
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphine-functionalised tris(pyrazolyl)methane ligands and their mono- and heterobimetallic complexes†‡
Hanna E.
Wagner
,
Silvia
Hohnstein
,
Max G.
Schußmann
,
Lukas A.
Steppe
and
Frank
Breher
 *
*
Institute of Inorganic Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstr. 15, 76131 Karlsruhe, Germany. E-mail: breher@kit.edu
First published on 5th June 2019
Abstract
The synthesis, characterisation and reactivity of new phosphine-functionalised tris(pyrazolyl)methane ligands (TpmPR2, 2a–c, with R = Ph, nBu, iPr) are presented. The reaction of 2a–c with [Rh(CO)2Cl]2 furnished N,P-heterochelate carbonyl complexes (3a–c), which were used to quantify the donor abilities of the ligands via IR spectroscopy. The coordination flexibility was demonstrated by treating representative members of this new ligand class with [CpRu(acn)3][PF6] (acn = acetonitrile), [(tht)AuCl] (tht = tetrahydrothiophene) and [Pd(allyl)Cl]2 providing either a N,N,P-heteroscorpionate complex (4) or the P-coordinated complexes (5,6) without any involvement of the pyrazolyl entities. With 2a and [Pd(cod)Cl2], another P,N-heterochelate complex (7) was obtained, which served as a precursor for a heterobimetallic complex containing palladium and copper (8). Detailed NMR spectroscopic and X-ray crystallographic investigations have been performed on all new complexes.
Introduction
Multidentate ligand systems have been of interest for several decades for the generation of transition metal complexes with specific coordination numbers, nuclearities, or structures. In particular, tripodal fac-coordinating N-donor ligands such as tris(pyrazolyl)hydroborates (TpR)1 or tris(pyrazolyl)methanes (RTpm)2 provide intrinsically well-defined coordination geometries. Scorpionate ligands of this type have long been of interest in the area of bioinorganic, organometallic or coordination chemistry.1–3 This ligand family has been extended some years ago to the anionic tris(pyrazolyl)methanide4 and -silanide4i,5 systems,6 which are isoelectronic with the hydroborate ligands, but feature ambidentate characteristics and two possible binding sites, i.e. the anionic carbon and silicon or the pyrazolyl ring nitrogen donors. Another possibility for increasing the number of donor atoms in scorpionate-type ligand scaffolds is to attach further donor-containing entities either in the 3-position of the pyrazolyl entities7 or at the apical boron (TpR) or carbon (RTpm) unit. Ligand systems that are functionalised at the non-coordinating back position of the scorpionate are commonly referred to as “third generation scorpionates”. As recently stated by Reger et al. in a comprehensive review article,8 the tris(pyrazolyl)methane ligands are particularly well suited for these studies as the chemistry is based on reaction chemistry at a C–H bond. Functional groups attached to this apical position can either affect the properties of the ligand, like its solubility, or increase the number of possible coordination modes.9 Selected examples of backbone-functionalised Tpm hybrid ligands are compiled in Chart 1.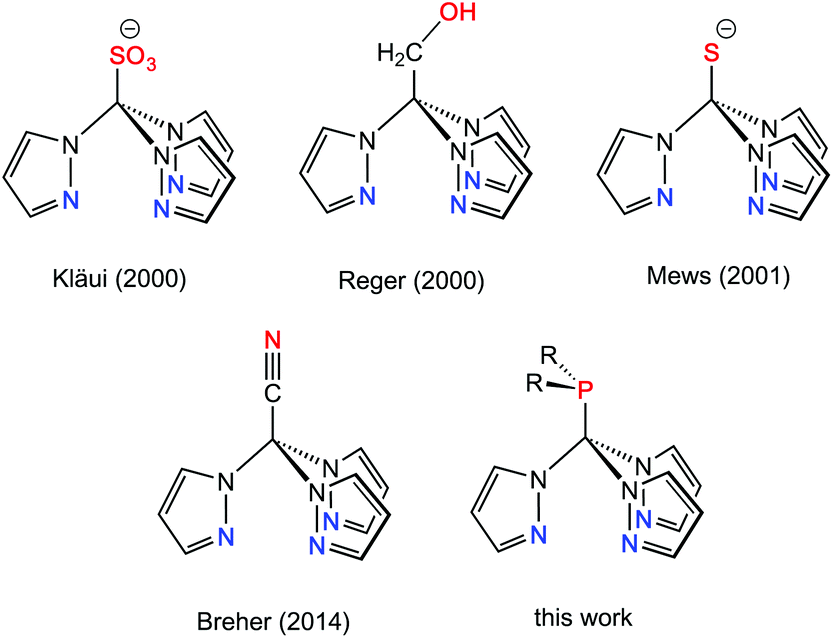 | ||
| Chart 1 Backbone-functionalised Tpm ligands from the literature. | ||
As part of our investigations on ambidentate ligands,10 we pursued the introduction of additional donor functions as additional binding sites in a Tpm-based ligand framework. We report here on neutral, backbone-functionalised Tpm hybrid ligands,11 namely tris(pyrazolyl)methyl phosphines. The latter were found to be versatile hybrid ligands for mono- and heterobimetallic transition metal complexes.
Results and discussion
Synthesis and characterisation of TpmPR2 (R = Ph, nBu, iPr)
Inspired by the pioneering work of Otero12 and Burzlaff13 on heteroscorpioante ligands based on bis(pyrazolyl)methanes2b or Reger9d and Kläui9a on tris(pyrazolyl)methanes, freshly prepared lithium tris(pyrazolyl)methanide (1) was reacted with R2PCl. Moisture and air sensitive colourless crystalline products 2a–c were obtained in analytically pure form in 65–71% isolated yield (Scheme 1).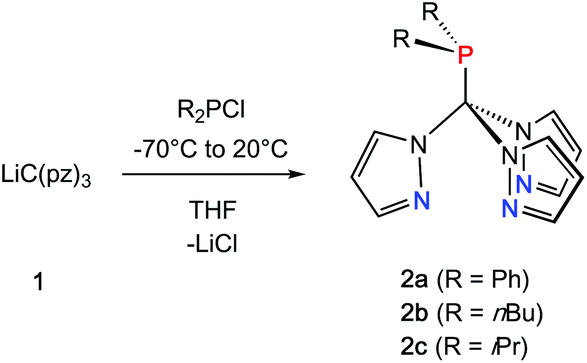 | ||
| Scheme 1 Synthesis of 2a–c (R = Ph, nBu, iPr). | ||
The 1H NMR spectra of the three ligands in benzene-d6 show the expected equivalence of all three pyrazolyl rings within one molecule. While the resonances of the phenyl groups in 2a appear in typical multiplets with a chemical shift of δ = 7.03 ppm (m-, p-position) and 7.98 ppm (o-position), 2b and 2c feature diastereotopic protons for the nBu and iPr substituents, as exemplified for 2c in Fig. 1 (see also ESI‡). Due to 1JP,C couplings, the carbon atoms in apical position of the tris(pyrazolyl)methyl entities in 2a–c appear as doublets with chemical shifts between δ = 95.2 and 96.0 ppm and coupling constants of 44.1–55.8 Hz.
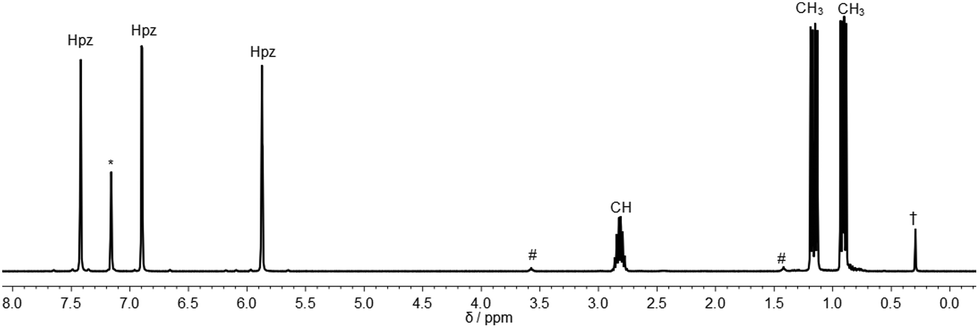 | ||
| Fig. 1 1H NMR spectrum of 2c (C6D6 (*), 300 MHz, # = THF, † = grease). | ||
As expected for alkyl and aryl phosphines, the 31P{1H} NMR appear at δ = 37.0 ppm for 2b, δ = 47.2 ppm for 2a and δ = 59.9 ppm for 2c, respectively.14
In order to unambiguously confirm the formation of the products, X-ray structure analyses were conducted on single crystals of 2a–c (Fig. 2). The compounds crystallise in the triclinic, monoclinic and orthorhombic space groups P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (2b), P21/c (2c) and Pna21 (2a), respectively (ESI‡). Relevant structural data are listed in the caption of Fig. 2. As already anticipated from the NMR data, the apical carbon atom of the C(pz)3 entities are in bound to the PR2 donor sets. In each case, the three pyrazolyl substituents adopt a “propeller”-like arrangement with the additional nitrogen donor atoms (Npz) pointing in direction of the PR2 group. Similar effects have been observed before for backbone-functionalised tris(pyrazolyl)silanides such as R3Si–Si(3,5-Me2pz)3 or R3Sn–Si(3,5-Me2pz)3.5a Although these structural aspects presumably can be attributed to steric repulsion between the individual pyrazolyl heterocycles, we cannot exclude additional P⋯Npz interactions in these compounds. In all three cases, however, The P1–C1 bond lengths of 194.6 pm (2a), 192.2 pm (2b) and 192.7 pm (2c) are slightly longer than regular C–P single bonds (ca. 184 pm). It is worth mentioning that these are the first phosphine-functionalised Tpm derivatives featuring additional Capical-bound PR2 donor sets.
(2b), P21/c (2c) and Pna21 (2a), respectively (ESI‡). Relevant structural data are listed in the caption of Fig. 2. As already anticipated from the NMR data, the apical carbon atom of the C(pz)3 entities are in bound to the PR2 donor sets. In each case, the three pyrazolyl substituents adopt a “propeller”-like arrangement with the additional nitrogen donor atoms (Npz) pointing in direction of the PR2 group. Similar effects have been observed before for backbone-functionalised tris(pyrazolyl)silanides such as R3Si–Si(3,5-Me2pz)3 or R3Sn–Si(3,5-Me2pz)3.5a Although these structural aspects presumably can be attributed to steric repulsion between the individual pyrazolyl heterocycles, we cannot exclude additional P⋯Npz interactions in these compounds. In all three cases, however, The P1–C1 bond lengths of 194.6 pm (2a), 192.2 pm (2b) and 192.7 pm (2c) are slightly longer than regular C–P single bonds (ca. 184 pm). It is worth mentioning that these are the first phosphine-functionalised Tpm derivatives featuring additional Capical-bound PR2 donor sets.
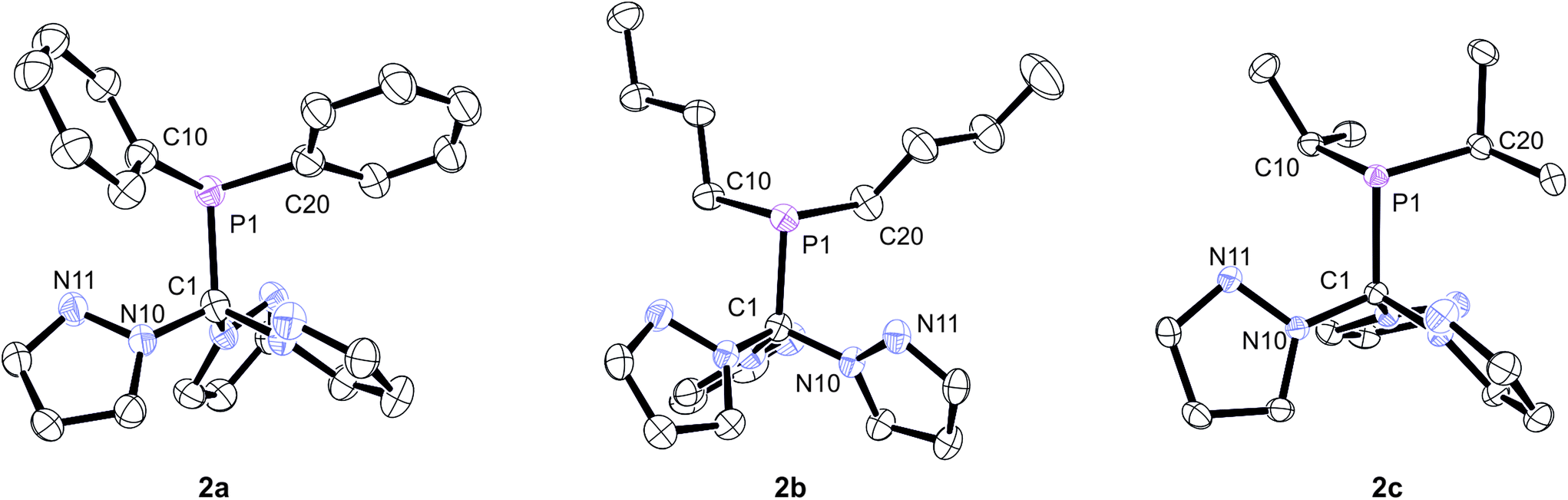 | ||
| Fig. 2 Molecular structures of the ligands 2a–c. The thermal ellipsoids are at 30% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths [pm] and angles [°]: (2a) C1–P1 194.6(5), C1–N10 145.4(6), C1–N20 146.4(6), C1–N30 144.2(6) P1–C10 182.8(5), P1–C20 183.1(6), P1–C1–N10 115.0(3), P1–C1–N20 107.0(3), P1–C1–N30 111.3(3). (2b) C1–P1 192.2(13), C1–N10 145.3(16), C1–N20 146.1(14), C1–N30 145.1(16) P1–C10 184.8(16), P1–C20 184.2(16), P1–C1–N10 108.5(8), P1–C1–N20 114.6(9), P1–C1–N30 111.3(9). (2c) C1–P1 192.7(14), C1–N10 145.5(18), C1–N20 145.7(17), C1–N30 146.5(16) P1–C10 186.4(15), P1–C20 187.2(16), P1–C1–N10 109.4(9), P1–C1–N20 109.1(9), P1–C1–N30 115.2(9). | ||
Rhodium carbonyl complexes of 2a–c
In order to shed some light on the coordination behaviour of this novel class of ligands, we investigated various d-block metal complexes. The mononuclear rhodium carbonyl complexes 3a–c were synthesised by treating the precursor [Rh(CO)2Cl]2 with 2a–c (Scheme 2). Analytically pure crystalline solids were obtained in very good yields of 80–87%. The microanalytical and IR spectral data provided evidence that only one carbonyl ligand is present in the complexes and that the overall composition should be [(2a–c)Rh(CO)Cl] with the hybrid ligands either acting as bi- or tridentate chelate. In the 1H NMR spectra, only three equivalent pyrazolyl rings are visible, which is most likely caused by a rapid exchange in solution. This phenomenon has already been observed for Tpms complexes (Chart 1), where the presence of a C3 symmetric κ3N-coordinated species alongside the heterochelate complexes could be verified in solution.15 In our case, however, the 31P{1H} spectra contradict the presence of a κ3N-coordinated species. Besides the coupling to the rhodium nucleus, the signals of the phosphorous atoms in the 31P{1H} NMR spectra show strong low-field shifts to δ = 112–144 ppm as compared to the free ligands. This would be in accordance with the assumption of having just a dynamic interchange between the three pz rings in solution and no presence of a κ3N-coordinated species.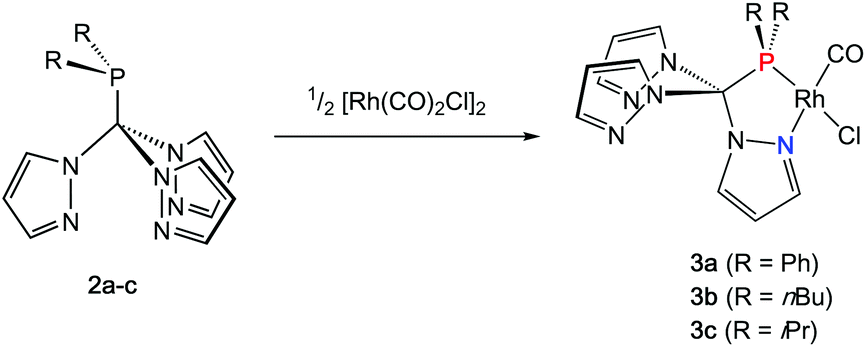 | ||
| Scheme 2 Synthesis of 3a–c. | ||
We were able to obtain crystals suitable for X-ray diffraction of all three complexes confirming that the TpmPR2 ligands act as a κ2N,P-hybrid ligand with two uncoordinated pyrazolyl moieties (Fig. 3, P21/c for 3a and 3b; P21 for 3c, ESI‡). The Rh1–P1 bond lengths range between 219–222 pm, whereas the Rh1–N11 bond lengths show values between 206 and 208 pm. The P1–C1 bond lengths are in the same range as observed for the uncoordinated ligands.
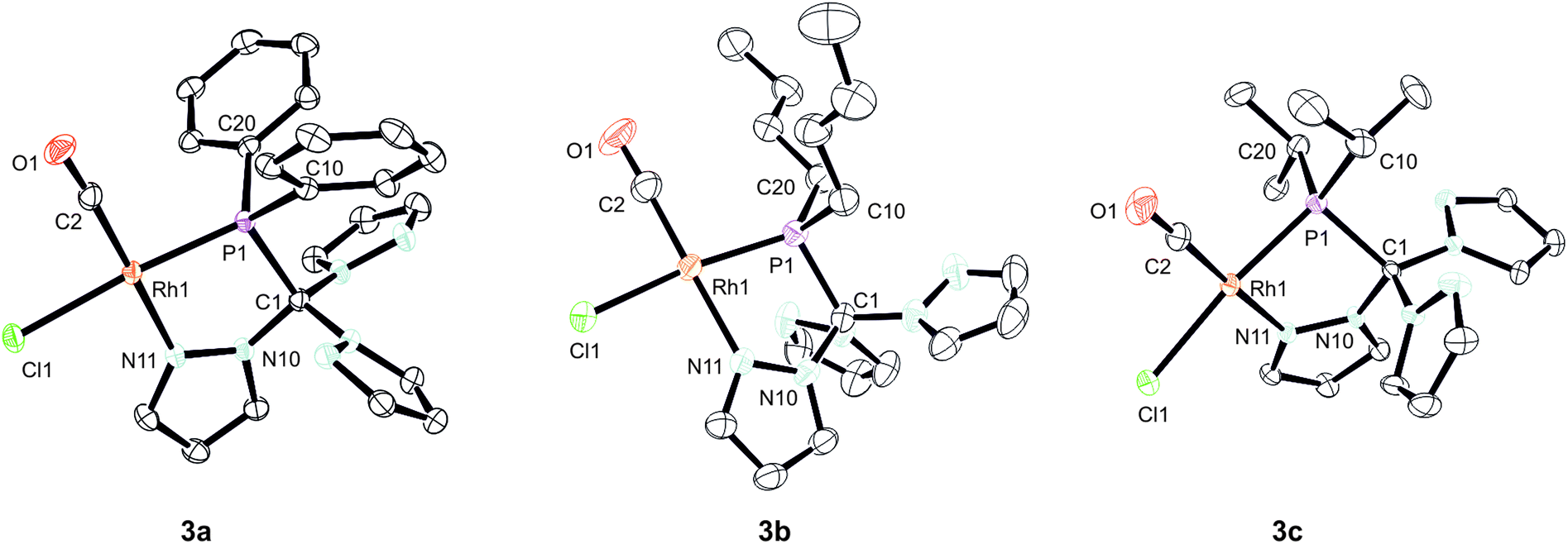 | ||
| Fig. 3 Molecular structures of complexes 3a–c. The thermal ellipsoids are at 30% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths [pm] and angles [°]: (3a) Rh1–P1 221.16(7), Rh1–Cl1 237.7(7), Rh1–N11 206.34(17), Rh1–C2 182.0(2), P1–C1 195.56(19), P1–Rh1–Cl1 174.195(19), N11–Rh1–P1 83.78(5), N11–Rh1–Cl1 90.44(6), C2–Rh1–P1 97.02(7), C2–Rh1–Cl1 88.76(7), C2–Rh1–N11 178.90(8). (3b) Rh1–P1 219.14(13), Rh1–Cl1 235.50(14), Rh1–N11 208.6(3), Rh1–C2 180.1(5), P1–C1 192.4(4), P1–Rh1–Cl1 168.96(5), N11–Rh1–P1 81.67(10), N11–Rh1–Cl1 89.81(10), C2–Rh1–P1 97.12(17), C2–Rh1–Cl1 91.59(16), C2–Rh1–N11 178.03(17). (3c) Rh1–P1 221.84(11), Rh1–Cl1 235.88(13), Rh1–N11 207.6(3), Rh1–C2 180.4(5), P1–C1 195.0(4), P1–Rh1–Cl1 169.85(4), N11–Rh1–P1 81.48(12), N11–Rh1–Cl1 88.39(12), C2–Rh1–P1 96.81(19), C2–Rh1–Cl1 93.30(19), C2–Rh1–N11 177.8(3). | ||
The IR spectra in solid state were recorded for compounds 3a–c and compared to the TEP values of the corresponding phosphines in CH2Cl2 solution (Table 1).16 As expected, the same tendencies in the donor capacities observed for the “classical” aryl- or alkyl phosphines can also be observed for the tris(pyrazolyl)methyl phosphines (Ph < nBu < iPr).
| Ligand | ν(CO) [cm−1] | Ligand | TEP [cm−1] |
|---|---|---|---|
| 3a | 1994 | PPh3 | 2069 |
| 3b | 1981 | PnBu3 | 2060 |
| 3c | 1978 | PiPr3 | 2059 |
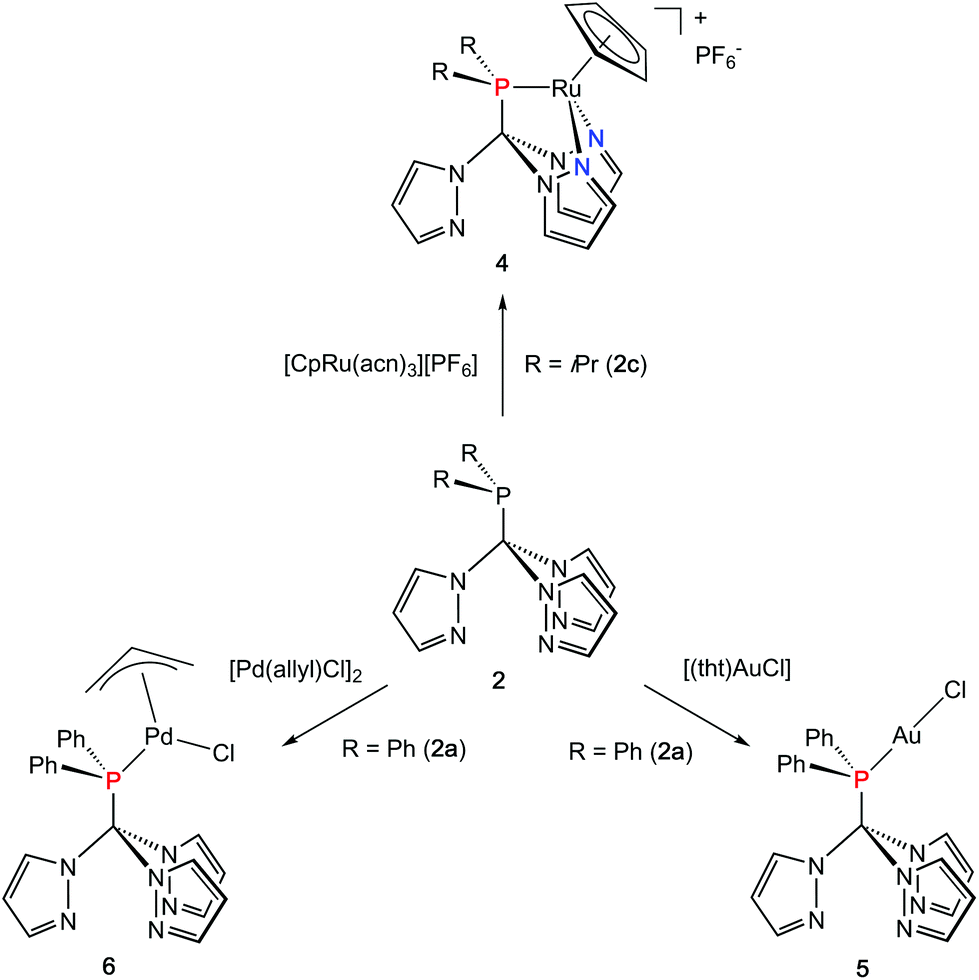
Coordination flexibility
As already mentioned before, different coordination modes are known from complexes containing Tpms as ligand. In Tpms, the oxygen atoms are very weak donor atoms and as a result, dynamic equilibria are observed in solution. We also became interested to studying the coordination flexibility of our new hybrid ligands 2a–c and, thus, investigated the coordination chemistry of 2a or 2c as representatives for the new ligand family in more detail.Since [CpRu(acn)3][PF6] (acn = acetonitrile) is a valuable precursor for transferring a {RuCp} entity to tripodal ligands such as Tpm or Tpmd,4n,17 we inspected its reactivity towards 2c (Scheme 3). Orange crystals of 4 were isolated in 91% yield. 1H NMR spectroscopic investigations revealed that the hybrid ligand is coordinated in a κ3N,N′,P fashion. The detection of one uncoordinated pyrazolyl ring indicated no interchange of the pz entities in solution in this case. The down field shift of the phosphorous signal in the 31P{1H} NMR spectrum of 4 is even stronger than for 3a–c (δ = 170.4 ppm). The molecular structure was established confirming the κ3N,N,P-heteroscorpionate structure (Fig. 4, space group Pbcn, ESI‡). The coordination sphere of the ruthenium atom is complemented by one η5-C5H5 ligand. The average Ru1–N bond length in 4 was found to amount to 209.1 pm, which is only slightly shorter than in comparable κ3N-Tpm (211.1 pm)17 and κ3N-Tpmd (210.6 pm)4n complexes published by our group. The Ru1–P1 bond length of 229.3 pm is in good agreement with those observed for other complexes from the literature.18
 | ||
| Scheme 3 Synthesis of 4–6. | ||
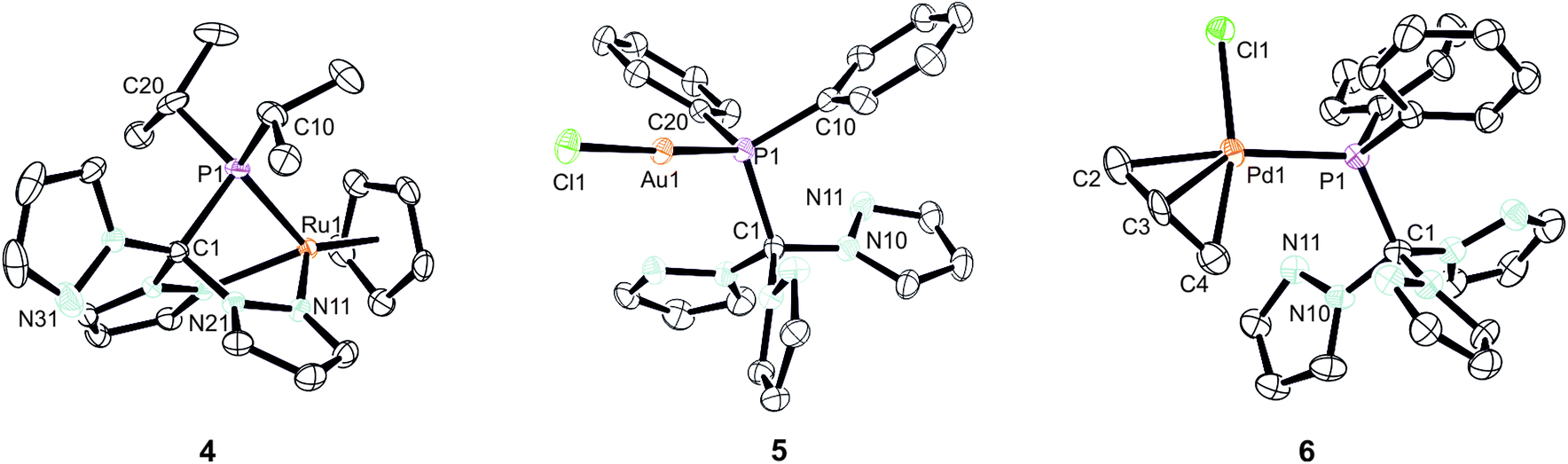 | ||
| Fig. 4 Molecular structures of 4–6. The thermal ellipsoids are at 30% probability level. Hydrogen atoms and the anion (4) have been omitted for clarity. Selected bond lengths [pm] and angles [°]: 4: Ru1–P1 229.27(6), Ru1–N11 209.26(18), Ru1–N21 209.00(17), Ru1–Cpcent 181.9 P1–C1 193.9(2), P1–Ru1–Cpcent 135.27, N11–Ru1–Cpcent 145.60, N21–Ru1–Cpcent 130.00, N11–Ru1–P1 78.93(5), N21–Ru1–P1 82.74(7); 5: Au1–P1 = 223.38(15), Au1–Cl1 = 227.66(15), C1–P1 = 194.2(6), P1–Au1–Cl1 176.46(5), C1–P1–Au1 = 111.54(18), Au1–P1–C200 = 114.93(19); 6: Pd1–P1 232.0(2), Pd1–Cl1 235.4(2), P1–C1 196.0(9), P1–Pd1–Cl1 93.97(8), Pd1–P1–C1 116.5(3). (Cpcent = centroid of the Cp ligand). | ||
Another metal precursor which was reacted with 2a was [Au(tht)Cl] (tht = tetrahydrothiophene) and complex 5 was obtained in 82% isolated yield as colourless crystalline solid. Considering that Au(I) is a rather soft metal ion and prefers linear coordination, we expected the P donor in 2a to solely act as 2-electron donor. Indeed, the NMR spectroscopic investigations reveal just one set of signals concerning the pyrazolyl protons. The coordination of the gold atom to the P donor causes a low field shift of the phosphorous atom in the 31P{1H} NMR spectra from δ = 47.2 ppm in 2a to δ = 78.7 ppm in 5. The molecular structure depicted in Fig. 4 (space group P21/n) illustrates the almost linear coordination of the gold atom (P1–Au1–Cl1 = 175.5°). Other structural parameters remain almost unchanged compared to the uncoordinated ligand.
As a representative of group 10 metals, 2a was reacted with [Pd(allyl)Cl]2 to provide a hardly soluble, pale yellow complex 6, which was isolated in rather low yields of 48% in an analytical pure form. Like in 5, the phosphorous atom is acting as donor ligand whereas the pyrazolyl rings stay uncoordinated. The downfield shift of the 31P{1H} chemical shift of δ = 89.4 ppm is even more distinct than for 5.
Crystals suitable for X-ray crystallography could be obtained from a THF/hexane mixture (Fig. 4, space group P212121, ESI‡). The Pd1–P1 bond length is 232.3 pm and in accordance to similar Pd–P bonds known from literature.18
Heterobimetallic complex of 2a
In an effort to prepare heterobimetallic complexes of the new hybrid ligands, various attempts have been made by treating 4–6 with various other metal sources, all of which were not successful. However, sequential treatment of 2a with [Pd(cod)Cl2] and CuCl2 provided the suitable entry point to the first heterobimetallic complex 8 (Scheme 4).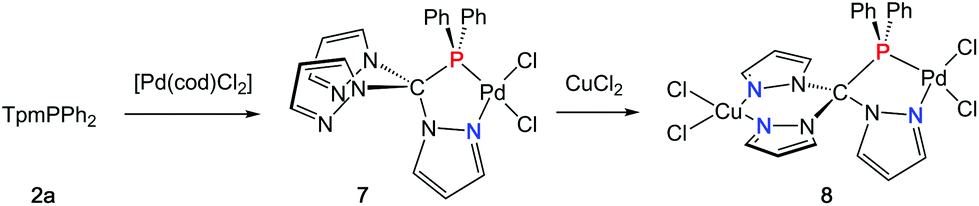 | ||
| Scheme 4 Synthesis of 7 and 8. | ||
The initial product (7) of this reaction is a yellow solid and was obtained in very good yields (87%). As evidenced by 1H and 31P NMR (δ = 90.7 ppm) spectroscopic investigations, the palladium atom is coordinated in a κ2P,N-coordination mode leaving two pyrazolyl moieties uncoordinated. The palladium atom in 7 is coordinated in a square planar coordination mode (Fig. 5, space group P21/c, ESI‡). Due to the different trans-influence of phosphorous and nitrogen donors, the Pd1–Cl1 bond length is slightly longer (237.0 pm) than Pd1–Cl2 (228.4 pm). The Pd1–P1 bond (218.3 pm) is remarkably shorter than the Pd1–P1 bond in complex 6 (232.3 pm).
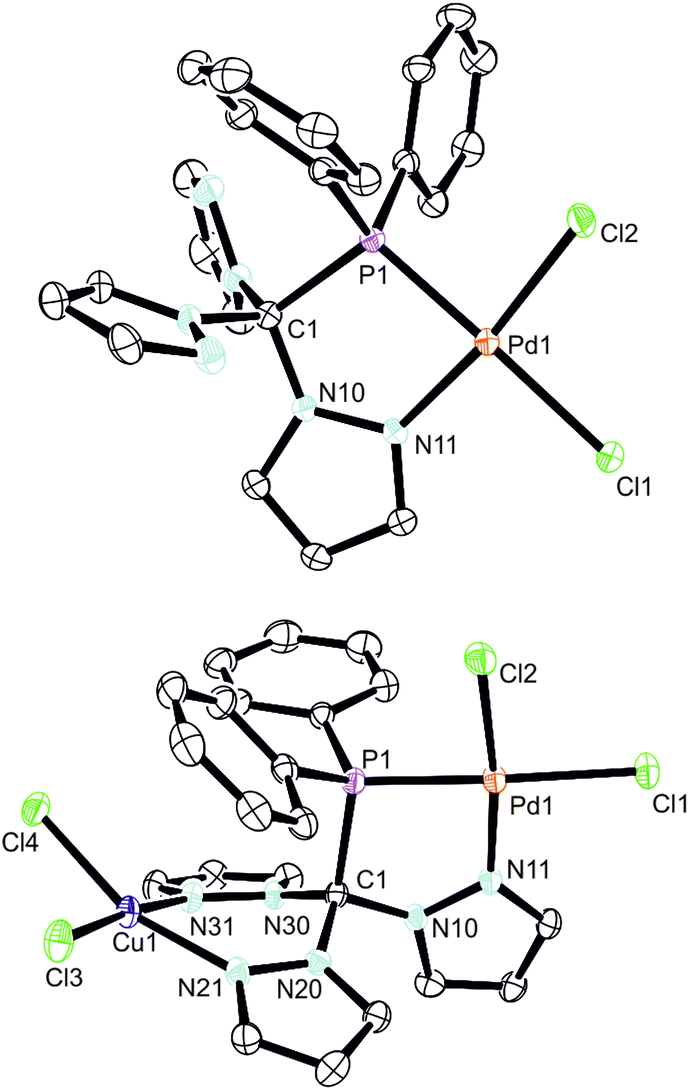 | ||
| Fig. 5 Molecular structures of 7 and 8. The thermal ellipsoids are at 30% probability level. Hydrogen atoms and solvent molecules (8: acetonitrile) have been omitted for clarity. Selected bond lengths [pm] and angles [*]: 7: Pd1–P1 = 218.3(6), Pd1–Cl1 = 237.0(6), Pd1–Cl2 = 228.42(6), Pd1–N11 = 201.41(16), P1–C1 = 196.2(2), P1–Pd1–Cl1 175.411(19), P1–Pd1–Cl2 87.87(2), Cl2–Pd1–Cl1 = 94.91(2), N11–Pd1–P1 = 84.8, N11–Pd1–Cl1 92.31(5), N11–Pd1–172.22(5). 8: Pd1–P1 219.56(17), Pd1–Cl1 236.21(17), Pd1–Cl2 227.67(18), Pd1–N11 200.5(5), Ci1–Cl3 221.46(19), Cu1–N21 202.6(5), Cu1–N31 201.4(5), P1–C1 195.2(6), Pd1⋯Cu1 603.61(14), P1–Pd1–Cl1 172.30(6), P1–Pd1–Cl2 87.97(7), Cl2–Pd1–Cl1 94.47(7), N11–Pd1–P1 84.84(15), N11–Pd1–Cl1 92.67(15), N11–Pd1–Cl2 172.81(15), Cl3–Cu1–Cl4 94.63(8), N21–Cu1–Cl3 91.32(15), N21–Cu1–Cl4 161.49(15), N31–Cu1–N21 84.8(2), N31–Cu1–Cl4 90.07(15), N31–Cu1–Cl3 174.86(15). | ||
By reacting 7 with CuCl2, the first heterobimetallic complex 8 was successfully isolated and characterised (Fig. 5, space group P21/c, ESI‡). The coordination sphere of the palladium atom is not affected by the coordination of Cu(II) since all bond lengths and angles stay almost the same. The average Cu–N and Cu–Cl bond lengths were found to amount to 223.6 pm and 202.1 pm, respectively. The square planar coordination mode at the copper atom is rather distorted as judged from the N21–Cu1–Cl4 angle of only 161.5(2) degrees. This distortion is also manifested in the boat-shaped arrangement of the six-membered chelate ring, which, however, has already been described for related copper complexes in the literature.19
Conclusions
In summary, we have introduced a novel class of phosphine-functionalised Tpm ligands (2a–c), which proved to be a very flexible class of hybrid ligands due to many different coordination modes observed for several transition metal complexes (3–7). Furthermore, we were able to synthesise the first heterobimetallic complex of a tris(pyrazolyl)methyl phosphine containing the metals palladium and copper. Further investigations regarding the cooperativity along the two metals within this complex and the synthesis of even more multimetallic complexes are part of ongoing research in our group.Experimental section
General methods and materials
All manipulations were carried out with standard Schlenk line and dry-box techniques in a dry argon atmosphere. Methylene chloride and acetonitrile were freshly distilled in an argon atmosphere from calcium hydride. Toluene, diethyl ether and tetrahydrofuran were dried using sodium/benzophenone ketyl. CD2Cl2 was vacuum transferred from calcium hydride while C6D6 and thf-d8 were vacuum transferred from sodium/benzophenone ketyl into thoroughly dried glassware equipped with Young Teflon valves.Tpm9b and nBu2PCl20 were synthesized in accordance to the literature. All other reagents were purchased from Aldrich or Roth and used without further purification.
1H, 13C, spectra were recorded with Bruker AV 300 and 400 spectrometers in dried deuterated solvents. The chemical shifts are expressed in parts per millions and 1H and 13C signals are given relative to TMS. Coupling constants J are given in hertz as positive values regardless of their real individual signs. The multiplicity of the signals is indicated as s, d, q, sept or m for singlets, doublets, quartets, septets or multiplets, respectively. The assignments were confirmed as necessary with the use of 2D NMR correlation experiments. Mass spectrometry measurements were performed on an Advion expressionL CMS mass spectrometer under atomic pressure chemical ionization (APCI) or under electrospray ionization (ESI). IR spectra were measured with a Bruker Alpha spectrometer using the attenuated total reflection (ATR) technique on powdered samples, and the data are quoted in wavenumbers (cm−1). The intensity of the absorption band is indicated as vw (very weak), w (weak), m (medium), s (strong), vs. (very strong) and br (broad). Melting points were measured with a Thermo Fischer melting point apparatus and are not corrected. Elemental analyses were carried out in the institutional technical laboratories of the Karlsruhe Institute of Technology (KIT).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2).
:2).
X-Ray crystallographic studies
A suitable crystal was covered in mineral oil (Aldrich) and mounted on a glass fiber or a mylar loop. The crystal was transferred directly to the cold stream of a STOE IPDS 2 diffractometer.All structures were solved by using the program SHELXS/T21 using Olex2.22 The remaining non-hydrogen atoms were located from successive difference Fourier map calculations. The refinements were carried out by using full-matrix least-squares techniques on F2 by using the program Olex2. In each case, the locations of the largest peaks in the final difference Fourier map calculations, as well as the magnitude of the residual electron densities, were of no chemical significance. Details on the data refinement are deposited in the ESI.‡
621 reflections measured, 11783 unique (Rint = 0.0935) which were used in all calculations. The final wR2 was 0.1631 (all data) and R1 was 0.0580 (I > 2(I)).
(no. 2), a = 8.1293(16) Å, b = 8.6311(17) Å, c = 15.582(3) Å, α = 77.76(3), β = 86.99(3)°, γ = 66.40(3), V = 978.5(4) Å3, T = 200 K, Z = 2, μ(MoKα) = 0.154, 18987 reflections measured, 5275 unique (Rint = 0.0458) which were used in all calculations. The final wR2 was 0.1196 (all data) and R1 was 0.0408 (I > 2(I)).
515 reflections measured, 4632 unique (Rint = 0.0911) which were used in all calculations. The final wR2 was 0.1014 (all data) and R1 was 0.0393 (I > 2(I)).
146 reflections measured, 4393 unique (Rint = 0.0375) which were used in all calculations. The final wR2 was 0.0648 (all data) and R1 was 0.0240 (I > 2(I)).
865 reflections measured, 4548 unique (Rint = 0.1361) which were used in all calculations. The final wR2 was 0.1632 (all data) and R1 was 0.0565 (I > 2(I)).
391 reflections measured, 5438 unique (Rint = 0.0528) which were used in all calculations. The final wR2 was 0.0687 (all data) and R1 was 0.0290 (I > 2(I)).
924 reflections measured, 6718 unique (Rint = 0.0415) which were used in all calculations. The final wR2 was 0.0821 (all data) and R1 was 0.0302 (I > 2(I)).
511 reflections measured, 4760 unique (Rint = 0.0548) which were used in all calculations. The final wR2 was 0.1086 (all data) and R1 was 0.0327 (I > 2(I)).
955 reflections measured, 6154 unique (Rint = 0.0289) which were used in all calculations. The final wR2 was 0.0817 (all data) and R1 was 0.0296 (I > 2(I)).
940 reflections measured, 6055 unique (Rint = 0.0960) which were used in all calculations. The final wR2 was 0.1639 (all data) and R1 was 0.0626 (I > 2(I)).
Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as a supplementary publication no. CCDC 1916045–1916055.‡
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the DFG-funded transregional collaborative research centre SFB/TRR 88 “Cooperative effects in homo- and heterometallic complexes (3MET)” (project B4). Support by the Federal Ministry of Education and Research (BMBF Project No. 02NUK020B) is also gratefully acknowledged.Notes and references
- (a) S. Trofimenko, Chem. Rev., 1993, 93, 943–980 CrossRef CAS; (b) S. Trofimenko, Scorpionates: The Coordination Chemistry of Polypyrazolylborate Ligands, Imperial College Press, London, 1999 CrossRef.
- (a) H. R. Bigmore, S. C. Lawrence, P. Mountford and C. S. Tredget, Dalton Trans., 2005, 635–651 RSC; (b) C. Pettinari and R. Pettinari, Coord. Chem. Rev., 2005, 249, 525–543 CAS.
- (a) G. Parkin, in Adv. Inorg. Chem, ed. A. G. Sykes, Academic Press, 1995, vol. 42, pp. 291–393 Search PubMed; (b) D. L. Reger, Coord. Chem. Rev., 1996, 147, 571–595 CrossRef CAS; (c) M. Etienne, Coord. Chem. Rev., 1996, 156, 201–236 CrossRef CAS; (d) C. Janiak, Coord. Chem. Rev., 1997, 163, 107–216 CrossRef CAS; (e) D. L. Reger, Comments Inorg. Chem., 1999, 21, 1–28 CrossRef CAS; (f) N. Kitajima and W. B. Tolman, in Progress in Inorganic Chemistry, ed. K. D. Karlin, John Wiley & Sons, Inc., 1995, vol. 43, p. 419 Search PubMed; (g) H. V. R. Dias and C. J. Lovely, Chem. Rev., 2008, 108, 3223–3238 CrossRef CAS; (h) Adv. Organomet. Chem, ed. R. West, A. F. Hill and M. J. Fink, Academic Press, 2008, vol. 56, pp. 1–347 Search PubMed; (i) S. Trofimenko, J. Chem. Educ., 2005, 82, 1715 CrossRef CAS; (j) A. Munetaka and H. Shiro, Bull. Chem. Soc. Jpn., 2002, 75, 1657–1679 CrossRef; (k) G. Parkin, Chem. Rev., 2004, 104, 699–768 CrossRef CAS PubMed.
- (a) P. K. Byers, N. Carr and F. G. A. Stone, J. Chem. Soc., Dalton Trans., 1990, 3701–3708 RSC; (b) P. K. Byers and F. G. A. Stone, J. Chem. Soc., Dalton Trans., 1991, 93–99 RSC; (c) S. C. Lawrence, M. E. G. Skinner, J. C. Green and P. Mountford, Chem. Commun., 2001, 705–706 RSC; (d) F. Breher, J. Grunenberg, S. C. Lawrence, P. Mountford and H. Rüegger, Angew. Chem., Int. Ed., 2004, 43, 2521–2524 CrossRef CAS; (e) H. R. Bigmore, S. R. Dubberley, M. Kranenburg, S. C. Lawrence, A. J. Sealey, J. D. Selby, M. A. Zuideveld, A. R. Cowley and P. Mountford, Chem. Commun., 2006, 436–438 RSC; (f) I. Krummenacher, H. Rüegger and F. Breher, Dalton Trans., 2006, 1073–1081 RSC; (g) H. R. Bigmore, J. Meyer, I. Krummenacher, H. Rüegger, E. Clot, P. Mountford and F. Breher, Chem. – Eur. J., 2008, 14, 5918–5934 CrossRef CAS PubMed; (h) I. Kuzu, I. Krummenacher, I. J. Hewitt, Y. Lan, V. Mereacre, A. K. Powell, P. Höfer, J. Harmer and F. Breher, Chem. – Eur. J., 2009, 15, 4350–4365 CrossRef CAS; (i) I. Fernández, P. Oña-Burgos, F. Armbruster, I. Krummenacher and F. Breher, Chem. Commun., 2009, 2586–2588 RSC; (j) M. G. Cushion, J. Meyer, A. Heath, A. D. Schwarz, I. Fernández, F. Breher and P. Mountford, Organometallics, 2010, 29, 1174–1190 CrossRef CAS; (k) D. Kratzert, D. Leusser, D. Stern, J. Meyer, F. Breher and D. Stalke, Chem. Commun., 2011, 47, 2931–2933 RSC; (l) J. Meyer, I. Kuzu, S. González-Gallardo and F. Breher, Z. Anorg. Allg. Chem., 2013, 639, 301–307 CrossRef CAS; (m) C. Müller, H. Görls, S. Krieck and M. Westerhausen, Eur. J. Inorg. Chem., 2013, 5679–5682 CrossRef; (n) S. González-Gallardo, I. Kuzu, P. Oña-Burgos, T. Wolfer, C. Wang, K. W. Klinkhammer, W. Klopper, S. Bräse and F. Breher, Organometallics, 2014, 33, 941–951 CrossRef; (o) C. Müller, A. Koch, H. Görls, S. Krieck and M. Westerhausen, Inorg. Chem., 2015, 54, 635–645 CrossRef PubMed; (p) C. Müller, S. Krieck, H. Görls and M. Westerhausen, Inorg. Chem., 2015, 54, 2100–2102 CrossRef PubMed; (q) R. Lalrempuia, A. Stasch and C. Jones, Chem. – Asian J., 2015, 10, 447–454 CrossRef CAS; (r) S. Krieck, A. Koch, K. Hinze, C. Müller, J. Lange, H. Görls and M. Westerhausen, Eur. J. Inorg. Chem., 2016, 2332–2348 CrossRef CAS; (s) I. Kuzu, A. Baldes, F. Weigend and F. Breher, Polyhedron, 2017, 125, 74–79 CrossRef CAS.
- (a) F. Armbruster, I. Fernandez and F. Breher, Dalton Trans., 2009, 5612–5626 RSC; (b) F. Armbruster, T. Augenstein, P. Oña-Burgos and F. Breher, Chem. – Eur. J., 2013, 19, 17899–17906 CrossRef CAS PubMed; (c) S. Styra, S. González-Gallardo, F. Armbruster, P. Oña-Burgos, E. Moos, M. Vonderach, P. Weis, O. Hampe, A. Grün, Y. Schmitt, M. Gerhards, F. Menges, M. Gaffga, G. Niedner-Schatteburg and F. Breher, Chem. – Eur. J., 2013, 19, 8436–8446 CrossRef CAS PubMed; (d) M. Simon and F. Breher, Dalton Trans., 2017, 46, 7976–7997 RSC; (e) C. Krempner, M. H. Chisholm and J. Gallucci, Angew. Chem., Int. Ed., 2008, 47, 410–413 CrossRef CAS PubMed; (f) H. Li, L. J. Hope-Weeks and C. Krempner, Chem. Commun., 2011, 47, 4117–4119 RSC; (g) B. Carlson, A. J. A. Aquino, L. J. Hope-Weeks, B. Whittlesey, B. McNerney, W. L. Hase and C. Krempner, Chem. Commun., 2011, 47, 11089–11091 RSC; (h) B. McNerney, B. Whittlesey and C. Krempner, Eur. J. Inorg. Chem., 2011, 1699–1702 CrossRef CAS; (i) H. Li, F. Hung-Low and C. Krempner, Organometallics, 2012, 31, 7117–7124 CrossRef CAS; (j) H. Li, A. J. A. Aquino, D. B. Cordes, W. L. Hase and C. Krempner, Chem. Sci., 2017, 8, 1316–1328 RSC.
- (a) A. Steiner and D. Stalke, J. Chem. Soc., Chem. Commun., 1993, 1702–1704 RSC; (b) A. Steiner and D. Stalke, Inorg. Chem., 1995, 34, 4846–4853 CrossRef CAS; (c) I. Krummenacher, C. Oschwald, H. Rüegger and F. Breher, Z. Anorg. Allg. Chem., 2007, 633, 2354–2361 CrossRef CAS; (d) I. Krummenacher, I. Fernández, H. Rüegger, F. Weigend and F. Breher, Dalton Trans., 2009, 5335–5347 RSC; (e) M.-Á. Muñoz-Hernández and V. Montiel-Palma, Inorg. Chim. Acta, 2009, 362, 4328–4339 CrossRef; (f) H. R. Simmonds and D. S. Wright, Chem. Commun., 2012, 48, 8617–8624 RSC; (g) K. Zeckert, Organometallics, 2013, 32, 1387–1393 CrossRef CAS; (h) K. Zeckert, S. Zahn and B. Kirchner, Chem. Commun., 2010, 46, 2638–2640 RSC; (i) M. A. Halcrow, Dalton Trans., 2009, 2059–2073 RSC; (j) J. Klingele, S. Dechert and F. Meyer, Coord. Chem. Rev., 2009, 253, 2698–2741 CrossRef CAS.
- M. D. Ward, J. A. McCleverty and J. C. Jeffery, Coord. Chem. Rev., 2001, 222, 251–272 CrossRef CAS.
- R. F. Semeniuc and D. L. Reger, Eur. J. Inorg. Chem., 2016, 2253–2271 CrossRef CAS.
- (a) W. Kläui, M. Berghahn, G. Rheinwald and H. Lang, Angew. Chem., Int. Ed., 2000, 39, 2464–2466 CrossRef; (b) D. L. Reger, T. C. Grattan, K. J. Brown, C. A. Little, J. J. S. Lamba, A. L. Rheingold and R. D. Sommer, J. Organomet. Chem., 2000, 607, 120–128 CrossRef CAS; (c) R. Wanke, P. Smoleński, M. F. C. Guedes da Silva, L. M. D. R. S. Martins and A. J. L. Pombeiro, Inorg. Chem., 2008, 47, 10158–10168 CrossRef CAS PubMed; (d) D. L. Reger and T. C. Grattan, Synthesis, 2003, 0350–0356 CrossRef CAS; (e) M. Müller, E. Lork and R. Mews, Angew. Chem., Int. Ed., 2001, 40, 1247–1249 CrossRef; (f) I. Trapp, S. González-Gallardo, S. Hohnstein, D. Garnier, P. Oña-Burgos and F. Breher, Dalton Trans., 2014, 43, 4313–4319 RSC; (g) T. Augenstein, F. Dorner, K. Reiter, H. E. Wagner, D. Garnier, W. Klopper and F. Breher, Chem. – Eur. J., 2016, 22, 7935–7943 CrossRef CAS PubMed.
- (a) I. Kuzu, I. Krummenacher, J. Meyer, F. Armbruster and F. Breher, Dalton Trans., 2008, 5836–5865 RSC; (b) F.-G. Fontaine, J. Boudreau and M.-H. Thibault, Eur. J. Inorg. Chem., 2008, 5439–5454 CrossRef CAS.
- (a) S. Maggini, Coord. Chem. Rev., 2009, 253, 1793–1832 CrossRef CAS; (b) W.-H. Zhang, S. W. Chien and T. S. A. Hor, Coord. Chem. Rev., 2011, 255, 1991–2024 CrossRef CAS.
- (a) A. Otero, J. Fernández-Baeza, J. Tejeda, A. Antiñolo, F. Carrillo-Hermosilla, E. Díez-Barra, A. Lara-Sánchez, M. Fernández-López, M. Lanfranchi and M. A. Pellinghelli, J. Chem. Soc., Dalton Trans., 1999, 3537–3539 RSC; (b) A. Otero, J. Fernández-Baeza, A. Antiñolo, F. Carrillo-Hermosilla, J. Tejeda, A. Lara-Sánchez, L. Sánchez-Barba, M. Fernández-López, A. M. Rodríguez and I. López-Solera, Inorg. Chem., 2002, 41, 5193–5202 CrossRef CAS PubMed; (c) A. Otero, J. Fernández-Baeza, A. Antiñolo, J. Tejeda, A. Lara-Sánchez, L. Sánchez-Barba, E. Martínez-Caballero, A. M. Rodríguez and I. López-Solera, Inorg. Chem., 2005, 44, 5336–5344 CrossRef CAS PubMed; (d) A. Otero, J. Fernández-Baeza, A. Antiñolo, J. Tejeda, A. Lara-Sánchez, L. Sánchez-Barba, M. Fernández-López and I. López-Solera, Inorg. Chem., 2004, 43, 1350–1358 CrossRef CAS PubMed; (e) A. Otero, J. Fernández-Baeza, A. Antiñolo, J. Tejeda, A. Lara-Sánchez, L. Sánchez-Barba and A. M. Rodríguez, Eur. J. Inorg. Chem., 2004, 260–266 CrossRef CAS; (f) A. Otero, J. Fernández-Baeza, A. Antiñolo, J. Tejeda and A. Lara-Sánchez, Dalton Trans., 2004, 1499–1510 RSC; (g) A. Otero, J. Fernández-Baeza, A. Lara-Sánchez, J. Tejeda and L. F. Sánchez-Barba, Eur. J. Inorg. Chem., 2008, 5309–5326 CrossRef CAS.
- (a) A. Beck, B. Weibert and N. Burzlaff, Eur. J. Inorg. Chem., 2001, 521–527 CrossRef CAS; (b) S. Tampier, R. Müller, A. Thorn, E. Hübner and N. Burzlaff, Inorg. Chem., 2008, 47, 9624–9641 CrossRef CAS PubMed; (c) H. Kopf, B. Holzberger, C. Pietraszuk, E. Hübner and N. Burzlaff, Organometallics, 2008, 27, 5894–5905 CrossRef CAS; (d) H. Kopf, C. Pietraszuk, E. Hübner and N. Burzlaff, Organometallics, 2006, 25, 2533–2546 CrossRef CAS; (e) R. Müller, E. Hübner and N. Burzlaff, Eur. J. Inorg. Chem., 2004, 2151–2159 CrossRef; (f) N. V. Fischer, F. W. Heinemann and N. Burzlaff, Eur. J. Inorg. Chem., 2009, 3960–3965 CrossRef CAS; (g) B. S. Hammes, M. T. Kieber-Emmons, J. A. Letizia, Z. Shirin, C. J. Carrano, L. N. Zakharov and A. L. Rheingold, Inorg. Chim. Acta, 2003, 346, 227–238 CrossRef CAS.
- S. Hohnstein, Vergleichende Studien zum Koordinationsverhalten tripodaler Liganden gegenüber f-Elementen sowie Synthese und Reaktivitätsstudien von neuartigen P,N-Hybridliganden, Dissertation, Logos Verlag Berlin GmbH, Berlin, 2016 Search PubMed.
- (a) W. Kläui, M. Berghahn, W. Frank, G. J. Reiß, T. Schönherr, G. Rheinwald and H. Lang, Eur. J. Inorg. Chem., 2003, 2003, 2059–2070 CrossRef; (b) C. Pettinari, F. Marchetti, G. Lupidi, L. Quassinti, M. Bramucci, D. Petrelli, L. A. Vitali, M. F. C. Guedes da Silva, L. M. D. R. S. Martins, P. Smoleński and A. J. L. Pombeiro, Inorg. Chem., 2011, 50, 11173–11183 CrossRef CAS PubMed; (c) F. Marchetti, C. Pettinari, R. Pettinari, A. Cerquetella, L. M. D. R. S. Martins, M. F. C. Guedes da Silva, T. F. S. Silva and A. J. L. Pombeiro, Organometallics, 2011, 30, 6180–6188 CrossRef CAS.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- I. Kuzu, D. Nied and F. Breher, Eur. J. Inorg. Chem., 2009, 872–879 CrossRef CAS.
- D. A. Fletcher, R. F. McMeeking and D. Parkin, J. Chem. Inf. Comput. Sci., 1996, 36, 746–749 CrossRef CAS.
- A. S. Potapov and A. I. Khlebnikov, Polyhedron, 2006, 25, 2683–2690 CrossRef CAS.
- P. Kübler and J. Sundermeyer, Dalton Trans., 2014, 43, 3750–3766 RSC.
- G. M. Sheldrick, Acta Cryst., 2015, A71, 3 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Annie Powell on the occasion of her 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: NMR data, X-ray crystal structure data. CCDC 1916045–1916055. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt02057h |
| This journal is © The Royal Society of Chemistry 2019 |