
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVoltammetric characterisation of diferrocenylborinic acid in organic solution and in aqueous media when immobilised into a titanate nanosheet film†
Martin
Konhefr
 *abc,
Adam C.
Sedgwick
a,
Tony D.
James
a,
Karel
Lacina
c,
Petr
Skládal
bc,
Budi Riza
Putra
ad,
Christian
Harito
e,
Dmitry V.
Bavykin
e,
Frank C.
Walsh
e,
Paul R.
Raithby
a,
Gabriele
Kociok-Köhn
af and
Frank
Marken
*a
*abc,
Adam C.
Sedgwick
a,
Tony D.
James
a,
Karel
Lacina
c,
Petr
Skládal
bc,
Budi Riza
Putra
ad,
Christian
Harito
e,
Dmitry V.
Bavykin
e,
Frank C.
Walsh
e,
Paul R.
Raithby
a,
Gabriele
Kociok-Köhn
af and
Frank
Marken
*a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: 379848@mail.muni.cz; F.Marken@bath.ac.uk
bDepartment of Biochemistry, Faculty of Science, Masaryk University, Kotlářská 2, 611 37 Brno, Czech Republic
cCentral European Institute of Technology, Masaryk University, Kamenice 5, 625 00 Brno, Czech Republic
dDepartment of Chemistry, Faculty of Mathematics and Natural Sciences, Bogor Agricultural University, Bogor, West Java, Indonesia
eEnergy Technology Research Group, Faculty of Engineering and the Environment, University of Southampton, SO17 1BJ, Southampton, UK
fMaterial and Chemical Characterisation Facility (MC2), University of Bath, Bath BA2 7AY, UK
First published on 24th May 2019
Abstract
Diferrocenylborinic acid (Fc2BOH, 1) has been synthesized in good yield via an improved synthetic path. Characterisation by nuclear magnetic resonance (NMR), mass spectrometry (HRMS), infrared spectroscopy (FTIR), X-ray crystallography, and by electrochemical methods reveal two one-electron oxidation processes for the two electronically coupled ferrocenyl moieties. The oxidation of 1 dissolved in organic media is contrasted to the oxidation of 1 in aqueous environments (by incorporation of 1 into a lamellar film of 2D titanate nanosheets on a glassy carbon electrode). Data from cyclic voltammetry and from square wave voltammetry suggest that the bridging boron can bind to nucleophiles (hydroxide, fluoride) upon oxidation of the ferrocenyl groups. A multi-pathway ECE reaction scheme is proposed. Potential applications in sensing are discussed.
1. Introduction
Ferroceneboronic acid as an electrochemically active boronic acid has been used previously in electroanalytical sensing protocols.1,2 Boronic acid moieties [R1B(OH)2] can bind nucleophiles and in particular vicinal diols or other polyols3,4 and importantly to glucose.5 From this, a range of sensing applications have been developed,6–8 including applications with electrochemical detection.9–13 In contrast, the borinic acid moiety, a chemical relative of boronic acid, consists of two alkyl substituents and only one hydroxyl linked to the boron atom [R1(R2)B–OH]. It has been suggested that borinic acids should have similar binding properties to nucleophilic analytes compared to those observed for boronic acids,14 but little attention has been paid to borinic acids. Diferrocenylborinic acid is an interesting system to study as it has the additional benefit of providing a two-electron redox system, where not only the formal potential for electron transfer, but also the potential gap between the first and second electron transfer, are accessible parameters for analytical purposes.Organometallic complexes containing boron-based pendants have been proven to offer fertile ground for the exploration of fundamental structure and reactivity issues.15 Trisferrocenylborane has been prepared and characterised.16 Also the zwitter-ionic ferricenyl(III)tris(ferrocenyl(II))borate has been obtained and characterised by cyclic voltammetry.17 Diferrocenylborinic acid (Fc2BOH, 1, Fig. 1), the simplest electroactive borinic acid, is an interesting material fundamentally and in conjunction with electrochemical sensing protocols. In contrast to trisferrocenylborane, it is hydrolytically stable and potentially useful as an analytical reagent in aqueous environments. Complex 1 was first prepared and reported by Post et al. in 1970,18 but its electrochemical properties have not been investigated in further detail. In this report, we describe the electrochemical properties of 1 in organic solution and in aqueous environments.
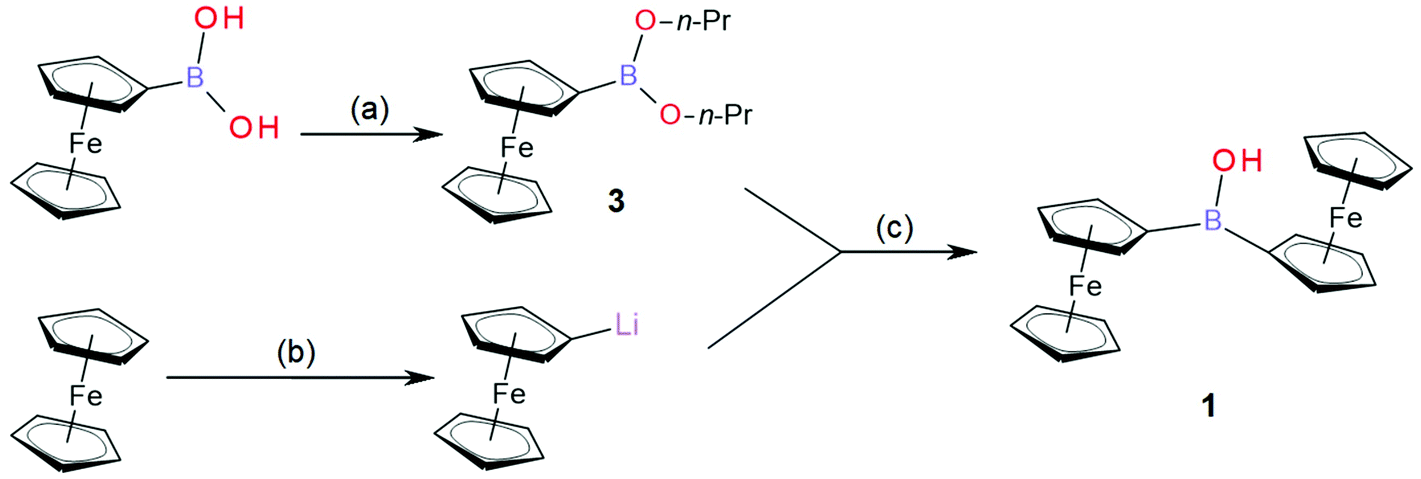 | ||
| Fig. 1 Synthesis of Fc2BOH (1); (a) in toluene, excess of n-propanol, molecular sieve 4 Å, 1 h azeotropic dehydration; (b) in THF under inert atmosphere, 1 mol. eq. of t-BuLi, 0 °C, 15 min; (c) in THF under inert atmosphere, 0 °C, 15 min, H2O quenching, further extraction and purification according to experimental details in section 2.3. | ||
In order to investigate water-based redox properties, water-insoluble redox systems can often be immobilised into films at electrode surfaces and immersed into an aqueous solution. In this way, metal complexes such as diferrocenylborinic acid 1 can be investigated for potential applications in electroanalysis in aqueous electrolyte environments. A versatile way of immobilising hydrophobic metal complexes at the surface of glassy carbon electrodes can be based on a 2D titanate nanosheet host,19 which is able to bind the redox active molecule from organic solvents. Nanomaterials such as titanate nanosheets have been widely investigated due to their catalytic, semiconducting, photo- and electrochemical properties and for their potential use in advanced, next-generation nanoelectronics.20,21 2D titanate nanosheets can be obtained as a colloidal solution by delaminating a layered titanate precursor.22 In previous studies, it has been shown that such titanate nanosheets can be beneficial as an additive to polymer composite materials to improve their mechanical properties.23 A report on potential applications of multi-layer titanate nanosheet film deposits in electroanalysis has also appeared.24 One of the first studies with modified electrodes based on titanate nanosheets with an embedded electrochemical probe (ferroceneboronic acid) was reported recently.25 It was shown that the ferroceneboronic redox signal in a titanate nanosheet film immersed in aqueous Na2SO4 was responsive for example to the anions in the electrolyte and to fructose binding. We therefore decided to explore the reactivity of diferrocenylborinic acid 1 under similar conditions in aqueous environments and exposed to nucleophiles such as fluoride.
In this study, diferrocenylborinic acid 1 is synthesized via an improved synthetic pathway (see Fig. 1), spectrally and structurally characterised, then utilized in a film modified electrode based on titanate nanosheets on a glassy carbon surface. The type of redox response and the potential for sensing applications are evaluated.
2. Experimental details
2.1. Chemical reagents
All chemicals were of reagent grade and were used without further purification unless otherwise noted. Ferrocene was purchased from Fluka, t-BuLi, tetrabutylammonium hexafluorophosphate (TBAH), sodium sulphate, and sodium fluoride from Sigma Aldrich, ferroceneboronic acid from TCI Europe, and all solvents were purchased from VWR International or Sigma Aldrich. Titanate nanosheet material was synthesized as a colloidal solution (2.56 g dm−3) as described previously by Sasaki26 and by Harito et al.23 Solutions were prepared under ambient condition in volumetric flasks with ultrapure water from ELGA Purelab Classic system (resistivity of 18.2 MΩ cm at 22 °C).2.2. Instrumentation
1H and 13C NMR spectra were recorded on a 300 MHz Bruker Avance spectrometer. 11B NMR spectra were recorded on a 500 MHz Agilent ProPulse spectrometer. All measurements were performed in chloroform-d1 (CDCl3) at 303 K. The chemical shifts are reported in ppm on the δ scale using signal of deuterated solvent residue as a reference. HRMS spectra were recorded on Agilent 6224 Accurate-Mass TOF LC-MS system with ESI + (MMI) ionisation. FTIR spectra were collected on Bruker Alpha FT-IR spectrometer (Platinum ATR). X-ray intensity data for 1 were collected at 150(2) K on Rigaku Xcalibur, EosS2 using monochromated graphite Mo-Kα radiation (λ = 0.71073 Å). A symmetry-related (multi-scan) absorption correction and an analytical numeric absorption correction had been applied. The structure was solved with SHELXT and refined by a full-matrix least-squares procedure based on F2 (SHELXL-2018/327). All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed in calculated positions and refined using a riding model except for the OH hydrogen atoms which had been located in the difference Fourier map and were refined with bond length restraints. Additional programmes used for analysing data and their graphical manipulation included: SHELXle28 and ORTEP 3 for Windows.29 The cif file of the structure has been deposited in the Cambridge Structural Database, deposition number CCDC 1894130.†Spectroelectrochemical data were recorded on an Autolab PGSTAT30 (Metrohm-Autolab, Netherlands) combined with Avantes spectrometer AvaSpec-ULS2048XL-EVO (Avantes, Netherlands) and optically transparent thin-layer spectro-electrochemical (OTTLE) cell with Ag wire as reference electrode and Pt grid as both working and counter electrode, respectively, and 1 mm optical path length. Electrochemical experiments were performed using three-electrode system controlled with a microAutolab III system (Metrohm-Autolab, Netherlands) in voltammetry mode. The scan rate was maintained at 100 mV s−1 or as otherwise noted. The platinum wire and KCl-saturated calomel electrode (SCE) were used as counter and reference electrodes, respectively. The working electrode was a 3.0 mm-diameter glassy carbon electrode (GCE) alternatively modified with TiO2 nanosheet film (or in some cases a 1 mm diameter Pt disk electrode). Working electrodes were polished with alumina slurry (50 nm diameter) and washed with ultrapure water before modification with titanate nanosheet films (see below for details).
2.3. Syntheses
Ferroceneboronic acid (0.38 g, 1.66 mmol) was dissolved in toluene (10 mL), a few drops of H2O and 1-propanol (1 mL) were added and the mixture was stirred with a 4 Å molecular sieve for 30 minutes at room temperature. All volatile solvents were then evaporated under continuous vacuum for 5 minutes to give a red oily intermediate containing mostly ferroceneboronic acid 1-propanol ester. The material was then dissolved in dry THF (3 mL) under inert atmosphere (flushed with N2) and stirred on an ice bath for further synthesis. Next, ferrocene (0.8 g, 4.3 mmol) was weighed into the Schlenk flask, flushed with N2 (inert atmosphere) and dissolved in dry THF (5 mL), maintaining 0 °C on an ice bath. t-BuLi (2.7 mL, 4.2 mmol) was added dropwise to the stirred solution of ferrocene. The formation of FcLi was accompanied by darkening from orange to red colour of solution. After 15 minutes of stirring, the batch of ferroceneboronic acid 1-propanol ester was transferred to the FcLi solution and stirred together for a further 15 minutes. The colour of the solution turned to dark red. Finally, the reaction was quenched with H2O (5 mL), brine (3 mL) was added, and the mixture was warmed to room temperature (22 °C). The crude product was extracted with Et2O (3 × 15 mL), organic layers were combined, dried using CaCl2 and evaporated to dryness under reduced pressure to give a dark red solid. This solid was slurried with hexane and placed on silica gel column (2 cm × 20 cm). The unreacted and quenched ferrocene was eluted with hexane or petroleum spirit at 40–60 °C (0.4 g, 2.15 mmol, recovered for further synthesis). The red band containing mainly Fc3B (2) was eluted with benzene (0.16 g, 0.28 mmol, 17% in respect to boron equivalent in reactants; note that Fc3B is easily hydrolysed to give Fc2BOH (1) in CDCl3 after 50 h or directly on the silica gel columns to give Fc2BOH). The desired Fc2BOH (1) was eluted as last orange-brown band with benzene and as main band slowly eluted with toluene as a brownish/ochre solid (0.25 g, 0.63 mmol, 38% in respect to boron equivalent in reactants).Note – Fc2BOH (small amounts e.g. 10 mg of Fc3B/Fc2BOH mixture) can be further purified by a Pasteur pipette flash LC (silica gel/hexane). Impurities stay either on the column or they are eluted with hexane (FcH). The pure Fc2BOH is then slowly eluted with benzene or toluene. The rest of less pure Fc2BOH is then eluted with diethylether.
2.4. Procedure for electrode preparation
An aliquot of 5 μL titanate nanosheet colloidal solution (2.56 g dm−3 in water) was deposited onto the glassy carbon electrode (GCE) surface by drop casting. The solution was dried at ambient temperature forming a transparent thin film of titanate nanosheets. Diferrocenylborinic acid (1) was immobilised into the titanate film by incubation of the modified electrode in 1 mM solution of Fc2BOH in chloroform overnight in a fridge/dark or at room temperature with ambient light conditions with the same results. The modified electrode was then potential-cycled in aqueous electrolyte solution (0.1 M Na2SO4 or 0.1 M NaF) until the stabilisation of the voltammetric response (typically from one to twelve potential cycles).3. Results and discussion
3.1. Synthesis and characterisation of diferrocenylborinic acid
The synthesis of diferrocenylborinic acid (1) was based on previously published procedures developed by Post et al.18 and by Rebiere30 (see Fig. 1). Ferroceneboronic acid or its anhydride or ester were used as the boron donor. Using only crude commercial ferroceneboronic acid (as purchased containing FcB(OH)2 and its anhydride boroxine (FcBO)3) gave the desired product with 15–20% isolated yield of 1. When employing the more reactive ferroceneboronic acid 1-propanol ester (3), the yield is improved to 38% (in respect to boron equivalent in reactants) with further product (1) being recovered from the hydrolysis of side product trisferrocenylborane (2).Fc2BOH (1) was structurally characterised using 1H, 11B and 13C NMR (ESI Fig. S1–S3, see ESI†), by HRMS (Fig. S9†), by FTIR (Fig. S11 and S12†), by spectroelectrochemistry (Fig. S15 and S16†), and by single crystal X-ray crystallography (Fig. S17†). In 11B NMR a characteristic peak is observed at 46.0 ppm consistent with sp2-boron, which compares to the 11B chemical shift for ferroceneboronic acid of 31.0 ppm.2 For further details and data, see ESI.† Fc3B (2) was also characterised (see ESI Fig. S4–S6, S10 and S13†). Ferroceneboronic acid 1-propanol ester (3) was identified only as an intermediate, as a part of the reaction mixture with (FcBO)3 (see NMR spectra ESI Fig. S7 and S8†).
Orange-red prism-shaped crystals of 1 for X-ray crystallography (needles growing from glass wall into solution) were slowly grown from saturated solutions in CDCl3 or toluene (50 mg cm−3, 1 week or to dryness). Single crystal X-ray crystallography analysis revealed that, in the monoclinic solid state, 1 exists as disordered Fc2BOH structure in the ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in space group P21/c (Fig. 2).
:1 in space group P21/c (Fig. 2).
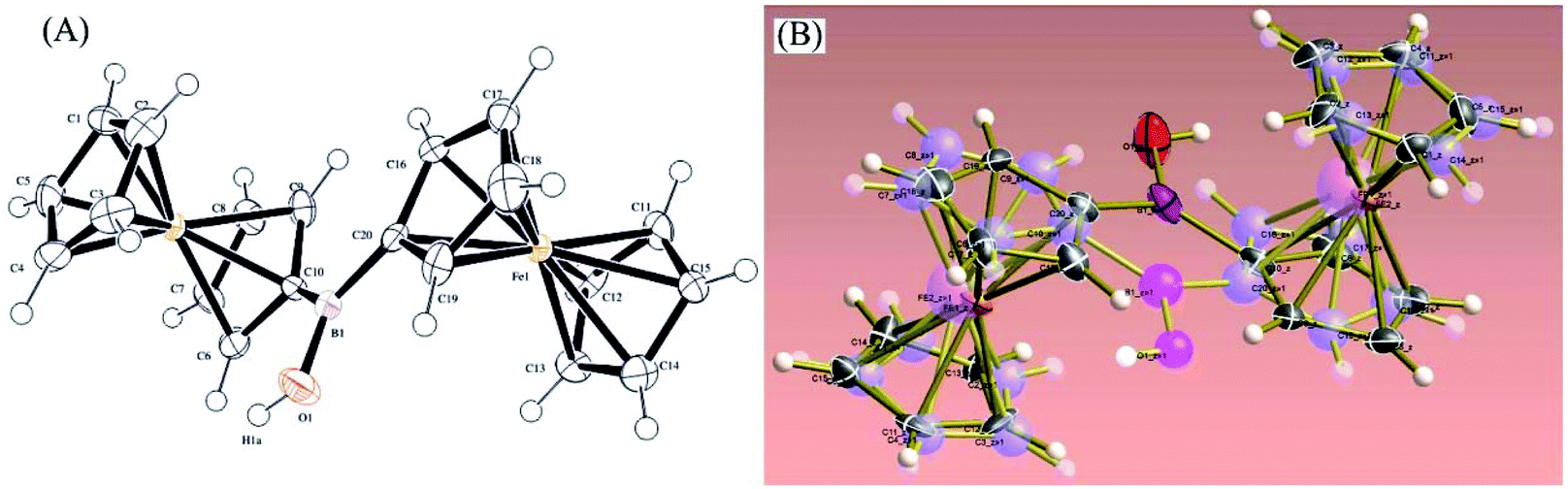 | ||
| Fig. 2 Molecular structure based on X-ray diffraction of 1 (A) and overlaid with both disordered structures of 1 (B). | ||
Both ferrocenyl moieties in Fc2BOH occupy places similarly to those observed in structurally related bridging borinic acid derivatives.31 The substituted cyclopentadienide rings are fairly co-planar. The B–Cipso bond distances are 1.563(4) Å and 1.571(4) Å. The B–O bond length is 1.375(4) Å, which is in line with bond length data for other related borinic acids as shown in Table 1. The B–O bond length is essentially identical to that for ferroceneboronic acid.32
| Compound | B–C (R1 and R2) bond lengths (Å) | B–O bond length (Å) |
|---|---|---|
| a This study. | ||
| Fc2BOHa | 1.563(4)/1.571(4) | 1.375(4) |
| 1,2-FcBOH32 | 1.539/1.550 | 1.379 |
| 1,2-FcBOMe32 | 1.556/1.556 | 1.361 |
| Diazulenylborinic acid33 | 1.568/1.571 | 1.348 |
| Bis(pentafluorophenyl)borinic acid34 | 1.615/1.614 | 1.528 |
| Dimesitylborinic acid35 | 1.574/1.588 | 1.365 |
| Mesityl(2,4,6-trimethoxyphenyl)borinic acid36 | 1.570/1.580 | 1.368 |
| 10H-Dibenzo[b,e][1,4]oxaborinin-10-ol (computed values)37 | 1.547/1.547 | 1.354 |
| 5-Methyldibenzo[b,e][1,4]azaborinin-10(5H)-ol (computed values)37 | 1.543/1.541 | 1.360 |
3.2. Voltammetric characterisation of diferrocenylborinic acid in solution
The electrochemical behaviour of 1 in solution is dependent on the type of electrolyte and the experimental conditions. For exploratory electrochemical characterisation, two non-aqueous solvents were used – acetonitrile and dichloromethane (with 50 mM TBAH as background electrolyte). Fig. 3A shows cyclic voltammetry data for 1 dissolved in acetonitrile. A separated pair of redox processes (see Ep,ox1 and Ep,ox2) consistent with the stepwise oxidation of the two ferrocenyl substituents can be seen with a gap in potentials ΔEp,ox = 160 mV. This observation is consistent with the oxidation of the first ferrocenyl unit causing a pull in electron density in the direction of the oxidised ferrocenyl moiety and a resulting shift in the oxidation of the second ferrocenyl moiety to higher potentials. Data from spectroelectrochemistry in Fig. 3D confirm an isosbestic point associated with predominantly two stable products, e.g. the Fe(II)Fe(II) starting material and the Fe(III)Fe(III) product. The shift of the main absorbance peak upon oxidation from 220 nm to 260 nm is consistent with published observations for similar materials.38 In dichloromethane (Fig. 3B), the same step-by-step oxidation of both ferrocenyl moieties is also observed (see scheme in Fig. 3F). A scan rate of approximately 0.4 V s−1 or higher is required to avoid the formation of new products (see new reduction peaks at 0.10 and −0.14 V vs. SCE, denoted by *, and ESI Fig. S14† for further detail). During lower scan rates in dichloromethane more complex changes in behaviour of 1 occur, which are tentatively assigned here to the formation of products with higher electron density on the boron, e.g. deprotonation and nucleophile adduct formation possibly leading to boroxines. The additional complexity is confirmed by the absence of an isosbestic point in data from spectroelectrochemical measurements (Fig. 3E).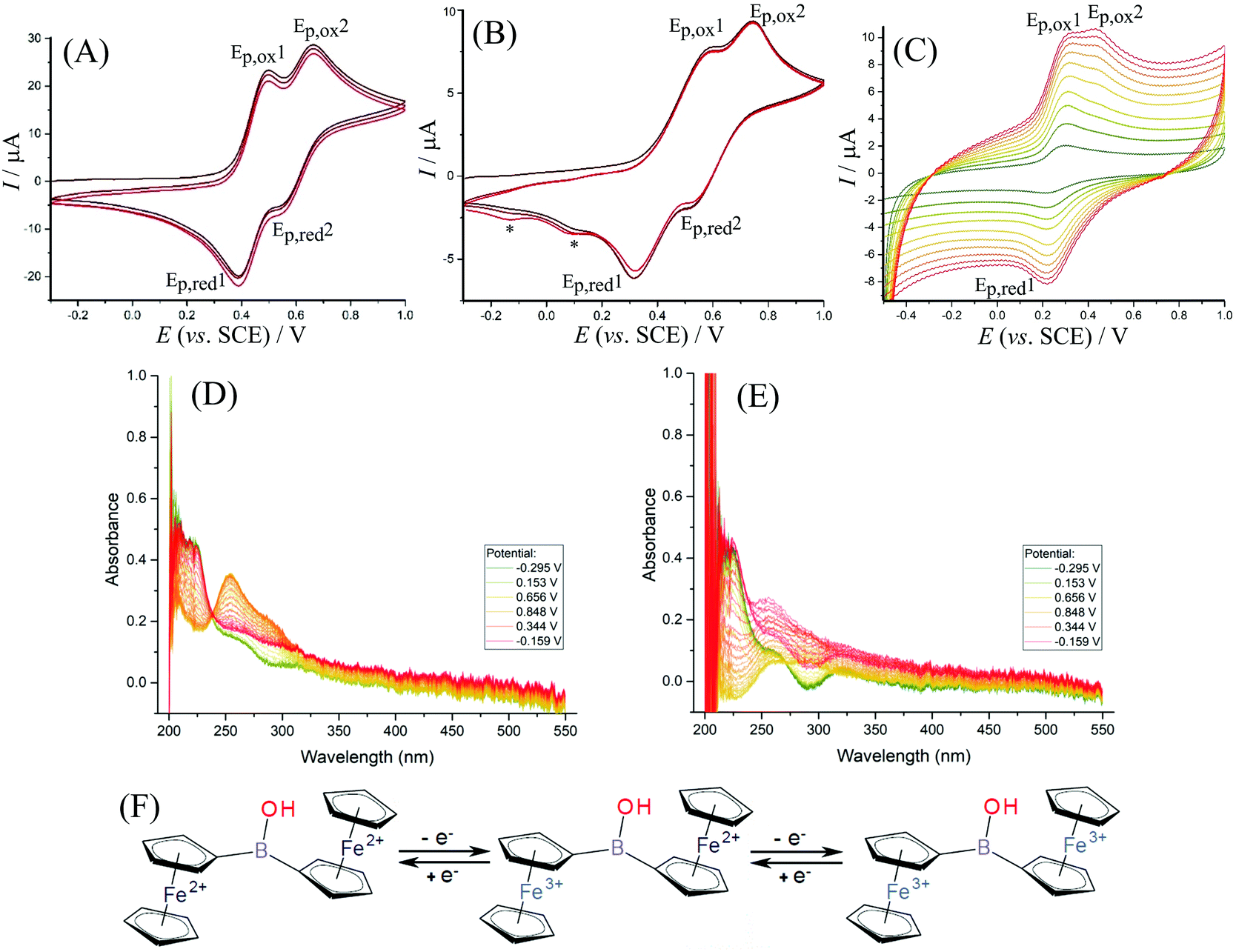 | ||
| Fig. 3 (A) Cyclic voltammograms (scan rate 100 mV s−1; 3 mm diameter glassy carbon electrode) for the oxidation of approx. 1 mM Fc2BOH in acetonitrile with 50 mM TBAH. (B) Cyclic voltammograms (scan rate 500 mV s−1; 1 mm diameter platinum electrode) for the oxidation of approx. 1 mM Fc2BOH in dichloromethane with 50 mM TBAH. (C) Cyclic voltammograms (scan rate 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9 and 1.0 V s−1; 3 mm diameter glassy carbon electrode) for the oxidation of Fc2BOH immobilised into a film of titanate nanosheets and immersed in 0.1 M Na2SO4. (D) Spectroelectrochemistry as a function of applied potential in acetonitrile and (E) in dichloromethane. (F) Proposed mechanism based on two consecutive one-electron transfer reactions. | ||
Data from cyclic voltammetry experiments are summarised in Table 2. The first oxidation in diferrocenylborinic acid occurs at 0.445 V vs. SCE in both acetonitrile and in dichloromethane. However, in the aqueous environment the oxidation occurs at a slightly lower potential of 0.265 V vs. SCE, which may be linked to the more polar solvent environment stabilising the charge. The separation of the first and second redox process, ΔEp,ox = Δ(Emid1 − Emid2), also decreases when going from organic to aqueous conditions. This may also suggest a solvent polarity effect and better compensation of the first Fe(III) ferrocenyl charge in the more polar aqueous environment.
| Environment | Potentials | ||||
|---|---|---|---|---|---|
| Diferrocenyl redox pair | E p,ox1 and Ep,ox2 (vs. SCE)/mV | E p,red1 and Ep,red2 (vs. SCE)/mV | E mid = ½(Eox + Ered) (vs. SCE)/mV | Δ(Emid1 − Emid2)/mV | |
| a Data from cyclic voltammograms in solution. | |||||
| Acetonitrilea | 1 | 500 | 390 | 445 | 160 |
| 2 | 660 | 550 | 605 | ||
| Dichloromethanea | 1 | 570 | 320 | 445 | 190 |
| 2 | 740 | 530 | 635 | ||
| Embedded in titanate composite immersed in 0.1 M aqueous Na2SO4 | 1 | 310 | 220 | 265 | 120 |
| 2 | 430 | n.o. | n.o. | ||
3.3. Voltammetric characterisation of diferrocenylborinic acid immobilised in a titanate nanosheet host
In aqueous electrolyte, 1 is essentially insoluble, thus immobilisation onto the electrode surface was necessary. Titanate nanosheet material has been shown recently to provide a good host for ferroceneboronic acid25 and for a range of other redox active and hydrophobic redox systems.19 Therefore, the glassy carbon electrode is firstly covered with a thin (ca. 5 μm) layer of 2D titanate nanosheets according to the previously published procedure. Following this, diferrocenylborinic acid 1 was immobilised by absorption from a chloroform solution (1 mM) into the titanate nanosheet film by immersion for 24 h. Cyclic voltammetry measurements were first performed in 0.1 M Na2SO4 as a background electrolyte, as this provided stable and reproducible voltammetric responses for both diferrocenylborinic and ferroceneboronic acid.25 The composite electrode containing 1 immobilised in a titanate nanosheet host was less suitable for work in organic media due to loss of the redox system into the surrounding solution.As in the case of data for diferrocenylborinic acid 1 in homogeneous organic phase, a split peak voltammetric response is observed in cyclic voltammograms in aqueous environments (Fig. 3C). Two distinct peaks for the oxidation of the ferrocenyl moieties are seen and the separation of the midpoint potentials can be estimated as ΔEmid = 120 mV (Table 2). The second oxidation step shows a peak response during the forward potential scan, but not during the backward potential scan. This observation can be explained by the occurrence of a follow-on chemical reaction step. The second oxidation peak during the forward potential scan is also diminished at lower scan rates (below 0.7 V s−1). The behaviour is reminiscent of that for an ECE scheme with a short-lived intermediate that exists after the first one-electron oxidation. In order to further explore this reaction, square wave voltammetry is considered.
Fig. 4A shows data for square wave voltammetry during oxidation and reduction of diferrocenylborinic acid 1 immobilized in titanate layer on the electrode. Data are consistent with those obtained for cyclic voltammetry (Fig. 3C) and indicate consecutive two-electron oxidation as long as the time for the measurement is short. The second oxidation peak at 0.4 V vs. SCE appears at a scan rate of approximately 0.5 V s−1 or higher consistent with a short-lived Fe3+–Fe2+ intermediate. The scheme in Fig. 4B shows a tentatively assigned reaction scheme with the oxidation to the Fe3+–Fe3+ intermediate only at higher scan rates (shorter times). During the reduction, there is no clear evidence for any Fe2+–Fe2+ intermediate being formed.
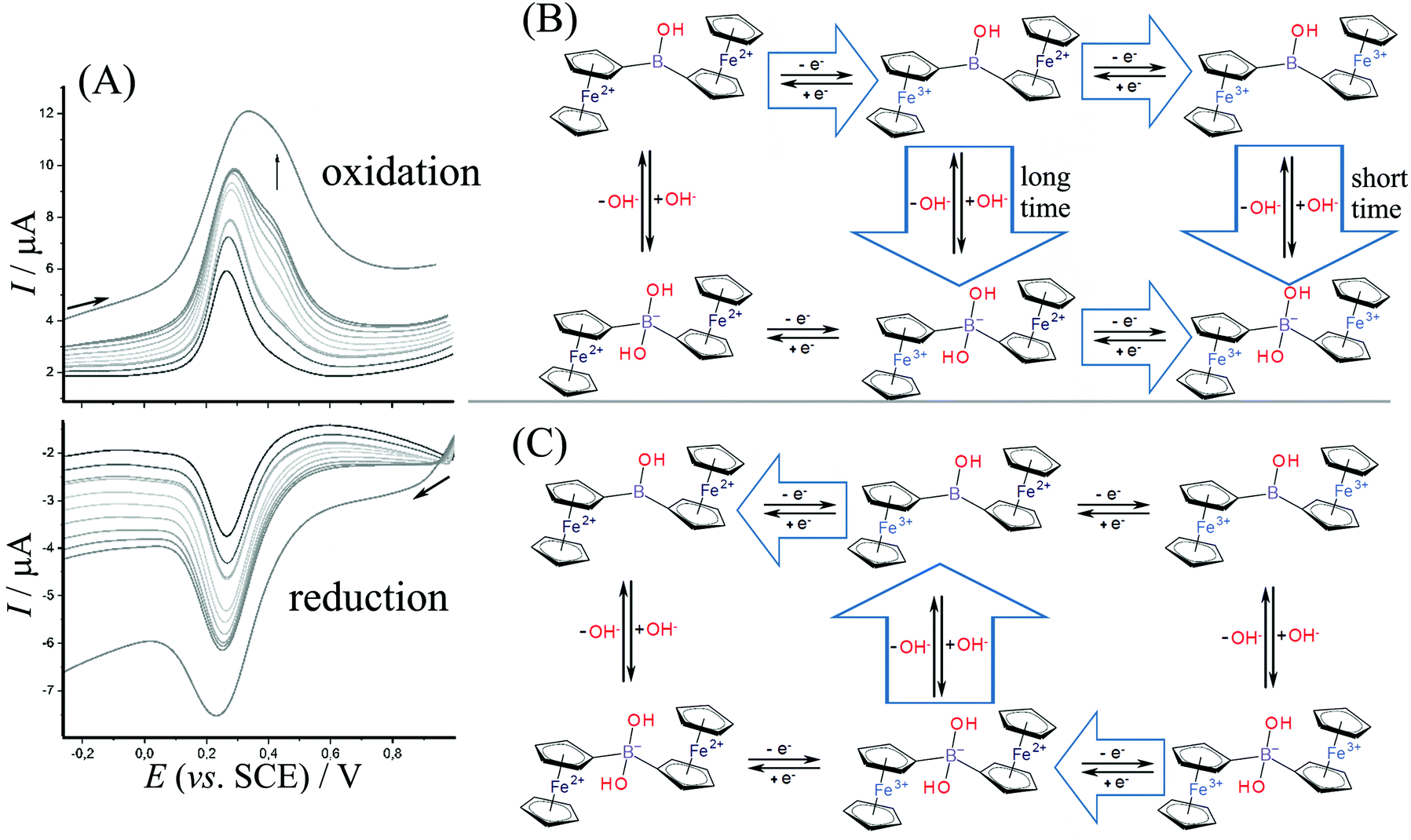 | ||
| Fig. 4 (A) Redox properties of 1 immobilized in a titanate nanosheet film immersed in aqueous 0.1 M Na2SO4. Square wave voltammograms (modulation amplitude 0.02 V, frequency 25 Hz, scan rate 0.12, 0.25, 0.37, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0, 2.0 V s−1), (B) reaction scheme for oxidation with a short-lived Fe3+/Fe2+ species only detected at scan rates higher than 0.4 V s−1. (C) Reaction scheme for reduction via a single reaction pathway. | ||
The oxidation reactions tentatively assumed in Fig. 4B includes the slow binding of a nucleophile (hydroxide). At short time scale, both the first and the second oxidation are observed before the nucleophile reacts (consistent with an EEC reaction type). At slower time scales, however, the first oxidation is rapidly followed by a chemical reaction step then by an immediate oxidation (due to the oxidation potential for the hydroxide adduct being lower, similar potential for the oxidation of the second Fe2+ centre as in the first oxidation step is assumed). The process can be considered as an ECE type reaction. Very similar reaction sequences have been reported for ferroceneboronic acid.13,25
Measurements were performed in aqueous 0.1 M NaF to investigate the electrochemically driven binding of fluoride as nucleophile to the borinic acid. Fig. 5 shows data for square wave voltammetry measurements based on diferrocenylborinic acid 1 immobilised in a titanate nanosheet film deposit on a glassy carbon electrode. Comparing the data obtained in Na2SO4 (Fig. 4) and in NaF electrolyte, the binding of fluoride at [Fc2BOH]2+ (EEC pathway during faster scan rates in Fig. 5B) appears to occur at a lower scan rate and seems slower compared to the tentative addition of hydroxide in Na2SO4 electrolyte. A more dramatic change is observed for the reduction where a new Fe2+–Fe2+ intermediate is detected at 0.0 V vs. SCE (see *). This suggests that there are two distinct pathways for reduction and that the corresponding Fe3+–Fe2+ intermediate is more long-lived with fluoride.
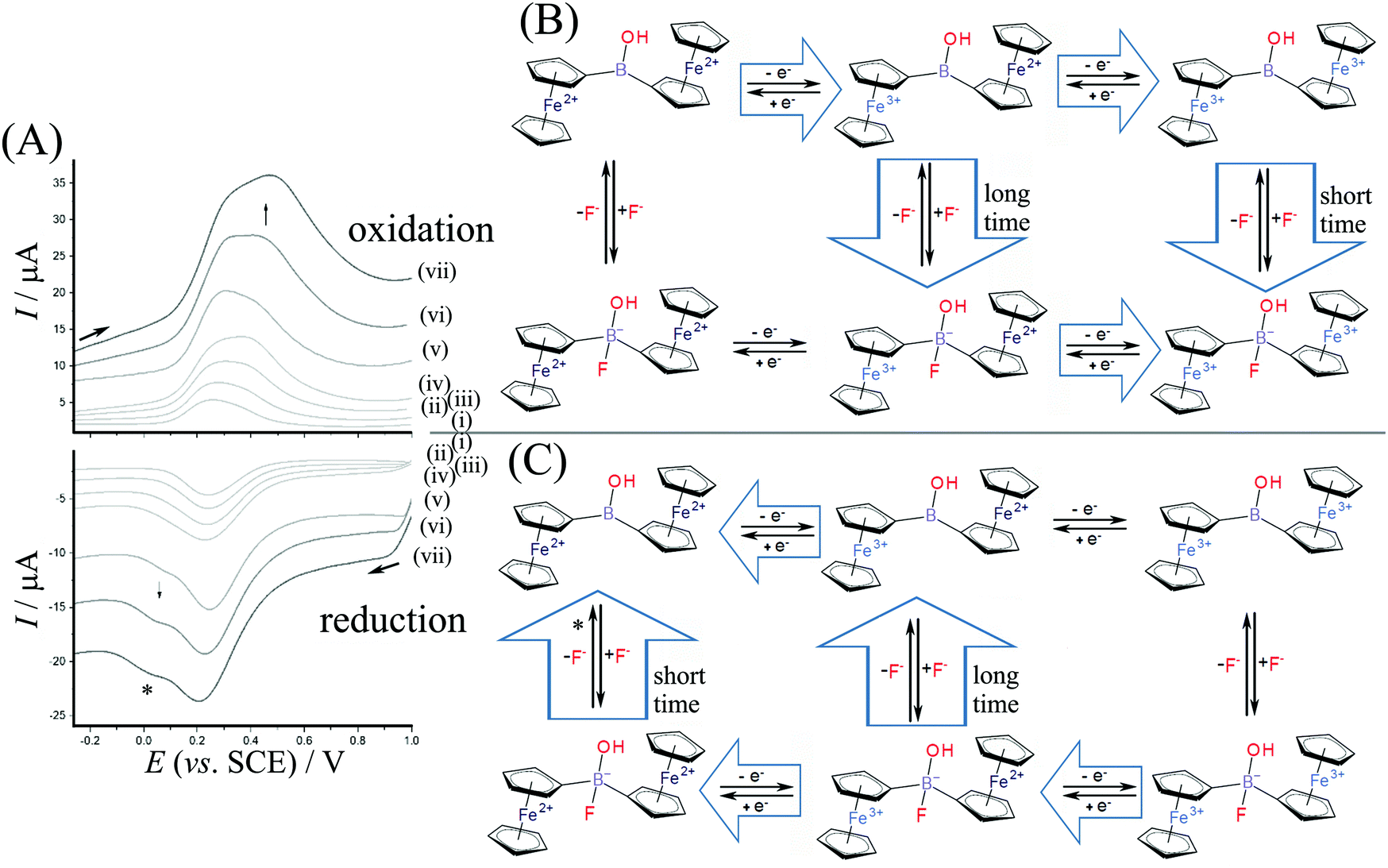 | ||
| Fig. 5 (A) Square wave voltammetry data for diferrocenylborinic acid 1 immobilised in a titanate nanosheet film on a glassy carbon immersed in aqueous 0.1 M NaF. Traces (i)–(iv) modulation amplitude 0.02 V, frequency 25 Hz, step 5–60 mV, scan rate 0.12, 0.25, 0.5, 1.5 V s−1; traces (v)–(vii) modulation amplitude 0.02 V, frequency 100 Hz, step 20–60 mV, scan rate 2.0, 4.0, 6.0 V s−1. (B) Reaction scheme for oxidation with a short-lived Fe3+–Fe2+ species only detected at scan rates higher than 0.1 V s−1. (C) Reaction scheme for reduction via a dual reaction pathway with a Fe2+–Fe2+ reaction intermediate. | ||
The implications of these data in terms of potential applications of diferrocenylborinic acid 1 are limited. The two sequential oxidation processes for the ferrocenyl moities are clearly observed, particularly in dry organic solvents new reactions occur that are currently unresolved. However, diferrocenylborinic acid is water stable and can be reversibly switched between Fe2+–Fe2+ to Fe3+–Fe2+ to Fe3+–Fe3+ state with a change in reactivity to nucleophiles. Here, only tentatively assigned reactions with fluoride are reported and more work will be required to explore reactivity towards other types of nucleophiles as well as reactivity in other types of immobilisation media. Diferrocenylborinic acid could be useful in electroanalytical detection protocols in cases where nucleophiles can bind more strongly or more selectively. More work in particular with saccharides (e.g. glucose) for sensing applications will be required.
4. Summary and conclusion
Diferrocenylborinic acid (Fc2BOH) was synthesized via an improved synthetic path and structurally characterised using NMR, HRMS, FTIR and X-Ray. Redox properties of Fc2BOH were investigated in both homogeneous and heterogeneous phase and redox activity of both ferrocenyl cores has been observed. A 2D-titanate nanosheet film was employed to host diferrocenylborinic acid redox probe molecules on a glassy carbon electrode immersed in aqueous electrolyte. In the presence of disodium sulfate, a stable reversible voltammetric response was observed. In the presence of sodium fluoride, “bound” and “unbound” states of the diferrocenylborinic acid were observed at higher scan rates (>0.1 V s−1). In comparison with the response of the related ferroceneboronic acid, diferrocenylborinic acid appears more stable during voltammetric cycling in the presence of aqueous electrolyte, but on the other hand, Fc2BOH in this titanate nanosheet film appears to be less sensitive towards binding of nucleophilic analytes such as fluoride.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
M. K. thanks the University of Bath for a fee waiver. M. K., K. L. and P. S. acknowledge the financial support from the Ministry of Education, Youth and Sports of the Czech Republic under the project CEITEC 2020 (LQ1601) and from the Czech Science Foundation, grant nr. 19-16273Y. TDJ wishes to thank the Royal Society for a Wolfson Research Merit Award. NMR characterisation facility was provided through the Material and Chemical Characterisation Facility (MC2) at the University of Bath (http://go.bath.ac.uk/mc2). Dr Bittová (Department of Chemistry, Masaryk University, Brno) is acknowledged for carrying out HRMS analysis.References
- F. Marken, Boron in Electroanalysis, in Boron: Sensing, Synthesis and Supramolecular Self-Assembly, ed. M. Li, J. S. Fossey and T. D. James, Book Series: Monographs in Supramolecular Chemistry, 2016, pp. 236–255 Search PubMed.
- W. Erb, J. P. Hurvois, T. Roisnel and V. Dorcet, Organometallics, 2018, 37, 3780–3790 CrossRef CAS.
- H. G. Kuivila, A. H. Keough and E. J. Soboczenski, J. Org. Chem., 1954, 19(5), 780–783 CrossRef CAS.
- T. D. James, K. R. A. S. Sandanayake, R. Iguchi and S. Shinkai, J. Am. Chem. Soc., 1995, 117, 8982–8987 CrossRef CAS.
- T. R. Jackson, J. S. Springall, D. Rogalle, N. Masurnoto, H. C. Li, F. D'Hooge, S. P. Perera, A. T. A. Jenkins, T. D. James, J. S. Fossey and J. M. H. van den Elsen, Electrophoresis, 2008, 29, 4185–4191 CrossRef CAS PubMed.
- T. D. James, P. Linnane and S. Shinkai, Chem. Commun., 1996, 281–282 RSC.
- T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Angew. Chem., Int. Ed., 1996, 35, 1910–1922 CrossRef.
- T. D. James, M. D. Phillips and S. Shinkai, Boronic Acids in Saccharide Recognition, ed. J. Fraser Stoddart, RSC, 2006 Search PubMed.
- A. Ori and S. Shinkai, J. Chem. Soc., Chem. Commun, 1995, 17, 1771–1772 RSC.
- C. Dusemund, K. R. A. S. Sandanayake and S. Shinkai, J. Chem. Soc., Chem. Commun, 1995, 3, 333–334 RSC.
- K. Lacina, M. Konhefr, J. Novotný, D. Potěšil, Z. Zdráhal and P. Skládal, Tetrahedron Lett., 2014, 55, 3235–3238 CrossRef CAS.
- K. Lacina, P. Skládal and T. D. James, Chem. Cent. J., 2014, 8, 60–77 CrossRef PubMed.
- M. Li, S.-Y. Xu, A. J. Gross, J. L. Hammond, P. Estrela, J. Weber, K. Lacina, T. D. James and F. Marken, ChemElectroChem, 2015, 2, 867–871 CrossRef CAS.
- Y. Sobue, T. Sugaya, S. Iwatsuki, M. Inamo, H. D. Takagi, A. Odani and K. Ishihara, J. Mol. Liq., 2016, 217, 29–34 CrossRef CAS.
- S. Aldridge and C. Bresner, Coord. Chem. Rev., 2003, 244, 71–92 CrossRef CAS.
- B. Wrackmeyer, U. Dorfler, W. Milius and M. Herberhold, Z. Naturforsch., B: J. Chem. Sci., 1995, 50, 201–204 CAS.
- D. O. Cowan, P. Shu, F. L. Hedberg, M. Rossi and T. J. Kistenmacher, J. Am. Chem. Soc., 1979, 101, 1304–1306 CrossRef.
- E. W. Post, R. G. Cooks and J. C. Kotz, Inorg. Chem., 1970, 9, 1670–1677 CrossRef CAS.
- W. Tri Wahyuni, B. Riza Putra, C. Harito, D. V. Bavykin, F. C. Walsh, P. J. Fletcher and F. Marken, Anal. Chim. Acta X, 2019, 1, 100001 Search PubMed.
- M. Osada and T. Sasaki, Adv. Mater., 2011, 24, 210–228 CrossRef PubMed.
- L. Wang and T. Sasaki, Chem. Rev., 2014, 114, 9455–9486 CrossRef CAS PubMed.
- T. Sasaki and M. Watanabe, J. Phys. Chem. B, 1997, 101, 10159–10161 CrossRef CAS.
- C. Harito, D. V. Bavykin, M. E. Light and F. C. Walsh, Composites, Part B, 2017, 124, 54–63 CrossRef CAS.
- K. Ahmad, A. Mohammad, R. Rajak and S. M. Mobin, Mater. Res. Express, 2016, 3, 074005 CrossRef.
- W. T. Wahyuni, B. R. Putra, C. Harito, D. V. Bavykin, F. C. Walsh, T. D. James, G. Kociok-Köhn and F. Marken, Electroanalysis, 2017, 30, 1303–1310 CrossRef.
- T. Sasaki, Y. Ebina, Y. Kitami, M. Watanabe and T. Oikawa, J. Phys. Chem. B, 2001, 105, 6116–6121 CrossRef CAS.
- G. M. Sheldrick, SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, ShelXle: a Qt graphical user interface for SHELXL, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- L. J. Farrugia, ORTEP3 for Windows, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- F. Rebiere, O. Samuel and H. Kagan, Tetrahedron Lett., 1990, 31, 3121–3124 CrossRef CAS.
- P. Thilagar, D. Murillo, J. Chen and F. Jäkle, Dalton Trans., 2013, 42, 665–670 RSC.
- C. Bresner, S. Aldridge, I. A. Fallis and L. L. Ooi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, M441–M443 CrossRef CAS.
- T. Murafuji, K. Shintaku, K. Nagao, Y. Mikata, K. Ishiguro and S. Kamijo, Heterocycles, 2017, 94, 676–690 CrossRef CAS.
- T. Beringhelli, G. D'Alfonso, D. Donghi, D. Maggioni, P. Mercandelli and A. Sironi, Organometallics, 2003, 22, 1588–1590 CrossRef CAS.
- C. D. Entwistle, A. S. Batsanov and T. B. Marder, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o2639–o2641 CrossRef CAS.
- S. Lulinski and J. Serwatowski, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, o1711–o1712 CrossRef CAS PubMed.
- E. Dimitrijevic and M. S. Taylor, Chem. Sci., 2013, 4, 3298–3299 RSC.
- H. H. Shah, R. A. Al-Balushi, M. K. Al-Suti, M. S. Khan, C. H. Woodall, K. C. Molloy, P. R. Raithby, T. P. Robinson, S. E. C. Dale and F. Marken, Inorg. Chem., 2013, 52, 4898–4908 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1894130. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt00881k |
| This journal is © The Royal Society of Chemistry 2019 |