands bonded to the lanthanide metal that have been structurally characterized have only one or two chlorido ligands bonded to the lanthanide,34,47–51 not three as in compounds 1–3. Also, two related compounds with three Cl− ligands bonded to the same lanthanide metal have been found, but with higher nuclearity, i.e., Tr2Ln2 compounds in which some of the chlorido ligands act as bridges between the lanthanide centers.47,56
There are four dinuclear molecules per unit cell of 1 (and likewise for 2 and 3). The packing of the complex molecules is governed by medium-strength hydrogen bonds of the O–H⋯Cl type yielding a chain-like supramolecular structure running along the a axis with additional, weaker Car–H⋯Cl, NC–H⋯Cl type hydrogen bonds and weak π–π stacking interactions of the aromatic rings of the neighbouring chains forming supramolecular layers in the ab plane (Fig. S3 and Table S2†). There are only close contacts of the C–H⋯Cl type between planes, involving methyl and methylene groups of the ligand.
DC magnetic data
In compounds 1–3 the central Ni(II) atom is in a square planar environment formed by four oxygen donor atoms. Thus, it is magnetically silent, except for some temperature-independent magnetism arising from the presence of low-lying excited states. The lanthanide Ln(III) centres bear orbital and spin angular momentum and the ground multiplet is 2F5/2 for Ce(III), 8S7/2 for Gd(III) and 6H15/2 for Dy(III), with the magnetogyric ratios gJ = 6/7, 2 and 4/3, respectively. Thus the magnetization per formula unit should saturate to M1 = Mmol/(NAμB) = gJ·J = 6/7 × 5/2 = 15/7, 2 × 7/2 = 7, and 4/3 × 15/2 = 10. The high-temperature limit of the effective magnetic moment, based upon the magnetic susceptibility, is μeff/μB = gJ[J(J + 1)]1/2 = (6/7)(5/2 × 7/2)1/2 = 2.54, 2 × (7/2 × 9/2)1/2 = 7.94, and (4/3)(15/2 × 17/2)1/2 = 10.6.The DC magnetic data for 1 are shown in Fig. 2. Clearly visible linear dependence of the effective magnetic moment between T = 9–300 K indicates some temperature-independent paramagnetism as expected for the Ni(II) centre in a square planar environment: χTIP = 6 × 10−9 m3 mol−1. With the above correction the room-temperature effective magnetic moment (the high-temperature limit) adopts a value of μeff = 2.36μB, that is not far from the single ion value (2.54). The magnetization data at T = 2.0 and B = 7 T, M1 = 1.0, is much lower relative to isolated multiplet 2F5/2 for Ce(III) (2.1).
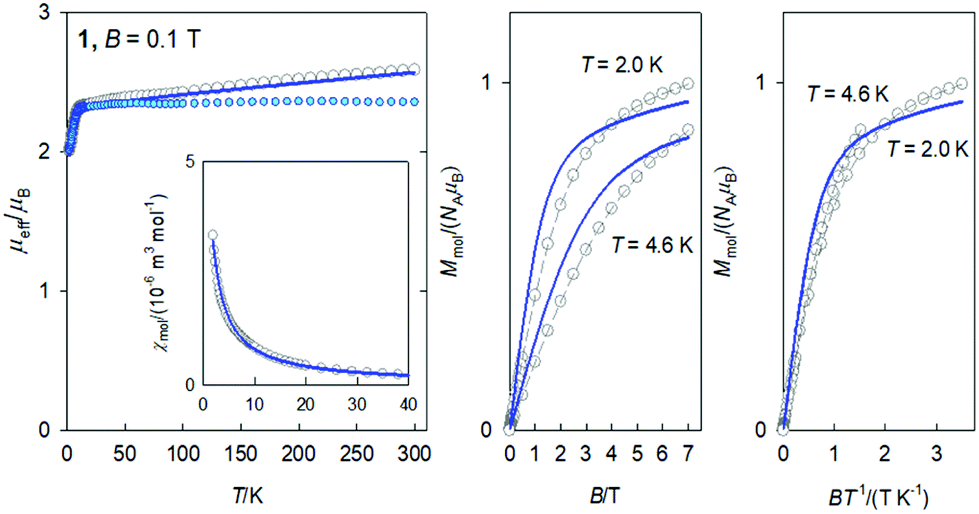 | ||
| Fig. 2 DC magnetic data for 1. Solid lines – fitted. Colored circles – TIP corrected data. | ||
Owing to the effect of the crystal field, the six-fold degenerate ground atomic multiplet 2F5/2 |R′3:J;MJ〉 = |5/2;±1/2,±3/2,±5/2〉 is split into three crystal-field multiplets (Kramers doublets) within the respective double group:  . The magnetic data was fitted assuming that only the lowest Kramers doublet is thermally populated. Then the effective Hamiltonian in use is
. The magnetic data was fitted assuming that only the lowest Kramers doublet is thermally populated. Then the effective Hamiltonian in use is
| Ĥeffa = μBB(geffzĴeffzcosθa + geffxyĴeffxsinθa) | (1) |
Ab initio calculations for 1 gave the values g{0.035, 0.251, 3.880} that support the magnetic data analysis. The first excited Kramers doublet lies 300 cm−1 above the ground one and its thermal population at T = 300 K is only 9%.
The DC magnetic data for 2 confirms the behaviour of the 8S7/2 ground term (Fig. 3): the effective magnetic moment is almost constant over the whole temperature range with μeff = 7.8–7.9μB; the magnetization per formula unit at B = 7.0 T and T = 2.0 K saturates to M1 = 6.6. This data is consistent with an isotropic g-factor slightly below 2.0. The data fitting to the Curie law and the Brillouin function gave g = 1.952(6). A superposition of the magnetization data vs. BT−1 confirms an invisible magnetic anisotropy of 2.
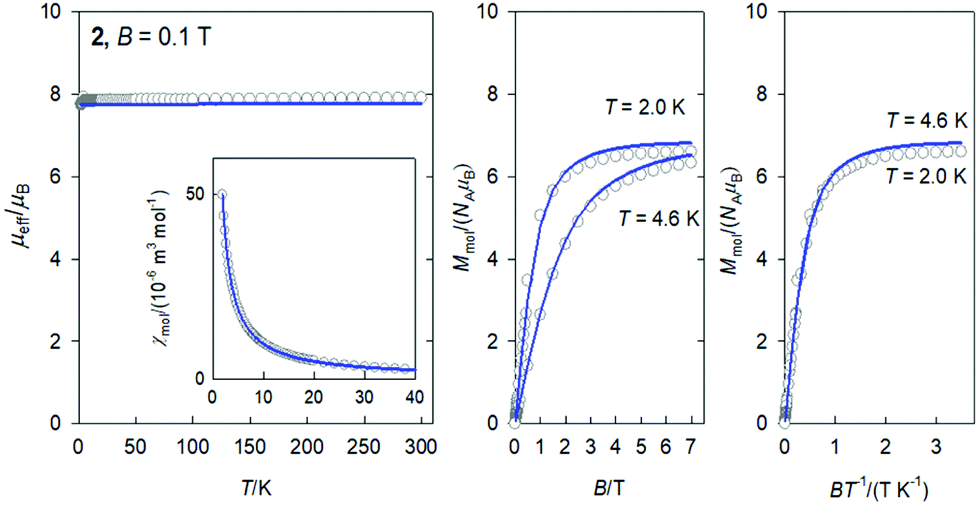 | ||
| Fig. 3 DC magnetic data for 2. Solid lines – fitted. | ||
The DC magnetic data for 3 is shown in Fig. 4. The effective magnetic moment displays the high-temperature (HT) limit of μeff = 12.15μB that decreases only slightly down to T ∼ 60 K; then it decreases more rapidly to 11.1μB at the lowest temperature of the measurements. The observed HT limit is somewhat higher than that expected for a single Dy(III) ion (10.6). This might be due to the presence of low-lying excited states of the cooperating Ni(II) centre. More surprising is the saturation value of the magnetization M1(J) = 7.9 at T = 2.0 and B = 7.0 T, which differs substantially from the single-ion expectation (M1 = 10). This can arise in a partial quenching of the orbital angular momentum of the Dy(III) centre by the asymmetric ligand field of the {DyO4O′Cl3} chromophore. Another reason could be the zero-field splitting of the ground crystal-field multiplet inherent to an anisotropic ion such as Dy(III). In such a case, the higher terms of the series of the crystal field potential will also contribute
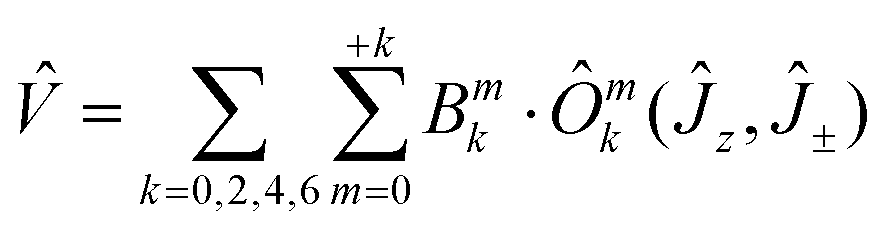 | (2) |
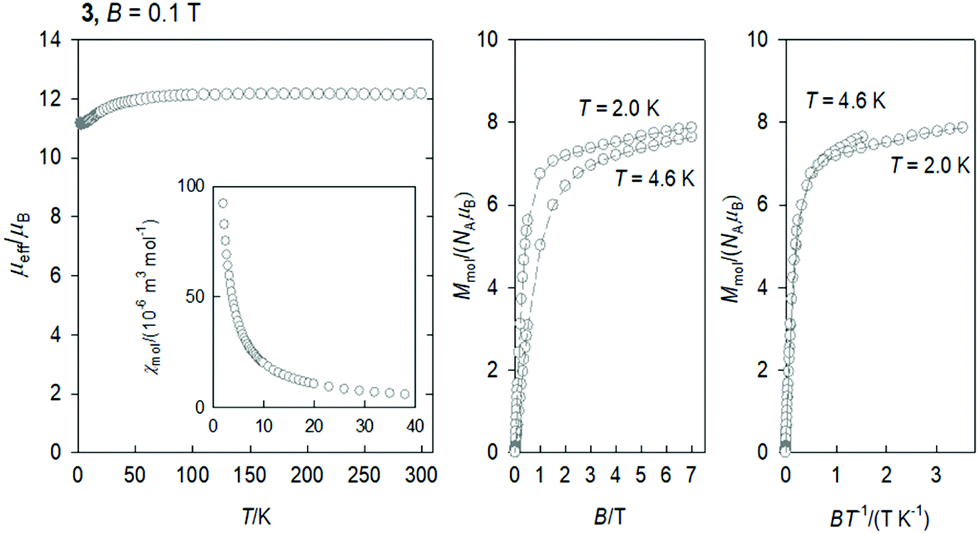 | ||
| Fig. 4 DC magnetic data for 3. Dashed lines are visual guides. | ||
Field scan of the AC susceptibility
The AC susceptibility data was acquired at the field amplitude of BAC = 0.38 mT and depending upon the external magnetic field BDC, temperature, and the frequency f of the oscillating field.The first scan for a variable magnetic field, fixed low temperature T = 2.0 K, and a set of four representative frequencies f = 1.1, 11, 111, and 1116 Hz is shown in Fig. 5. It is seen that the out-of-phase susceptibility χ′′ is zero at BDC = 0, which is traditionally explained as an effect of the fast magnetic tunnelling. With the applied external field, the out-of-phase susceptibility rises to a maximum and then attenuates at higher fields. The position of the maximum slightly depends upon the frequency of the oscillating field. The subsequent data measurements were done at the external field for which the out-of-phase susceptibility component displays a maximum: BDC = 0.1 or 0.5, 0.3, and 0.15 T for 1 through 3.
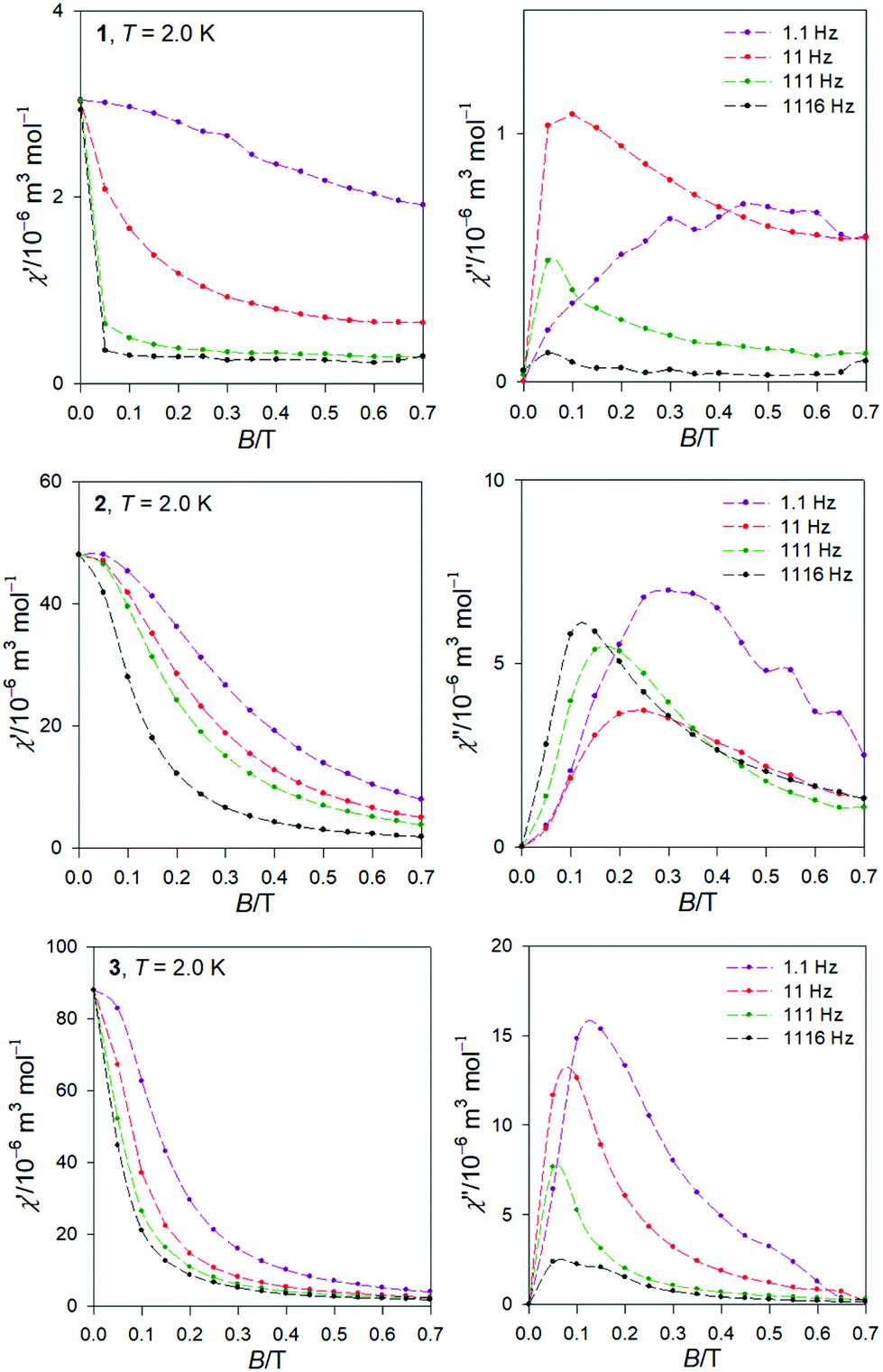 | ||
| Fig. 5 AC magnetic data for 1, 2, and 3. Field scans for a set of frequencies at low temperature. | ||
Temperature scan of the AC susceptibility
Temperature evolution of the AC susceptibility components for a set of frequencies varying between f = 0.1 and 1500 Hz at a selected external field BDC is shown in Fig. S4 (see ESI†). It can be concluded that the samples 1 through 3 show field induced slow magnetic relaxation typical for single ion magnets (SMM). On heating, the in-phase susceptibility components for various frequencies tend to merge at the critical temperature (ca. 10, >7, and 7 K); at the same time the out-of-phase component vanishes since then the system behaves as an ordinary paramagnet.Frequency scan of the AC susceptibility for 3, Ni–Dy complex
The same data sets have been rearranged to the frequency dependence of the AC susceptibility components for a set of temperatures.For 3, it is evident (Fig. 6) that the out-of-phase component consists of three contributions: low-frequency (LF), intermediate-frequency (IF), and the high-frequency (HF). For the lowest-temperature measurements (T = 1.9 K) the LF component dominates with the peak-position at fmax ∼ 0.16 Hz. Then the corresponding relaxation time is estimated to be as long as τ(LF) = 1/(2πfmax) ∼ 0.995 s. Both, in-phase and out-of-phase susceptibilities were fitted simultaneously by employing the three-set Debye model, as explained in the ESI.† Using the resulting relaxation parameters (thermal susceptibilities χTk, distribution parameters αk, and relaxation times τk, k = 1–3), the convoluted susceptibility components are drawn as solid lines in Fig. 6. It is evident that these lines pass through the experimental points and this is also supported by the statistical analysis in the ESI.†
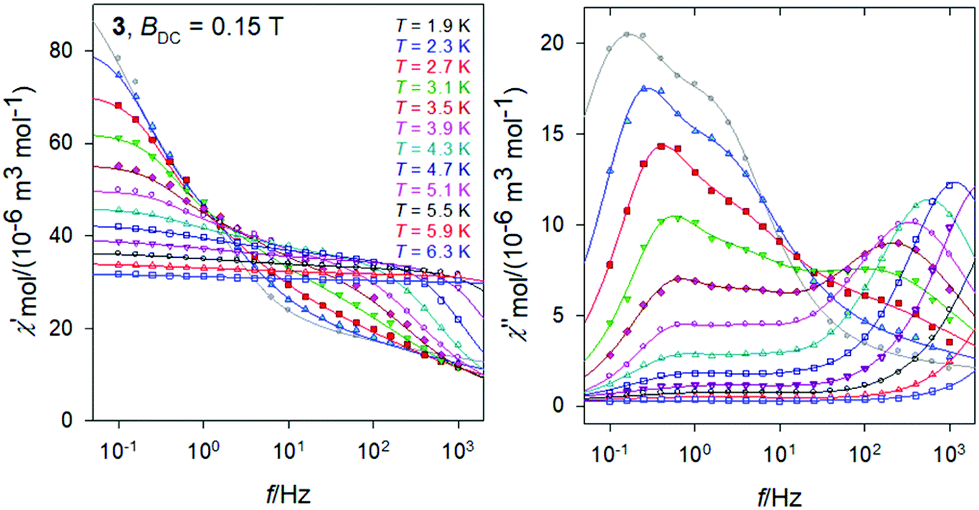 | ||
| Fig. 6 AC magnetic data for 3. Solid lines – fitted by the extended three-set Debye model. | ||
At low temperature the HF component recovers only a tail; however, with increasing temperature the LF component decays progressively in favor of the HF one; the IF component balances in-between. At temperature T > 5 K, the IF component cannot be fitted satisfactorily and then only a two-set Debye model is applicable. At the lowest measurement temperature, T = 1.9 K, the mole fractions of the individual relaxation species are x(LF) = 0.49, x(IF) = 0.28, and x(HF) = 0.23, respectively. At the same time, the three relaxation times are τ(LF) = 1.29 s, τ(IF) = 74 ms, and τ(HF) = 964 μs. In contrast, at T = 6.3 K, x(LF) = 0.13 and x(HF) = 0.87 hold true.
In Fig. 7, the Argand diagram is shown; it is a convolution of three distorted semicircles (arcs) of different height, position and width. Also an Arrhenius-like plot lnτ vs. T−1 is shown on the right for the individual relaxation processes. The three highest-temperature points for the HF relaxation channel have been fitted by a straight line in order to get parameters for the Orbach relaxation process τ = τ0exp(Ueff/kBT): Ueff/kB = 65 K and τ0 = 1.9 × 10−10 s.
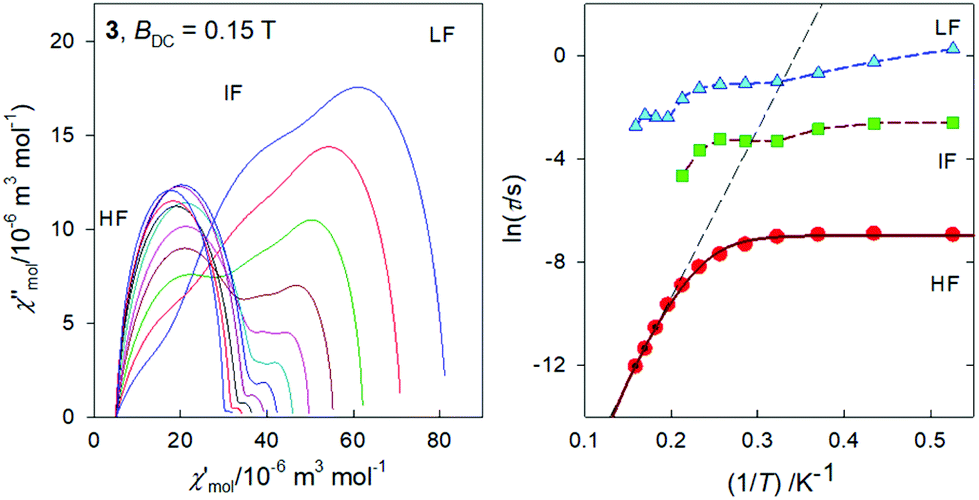 | ||
| Fig. 7 Argand diagram (left) and the Arrhenius-like plot (right) for 3. Straight line – high-temperature limit of the Arrhenius equation referring to the Orbach process. Solid curve – fitted using Raman and quantum tunneling processes. | ||
The temperature evolution of the HF relaxation time can be represented by various graphs as shown in Fig. S5 (see ESI†). The dependence lnτ vs. lnT at its two edges can be recovered by straight lines: lnτ = –8.81 + 11.3(lnT) at the highest temperatures, and lnτ = –Dτ at the lowest temperatures. Therefore, the whole dataset has been fitted via the equation
| τ−1 = CTn + Dτ | (3) |
Frequency scan of the AC susceptibility for 1, Ni–Ce complex
The frequency dependence of the AC susceptibility components for 1 is shown in Fig. 8. For T = 1.9 K the well-developed peak of χ′′ at f ∼ 10 Hz implies the relaxation time τ(LF) ∼ 16 ms. This is much shorter relative to 3. However, the peak is slightly asymmetric with an arm, and its reliable fit requires a two-set Debye model where the HF component is also considered. On heating the peak is shifted to higher frequencies but for T > 4 K the two-set model becomes unstable; then the single-set model has been applied for higher temperatures. The resulting relaxation parameters are listed in the ESI† along with statistical analysis.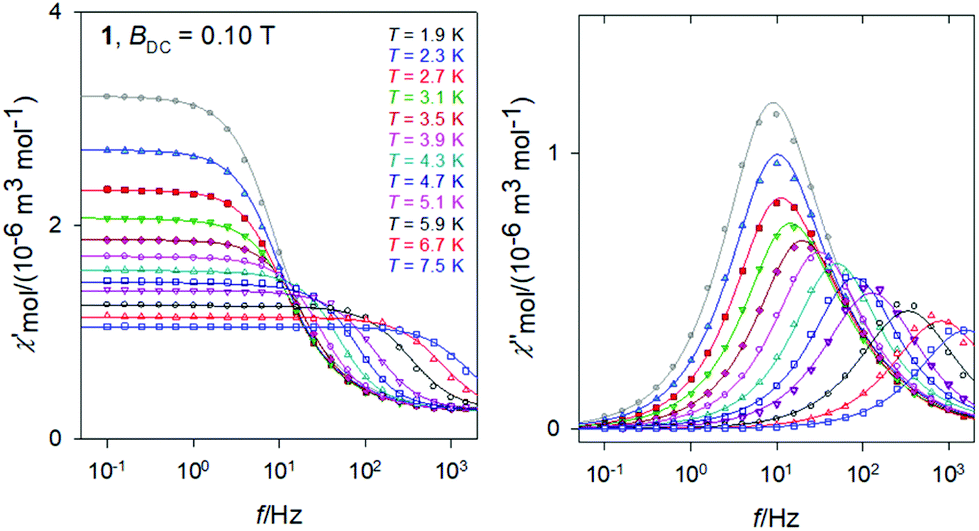 | ||
| Fig. 8 AC magnetic data for 1. Solid lines – fitted by the extended two-set Debye model. | ||
The Argand diagram and the Arrhenius-like plot for 1 are displayed in Fig. 9. In this case the LF relaxation time was fitted viaeqn (3) giving n = 6.59(7), C = 174(22) × 10−4 K−n s−1, and Dτ = 51(1) s−1. At the same time the parameters of the Orbach relaxation process are Ueff/kB = 43 K and τ0 = 3.1 × 10−7 s.
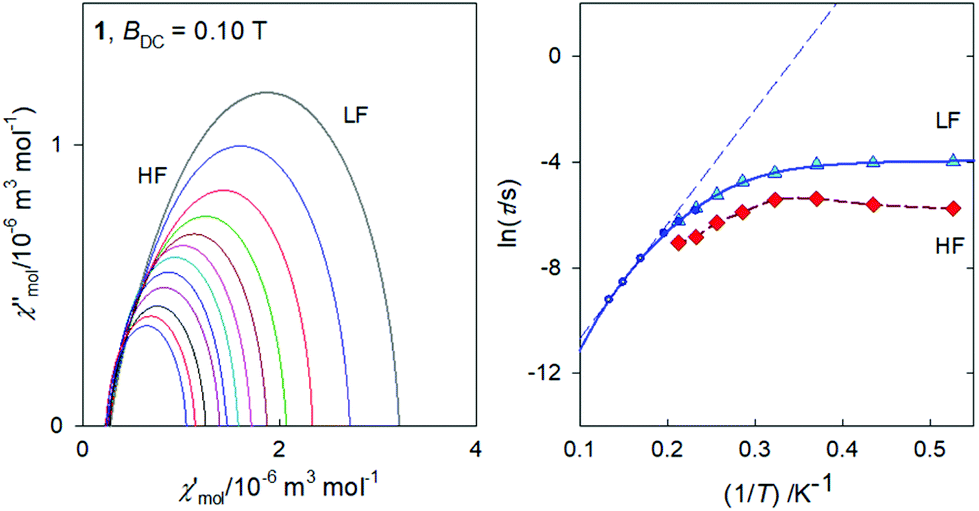 | ||
| Fig. 9 Argand diagram (left) and the Arrhenius-like plot (right) for 1. Full circles – single-set Debye model; solid curve – fitted with Raman & tunneling process; dashed – three-point linear extrapolation for the Orbach process. | ||
As expected, the external magnetic field has a visible effect on the relaxation behaviour of 1 (see ESI†): the relaxation time at T = 1.9 K and BDC = 0.5 T becomes τ(LF) = 60.3(5) ms as compared to 18.7(11) ms at BDC = 0.1 T. A successful data fitting requires the two-set Debye model for T > 3.5 K. SIM behaviour of the Ce(III) ions has been reviewed recently.14 For instance, AC susceptibility studies of single-molecule magnet behaviour in polynuclear assembly of Ce(III) with polymolybdates reveal a two-channel slow relaxation with the relaxation times τ = 6009 and 217 μs at T = 1.9 K and BDC = 0.02 T.18 There is a sizable field influence since at BDC = 0.14 T the relaxation times merge to τ = 605 μs.
Frequency scan of the AC susceptibility for 2, Ni–Gd complex
The frequency dependence of the AC susceptibility components for the Gd compound 2 is shown in Fig. 10. In this case the two-set Debye model reproduces the experimental data with well separated branches. The existence of slow relaxation is unexpected in the case of the isotropic gadolinium centre. Even more surprising is the Arrhenius-like plot drawn in Fig. 11 where the HF relaxation time is only slightly temperature dependent between 1.9 and 6.5 K. Moreover, the HF branch shows a “strange” behaviour (Fig. S6†): on cooling the relaxation time passes through a maximum and then it is shortened: τ(HF) = 223 μs at T = 3.9 as compared to τ(HF) = 120 μs at T = 1.9 K.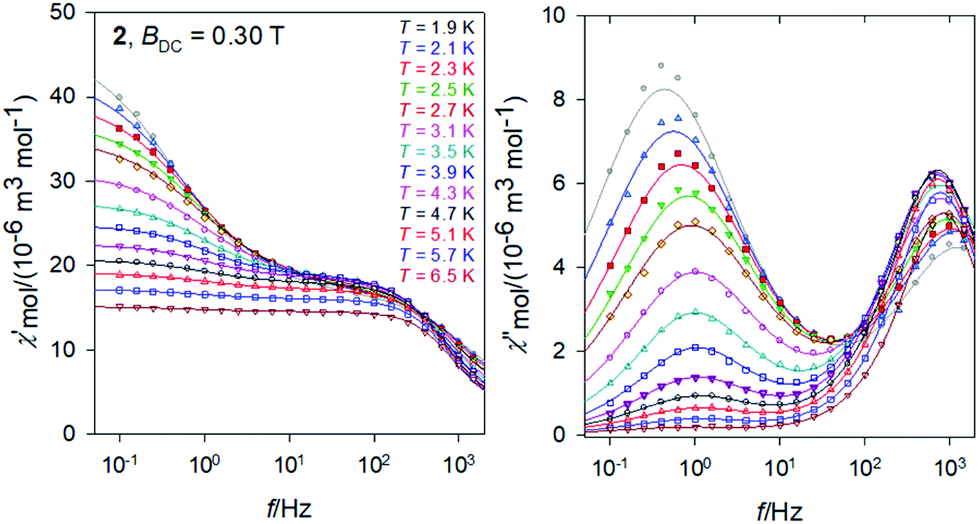 | ||
| Fig. 10 AC magnetic data for 2. Solid lines – fitted by the extended two-set Debye model. | ||
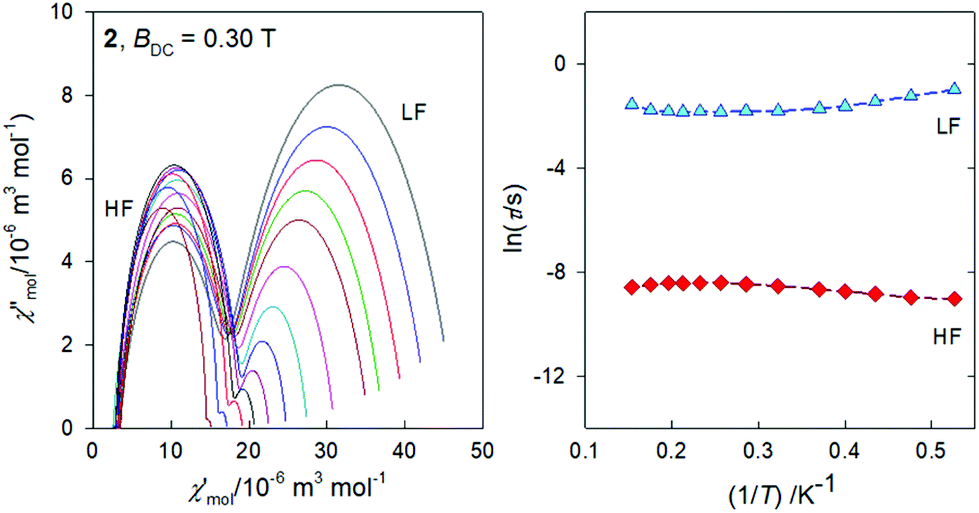 | ||
| Fig. 11 Argand diagram (left) and the Arrhenius-like plot (right) for 2. | ||
The strange behaviour has been observed recently in Cu(II), Ni(II), Co(II), and Mn(II) complexes showing distorted octahedral geometry.58–61 It was possible to reproduce such data by using a phenomenological equation
| τ−1 = CTn + EτT−k | (4) |
The geometry and the electron density of the coordination sites has been proved to exert a very important influence on the SMM behaviour of lanthanide ions. There are few studies of the influence of ions and since the number of chlorido derivatives is smaller than for instance, nitrato, the studies reported about the influence of the chlorido ligands in the SMM behaviour are very rare and it may require more research.
The magnetic study on the [Ln2(mq)4X6](EtOH)2 (Fig. 12, left) (mq = 8-hydroxy-2-methylquinoline) compounds which have been prepared for Ln = Dy, with X = NO3− or Cl− shows that for these compounds the energy barrier is higher when X = Cl− (NO3−: Δ/kB = 40.0 K and Cl−: Δ/kB = 102.4 K).62 We note that for Ln = Gd, X = NO3−, Cl−, no slow relaxation was observed. However, a recent study on [Ln(tpm)X3]·yMeCN63 (tpm = tris(3,5-dimethyl-pyrazolyl)methane) shows that at zero DC field, no significant out-of-phase susceptibility was detected for Ln = Dy or Er, none for X = NO3− nor X = Cl−, but under applied fields, it was observed for Ln = Dy and NO3− but not for Cl−, while for Ln = Er, the situation was the opposite, indicating that NO3− in this case is a more suitable anion for obtaining SMM with Dy while Cl− is a better anion for Er. No such comparative information for Ln = Gd has been reported, to our knowledge.
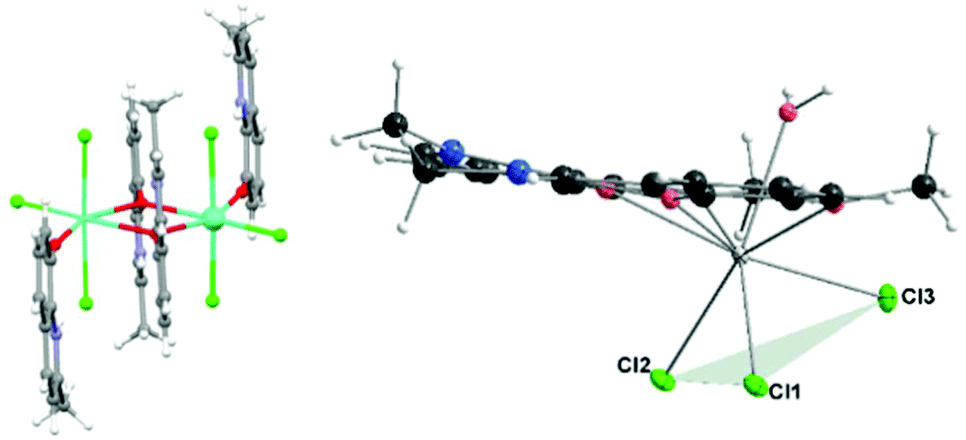 | ||
| Fig. 12 Left: [Ln2(mq)4Cl6] showing the disposition of the Cl− ligands. Right: Molecular drawing of compounds 1–3 showing the “fac” disposition of the three clorido ligands. | ||
All three of our compounds, including the Gd(III) one, with three Cl− ligands and under applied external field, show slow relaxation. We note that the location of the three coordinated chlorido atoms in our compounds (Fig. 12, right) is different than in the above-mentioned compounds [Ln2(mq)4Cl6]. From Fig. 12, it can be seen that in compounds 1–3 the chlorido ligands are in what can be called fac-configuration – that is, concentrated in the same part of the coordination sphere, while the other compounds present a more symmetrical distribution; that has to affect the electron distribution in the coordination sphere.
Conclusions
Using mild chemical conditions a new kind of 3d–4f dinuclear NiLn Schiff base compound, [Ni(o-van-en)LnCl3(H2O)] (Ln = Ce, Gd, Dy) was prepared and structurally characterized. In all three complexes the square four-coordinated nickel(II) atom and octacoordinated Ln(III) atom (donor set O5Cl3) are located in the neighbouring compartments provided by the Schiff base ligand and linked by two monatomic O bridges. The coordination of the Ln atom is highly unsymmetrical with all chlorido ligands situated in a “fac” fashion. AC susceptibility measurements reveal that all three complexes show field supported slow magnetic relaxation. This result in the case of the Gd(III) compound suggests a need for further detailed magnetic studies, in the field of SMM, of complexes with the chlorido ligands concentrated in a face of the coordination polyhedron. AC magnetic measurements revealed that all three complexes, including the nominally isotropic Gd(III) system, show field induced slow magnetic relaxation with two or three relaxation channels: at T = 1.9 K the low-frequency relaxation time is τLF(1) = 0.060 s at BDC = 0.5 T, τLF(2) = 0.37 s at BDC = 0.3 T, and τLF(3) = 1.29 s at BDC = 0.15 T.Experimental
Syntheses
Ethylenediamine, o-vanillin, NiCO3, GdCl3·6H2O, CeCl3·7H2O, DyCl3·6H2O, ethanol and diethyl ether, all of analytical grade, were purchased from commercial sources and used as received.Physical measurements
Elemental analyses (C, H, N) were performed on a PerkinElmer 2400 Series II CHNS/O analyser. Infrared spectra were recorded on a PerkinElmer Spectrum 100 CsI DTGS FTIR Spectrometer with UATR 1 bounce-KRS-5 in the range of 4000–300 cm−1. Powder X-Ray data were collected using a RIGAKU D/max 2500 diffractometer equipped with a copper rotating anode, operating at 40 kV and 80 mA, with a graphite monochromator. Measurements were made for 2θ from 2.5° to 40° in steps of 0.02° with a rate of 1 s per step. The magnetic susceptibility (determined as χ = M/H) was taken at B = 0.1 T using a SQUID magnetometer (Quantum Design, MPMS-XL7) in the temperature range from 1.9 to 300 K. Raw susceptibility data was corrected for the diamagnetic contribution. The magnetization data was acquired at low temperature T = 2.0 and 4.6 K until BDC = 7.0 T. The AC susceptibility data was taken at the applied external fields using the amplitude of the oscillating field BAC = 0.38 mT for a set of temperatures and for 22 frequencies between f = 0.1 to 1500 Hz.Crystallography
X-ray experiments were carried out on a four-circle κ-axis Xcalibur2 diffractometer equipped with a Sapphire3 CCD detector (Oxford Diffraction). The CrysAlis software package [Oxford Diffraction 2006 CrysAlisPro CCD and CrysAlisPro RED] was used for data collection and reduction. Crystals of compound 1 were found to exhibit multiple domains. For the crystal used for data collection, two domains were indexed and data integration was conducted simultaneously for both. The structures for 1, 2 and 3 were solved using SHELXT.64 Refinement based on intensities was performed using SHELXL-2014/7.65 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were located in difference Fourier maps. For the final refinement, H atoms bound to C were placed at calculated positions and refined as riders with Uiso(H) = 1.2Ueq(C) for non-methyl H and with Uiso(H) = 1.5Ueq(C) for methyl H. Using the program CALC-OH the positions of the water hydrogen atoms for the O1 aqua ligand were found; their isotropic thermal parameters were tied with the parent atom [Uiso(H) = 1.2Ueq(O)].66 For compound 1 the final refinement was conducted using data from both domains. The structure figures were drawn using Diamond.67 Crystal data and final parameters for the structure refinements are summarized in Table S1† and possible hydrogen bonds are given in Table S2.†Ab initio calculations
Ab initio calculations were performed with ORCA 4.1.2 computational package using the experimental geometry of complex under study.68 The relativistic effects were included in the calculations with second-order Douglas–Kroll–Hess (DKH)69 procedure together with the scalar relativistic contracted version of def2-SV(P) basis function for all elements, except Ce atom. For Ce, the SARC2-DKH-QZVP basis function was used.70 The calculations were based on state average complete active space self-consistent field (SA-CASSCF) wave functions. The active space of the CASSCF calculations comprised of one electron in seven metal-based f-orbitals. The state averaged approach was used, in which 7 doublet states were equally weighted. The spin–orbit effects have been included through quasi-degenerate perturbation theory in which an approximation to the Breit–Pauli form of the spin–orbit coupling operator (SOMF) was utilized.71Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Slovak grant agencies (APPV-18-0016 and VEGA 1/0063/17) is gratefully acknowledged, as is support from the Ministerio de Ciencia, Innovación y Universidades (Spain, Grants MAT2015-68200-C2-1-P and PGC2018-093451-B-I00), the European Union Regional Development Fund, FEDER), the Diputación General de Aragón, Project M4, E11_17R and the support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI). Also, this work was supported by the project NFP313010V954 of call OPVaI-VA/DP/2018/1.1.3-07. AV thanks the National Scholarship Programme of the Slovak Republic and grant VVGS-PF-2018-777.References
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141 CrossRef CAS.
- N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694 CrossRef CAS PubMed.
- S. Osa, T. Kido, N. Matsumoto, N. Re, A. Pochaba and J. Mrozinski, J. Am. Chem. Soc., 2004, 126, 420 CrossRef CAS PubMed.
- A. Bencini, C. Benelli, A. Caneschi, R. L. Carlin, A. Dei and D. Gatteschi, J. Am. Chem. Soc., 1985, 107, 8128 CrossRef CAS.
- S. K. Langley, D. P. Wielechowski, V. Vieru, N. F. Chilton, B. Moubaraki, B. F. Abrahams, L. F. Chibotaru and K. S. Murray, Angew. Chem., Int. Ed., 2013, 52, 12014 CrossRef CAS PubMed.
- M. Fondo, J. Corredoira-Vázquez, A. Herrera-Lanzós, A. M. García-Deibe, J. Sanmartín-Matalobos, J. M. Herrera, E. Colacio and C. Nuñez, Dalton Trans., 2017, 46, 17000 RSC.
- S. K. Langley, N. F. Chilton, L. Ungur, B. Moubaraki, L. F. Chibotaru and K. S. Murray, Inorg. Chem., 2012, 51, 11873 CrossRef CAS PubMed.
- A. Upadhyay, S. K. Singh, C. Das, R. Mondol, S. K. Langley, K. S. Murray, G. Rajaraman and M. Shanmugam, Chem. Commun., 2014, 50, 8838 RSC.
- A. Upadhyay, C. Das, S. Vaidya, S. K. Singh, T. Gupta, R. Mondol, S. K. Langley, K. S. Murray, G. Rajaraman and M. Shanmugam, Chem. – Eur. J., 2017, 23, 4903 CrossRef CAS.
- W.-B. Sun, P.-F. Yan, S.-D. Jiang, B.-W. Wang, Y.-Q. Zhang, H.-F. Li, P. Chen, Z.-M. Wang and S. Gao, Chem. Sci., 2016, 7, 684 RSC.
- Z. Jin, J. Bai, T. Wei, F. Li, C. Song, X. Luo and L. Xu, New J. Chem., 2017, 41, 13490 RSC.
- G. A. Kostin, A. O. Borodin, N. V. Kuratieva, A. S. Bogomyakov and A. A. Mikhailov, Inorg. Chim. Acta, 2018, 479, 135 CrossRef CAS.
- S. P. Petrosyants, A. B. Ilyukhin, N. N. Efimov, A. V. Gavrikov and V. M. Novotortsev, Inorg. Chim. Acta, 2018, 482, 813 CrossRef CAS.
- F. Pointillart, O. Cador, B. Le Guennic and L. Ouahab, Coord. Chem. Rev., 2017, 346, 150 CrossRef CAS.
- S. Hino, M. Maeda, K. Yamashita, Y. Kataoka, M. Nakano, T. Yamamura, H. Nojiri, M. Kofu, O. Yamamuro and T. Kajiwara, Dalton Trans., 2013, 42, 2683 RSC.
- S. Hino, M. Maeda, Y. Kataoka, M. Nakano, T. Yamamura and T. Kajiwara, Chem. Lett., 2013, 42, 1276 CrossRef CAS.
- J. J. Le Roy, I. Korobkov, J. E. Kim, E. J. Schelter and M. Murugesu, Dalton Trans., 2014, 43, 2737 RSC.
- A. B. Khélifa, M. S. Belkhiria, G. Huang, S. Freslon, O. Guillou and K. Bernot, Dalton Trans., 2015, 44, 16458 RSC.
- S. K. Singh, T. Gupta, L. Ungur and G. Rajaraman, Chem. – Eur. J., 2015, 21, 13812 CrossRef CAS PubMed.
- D. C. Izuogu, T. Yoshida, H. Zhang, G. Cosquer, K. Katoh, S. Ogata, M. Hasegawa, H. Nojiri, M. Damjanović, W. Wernsdorfer, T. Uruga, T. Ina, B. K. Breedlove and M. Yamashita, Chem. – Eur. J., 2018, 24, 9285 CrossRef CAS PubMed.
- T. Kanetomo, T. Kihara, A. Miyake, A. Matsuo, M. Tokunaga, K. Kindo, H. Nojiri and T. Ishida, Inorg. Chem., 2017, 56, 3310 CrossRef CAS PubMed.
- A. J. Calahorro, I. Oyarzabal, B. Fernández, J. M. Seco, T. Tian, D. Fairen-Jimenez, E. Colacio and A. Rodríguez-Diéguez, Dalton Trans., 2016, 45, 591 RSC.
- V. Tkáč, A. Orendáčová, R. Tarasenko, E. Čižmár, M. Orendáč, K. Tibenská, A. G. Anders, S. Gao, V. Pavlík and A. Feher, J. Phys.: Condens. Matter, 2013, 25, 506001 CrossRef.
- M. J. Martínez-Pérez, S. Cardona-Serra, C. Schlegel, F. Moro, P. J. Alonso, H. Prima-García, J. M. Clemente-Juan, M. Evangelisti, A. Gaita-Ariño, J. Sesé, J. van Slageren, E. Coronado and F. Luis, Phys. Rev. Lett., 2012, 108, 247213 CrossRef.
- V. Chandrasekhar, B. M. Pandian, R. Azhakar, J. J. Vittal and R. Clérac, Inorg. Chem., 2007, 46, 5140 CrossRef CAS PubMed.
- M. Orendáč, L. Sedláková, E. Čižmár, A. Orendáčová, A. Feher, S. A. Zvyagin, J. Wosnitza, W. H. Zhu, Z. M. Wang and S. Gao, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 214410 CrossRef.
- P. I. Girginova, L. C. J. Pereira, J. T. Coutinho, I. C. Santos and M. Almeida, Dalton Trans., 2014, 43, 1897 RSC.
- A. Arauzo, A. Lazarescu, S. Shova, E. Bartolomé, R. Cases, J. Luzón, J. Bartolomé and C. Turta, Dalton Trans., 2014, 43, 12342 RSC.
- R. J. Holmberg, L. T. A. Ho, L. Ungur, I. Korobkov, L. F. Chibotaru and M. Murugesu, Dalton Trans., 2015, 44, 20321 RSC.
- T. D. Pasatoiu, J.-P. Sutter, A. M. Madalan, F. Z. C. Fellah, C. Duhayon and M. Andruh, Inorg. Chem., 2011, 50, 5890 CrossRef CAS PubMed.
- T. D. Pasatoiu, C. Tiseanu, A. M. Madalan, B. Jurca, C. Duhayon, J.-P. Sutter and M. Andruh, Inorg. Chem., 2011, 50, 5879 CrossRef CAS PubMed.
- J.-P. Costes, F. Dahan, C. Duhayon and A. J. Mota, Polyhedron, 2015, 96, 51 CrossRef CAS.
- A. S. Dinca, S. Shova, A. E. Ion, C. Maxim, F. Lloret, M. Julve and M. Andruh, Dalton Trans., 2015, 44, 7148 RSC.
- X. Feng, W. Zhou, Y. Li, H. Ke, J. Tang, R. Clérac, Y. Wang, Z. Su and E. Wang, Inorg. Chem., 2012, 51, 2722 CrossRef CAS PubMed.
- X.-C. Huang, C. Zhou, H.-Y. Wei and X.-Y. Wang, Inorg. Chem., 2013, 52, 7314 CrossRef CAS PubMed.
- L. Jiang, B. Liu, H.-W. Zhao, J.-L. Tian, X. Liu and S.-P. Yan, CrystEngComm, 2017, 19, 1816 RSC.
- H. J. Im and S. W. Lee, Polyhedron, 2015, 101, 48 CrossRef CAS.
- D. Visinescu, A. M. Madalan, M. Andruh, C. Duhayon, J.-P. Sutter, L. Ungur, W. Van den Heuvel and L. F. Chibotaru, Chem. – Eur. J., 2009, 15, 11808 CrossRef CAS PubMed.
- D. Visinescu, I.-R. Jeon, A. M. Madalan, M.-G. Alexandru, B. Jurca, C. Mathonière, R. Clérac and M. Andruh, Dalton Trans., 2012, 41, 13578 RSC.
- M. Andruh, Dalton Trans., 2015, 44, 16633 RSC.
- J.-P. Costes, F. Dahan, A. Dupuis and J.-P. Laurent, Inorg. Chem., 1997, 36, 3429 CrossRef CAS PubMed.
- B. N. Ghose, J. Chem. Eng. Data, 1984, 29, 237 CrossRef CAS.
- Z. Saedi, E. Hoveizi, M. Roushani, S. Massahi, M. Hadian and K. Salehi, J. Mol. Struct., 2019, 1176, 207 CrossRef CAS.
- M. Llunell, D. Casanova, J. Cirera, P. Alemany and S. Alvarez, SHAPE Program, version 2.1, Universitat de Barcelona, 2013.
- G. Xiao, B. Yan, R. Ma, W. J. Jin, X. Q. Lü, L. Q. Ding, C. Zeng, L. L. Chen and F. Bao, Polym. Chem., 2011, 2, 659 RSC.
- W.-J. Jin, L.-Q. Ding, Z. Chu, L.-L. Chen, X.-Q. Lü, X.-Y. Zheng, J.-R. Song and D.-D. Fan, J. Mol. Catal. A: Chem., 2011, 337, 25 CrossRef CAS.
- P. Chen, H. Chen, P. Yan, Y. Wang and G. Li, CrystEngComm, 2011, 13, 6237 RSC.
- X. Yang, D. Lam, C. Chan, J. M. Stanley, R. A. Jones, B. J. Holliday and W.-K. Wong, Dalton Trans., 2011, 40, 9795 RSC.
- Y. Wang, Q. Zhang, P.-F. Yan, G.-F. Hou and H.-F. Li, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m589 CrossRef CAS PubMed.
- L. Xu, Q. Zhang, G. Hou, P. Chen, G. Li, D. M. Pajerowski and C. L. Dennis, Polyhedron, 2013, 52, 91 CrossRef CAS.
- J.-W. Yang, Y.-M. Tian, J. Tao, P. Chen, H.-F. Li, Y.-Q. Zhang, P.-F. Yan and W.-B. Sun, Inorg. Chem., 2018, 57, 8065 CrossRef CAS PubMed.
- S. A. Güngör and M. Kose, J. Mol. Struct., 2017, 1150, 274 CrossRef.
- T. Gao, P.-F. Yan, G.-M. Li, J.-W. Zhang, W.-B. Sun, M. Suda and Y. Einaga, Solid State Sci., 2010, 12, 597 CrossRef CAS.
- S. Hazra, J. Titiš, D. Valigura, R. Boča and S. Mohanta, Dalton Trans., 2016, 45, 7510 RSC.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS PubMed.
- W. Wong, X. Yang, R. A. Jones, J. H. Rivers, V. Lynch, W.-K. Lo, D. Xiao, M. M. Oye and A. L. Holmes, Inorg. Chem., 2006, 45, 4340 CrossRef CAS PubMed.
- A. Vráblová, J. Černák, C. Rajnák, Ľ. Dlháň, M. Tomás, L. R. Falvello and R. Boča, Dalton Trans., 2018, 47, 15523 RSC.
- R. Boča, C. Rajnák, J. Titiš and D. Valigura, Inorg. Chem., 2017, 56, 1478 CrossRef PubMed.
- J. Titiš, C. Rajnák, D. Valigura and R. Boča, Dalton Trans., 2018, 47, 7879 RSC.
- R. Boča, C. Rajnák, J. Moncoľ, J. Titiš and D. Valigura, Inorg. Chem., 2018, 57, 14314 CrossRef PubMed.
- C. Rajnák, J. Titiš, J. Moncoľ, R. Mičová and R. Boča, Inorg. Chem., 2019, 58, 991 CrossRef PubMed.
- F. Yang, Q. Zhou, G. Zeng, G. Li, L. Gao, Z. Shi and S. Feng, Dalton Trans., 2014, 43, 1238 RSC.
- J. Long, D. M. Lyubov, T. V. Mahrova, A. V. Cherkasov, G. K. Fukin, Y. Guari, J. Larionova and A. A. Trifonov, Dalton Trans., 2018, 47, 5153 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- M. Nardelli, J. Appl. Crystallogr., 1999, 32, 563 CrossRef CAS.
- K. Brandenburg and H. Putz, Diamond: Crystal Impact, GbR, Postfach 1251, D-53002 Bonn, Germany, 2008 Search PubMed.
- (a) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 CAS; (b) F. Neese, ORCA – An Ab Initio, Density Functional and Semi-empirical Program Package, Version 4.1.2 Search PubMed.
- M. Reiher and A. Wolf, J. Chem. Phys., 2004, 121, 2037 CrossRef CAS PubMed.
- (a) D. Aravena, F. Neese and D. A. Pantazis, J. Chem. Theory Comput., 2016, 12, 1148 CrossRef CAS PubMed; (b) G. L. Stoychev, A. A. Auer and F. Neese, J. Chem. Theory Comput., 2017, 13, 554 CrossRef CAS PubMed.
- M. Atanasov, D. Aravena, E. Suturina, E. Bill, D. Maganas and F. Neese, Coord. Chem. Rev., 2015, 177, 289 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray structure analysis, synthetic, crystallographic and physical data. CCDC 1916431–1916433. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt02122a |
| This journal is © The Royal Society of Chemistry 2019 |